

Abstract

Since the problem of transporter-mediated multidrug resistance of tumor cells is becoming increasingly important in cancer therapy, it is necessary to modulate the activity of efflux transporters of the ABC family, among which P-glycoprotein is the best known. We consider the nucleotide binding domain, a universal fragment of these transporters, as a target for the rational design of small molecule compounds capable of preventing ATP-dependent drug efflux. Using various ATP mimetics, we showed that they suppress the efflux of fluorescent substrates and paclitaxel from the cells due to suppressing the ATPase activity of the transporters. The combined use of paclitaxel and ATP mimetics significantly increases its antitumor efficacy, including in cells with the multidrug resistance phenotype. The considered compounds are promising agents for the development of therapeutic efflux modulators, since they are not toxic at the given concentrations and do not induce the transporter overexpression. Moreover, the compounds overcome not only P-gp-mediated but also BCRP-mediated resistance of tumor cells.
Keywords: multidrug resistance, P-glycoprotein, breast cancer resistance protein, nucleotide binding domain, nucleotide mimetic, paclitaxel
The phenomenon of multidrug resistance is one of the serious problems of tumor chemotherapy.1 The efflux provided by the members of the ATP-binding cassette (ABC) transporter family is the major cause for the development of tumor resistance to a wide range of structurally and functionally diverse antineoplastic agents.2−4 Each transporter has its own wide specificity with respect to small molecule substrates, and acting together, the transporters are able to neutralize the activity of almost the entire variety of synthetic anticancer drugs. Two of the most studied members of this protein family are MDR1 (P-glycoprotein, P-gp, ABCB1) and BCRP (breast cancer resistance protein, ABCG2). For these transporters, small molecule inhibitors were developed and continue to be developed with widely varying selectivity for the target protein;5−7 in particular, zosuquidar, elaquidar, laniquidar, and tariquidar were proposed as the third generation P-gp inhibitors. However, none of them has yet been able to show convincing results in clinical trials.5,8
In clinical conditions, one of the factors that reduces the effectiveness of selective small molecule efflux inhibitors is the high frequency of coexpression of several exporters in one cancer cell line.9,10 Thus, taking into account the difference in the structure of the main transport proteins, it seems reasonable to develop a drug capable of inhibiting the majority of overexpressed proteins responsible for the efflux.
All modern inhibitors that have reached clinical trials are united by a common approach to the design of the structure: they target the internal cavity of a specific transporter, mostly P-gp (Figure S1), that is formed by the helices of the transmembrane domains (TMDs).11−13 At the same time, highly conserved nucleotide binding domains (NBDs) that provide not only ATP binding, but also energetically significant hydrolysis of the phosphate bond, which is accompanied by conformational rearrangements in the TMDs and substrate transfer into the extracellular space, are an integral element of the structure of the ABC transporters.14,15 Previously, it was shown that a number of substances can reduce the activity of the transporter due to the interaction with the NBD;16,17 however, modeling was used solely to explain the observed effects in these studies.
In this report, we consider the nucleotide binding domain as a target for the rational design of small molecule compounds capable of preventing ATP-dependent drug efflux. It is assumed that such an interaction will effectively suppress not only P-gp-mediated efflux, but also the functioning of BCRP and other exporter proteins.
Since the NBD structure implies ATP binding, we built on structurally diverse nucleoside mimetics in our work (Figure 1). To initially verify the adequacy of the hypothesis, we simulated the interaction of the NBD with a number of compounds that have the ability to inhibit P-gp-mediated efflux (see the Supporting Information).18 The NBD model was built based on P-gp structures presented in the Protein Data Bank: 4Q9H(19) and 6C0V.20 The compounds were divided into three groups: (A) ligands known to interact with the NBD: ATP and AMP-PNP (a nonhydrolyzable ATP analogue);21 (B) ligands known to interact with the TMDs: doxorubicin, Hoechst 33342 (P-gp substrates), and tariquidar (a P-gp inhibitor), (Figure S2);22,23 (C) drugs of other classes, for which the ability to inhibit P-gp-mediated efflux was found: nutlin-3a (an inhibitor of p53-Mdm2 protein–protein interaction),24 nilotinib and imatinib (tyrosine kinase inhibitors), (Figure S3).25
Figure 1.

Main considered structures (see also the Supporting Information): adenine nucleoside triphosphate ATP, its nonhydrolyzable analogue AMP-PNP, adenine nucleoside monophosphate AMP, 9-[(2-hydroxyethoxy)methyl]-substituted adenine SN202, nucleoside analogues AICAR and Ribavirin, and adenosine-like diketopiperazine DKPPVal.
The results of the calculations showed both the agreement with the initial assumptions and the possibility of forming a pharmacophore hypothesis for the NBD inhibitors. ATP and its analogue (group A) showed low scores when interacting with the substrate-oriented protein cavity and high scores with the NBD; substances that bind to the TMDs of P-gp (group B) showed weak affinities to the NBD. The inferior transport inhibitors, nutlin-3a, imatinib, and nilotinib (group C), acted similarly to the transported substrates and tariquidar. The data obtained indicate that compounds from group C have an ability to inhibit the transporter due to the competition for the binding with efflux substrates in the TMDs, and not due to the interaction with the NBD (Table 1).
Table 1. Docking Score (kcal/mol) of the Top Scoring Poses (Protein–Ligand Complexes) for the Three Groups of Compounds.
| ligands |
||||||||
|---|---|---|---|---|---|---|---|---|
| group
A |
group
B |
group
C |
||||||
| binding site | ATP | AMP-PNP | tariquidar | Hoechst 33342 | doxorubicin | imatinib | nilotinib | nutlin-3a |
| NBD | –7.3 | –7.9 | –5.0 | –2.9 | –4.4 | –3.1 | –4.3 | –4.1 |
| TMD | –6.8 | –6.4 | –7.3 | –6.3 | –6.5 | –6.3 | –8.3 | –7.6 |
It should be noted that although the mentioned nilotinib and imatinib inhibit the catalytic activity of tyrosine kinases, they did not tend to interact with the ATP-binding site of P-gp (Table 1). Such data indicate that it is possible to provide a sufficient selectivity with respect to other ATP-binding targets and, accordingly, to reduce the risk of undesirable effects. Similarly to the three groups of substances, the interaction of a number of AMP mimetics that inhibit the ATPase activity of the transport proteins with the NBD was simulated (Table 2; the structures were taken from the paper;26 see Supporting Information, Figure S4). Among them, different diketopiperazines (DKPPs) based on both l- and d-amino acids (Figure S5) were used since the activity of optical isomers can vary significantly. As expected, the best results were shown by the closest analogues of AMP: compounds with a purine base, ribose moiety, and phosphate group (for example, AMP-PNP, −7.9 kcal/mol). However, structures with less similarity (such as AICAR, −5.3 kcal/mol) also showed the binding that is different from that of compounds with the affinity to the TMD. The MD simulations showed similar results: AICAR and AMP-PNP formed a stable complexes with P-gp (RMSD < 2.5 Å) with binding free energy values MM-GBSA ΔGBind = −24.1 and −53.9 kcal/mol (Figure 2A).
Table 2. Docking Score (kcal/mol) of the Top Scoring Poses (Protein–Ligand Complexes) of ATP Mimetics.
| compound | docking score, kcal/mol |
|---|---|
| ATP | –7.3 |
| GTP | –7.2 |
| ADP | –7.1 |
| AMP | –6.0 |
| AMP-PNP | –7.9 |
| cAMP | –6.3 |
| 1,N6-etheno-AMP | –6.0 |
| cZMP | –5.8 |
| ZMP | –5.4 |
| AICAR | –5.3 |
| SAICAR | –5.7 |
| 6-SH-AMP | –5.3 |
| dAMP | –5.5 |
| IMP | –5.3 |
| Ara-AMP | –5.2 |
| N6-methyl-AMP | –5.1 |
| DKPPSer | –5.2 |
| DKPPVal | –5.3 |
| SN202 | –5.4 |
| Ribavirin | –5.6 |
Figure 2.

Docking poses and 100 ns MD simulations of the compounds in the NBD cavity of P-gp: (A) AICAR (violet) and AMP-PNP (turquoise), RMSD plots of proteins (RMSDprotein; gray plots) and ligand heavy atoms (RMSDligand; violet and light blue plots); (B) tariquidar (green); (C) DKPPSer (purple) and DKPPVal (lilac). The MD simulations showed that the compounds formed a stable complex with P-gp. In contrast to tariquidar, the studied mimetics reproduce well the binding of ATP/ADP/AMP-PNP in the NBD.
Although tariquidar showed a relatively high affinity to the NBD (score = −5.0 kcal/mol, Table 1), its binding pose is fundamentally different from that of ATP (Figure 2B). The studied mimetics reproduce well the binding of ATP/ADP/AMP-PNP in the NBD. Moreover, DKPPs, having comparable scores (from −5 to −6 kcal/mol), can occupy the subpockets that bind the purine base and the phosphate group (Figure 2C). However, the practically planar bicyclic structure of DKPPs allows any isomer to occupy the optimal position in the binding site, which makes it rational to use derivatives of available l-amino acids (in particular, DKPPVal), while modifying the structure with lateral substituents will lead to significant differences in the geometry of optical isomers, which could not be ignored.27,28
On the next step, HCT116tax cells with induced resistance29 as well as cells transfected with plasmids expressing MDR130 and BCRP31 (HCT116MDR1 and HCT116BCRP) were used to study the effect of nucleoside mimetics on the transport activity (see the Supporting Information).
The amount of the fluorescent substrate, Hoechst 33342, which is actively released from the cells by the ABC transporters under normal conditions,32 was evaluated. The advantage of this substrate is the possibility to exclude time-consuming washing steps since fluorescence intensity increases enormously if Hoechst 33342 is bound to DNA or is localized in a lipophilic environment like the plasma membrane.32 The use of transport inhibitors leads to the accumulation of the dye in the cells, which is reflected in the enhancement of blue fluorescence. The known inhibitors tariquidar (1 μM) and sodium orthovanadate (500 μM) were used as controls.
All the considered compounds have a significant effect on the efflux in the micromolar concentration range (Figures 3 and S6), which is consistent with the simulation results, while AICAR and ribavirin showing the best results.
Figure 3.
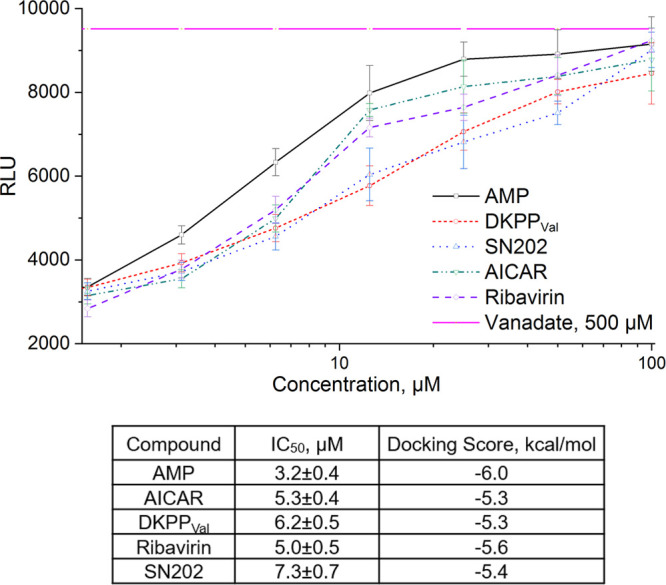
Effect of the studied compounds. (A) Intensity of the fluorescence of Hoechst 33342 accumulated by HCT116tax cells in the presence of the studied compounds. The data were used to determine IC50. (B) IC50 values and Docking Scores for selected compounds.
In the cell, AICAR is metabolized to ZMP phosphorylated at the ribose fragment, which interacts with the γ-regulatory subunit of AMP-activated protein kinase (AMPK), which leads to the structural rearrangement of the γ-subunit and AMPK activation.33−35 To make sure that the observed effect is not associated with the activation of AMPK, we used an AMPK activator of another chemical class, C24 (Figure S7),36 which interacts not with the AMP-binding cavity of the γ-subunit, but with the autoinhibitory domain of AMPK and activates the kinase by a different mechanism.35 Accordingly, C24 should not affect ATP-dependent efflux. Indeed, no effect of C24 on the substrate accumulation was observed (Figure 4).
Figure 4.

Hoechst 33342 accumulation by HCT116tax cells in the presence of AMPK activators: (A) Relative Hoechst 33342 accumulation. The difference in cell fluorescence in the presence of vanadate is taken as 100%. (B) Operetta (PerkinElmer) snapshots. In contrast to AICAR, C24 did not affect ATP-dependent efflux, which is clearly seen from the unprocessed snapshots.
The effect of the compounds on the sensitivity of HCT116 cells to paclitaxel (brand name Taxol) was studied by the accumulation of violet blue, the water-insoluble product of MTT metabolism in the cells (see the Supporting Information). All considered substances showed a tendency to enhance the effect of paclitaxel (Figures 5 and S8), while the substances themselves, unlike tariquidar, did not have a cytotoxic effect at the considered concentrations. Moreover, the compounds proved to be nontoxic (EC50 > 100 μM) in different sensitive, selected, and transfected cell lines (HCT116, HCT116tax, HEK293, HCT116MDR1, HCT116BCRP). At relevant concentrations, none of the mimetics had a negative influence on cell growth or viability.
Figure 5.

Effect of the substances on wild type HCT116 survival. (A) Relative HCT116 viability determined by 48 h MTT test in OptiMEM. The value obtained in the presence of 0.5% DMSO is taken as 100%. (B) Schematic representation of the action of a potential inhibitor: in the presence of AICAR, more antitumor drug (paclitaxel) enters the tumor cell, which enhances cell death.
Since the HCT116tax cell line was routinely cultivated in the presence of paclitaxel, in this case efflux inhibitors were added to their usual medium. It turned out that the presence of the substances at concentrations that are safe for wild-type cells leads to a decrease in the survival of resistant cells, i.e., there is a restoration of their sensitivity to paclitaxel (Figure 6).
Figure 6.

Effect of the substances on HCT116tax survival. (A) Relative HCT116tax viability determined by 48 h MTT test in OptiMEM with 60 nM of paclitaxel. The value obtained in the presence of 0.5% DMSO is taken as 100%. (B) Schematic representation of the action of a potential inhibitor. Unlike wild-type HCT116, HCT116tax has evolved a mechanism to prevent paclitaxel from entering the cell and killing it. The addition of AICAR enhances the permeation of the antitumor drug into the cell and, accordingly, cell death.
The efflux process is associated with hydrolysis of ATP molecules, which is necessary for the implementation of conformational rearrangements of the transporter that determine the transfer of a substance through the cell membrane. The binding of substances to the transporter can either stimulate or suppress the ATPase activity of the transporter.
Effects of the compounds on the ATPase activity of recombinant human P-gp were evaluated using the Pgp-Glo assay system (Promega, United States). Sodium orthovanadate is a P-gp inhibitor, and samples treated with Na3VO4 have no P-gp ATPase activity.37 Accordingly, the difference in luminescence of untreated samples and samples treated with orthovanadate reflects the basal level of P-gp activity. The inhibitor, tariquidar, blocks the transition to the open state during the catalytic cycle of P-gp; however, it should activate the ATPase activity of P-gp,38 which is confirmed by our results (Figure 7). In turn, AICAR, DKPPVal, SN202, and ribavirin suppress the ATPase activity of P-gp, although not as actively as orthovanadate (taken as 0% or zero level) (Figure 7), which confirms the proposed hypothesis about the binding of these substances to the NBD.
Figure 7.
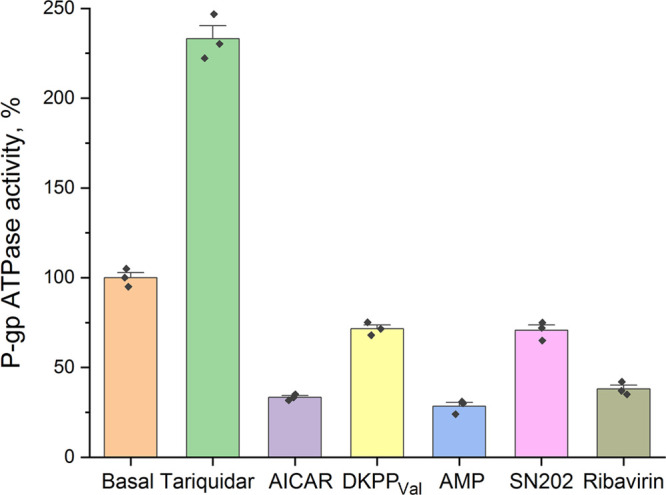
ATPase activity of P-gp. The difference between the luminescence of samples in the presence of 0.5% DMSO and 100 μM sodium orthovanadate is defined as basal level and taken as 100%. Tariquidar increases the ATPase activity of the transporter relative to the basal level, while all the considered ATP mimetics decrease it.
An important point is the assessment of the effect of the studied ATP mimetics on expression of the transport proteins. Obviously, expression stimulation in response to the treatment should lead to the development of cell tolerance to a potential inhibitor and, moreover, to an increased release of other substrates from the cell. On the other hand, expression inhibition is a powerful, but not yet explained factor that reduces cell resistance. In the case of AICAR, no such negative effect was found (Figure 8).
Figure 8.
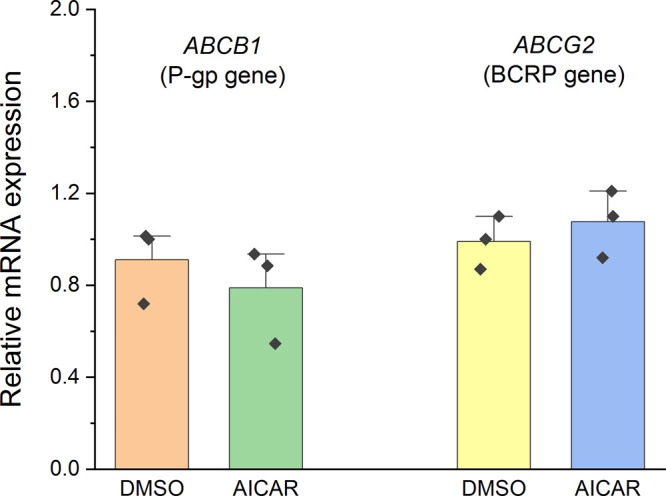
Effect of AICAR on ABCB1 (P-gp) and ABCG2 (BCRP) gene expression. The results by the 2–ΔΔCt method relative to expression of the reference GAPDH gene are presented.
Clinical trials confirm that combined therapies using the proposed type of compounds are effective.
Thus, ribavirin synergizes the effects of many chemotherapeutic agents;39 it was shown to restore the sensitivity to docetaxel in patients with prostate cancer.40,41 AICAR, in turn, is capable of potentiating the antiproliferative effects of methotrexate in MCF-7 cells42 and 5-fluorouracil in HCT116 cells.43 Considering the previously noted transport-associated nature of 5-fluorouracil resistance,44 the effect of AICAR can be reasonably assumed as transport-associated.
Representatives of the considered classes showed their effectiveness in the combined therapy of various cancer types; however, the observed effects were previously associated with the interference with intracellular mechanisms. Thus, the effect of ribavirin is associated with its direct effects on the eIF4E and IMPDH pathways; ribavirin treatment also appears to modulate other pathways central to carcinogenesis,39 while the effect of AICAR is associated with AMPK activation and the following metabolic rearrangements in cells.42
Our results suggest that, in addition to interfering with intracellular processes, the proposed compounds can increase the sensitivity of tumor cells, including resistant ones, to the therapy by suppressing efflux. ATP mimetics interact with the NBD (Table 2, Figure 2), inhibiting the ATPase activity of the transporter (Figure 7), which is expressed in reduced efflux of substrates such as Hoechst 33342 (Figure 3) and paclitaxel. Enhanced transport of paclitaxel, in turn, promotes tumor cell death (Figures 5 and 6). The studied compounds do not exhibit their own cytotoxicity and do not activate expression of efflux transporters (Figures 5 and 8), which makes them promising agents for increasing the bioavailability of a wide range of drugs, including antitumor agents, whose activity is reduced in efflux-mediated resistance.
Thus, we suggest not only drug repurposing, the interest in which has grown during the past decade,39 but also a way to the development of new drugs. The concept of designing efflux inhibitors targeted at the nucleotide-binding site, proposed in this work, showed its validity: the results obtained in in vitro tests and in cell models are in good agreement with the hypothesis and correlate with the results of the computer simulations.
Acknowledgments
The authors are grateful to Gureev Maxim (https://orcid.org/0000-0002-0385-922X) for technical support during computational studies.
Glossary
Abbreviations
- ABC
ATP-binding cassette
- MDR
multidrug resistance
- P-gp
P-glycoprotein
- BCRP
breast cancer resistance protein
- NBD
nucleotide binding domain
- TMD
transmembrane domain
- MD
molecular dynamics
- AMPK
AMP-activated protein kinase
- DKPP
diketopiperazine
- AICAR
5-aminoimidazole-4-carboxamide ribonucleoside
- eIF4E
eukaryotic translation initiation factor 4E
- IMPDH
inosine 5′-monophosphate dehydrogenase
- RLU
relative light unit
- MTT
3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl tetrazolium bromide
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsmedchemlett.2c00196.
Structures of assayed compounds, additional data, materials and methods (PDF)
Author Contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.
The work was financially supported by the Russian Science Foundation (project no. 19-73-10150).
The authors declare no competing financial interest.
Supplementary Material
References
- Nanayakkara A. K.; Follit C. A.; Chen G.; Williams N. S.; Vogel P. D.; Wise J. G. Targeted inhibitors of P-glycoprotein increase chemotherapeutic-induced mortality of multidrug resistant tumor cells. Sci. Rep. 2018, 8, 967. 10.1038/s41598-018-19325-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmitt S. M.; Stefan K.; Wiese M. Pyrrolopyrimidine derivatives and purine analogs as novel activators of Multidrug Resistance-associated Protein 1 (MRP1, ABCC1). Biochim. Biophys. Acta Biomembr. 2017, 1859, 69–79. 10.1016/j.bbamem.2016.10.017. [DOI] [PubMed] [Google Scholar]
- Namasivayam V.; Silbermann K.; Pahnke J.; Wiese M.; Stefan S. M. Scaffold fragmentation and substructure hopping reveal potential, robustness, and limits of computer-aided pattern analysis (C@PA). Comput. Struct. Biotechnol. J. 2021, 19, 3269–3283. 10.1016/j.csbj.2021.05.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grigoreva T.; Sagaidak A.; Novikova D.; Tribulovich V. Implication of ABC transporters in non-proliferative diseases. Eur. J. Pharmacol. 2022, 935, 175327. 10.1016/j.ejphar.2022.175327. [DOI] [PubMed] [Google Scholar]
- Palmeira A.; Sousa E.; Vasconcelos M. H.; Pinto M. M. Three decades of P-gp inhibitors: skimming through several generations and scaffolds. Curr. Med. Chem. 2012, 19, 1946–2025. 10.2174/092986712800167392. [DOI] [PubMed] [Google Scholar]
- Stefan S. M.; Wiese M. Small-molecule inhibitors of multidrug resistance-associated protein 1 and related processes: A historic approach and recent advances. Med. Res. Rev. 2019, 39, 176–264. 10.1002/med.21510. [DOI] [PubMed] [Google Scholar]
- Namasivayam V.; Silbermann K.; Wiese M.; Pahnke J.; Stefan S. M. C@PA: Computer-aided pattern analysis to predict multitarget ABC transporter inhibitors. J. Med. Chem. 2021, 64, 3350–3366. 10.1021/acs.jmedchem.0c02199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lai J. I.; Tseng Y. J.; Chen M. H.; Huang C. F.; Chang P. M. Clinical perspective of FDA approved drugs with P-glycoprotein inhibition activities for potential cancer therapeutics. Front. Oncol. 2020, 10, 561936. 10.3389/fonc.2020.561936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robey R. W.; Pluchino K. M.; Hall M. D.; Fojo A. T.; Bates S. E.; Gottesman M. M. Revisiting the role of ABC transporters in multidrug-resistant cancer. Nat. Rev. Cancer 2018, 18, 452–464. 10.1038/s41568-018-0005-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stefan S. M. Multi-target ABC transporter modulators: what next and where to go?. Future Med. Chem. 2019, 11, 2353–2358. 10.4155/fmc-2019-0185. [DOI] [PubMed] [Google Scholar]
- Jara G. E.; Vera D. M.; Pierini A. B. Binding of modulators to mouse and human multidrug resistance P-glycoprotein. A computational study. J. Mol. Graph. Model. 2013, 46, 10–21. 10.1016/j.jmgm.2013.09.001. [DOI] [PubMed] [Google Scholar]
- Nosol K.; Romane K.; Irobalieva R. N.; Alam A.; Kowal J.; Fujita N.; Locher K. P. Cryo-EM structures reveal distinct mechanisms of inhibition of the human multidrug transporter ABCB1. Proc. Natl. Acad. Sci. U.S.A. 2020, 117, 26245–26253. 10.1073/pnas.2010264117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kowal J.; Ni D.; Jackson S. M.; Manolaridis I.; Stahlberg H.; Locher K. P. Structural basis of drug recognition by the multidrug transporter ABCG2. J. Mol. Biol. 2021, 433, 166980. 10.1016/j.jmb.2021.166980. [DOI] [PubMed] [Google Scholar]
- Sauna Z. E.; Ambudkar S. V. About a switch: how P-glycoprotein (ABCB1) harnesses the energy of ATP binding and hydrolysis to do mechanical work. Mol. Cancer Ther. 2007, 6, 13–23. 10.1158/1535-7163.MCT-06-0155. [DOI] [PubMed] [Google Scholar]
- Zoghbi M. E.; Mok L.; Swartz D. J.; Singh A.; Fendley G. A.; Urbatsch I. L.; Altenberg G. A. Substrate-induced conformational changes in the nucleotide-binding domains of lipid bilayer-associated P-glycoprotein during ATP hydrolysis. J. Biol. Chem. 2017, 292, 20412–20424. 10.1074/jbc.M117.814186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wongrattanakamon P.; Lee V. S.; Nimmanpipug P.; Sirithunyalug B.; Chansakaow S.; Jiranusornkul S. Insight into the molecular mechanism of P-glycoprotein mediated drug toxicity induced by bioflavonoids: an integrated computational approach. Toxicol. Mech. Methods 2017, 27, 253–271. 10.1080/15376516.2016.1273428. [DOI] [PubMed] [Google Scholar]
- Martins E.; Silva V.; Lemos A.; Palmeira A.; Puthongking P.; Sousa E.; Rocha-Pereira C.; Ghanem C. I.; Carmo H.; Remiao F.; Silva R. Newly synthesized oxygenated xanthones as potential P-glycoprotein activators: in vitro, ex vivo, and in silico studies. Molecules 2019, 24, 707. 10.3390/molecules24040707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gureev M.; Novikova D.; Grigoreva T.; Vorona S.; Garabadzhiu A.; Tribulovich V. Simulation of MDM2 N-terminal domain conformational lability in the presence of imidazoline based inhibitors of MDM2-p53 protein-protein interaction. J. Comput. Aided Mol. Des. 2020, 34, 55–70. 10.1007/s10822-019-00260-6. [DOI] [PubMed] [Google Scholar]
- Szewczyk P.; Tao H.; McGrath A. P.; Villaluz M.; Rees S. D.; Lee S. C.; Doshi R.; Urbatsch I. L.; Zhang Q.; Chang G. Snapshots of ligand entry, malleable binding and induced helical movement in P-glycoprotein. Acta Crystallogr. D Biol. Crystallogr. 2015, 71, 732–741. 10.1107/S1399004715000978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim Y.; Chen J. Molecular structure of human P-glycoprotein in the ATP-bound, outward-facing conformation. Science 2018, 359, 915–919. 10.1126/science.aar7389. [DOI] [PubMed] [Google Scholar]
- Futamata R.; Ogasawara F.; Ichikawa T.; Kodan A.; Kimura Y.; Kioka N.; Ueda K. In vivo FRET analyses reveal a role of ATP hydrolysis-associated conformational changes in human P-glycoprotein. J. Biol. Chem. 2020, 295, 5002–5011. 10.1074/jbc.RA119.012042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharom F. J. Complex interplay between the P-glycoprotein multidrug efflux pump and the membrane: Its role in modulating protein function. Front. Oncol. 2014, 4, 41. 10.3389/fonc.2014.00041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loo T. W.; Clarke D. M. Mapping the binding site of the inhibitor tariquidar that stabilizes the first transmembrane domain of P-glycoprotein. J. Biol. Chem. 2015, 290, 29389–29401. 10.1074/jbc.M115.695171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grigoreva T.; Romanova A.; Sagaidak A.; Vorona S.; Novikova D.; Tribulovich V. Mdm2 inhibitors as a platform for the design of P-glycoprotein inhibitors. Bioorg. Med. Chem. Lett. 2020, 30, 127424. 10.1016/j.bmcl.2020.127424. [DOI] [PubMed] [Google Scholar]
- Dohse M.; Scharenberg C.; Shukla S.; Robey R. W.; Volkmann T.; Deeken J. F.; Brendel C.; Ambudkar S. V.; Neubauer A.; Bates S. E. Comparison of ATP-binding cassette transporter interactions with the tyrosine kinase inhibitors imatinib, nilotinib, and dasatinib. Drug Metab. Dispos. 2010, 38, 1371–1380. 10.1124/dmd.109.031302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Novikova D. S.; Garabadzhiu A. V.; Melino G.; Barlev N. A.; Tribulovich V. G. Small-molecule activators of AMP-activated protein kinase as modulators of energy metabolism. Russ. Chem. Bull., Int. Ed. 2015, 64, 1497–1517. 10.1007/s11172-015-1036-x. [DOI] [Google Scholar]
- Grigoreva T. A.; Novikova D. S.; Gureev M. A.; Garabadzhiu A. V.; Tribulovich V. G. Amino acids as chiral derivatizing agents for antiproliferative substituted N-benzyl isoindolinones. Chirality 2018, 30, 785–797. 10.1002/chir.22854. [DOI] [PubMed] [Google Scholar]
- Krasavin M.; Gureyev M. A.; Dar’in D.; Bakulina O.; Chizhova M.; Lepikhina A.; Novikova D.; Grigoreva T.; Ivanov G.; Zhumagalieva A.; Garabadzhiu A. V.; Tribulovich V. G. Design, in silico prioritization and biological profiling of apoptosis-inducing lactams amenable by the Castagnoli-Cushman reaction. Bioorg. Med. Chem. 2018, 26, 2651–2673. 10.1016/j.bmc.2018.04.036. [DOI] [PubMed] [Google Scholar]
- Grigoreva T.; Sagaidak A.; Romanova A.; Novikova D.; Garabadzhiu A.; Tribulovich V. Establishment of drug-resistant cell lines under the treatment with chemicals acting through different mechanisms. Chem. Biol. Interact. 2021, 344, 109510. 10.1016/j.cbi.2021.109510. [DOI] [PubMed] [Google Scholar]
- Pastan I.; Gottesman M. M.; Ueda K.; Lovelace E.; Rutherford A. V.; Willingham M. C. A retrovirus carrying an MDR1 cDNA confers multidrug resistance and polarized expression of P-glycoprotein in MDCK cells. Proc. Natl. Acad. Sci. U.S.A. 1988, 85, 4486–4490. 10.1073/pnas.85.12.4486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeng H.; Park J. W.; Guo M.; Lin G.; Crandall L.; Compton T.; Wang X.; Li X. J.; Chen F. P.; Xu R. H. Lack of ABCG2 expression and side population properties in human pluripotent stem cells. Stem Cells 2009, 27, 2435–2445. 10.1002/stem.192. [DOI] [PubMed] [Google Scholar]
- Muller H.; Klinkhammer W.; Globisch C.; Kassack M. U.; Pajeva I. K.; Wiese M. New functional assay of P-glycoprotein activity using Hoechst 33342. Bioorg. Med. Chem. 2007, 15, 7470–7479. 10.1016/j.bmc.2007.07.024. [DOI] [PubMed] [Google Scholar]
- Novikova D. S.; Ivanov G. S.; Garabadzhiu A. V.; Tribulovich V. G.. Compounds of plant origin as AMP-activated protein kinase activators. In Chemistry and Technology of Plant Substances: Chemical and Biochemical Aspects; Kutchin A. V., Shishkina L. N.; Weisfeld L. I., Eds.; Apple Academic Press: Waretown, NJ, 2017; pp 181–209. [Google Scholar]
- Novikova D. S.; Grigoreva T. A.; Ivanov G. S.; Melino G.; Barlev N. A.; Tribulovich V. G. Activating effect of 3-benzylidene oxindoles on AMPK: From computer simulation to high-content screening. ChemMedChem. 2020, 15, 2521–2529. 10.1002/cmdc.202000579. [DOI] [PubMed] [Google Scholar]
- Visnjic D.; Lalic H.; Dembitz V.; Tomic B.; Smoljo T. AICAr, a widely used AMPK activator with important AMPK-independent effects: A systematic review. Cells 2021, 10, 1095. 10.3390/cells10051095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Novikova D. S.; Grigoreva T. A.; Zolotarev A. A.; Garabadzhiu A. V.; Tribulovich V. G. Advanced palladium free approach to the synthesis of substituted alkene oxindoles via aluminum-promoted Knoevenagel reaction. RSC Adv. 2018, 8, 34543–34551. 10.1039/C8RA07576J. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ambudkar S. V.; Dey S.; Hrycyna C. A.; Ramachandra M.; Pastan I.; Gottesman M. M. Biochemical, cellular, and pharmacological aspects of the multidrug transporter. Annu. Rev. Pharmacol. Toxicol. 1999, 39, 361–398. 10.1146/annurev.pharmtox.39.1.361. [DOI] [PubMed] [Google Scholar]
- Loo T. W.; Clarke D. M. Tariquidar inhibits P-glycoprotein drug efflux but activates ATPase activity by blocking transition to an open conformation. Biochem. Pharmacol. 2014, 92, 558–566. 10.1016/j.bcp.2014.10.006. [DOI] [PubMed] [Google Scholar]
- Pfab C.; Schnobrich L.; Eldnasoury S.; Gessner A.; El-Najjar N. Repurposing of antimicrobial agents for cancer therapy: What do we know?. Cancers (Basel) 2021, 13, 3193. 10.3390/cancers13133193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kosaka T.; Maeda T.; Shinojima T.; Nagata H.; Mizuno R.; Oya M. A clinical study to evaluate the efficacy and safety of docetaxel with ribavirin in patients with progressive castration resistant prostate cancer who have previously received docetaxel alone. J. Clin. Oncol. 2017, 35, e14010 10.1200/JCO.2017.35.15_suppl.e14010. [DOI] [Google Scholar]
- Kosaka T.; Shinojima T.; Kikuchi K.; Hagiwara S.; Kojima S.; Hongo H.; Ueda K.; Miyake S.; Oya M. A phase 1/2a trial of docetaxel plus ribavirin for reprogramming efficacy in patients with progressive metastatic castration resistant prostate cancer who have previously received docetaxel alone: DRREEM trial. J. Clin. Oncol. 2018, 36, 329. 10.1200/JCO.2018.36.6_suppl.329. [DOI] [Google Scholar]
- Fodor T.; Szanto M.; Abdul-Rahman O.; Nagy L.; Der A.; Kiss B.; Bai P. Combined treatment of MCF-7 cells with AICAR and methotrexate, arrests cell cycle and reverses Warburg metabolism through AMP-activated protein kinase (AMPK) and FOXO1. PLoS One 2016, 11, e0150232 10.1371/journal.pone.0150232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee K. C.; Lin C. T.; Chang S. F.; Chen C. N.; Liu J. L.; Huang W. S. Effect of AICAR and 5-fluorouracil on X-ray repair, cross-complementing group 1 expression, and consequent cytotoxicity regulation in human HCT-116 colorectal cancer cells. Int. J. Mol. Sci. 2017, 18, 2363. 10.3390/ijms18112363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marin J. J. G.; Macias R. I. R.; Monte M. J.; Herraez E.; Peleteiro-Vigil A.; Blas B. S.; Sanchon-Sanchez P.; Temprano A. G.; Espinosa-Escudero R. A.; Lozano E.; Briz O.; Romero M. R. Cellular mechanisms accounting for the refractoriness of colorectal carcinoma to pharmacological treatment. Cancers (Basel) 2020, 12, 2605. 10.3390/cancers12092605. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.