

Abstract
Lazertinib (YH25448) is a novel third-generation tyrosine kinase inhibitor (TKI) developed as a treatment for EGFR mutant non-small cell lung cancer. To better understand the nature of lazertinib inhibition, we determined crystal structures of lazertinib in complex with both WT and mutant EGFR and compared its binding mode to that of structurally related EGFR TKIs. We observe that lazertinib binds EGFR with a distinctive pyrazole moiety enabling hydrogen bonds and van der Waals interactions facilitated through hydrophilic amine and hydrophobic phenyl groups, respectively. Biochemical assays and cell studies confirm that lazertinib effectively targets EGFR(L858R/T790M) and to a lesser extent HER2. The molecular basis for lazertinib inhibition of EGFR reported here highlights previously unexplored binding interactions leading to improved medicinal chemistry properties compared to clinically approved osimertinib (AZD9291) and offers novel strategies for structure-guided design of tyrosine kinase inhibitors.
Keywords: Lung cancer, targeted therapy, kinase inhibitor, mutant, epidermal growth factor receptor, structural biology
Activating mutations within the epidermal growth factor receptor (EGFR) kinase domain, most prevalently L858R and exon19del, are common causes of non-small cell lung cancer (NSCLC) and serve as predictive markers for the selection of EGFR tyrosine kinase inhibitors (TKIs) as effective targeted therapies.1,2 Prolonged efficacy of first-generation TKIs (gefitinib and erlotinib) is eventually made limited due to drug resistance as a result of patients acquiring a second T790M “gatekeeper” mutation.3 To produce a viable treatment option for T790M-positive NSCLC tumors, drug development efforts have yielded the clinically approved drug AZD9291 (osimertinib), which is selective for T790M-containing EGFR and made potent by forming an irreversible covalent bond to C797.4,5 Despite promising indications, drug resistance to osimertinib is inevitable and caused in part by the acquisition of a third kinase domain mutation (C797S) that prevents formation of the osimertinib potency-enabling covalent bond. The emergence of further drug resistance by the C797S mutation has given way to several next-generation TKIs,6,7 including structurally related trisubstituted imidazole scaffolds (e.g., LN2057), which are more effective inhibitors of C797S-containing EGFR kinases.8−10 Recently, osimertinib has been shown effective, and clinically approved, as a front-line therapy in untreated patients harboring EGFR L858R and exon19del activating mutations.11
As osimertinib is the only approved third-generation EGFR TKI for L858R and exon19del EGFR mutant tumors, development efforts from Yuhan and Janssen sought to produce a drug with improved medicinal chemistry properties. These efforts resulted in YH25448 (lazertinib), which is structurally related to osimertinib comprising an aminopyrimidine core and acrylamide warhead but is distinct with respect to the substituted pyrazole as well as morpholine groups (Figure 1).12 Preclinical head-to-head experiments confirm that lazertinib is superior to osimertinib in several key respects including in vivo efficacy against H1975 (L858R/T790M) xenograft mouse models, brain penetrance, target specificity, and dose-limiting toxicity.13 These improvements have led way to clinical evaluation of lazertinib in clinical trials, for example with lazertinib being assessed as front-line therapy (NCT04248829) or in combination with the antibody amivantamab (NCT04077463), and is currently approved to treat T790M-containing NSCLC in the Republic of Korea.12 Regardless of these improvements, drug resistance to lazertinib has been shown to be due to the acquisition of C797S mutation.14,15 Despite the improved properties and positive clinical outlook, no crystal structures or time-dependent kinetic studies have been reported detailing the molecular basis for lazertinib inhibition of mutant EGFR.
Figure 1.

Chemical structures of third-generation EGFR inhibitors osimertinib, lazertinib, and LN2057.
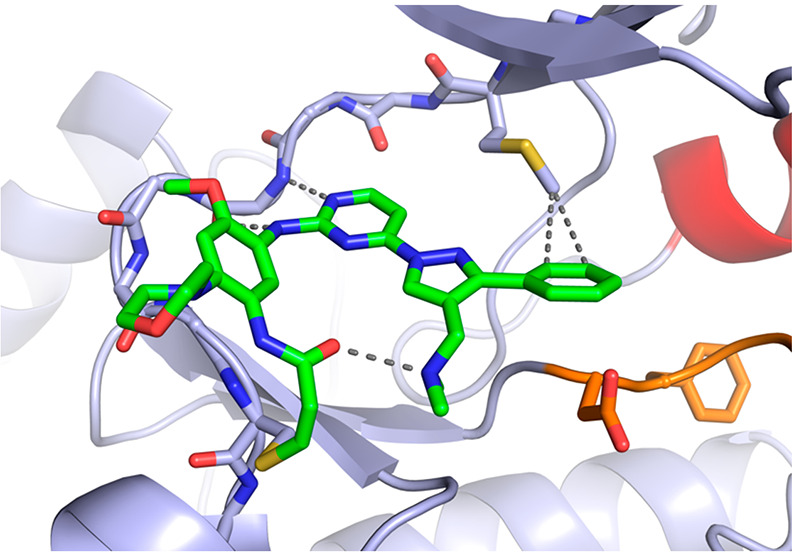
To characterize the binding mode of lazertinib in complex with the EGFR kinase domain, we determined a 2.4 Å resolution X-ray cocrystal structure of lazertinib soaked into WT EGFR crystals (Figures 2 & S1, PDB ID 7UKV). WT kinase domains crystallize in the active “αC-helix in” conformation due to crystal packing of the kinase domains as asymmetric dimers.16 As expected, lazertinib binds with the aminopyrimidine anchored to the hinge region by H-bonds to M793 and a covalent bond formed at C797 as generally observed for third-generation TKIs.17 Importantly, the unique pyrazole group extends away from the hinge in a conformation that positions the phenyl ring toward the K745-E762 salt bridge and the N,N-dimethylmethyleneamine (methyleneamine) positioned as an H-bond donor with the DFG-motif D855 carboxylate.
Figure 2.
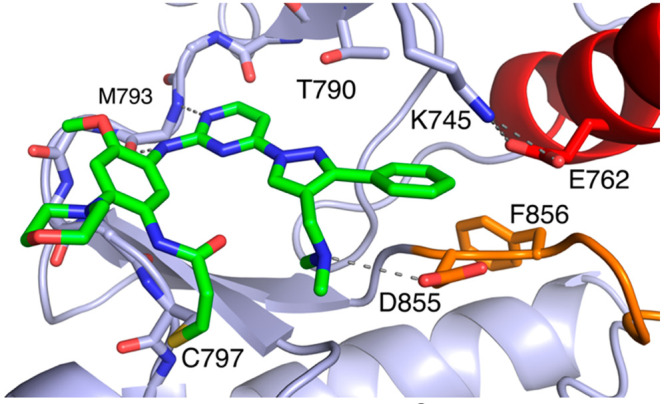
Lazertinib bound to WT EGFR. Binding mode of lazertinib in complex with WT EGFR with the kinase domain in the active αC-helix in conformation. P-loop cartoon removed for clarity. (PDB ID 7UKV).
Additionally, we determined a 2.6 Å resolution X-ray cocrystal structure of lazertinib in complex with EGFR containing the T790M mutation. This was accomplished by soaking crystals of EGFR(T790M/V948R), where the V948R variant prevents asymmetric dimer packing and stabilization of the inactive “αC-helix out” conformation. We succeeded in modeling lazertinib in two of the four copies of the asymmetric unit. Despite extensive efforts, we were unable to model the electron density observed in two kinase domain chains (A and D), which we have elected to leave without bound ligands. In one protein chain, we observe lazertinib binding in an identical conformation to the WT EGFR structure where the phenyl ring of the pyrazole binds near T790M exhibiting close contacts to the carbon of the T790M methionine (Figures 3A & S2, PDB ID 7UKW). Distinctly, the methyleneamine is observed in an intramolecular H-bond with the acrylamide carbonyl suggesting a role for this group in stabilizing the drug conformation. In the other chain, lazertinib is observed bound in an alternative “flipped” conformation, marked by a 180° rotation of the pyrazole moiety (Figures 3B & S2). This conformation shows the methyleneamine simultaneously H-bonding with D855 and contacting the methionine thioether. Additionally, the pyrazole nitrogen is positioned toward the acrylamide carbonyl potentially indicating an intramolecular dipole–dipole interaction. The overall picture revealed by these cocrystal structures defines the molecular basis for lazertinib binding to EGFR where the pyrazole substitutions, a hydrophobic phenyl and positively charged methyleneamine, produce distinct intra- and intermolecular interactions.
Figure 3.

Lazertinib bound to inactive EGFR(T790M/V948R) with distinct conformations. (A) Lazertinib bound with phenyl ring anchored within van der Waals distance to the T790M methionine (∼3.8 Å) and methane amine involved in intramolecular H-bonding. (B) A “flipped” conformation with outward phenyl and methyleneamine donating an H-bond to D855. Other interactions seen in this structure include an intramolecular dipole–dipole pyrazole to carbonyl (3.2 Å) as well as a van der Waals methyleneamine methyl to thioether (3.8 Å). P-loop cartoon removed for clarity. (PDB ID 7UKW).
The substituted pyrazole of lazertinib is unique among third-generation EGFR TKIs, containing a hydrophilic methyleneamine and hydrophobic phenyl group, and most likely the basis for mutant selectivity and improved medicinal chemistry properties. A comparison of these lazertinib cocrystal structures from the “αC-helix in” active WT (Figure 2) and “αC-helix out” inactive T790M EGFR (Figure 3) show very similar binding positions indicating that lazertinib is anchored to the EGFR kinase domain identically in both active and inactive states and invariable based on the status of the 790 gatekeeper residue being either a Thr versus Met (Figure 4A). We expect that this conformation of lazertinib is most relevant compared to the “flipped” conformation (Figure 3B) in the “αC-helix in” active state due to the apparent electrostatic incompatibility due to the proximity of the positive methyleneamine adjacent to the K745-E762 salt bridge (Figure S3) and correspondence to the binding mode observed in WT EGFR (Figure 4A). Investigations of the role of residues involved in this “flipped” confirmation are difficult due to many of these interactions being maintained by catalytically required residue side chains (e.g., D855). Recent structural studies of EGFR(T790M) X-ray cocrystal structures with bound osimertinib revealed a novel binding mode where the N-methylindole directly contacts the T790M side chain through van der Waals interactions, which causes osimertinib mutant selectivity.18 We observe analogous intermolecular interactions in the case of the bound lazertinib phenyl showcasing that both inhibitors are made selective for T790M-containing EGFR through van der Waals interactions (Figure 4B). Additionally, H-bonding, both observed intra- and intermolecular interactions, by the lazertinib methyleneamine moiety is distinctive, likely enabling improvements in binding compared to osimertinib. Another informative comparison is the structurally related imidazole-based covalent inhibitor LN2057 (Figure S4) that forms an H-bond with the catalytic lysine (K745), which is conserved in kinases, enabling C797S mutant inhibition at the expense of T790M mutant selectivity, i.e., enhanced binding to WT EGFR.8,10 By comparison, the lazertinib pyrazole substituents sterically block K745, causing the extended side chain to reposition under the P-loop, potentially diminishing binding to WT EGFR (Figure 4C). The correlation of these differences in binding mode demonstrates how the pyrazole moiety of lazertinib affords distinct interactions with EGFR to enable T790M mutant selectivity.
Figure 4.

Distinct binding mode features of third-generation EGFR TKIs. (A) Superposition of lazertinib in complex with active WT (magenta, PDB ID 7UKV) and inactive T790M/V948R (green, PDB ID 7UKW). (B) Overlay of lazertinib in complex with T790M/V948R (green, PDB ID 7UKW) and osimertinib in T790M (cyan, PDB ID 6JX0) showing T790M van der Waals intermolecular interactions. (C) Overlay of lazertinib (green PDB ID 7UKW) compared with LN2057 (forest green, PDB ID 6V6K) in complex with T790M/V948R highlighting the contrasting impact on steric repulsion versus H-bonding with K745, respectively.
To characterize the functional significance of the binding modes of lazertinib and their comparisons to osimertinib and LN2057, we conducted time-dependent inhibition biochemical assays with purified kinase domains. Comparing two-step irreversible inhibitor rate constants defining inhibitor potency (kinact/KI),19,20 we observed that lazertinib is the most potent inhibitor of EGFR(L858R/T790M) and exhibits mutant selectivity as seen by the 3.2-fold enhancement of kinact/KI for the mutant over WT EGFR (Table 1). Osimertinib is also selective for EGFR(L858R/T790M), although to a lesser extent compared to lazertinib (∼1.6-fold).9,21 LN2057 is found to be distinct from the other two TKIs, exhibiting comparable potencies against EGFR(L858R/T790M) and WT EGFR, as consistent with previous functional studies and binding modes from crystal structures.9,10 Overall, time-dependent inhibitor potencies for these three inhibitors showcase that lazertinib exhibits superior potency and selectivity when compared to these structurally related irreversible inhibitors.
Table 1. Time-Dependent Inhibition Kinetic Parameters for Lazertinib, Osimertinib, and LN2057a.
| WT EGFR |
EGFR(L858R/T790M) |
WT
HER2 |
|||||||
|---|---|---|---|---|---|---|---|---|---|
| Compound | kinact (min–1) | KI (μM) | kinact/KI (M–1 s–1) | kinact (min–1) | KI (μM–1) | kinact/KI (M–1 s–1) | kinact (min–1) | KI (μM–1) | kinact/KI (M–1 s–1) |
| Lazertinib | 0.648 ± 0.036 | 0.271 ± 0.015 | 35,800 ± 400 | 0.235 ± 0.0042 | 0.0340 ± 0.0011 | 115,000 ± 2,000 | 0.166 ± 0.0130 | 0.437 ± 0.059 | 6,310 ± 440 |
| Osimertinib | 0.690 ± 0.042 | 0.434 ± 0.032 | 26,400 ± 400 | 0.648 ± 0.036 | 0.256 ± 0.016 | 42,400 ± 600 | n.d.b | n.d.b | 14,000 ± 600 |
| LN2057 | 0.335 ± 0.011 | 0.0989 ± 0.0039 | 56,500 ± 600 | 0.561 ± 0.041 | 0.224 ± 0.019 | 41,800 ± 700 | 0.924 ± 0.30 | 0.142 ± 0.013 | 42,000 ± 2,000 |
All values were measured from a single experiment in duplicate. The ATP concentration was 1000 μM, and the Sox-based substrate sensor was 15 μM. Errors are reported as ± the standard error from the fit. n.d. – not determined.
A sufficient two-step fit was not possible due to the inability to reach the maximal inactivation rate at high osimertinib concentration (kinact).
To better understand the potency differences between these compounds, we determined the individual rates of maximal inactivation rate (kinact) and the inactivation constant (KI), where the latter captures noncovalent affinity prior to covalent bond formation.19 We find that inhibitor potency (kinact/KI) differences are principally due to differences in changes to noncovalent binding. Specifically, lazertinib inhibition of EGFR(L858R/T790M) is observed as an 8-fold diminished KI and 2.8-fold decreased rate of the maximal inactivation rate (kinact) compared to WT EGFR accounting for the mutant-selective activity of lazertinib and demonstrating that mutant selectivity is enabled by stronger noncovalent binding to EGFR(L858R/T790M). The enhanced potency for osimertinib against EGFR(L858R/T790M) is more subtle being driven entirely by a 2-fold decrease in KI and no change in kinact. From our structural analysis, it is clear that the combined effects of the unique pyrazole phenyl and methyleneamine of lazertinib are dually responsible for the improved potency and selectivity when compared to the N-methylindole of osimertinib (Figure 4B). Additionally, we suspect that the presence of the hydrophobic methionine side chain in the T790M mutant is primarily responsible for directing the mutant selectivity as seen in the lazertinib T790M/V948R binding modes (Figure 3). Indeed, the N-methylindole of osimertinib and phenyl of lazertinib act similarly in their intermolecular interactions with T790M through van der Waals contacts, as consistent with their mutant selectivity (Figure 4B).18 Lazertinib is further unique in its capacity to rigidify this binding mode through the intramolecular H-bond between the methyleneamine and the acrylamide carbonyl. We speculate that the enhanced binding to L858R/T790M with lazertinib is driven by stronger noncovalent binding afforded by intramolecular H-bonding that reinforces the productive binding mode to EGFR. Osimertinib is incapable of “rigidifying” in a similar fashion, and the methylindole moiety has been observed to exhibit rotational flexibility.18 Our combined structural and kinetics analysis on LN2057 showcases another source of selectivity due to the lack of strong H-bonding interactions with the catalytic K745 (Figure 4C), which actually shows slightly weaker binding KI and overall potency for EGFR(L858R/T790M) versus WT. The enhanced potency and mutant selectivity of lazertinib can therefore be attributed to rigidification through a methleneamine-mediated intramolecular H-bond and intermolecular van der Waals interactions of the phenyl groups, respectively.
We also assessed inhibition kinetics of these three inhibitors against WT HER2 as lazertinib has been reported to exhibit unique diminished activity against this off-target ErbB-family member.13 In line with earlier studies, our kinetic parameters show that lazertinib exhibits significantly lower potency (kinact/KI) against HER2 and is significantly lower when compared to osimertinib and LN2057 (Table 1). It is difficult to know which comparison (WT or L858R/T790M EGFR) is most meaningful to understanding the differences as they relate to WT HER2 kinact and KI values, although decreased potency appears to be due to both diminished kinact and increased KI. We speculate that changes in kinact reactivity of the lazertinib Michael acceptor toward C805 in HER2 versus C797 in EGFR may be altered by the observed intramolecular H-bond; however, the conserved sequence and structural similarity around this residue most likely suggest that the rate of formation of the covalent bond to be largely comparable. Alternatively, the lower lazertinib potency is also likely due to less favorable noncovalent binding due to structural differences in the kinase domains, which remains unclear due the absence of a cocrystal structure of lazertinib bound to WT HER2. For completeness, we confirmed lazertinib forms a covalent bond at Cys-805 within the HER2 purified kinase domain with LC-MS/MS (Figure S4). These accumulated kinetic findings are in agreement with IC50 values obtained by preincubating EGFR and HER2 kinase domains with inhibitors for 30 min prior initiating activity with added ATP substrate (Table S2). Overall, our biochemical assays confirm the marked lower potency against WT HER2 for lazertinib as compared to osimertinib and LN2057.
Since potency and selectivity against EGFR(L858R/T790M) with limited potency for HER2 is an advantage for treatment of mutant EGFR NSCLC with lazertinib,13 we assessed lazertinib inhibition in cellular contexts. First, we carried out dose-dependence inhibition experiments of EGFR(L858R/T790M) in H1975 NSCLC cells by osimertinib, lazertinib, and LN2057 by Western blotting for active EGFR (pY1068). After dosing 5 or 50 nM of these drugs for 2 h, we observed that lazertinib suppressed pY1068 to a greater extent compared to equivalent dosing of osimertinib and LN2057, as consistent with previous studies (Figure 5A).13 Uniquely, lazertinib is observed to be notably less effective at inhibiting active HER2 (pY1221/1222) in BT474 HER2 overexpressing breast cancer cells, confirming the selective targeting of EGFR by lazertinib compared to osimertinib and LN2057 (Figure 5B). To quantify cell-based inhibitor potency at longer dosing times, we conducted experiments with Ba/F3 cells expressing WT HER2, EGFR(L858R), and EGFR(L858R/T790M) dosed with inhibitors for 72 h.22 These experiments show that lazertinib is less effective against HER2 while maintaining superior cellular potency against EGFR(L858R/T790M) and EGFR(L858R) (Table 2) as consistent with previous studies.13,23 Overall, these cellular experiments recapitulate the differences in potency observed in biochemical assays (Table 1) and show that lazertinib exhibits effective targeting of EGFR(L858R/T790M) including the lack of potency of lazertinib against HER2, especially at longer time scales, indicating that lazertinib has inhibitory selectivity for EGFR and less so against HER2 relevant for lazertinib efficacy and safety in vivo.
Figure 5.

Lazertinib effectively ablates EGFR(L858R/T790M) phosphorylation in NSCLC H1975 cells and marginal inhibition of HER2 in BT474 breast cancer cells. (A) Western blot analysis of phosphorylated EGFR (pY1068) after dose-dependent treatment of H1975 (L858R/T790M) lung adenocarcinoma cell lines with osimertinib, lazertinib, and LN2057 for 2 h. (B) Western blot analysis of phosphorylated HER2 (pY1221/1222) after dose-dependent treatment of BT474 breast cancer cells with osimertinib, lazertinib, and LN2057 for 6 h. Western blots are representative examples of three independent experiments.
Table 2. Antiproliferative Activities on the Proliferation of Ba/F3 Cell Lines of EGFR(L858R), EGFR(L858R/T790M), and Wild-Type HER2a.
| Cellular
(Ba/F3) antiproliferative activity EC50 (nM) |
|||
|---|---|---|---|
| Compound | EGFR(L858R) | EGFR(L858R/T790M) | WT HER2 |
| Lazertinib | 4.8 ± 2 | 7.4 ± 1 | >100 |
| Osimertinib | 3.2 ± 0.3 | 14 ± 4 | 34 ± 8 |
| LN2057 | 0.51 ± 0.08 | 21 ± 5 | 2.7 ± 1 |
EC50 values are averages of at least three independent experiments with each experiment performed in triplicate. Errors are reported as ± standard deviation.
In conclusion, we have determined the molecular basis of the novel third-generation EGFR TKI lazertinib bound to EGFR in X-ray cocrystal structures showcasing that the lazertinib pyrazole ring facilitates H-bonds and van der Waals interactions consistent with drug efficacy and T790M mutant selectivity. Structural and functional correlations to osimertinib demonstrate the importance of lazertinib activity enabled through productive intermolecular interactions with the T790M side chain18 coupled to rigidification through intramolecular H-bonding. Additionally, we find that lazertinib does not form productive intermolecular interactions with the catalytic lysine K745, as is a principal feature of LN2057 binding and likely contributes to lower potency of lazertinib for WT EGFR and HER2. Another important feature to lazertinib is the lack of potency on HER2 that is associated with dose-limiting adverse events,24 as confirmed here in time-dependent enzyme inhibitor assay and cell-based studies. The origin of the lower potency of lazertinib against HER2 is challenging to assign to specific structural elements since the ATP binding site is practically identical to EGFR (Figure S5). Our kinetic analysis suggests that lower HER2 potency, as compared to WT and L858R/T790M, may be a combination of slower maximal rates of inhibition and/or weaker noncovalent interactions, motivating future studies to determine lazertinib crystal structures in HER2 and other kinases. Results from these studies demonstrate how the productive combination of hydrophobic phenyl and hydrophilic amine groups of lazertinib leads to superior EGFR(L858R/T790M) inhibition by impacting reversible binding properties of this irreversible inhibitor and serve as a noteworthy example for inspiring the development of highly effective next-generation kinase inhibitors.
Acknowledgments
We acknowledge startup funds from The State University of New York (DEH), National Institutes of Health Grants R01CA201049, R01CA116020, and R35CA242461 (to M.J.E.), Roswell Park Alliance Foundation (PAH). Research reported in this publication was supported by the National Center for Advancing Translational Sciences of the National Institutes of Health under award Number UL1TR001412-07. The content is solely the responsibility of the authors and does not necessarily represent the official views of the NIH. T.S.B. is supported by a Ruth L. Kirschstein National Research Service Award (5F32CA247198-02). This work was partially done in the Drug Discovery Core Facility of Roswell Park Comprehensive Cancer Center supported by National Cancer Institute (R01CA197967 to K.V. Gurova and P30CA016056 to Roswell Park Cancer Center) S.A.L. and iFIT are funded by the Deutsche Forschungsgemeinschaft (DFG, German Research Foundation) under Germany’s Excellence Strategy (EXC 2180-390900677). TüCAD2 is funded by the Federal Ministry of Education and Research (BMBF) and the Baden-Württemberg Ministry of Science as part of the Excellence Strategy of the German Federal and State Governments. This work is based on research utilizing resources of the Frontier Microfocusing Macromolecular Crystallography (17-ID-2, FMX) beamline at National Synchrotron Light Source II at Brookhaven National Laboratory to Block Allocation Group 308246.25 This work is also based upon research conducted at the Northeastern Collaborative Access Team beamlines (P30 GM124165, P41 GM103403) utilizing resources of the Advanced Photon Source at the Argonne National Laboratory (DE-AC02-06CH11357). We thank Dr. Ekin Atilla, Dr. Andrew Murkin, and Dr. Steven Diver for insightful comments and discussions. We thank Dr. Michael Malkowski and Dr. Liang Dong for SF21 cells and access to their tissue culture lab. We acknowledge Dr. Diana Monteiro and Dr. Edward Snell (Hauptman-Woodward Medical Research Institute) for access to laboratory space and equipment for protein purification and crystallization resources. We also acknowledge Dr. Yuesheng Zhang (Roswell Park) for the BT474 cells and Dr. Pasi Jänne (Dana-Farber Cancer Institute) for providing the Ba/F3 cells.
Glossary
Abbreviations
- H-bond
hydrogen bond
- EGFR
epidermal growth factor receptor
- methyleneamine
N,N-dimethylmethyleneamine
- HER2
human epidermal growth factor receptor 2
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsmedchemlett.2c00213.
Materials and methods, X-ray crystallography data and statistics, electron density maps, and LC-MS/MS images (PDF)
The authors declare no competing financial interest.
Supplementary Material
References
- Paez J. G.; Jänne P. A.; Lee J. C.; Tracy S.; Greulich H.; Gabriel S.; Herman P.; Kaye F. J.; Lindeman N.; Boggon T. J.; et al. EGFR mutations in lung cancer: correlation with clinical response to gefitinib therapy. Science 2004, 304 (5676), 1497–1500. 10.1126/science.1099314. [DOI] [PubMed] [Google Scholar]
- Lynch T. J.; Bell D. W.; Sordella R.; Gurubhagavatula S.; Okimoto R. A.; Brannigan B. W.; Harris P. L.; Haserlat S. M.; Supko J. G.; Haluska F. G.; et al. Activating mutations in the epidermal growth factor receptor underlying responsiveness of non–small-cell lung cancer to gefitinib. New England Journal of Medicine 2004, 350 (21), 2129–2139. 10.1056/NEJMoa040938. [DOI] [PubMed] [Google Scholar]
- Yun C.-H.; Mengwasser K. E.; Toms A. V.; Woo M. S.; Greulich H.; Wong K.-K.; Meyerson M.; Eck M. J. The T790M mutation in EGFR kinase causes drug resistance by increasing the affinity for ATP. Proc. Natl. Acad. Sci. U. S. A. 2008, 105 (6), 2070–2075. 10.1073/pnas.0709662105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou W.; Ercan D.; Chen L.; Yun C.-H.; Li D.; Capelletti M.; Cortot A. B.; Chirieac L.; Iacob R. E.; Padera R.; Engen J. R.; Wong K.-K.; Eck M. J.; Gray N. S.; Jänne P. A. Novel mutant-selective EGFR kinase inhibitors against EGFR T790M. Nature 2009, 462 (7276), 1070–1074. 10.1038/nature08622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wittlinger F.; Laufer S. A. The pre-clinical discovery and development of osimertinib used to treat non-small cell lung cancer. Expert Opinion on Drug Discovery 2021, 16 (10), 1091–1103. 10.1080/17460441.2021.1936496. [DOI] [PubMed] [Google Scholar]
- Chen L.; Fu W.; Zheng L.; Liu Z.; Liang G. Recent progress of small-molecule epidermal growth factor receptor (EGFR) inhibitors against C797S resistance in non-small-cell lung cancer: miniperspective. Journal of medicinal chemistry 2018, 61 (10), 4290–4300. 10.1021/acs.jmedchem.7b01310. [DOI] [PubMed] [Google Scholar]
- Zhao H.-Y.; Xi X.-X.; Xin M.; Zhang S.-Q. Overcoming C797S mutation: The challenges and prospects of the fourth-generation EGFR-TKIs. Bioorganic Chemistry 2022, 128, 106057. 10.1016/j.bioorg.2022.106057. [DOI] [PubMed] [Google Scholar]
- Günther M.; Juchum M.; Kelter G.; Fiebig H.; Laufer S. Lung Cancer: EGFR Inhibitors with Low Nanomolar Activity against a Therapy-Resistant L858R/T790M/C797S Mutant. Angew. Chem., Int. Ed. 2016, 55 (36), 10890–10894. 10.1002/anie.201603736. [DOI] [PubMed] [Google Scholar]
- Günther M.; Lategahn J.; Juchum M.; Döring E.; Keul M.; Engel J.; Tumbrink H. L.; Rauh D.; Laufer S. Trisubstituted pyridinylimidazoles as potent inhibitors of the clinically resistant L858R/T790M/C797S EGFR mutant: targeting of both hydrophobic regions and the phosphate binding site. J. Med. Chem. 2017, 60 (13), 5613–5637. 10.1021/acs.jmedchem.7b00316. [DOI] [PubMed] [Google Scholar]
- Heppner D. E.; Günther M.; Wittlinger F.; Laufer S. A.; Eck M. J. Structural Basis for EGFR Mutant Inhibition by Trisubstituted Imidazole Inhibitors. J. Med. Chem. 2020, 63 (8), 4293–4305. 10.1021/acs.jmedchem.0c00200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soria J.-C.; Ohe Y.; Vansteenkiste J.; Reungwetwattana T.; Chewaskulyong B.; Lee K. H.; Dechaphunkul A.; Imamura F.; Nogami N.; Kurata T.; et al. Osimertinib in untreated EGFR-mutated advanced non–small-cell lung cancer. New England journal of medicine 2018, 378 (2), 113–125. 10.1056/NEJMoa1713137. [DOI] [PubMed] [Google Scholar]
- Dhillon S. Lazertinib: First Approval. Drugs 2021, 81 (9), 1107–1113. 10.1007/s40265-021-01533-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yun J.; Hong M. H.; Kim S.-Y.; Park C.-W.; Kim S.; Yun M. R.; Kang H. N.; Pyo K.-H.; Lee S. S.; Koh J. S.; et al. YH25448, an irreversible EGFR-TKI with potent intracranial activity in EGFR mutant non–small cell lung cancer. Clin. Cancer Res. 2019, 25 (8), 2575–2587. 10.1158/1078-0432.CCR-18-2906. [DOI] [PubMed] [Google Scholar]
- Thress K. S.; Paweletz C. P.; Felip E.; Cho B. C.; Stetson D.; Dougherty B.; Lai Z.; Markovets A.; Vivancos A.; Kuang Y.; Ercan D.; Matthews S. E.; Cantarini M.; Barrett J. C.; Jänne P. A.; Oxnard G. R. Acquired EGFR C797S mutation mediates resistance to AZD9291 in non–small cell lung cancer harboring EGFR T790M. Nature Medicine 2015, 21 (6), 560–562. 10.1038/nm.3854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park S.; Ku B. M.; Jung H. A.; Sun J.-M.; Ahn J. S.; Lee S.-H.; Park K.; Ahn M.-J. EGFR C797S as a Resistance Mechanism of Lazertinib in Non-small Cell Lung Cancer with EGFR T790M Mutation. Cancer Res Treat 2020, 52 (4), 1288. 10.4143/crt.2020.278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X.; Gureasko J.; Shen K.; Cole P. A.; Kuriyan J. An allosteric mechanism for activation of the kinase domain of epidermal growth factor receptor. Cell 2006, 125 (6), 1137–1149. 10.1016/j.cell.2006.05.013. [DOI] [PubMed] [Google Scholar]
- Heppner D. E.; Eck M. J. A structural perspective on targeting the RTK/Ras/MAP kinase pathway in cancer. Protein Sci. 2021, 30 (8), 1535–1553. 10.1002/pro.4125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan X.-E.; Ayaz P.; Zhu S.-J.; Zhao P.; Liang L.; Zhang C. H.; Wu Y.-C.; Li J.-L.; Choi H. G.; Huang X.; Shan Y.; Shaw D. E.; Yun C.-H. Structural Basis of AZD9291 Selectivity for EGFR T790M. J. Med. Chem. 2020, 63 (15), 8502–8511. 10.1021/acs.jmedchem.0c00891. [DOI] [PubMed] [Google Scholar]
- Copeland R. A.Evaluation of enzyme inhibitors in drug discovery: a guide for medicinal chemists and pharmacologists, 2nd ed.; John Wiley & Sons, 2013. [PubMed] [Google Scholar]
- Mons E.; Roet S.; Kim R. Q.; Mulder M. P. C. A Comprehensive Guide for Assessing Covalent Inhibition in Enzymatic Assays Illustrated with Kinetic Simulations. Current Protocols 2022, 2 (6), e419. 10.1002/cpz1.419. [DOI] [PubMed] [Google Scholar]
- Zhai X.; Ward R. A.; Doig P.; Argyrou A. Insight into the Therapeutic Selectivity of the Irreversible EGFR Tyrosine Kinase Inhibitor Osimertinib through Enzyme Kinetic Studies. Biochemistry 2020, 59 (14), 1428–1441. 10.1021/acs.biochem.0c00104. [DOI] [PubMed] [Google Scholar]
- Warmuth M.; Kim S.; Gu X.-j.; Xia G.; Adrián F. Ba/F3 cells and their use in kinase drug discovery. Current opinion in oncology 2007, 19 (1), 55–60. 10.1097/CCO.0b013e328011a25f. [DOI] [PubMed] [Google Scholar]
- Wittlinger F.; Heppner D. E.; To C.; Günther M.; Shin B. H.; Rana J. K.; Schmoker A. M.; Beyett T. S.; Berger L. M.; Berger B.-T.; Bauer N.; Vasta J. D.; Corona C. R.; Robers M. B.; Knapp S.; Jänne P. A.; Eck M. J.; Laufer S. A. Design of a “Two-in-One” Mutant-Selective Epidermal Growth Factor Receptor Inhibitor That Spans the Orthosteric and Allosteric Sites. J. Med. Chem. 2022, 65 (2), 1370–1383. 10.1021/acs.jmedchem.1c00848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Copeland-Halperin R. S.; Liu J. E.; Yu A. F. Cardiotoxicity of HER2-targeted therapies. Current Opinion in Cardiology 2019, 34 (4), 451–458. 10.1097/HCO.0000000000000637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneider D. K.; Shi W.; Andi B.; Jakoncic J.; Gao Y.; Bhogadi D. K.; Myers S. F.; Martins B.; Skinner J. M.; Aishima J.; Qian K.; Bernstein H. J.; Lazo E. O.; Langdon T.; Lara J.; Shea-McCarthy G.; Idir M.; Huang L.; Chubar O.; Sweet R. M.; Berman L. E.; McSweeney S.; Fuchs M. R. FMX - the Frontier Microfocusing Macromolecular Crystallography Beamline at the National Synchrotron Light Source II. Journal of Synchrotron Radiation 2021, 28 (2), 650–665. 10.1107/S1600577520016173. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.