Effects of 6-Aminonicotinic Acid Esters on the Reprogrammed Epigenetic State of Distant Metastatic Pancreatic Carcinoma
Among the various NAD-derived inhibitors explored for treating
pancreatic cancer, 6-aminonicotinamide (6AN) is a widely used heterocycle
that is metabolized by an NAD salvage pathway to form 6-amino-NADP+. This metabolite inhibits 6-phosphogluconate dehydrogenase
(6PDG) with moderate selectivity over other NADP+/NADPH-dependent
enzymes and reverses chromatin methylation and gene expression in
metastatic pancreatic ductal adenocarcinoma. However, dose-limiting
neurotoxic effects render 6AN unacceptable for clinical progression.
In this issue, Tsukamoto and colleagues from Johns Hopkins University
disclose a novel prodrug strategy that masks the 2-aminonicotinic
acid precursor as an ester. This strategy leads to antiproliferative
activity, as indicated by histone methylation, and cell viability
while also reducing neurotoxic effects in rat hippocampal neurons
(DOI: 10.1021/acsmedchemlett.2c00404). Notably,
metabolomic analyses showed that, upon entry into a cell, the leading
prodrugs undergo ester hydrolysis and bioconversion to generate the
active compound 6-amino-NADP+ via the Preiss–Handler
pathway, which is overactive in pancreatic cancers and which differs
from the previously exploited NAD salvage pathway. Thus, the selective
activation of the prodrug in carcinoma cells, combined with the low
activity level of the Preiss–Handler pathway in neurons, might
contribute to the improved in vitro therapeutic window.
Though ADMET characterization remains to be conducted, future studies
will hopefully establish whether the promising prodrug strategy that
exploits an alternate biosynthetic activation pathway will ultimately
translate to providing in vivo benefits.
Structure–Activity Relationship Study of Tertiary Alcohol Hsp90α-Selective Inhibitors with Novel Binding Mode
The 90 kDa heat shock chaperone proteins
(Hsp90) fold and activate
client proteins associated with all 10 hallmarks of cancer, and nearly
20 Hsp90 inhibitors have reached clinical trials for the treatment
of various cancers. These candidates, however, have all failed in
the clinic because of unacceptable toxicities that likely arise from
the pan-inhibition of all four Hsp90 isoforms (Hsp90α, Hsp90β,
Trap1, and GRP94), which disrupts proper folding of many essential
proteins. In contrast, isoform-selective inhibition has the potential
to block the function of a subset of oncogenic proteins while minimizing
pan-interference with protein homeostasis. Specifically, the stress-inducible
Hsp90α isoform resides in both cytosolic and extracellular compartments
and regulates the folding of several surface receptors, extracellular
clients, and regulators of cell signaling. Further, this isoform is
associated with aggressive cancer phenotypes, which suggests that
the selective inhibition of Hsp90α should reduce the progression
of cancers that rely on Hsp90α-dependent client proteins. In
this issue, Blagg (University of Notre Dame), Matts (Oklahoma State
University), and co-workers exploit crystallographic information to
guide the design of Hsp90α-selective inhibitors (DOI: 10.1021/acsmedchemlett.2c00327). Notably, the team
identifies previously underexploited pockets and contacts to generate
Hsp90α-selective inhibitors. Their lead inhibitors can induce
a novel conformational change in Hsp90α, leading to >300-fold
selectivity over Trap1 and GRP94 and >13-fold selectivity over
the
Hsp90β isoform, which has 95% homology and differs by just two
amino acids in the active site. Though the lead inhibitors do not
manifest significant antiproliferative activity, improved analogs
should provide opportunities to develop potent isoform-selective Hsp90α-selective
inhibitors to explore basic cancer biology, synergistic therapies,
and/or synthetic lethal combinations of therapeutics with Hsp90α-selective
inhibitors.
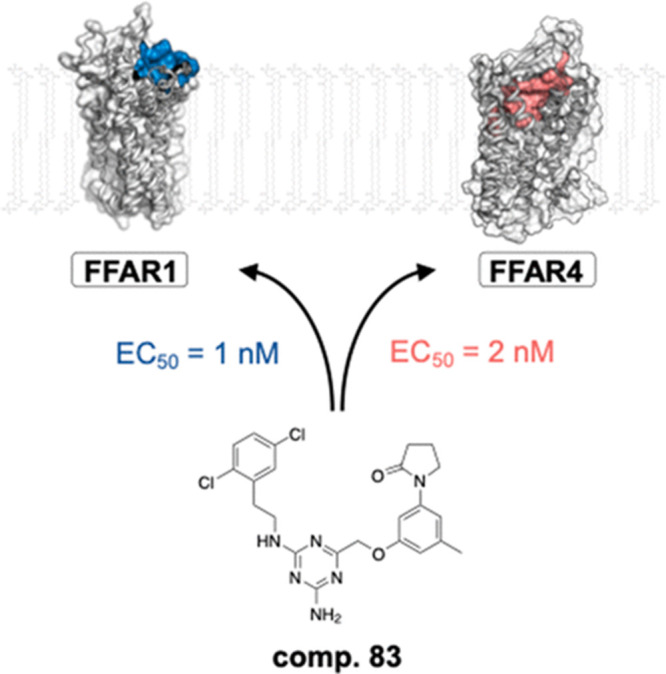
Optimization of First-in-Class Dual-Acting FFAR1/FFAR4 Allosteric Modulators with Novel Mode of Action
The GPCRs GPR40 and
GRP120, also known as free fatty acid receptors
(FFARs) 1 and 4, play important roles in glucose and energy homeostasis
and have been long targeted for treating type 2 diabetes mellitus
(T2DM). Notably, orthosteric agonists of FFAR1 have shown clinical
efficacy, though both the clinical candidates and next-generation
agonists displayed hepatic and pancreatic β-cell toxicities,
respectively, which are likely linked to the highly lipophilic scaffolds
required to engage the orthosteric site. Additionally, dual FFAR1/FFAR4
agonists have also shown preclinical promise in decreasing insulin
resistance and ultimately β-cell failure, though the previously
reported dual agonists require further development. In this issue,
Frimurer and collaborators at the University of Copenhagen and King’s
College London report a series of novel allosteric agonists that activate
both FFAR1 (full agonist) and FFAR4 (partial agonist) with nanomolar
potency (DOI: 10.1021/acsmedchemlett.2c00160). Notably, engagement of the allosteric site enables activation
by less lipophilic scaffolds that are not tolerated at the hydrophobic
orthosteric site that normally recognizes medium- and long-chain fatty
acids. Preliminary ADME studies indicate that the lead compounds only
display modest metabolic stability, and future lead optimization will
be required to deliver probes for in vivo studies
that may ultimately validate this allosteric site for therapeutic
intervention.