

Chronic obstructive pulmonary disease (COPD) is characterized by aberrant inflammatory and repair responses, leading to bronchitis, small airway remodeling, and emphysema. It is the third leading cause of death worldwide and is caused by exposure to tobacco smoke or environmental pollutants (1). Importantly, the inflammatory processes in COPD are known to persist long after smoking cessation (2). However, the underlying mechanisms that initiate and drive inflammation are still not clear, and currently, no pharmacological treatments are available to alter the course of the disease. These mechanisms include defective epithelial repair in the airways and alveoli, suggesting that the endogenous lung stem cells may be the initial drivers of the disease. In line with that, research has demonstrated that basal (stem) cells in the airway are among the first cells to be affected by inhaled toxins, resulting in epithelial damage, defective repair, and subsequent inflammation (3, 4). The mechanisms triggering the initiation of this inflammation are not understood, but it is clear that the epithelial cells are in constant communication with the inflammatory cells. It was recently shown that a subpopulation of basal cells can drive the inflammatory processes observed in COPD, suggesting that these inflammatory signals represent a potential therapeutic target to inhibit tissue destruction and promote regeneration of the normal cells that still exist in the airways at end-stage COPD (5, 6). In the alveolar compartment, the alveolar type 2 (AT2) cells are the stem cells responsible for homeostasis and regeneration (Figure 1A). Even though AT2 cells have been evaluated in multiple single-cell OMICS studies (7–9), the mechanistic alterations in AT2 cells in COPD are unknown, and the question remains as to whether a disease-propagating subpopulation of AT2 cells exists in COPD. Hence, a deeper understanding of the AT2 cells in patients with COPD is needed to understand their contribution to disease.
Figure 1.
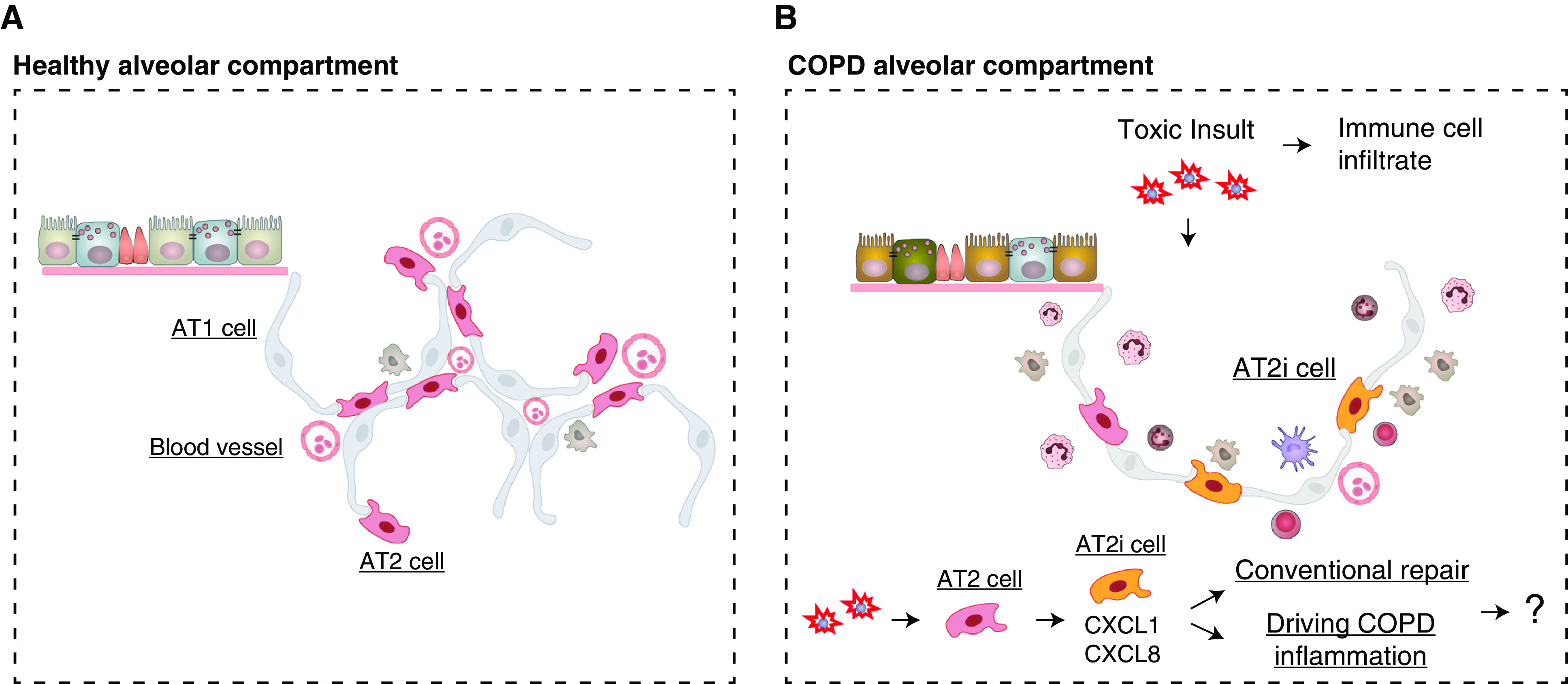
Schematic of the lung alveolar compartment during health and chronic obstructive pulmonary disease (COPD). (A) Representative schematic of the distal airway epithelium and the alveolar compartment in a healthy individual showing healthy alveolar epithelial type 1 (AT1) and AT2 cells together with their interaction with blood vessels. Included are a few representative resident immune cells (khaki). (B) The representative COPD alveolar compartment, because of repeated toxic insult, is characterized with increased alveolar destruction (increased alveolar space), increased immune cell infiltrate, and a more heterogenous alveolar epithelium. Part of this heterogeneity is the AT2 cell subpopulation, the AT2 inflammatory cells (AT2i) (orange). This newly identified AT2i population increases in relation to disease severity and correlates to increased inflammatory genes and chemokines. However, whether this AT2 cell subpopulation is part of conventional repair or the driving force behind COPD inflammation is not known.
In this issue of the Journal (pp. 708–719), Watanabe and colleagues (10) describe their approach to this problem, which included single-cell RNA sequencing of patients with COPD and yielded interesting findings with regard to the alterations in AT2 cells in COPD. By leveraging the power of single-cell RNA sequencing, they show a comprehensive molecular profile of the distal COPD lung, including the identification of a potential new AT2 cell subpopulation, labeled AT2i (inflammatory) cells, that the authors present as the potential subpopulation responsible for driving distal lung inflammation in COPD. The first question that arises in any article with a focus on single-cell RNA sequencing relates to quality control and sample distribution among all the sequencing-derived clusters and specifically in the population of interest. They produced an unsupervised uniform manifold approximation and projection of all their cells attained from COPD, healthy smokers, and never-smokers. In the uniform manifold approximation and projection, they show that they have a good distribution of their cells with respect to the known markers for distal lung cells in the three sample cohorts. This verification is important, as they proceeded to explore the COPD epithelial cell heterogeneity in greater depth. They explored epithelial cell heterogeneity using a predictive “closeness centrality” analysis that determines how “similar” the cells are to each other. Strikingly, they show that the epithelial cells are more heterogenous in subjects with COPD when compared with smokers without COPD or the never-smoker populations. An immediate question that arises is “what are the cells that are different in COPD when compared with the other populations?”
Thus, the authors next looked more closely at the AT2 cells, because of their role in maintaining alveolar homeostasis. They noted that three AT2 cell clusters could be identified in patients with COPD. In these three clusters, they focused on the AT2 cell fraction that increased in correlation to COPD severity and in relation to the expression of inflammatory chemokine-related genes, such as CXCL1, CXCL1, CXCL2, CXCL3, CXCL8, CCL2, and CCL20. These genes were verified at the protein level through coimmunostaining with surfactant protein C. They labeled this population as AT2i cells (Figure 1B). To further validate their results, they integrated their data with publicly available datasets and found cells in one cluster that expressed similar chemokine markers to those associated with AT2i cells, thereby confirming their findings.
However, it remains unclear which factors trigger activation of quiescent AT2 cells to AT2i cells and which differentiation trajectory they follow during lung regeneration. To answer that, in part, they used a pseudotime analysis to study gene expression as a function of an artificial time vector, which indicated that the AT2i population undergoes dysfunctional differentiation, with an enrichment of genes driving inflammatory pathways. In addition, to further scrutinize the role of the AT2i cells, Watanbe and colleagues looked into potential immune–response network interactions that could exist between epithelial and immune cells. They found that the strongest immune–epithelial interaction in the AT2i cell population was in COPD when compared with smokers without COPD and never-smokers.
Although the work from Watanabe and colleagues provides intriguing results, several drawbacks remain. One drawback, and this could be said of other single-cell–based analyses, is the small sample size (number of donors and cells per donor). Another drawback is the representation of COPD classifications within this analysis (Global Initiative for Chronic Obstructive Lung Disease I–IV). This would provide further subclassification and context for their findings indicating that the percentage of AT2i population increases with disease severity, suggesting that AT2i cells may correlate with the increased inflammation. Another important consideration is whether the AT2i subpopulation is a distinct population with a different functional profile than AT2 cells and whether this population is unique to COPD. It is important to understand whether this population is also present in other diseases as a regenerative response. Another limitation of their work is the lack of exploration into whether the AT2i cells differentiate into aberrant or normal AT1 cells. This finding would have implications into the purpose of AT2i cells and whether their presence indicates aberrant regenerative processes (Figure 1B), because we know that immune cells are also crucial for conventional lung regeneration (11). Hence, in future studies, it will be important to understand the balance between inflammatory signals that promote regeneration while inhibiting destruction. As a step toward the future, the single-cell RNA analysis from Watanabe and colleagues acts as a guide directing deeper dives into previously unknown cell populations and interactive mechanisms. In this way, we will progress toward a treatment for COPD.
Acknowledgments
Acknowledgment
The authors thank Dr. T. Koopmans for help with the figure.
Footnotes
Supported by Swedish Cancer foundation grant 20 1326 PjF and Swedish Heart-Lung foundation grant 2021/0289.
Originally Published in Press as DOI: 10.1165/rcmb.2022-0371ED on October 12, 2022
Author disclosures are available with the text of this article at www.atsjournals.org.
References
- 1. Hogg JC, Timens W. The pathology of chronic obstructive pulmonary disease. Annu Rev Pathol . 2009;4:435–459. doi: 10.1146/annurev.pathol.4.110807.092145. [DOI] [PubMed] [Google Scholar]
- 2. Shapiro SD. End-stage chronic obstructive pulmonary disease: the cigarette is burned out but inflammation rages on. Am J Respir Crit Care Med . 2001;164:339–340. doi: 10.1164/ajrccm.164.3.2105072c. [DOI] [PubMed] [Google Scholar]
- 3. Ryan DM, Vincent TL, Salit J, Walters MS, Agosto-Perez F, Shaykhiev R, et al. Smoking dysregulates the human airway basal cell transcriptome at COPD risk locus 19q13.2. PLoS One . 2014;9:e88051. doi: 10.1371/journal.pone.0088051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Ghosh M, Miller YE, Nakachi I, Kwon JB, Barón AE, Brantley AE, et al. Exhaustion of airway basal progenitor cells in early and established chronic obstructive pulmonary disease. Am J Respir Crit Care Med . 2018;197:885–896. doi: 10.1164/rccm.201704-0667OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Rao W, Wang S, Duleba M, Niroula S, Goller K, Xie J, et al. Regenerative metaplastic clones in COPD lung drive inflammation and fibrosis. Cell . 2020;181:848–864.e18. doi: 10.1016/j.cell.2020.03.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Wijk SC, Prabhala P, Michaliková B, Sommarin M, Doyle A, Lang S, et al. Human primary airway basal cells display a continuum of molecular phases from health to disease in chronic obstructive pulmonary disease. Am J Respir Cell Mol Biol . 2021;65:103–113. doi: 10.1165/rcmb.2020-0464OC. [DOI] [PubMed] [Google Scholar]
- 7. Sauler M, McDonough JE, Adams TS, Kothapalli N, Barnthaler T, Werder RB, et al. Characterization of the COPD alveolar niche using single-cell RNA sequencing. Nat Commun . 2022;13:494. doi: 10.1038/s41467-022-28062-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Travaglini KJ, Nabhan AN, Penland L, Sinha R, Gillich A, Sit RV, et al. A molecular cell atlas of the human lung from single-cell RNA sequencing. Nature . 2020;587:619–625. doi: 10.1038/s41586-020-2922-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Adams TS, Schupp JC, Poli S, Ayaub EA, Neumark N, Ahangari F, et al. 2020. [DOI] [PMC free article] [PubMed]
- 10. Watanabe N, Fujita Y, Nakayama J, Mori Y, Kadota T, Hayashi Y, et al. Anomalous epithelial variations and ectopic inflammatory response in chronic obstructive pulmonary disease. Am J Respir Cell Mol Biol . 2022;67:708–719. doi: 10.1165/rcmb.2021-0555OC. [DOI] [PubMed] [Google Scholar]
- 11. Lechner AJ, Driver IH, Lee J, Conroy CM, Nagle A, Locksley RM, et al. Recruited monocytes and type 2 immunity promote lung regeneration following pneumonectomy. Cell Stem Cell . 2017;21:120–134.e7. doi: 10.1016/j.stem.2017.03.024. [DOI] [PMC free article] [PubMed] [Google Scholar]