

Abstract
The free fatty acid receptors FFAR1 and FFAR4 are considered promising therapeutic targets for management of metabolic and inflammatory diseases. However, there is a need for entirely novel chemical scaffolds, since many of the highly similar lipophilic chemotypes in development have been abandoned by the pharmaceutical industry, due to toxic effects on hepatocytes and β-cells. Our group has recently reported the discovery of a 1,3,5-triazine-2-amine-based compound that acts as an allosteric agonist on FFAR1. Here, we present the synthesis and investigation of the structure–activity relationship of an extensive set of analogues of which many display dual-acting agonist properties for both FFAR1 and FFAR4. In several rounds of optimization, we discovered multiple analogues with single-digit nanomolar potency on FFAR1. Pending additional optimization for metabolic stability, the compounds in this study present novel ways of providing beneficial glycemic control while avoiding the notorious toxicity challenges associated with previously identified chemotypes.
Keywords: free fatty acid receptor, FFAR1, FFAR4, GPR40, GPR120, G-protein-coupled receptor
The free fatty acid (FFA) receptors FFAR1 and FFAR4 (previously known as GPR40 and GPR120) are important players in glucose and energy homeostasis. Although they share just 15% sequence homology, both are medium- and long-chain fatty acid sensing G-protein-coupled receptors (GPCRs).1 Their key role in mediation of glucose-dependent insulin secretion and sensitivity has moved them into the spotlight as potential targets for treatment of type-2 diabetes mellitus (T2DM). Synthetic small-molecule compounds activating FFAR1 via the Gq-signaling pathway have proceeded furthest in the development, spearheaded by Takeda’s TAK-875 (Figure 1) which demonstrated good glycemic control in clinical phase-3 studies.2 Many other pharmaceutical companies have developed highly potent agonists based on similar fatty acid mimicking chemotypes, but abandoned further efforts after TAK-875 showed hepatotoxic effects, which is likely caused by inhibition of certain bile-acid transporters.3 It has generally proven difficult to move away from these lipophilic scaffolds which appear to be associated with hepatotoxicity.4,5 Second generation compounds with an alternative pharmacological profile (both Gq- and Gs-based signaling), such as AM-5262 (Figure 1), showed increased efficacy, but were chemically still highly similar to TAK-875 and, moreover, showed β-cell toxicity.6
Figure 1.

Chemical structures of the first- and second-generation FFAR1 reference compounds TAK-875 and AM-5262 (left-hand side) in comparison with compound 1 (right-hand side) and binding site overview for FFAR1 (middle panel, PDB 5TZY). MW, molecular weight; tPSA, topological polar surface area (calculated with ChemDraw, v. 19.1.0.5).
Besides pursuing FFAR1 selective ligands, many groups have developed dual-acting FFAR1/FFAR4 agonist compounds.7,8 The added effect of FFAR4 activation has been associated with increased insulin sensitization through anti-inflammatory effects,9−11 while maintaining a similarly beneficial control of glucose homeostasis. FFAR1/FFAR4 dual agonism thereby appears to address two fundamental pathological defects of T2DM: β-cell failure and insulin resistance.
Our group has previously utilized molecular dynamics simulations to study the dynamics of a new solvent-exposed binding site in FFAR1 (termed “site 3” in ref (12)) and consequently used structure-based design to discover a ligand (1) that binds this pocket and activates FFAR1 in an allosteric fashion13 (Figure 1). In contrast, X-ray crystallography12 has confirmed the location of the reference compounds TAK-875 to be the outer-leaflet pocket, with the entry site located between TM-III and TM-IV, while the second-generation compound AM-5262 has been shown to bind to the inner-leaflet pocket,13 located between the intracellular ends of TM-III, TM-IV, and ICL2.14 Ligand 1, based on a 1,3,5-triazine-2-amine scaffold, represents the first and currently only FFAR1 small-molecule agonist with a chemotype that is not based on the lipophilic fatty acid mimicking scaffolds that have previously been described.5 During follow-up studies, we discovered that several analogues of 1 were dual-acting FFAR1/FFAR4 agonists while maintaining high selectivity over a panel of other metabolite-sensing GPCR targets.
In this study, we report the design and synthesis of a new class of dual-acting FFAR1/FFAR4 agonists based on 1. We systematically explored the structure–activity relationship (SAR) of this chemical series and conducted mutagenesis and molecular modeling studies to describe the underlying structural basis. Our efforts have produced a set of highly potent (EC50 ∼single-digit nM) and selective compounds that show the possibility of targeting free fatty acid receptors through a novel binding site and distinct chemotype.
Our general strategy for the medicinal chemistry work was to iteratively explore specific parts of the molecule while maintaining the most beneficial combination of moieties from the previous optimization round. The aim was to conduct a systematic exploration; however, in some cases the optimization was guided by synthetic feasibility, i.e., readily available building blocks. ADME and solubility properties were evaluated between each optimization round and were initially used to highlight major problems associated with certain chemotypes. Molecular docking studies were used as a guidance to identify general special constraints and potential areas to expand the compounds in size. After nanomolar potency had been reached, we shifted focus on improving ADME and solubility while keeping an acceptable level of bioactivity.
The primary focus in the first rounds of optimization was improvement of potency and efficacy on FFAR1 based on variations of the left-hand phenyl ring and right-hand 4-methylchroman-2-one motif (Table 1). 4-Cl (2) and 4-F (3) did not yield improved potency, whereas 4-Me (4) resulted in a modest improvement compared to the original 2-Me.13 Me (5) or Cl (6) substitution in the 3-position also improved potency slightly, albeit with a marked drop in efficacy. 4-cPr (7) and 4-iPr (8) showed similar potency improvements while maintaining high efficacy, whereas potency gains for the bulkier 1,3-dihydroisobenzofuran (9) and 2-naphthyl (10) were accompanied by significant loss of efficacy. Based on the increase in potency we observed for single Me and Cl substitutions in the 3- and 4- position, we then tested several double and triple 3,4-substituted compounds (11–16), all of which had potencies in the sub-micromolar range. No further improvements could be made by introducing 3,5-Cl-4-Me (15) or 3-Cl-4-Et (16) moieties at those positions. Attempts to increase polarity by introducing a hydroxyl group in the 3- (17) or 4-position (18) led to inactive compounds. Insertion of a methylene group between the amine linker and phenyl ring (19) was not tolerated.
Table 1. SAR Exploration of the 4-Methylchroman-2-one-Based Chemical Scaffolda.
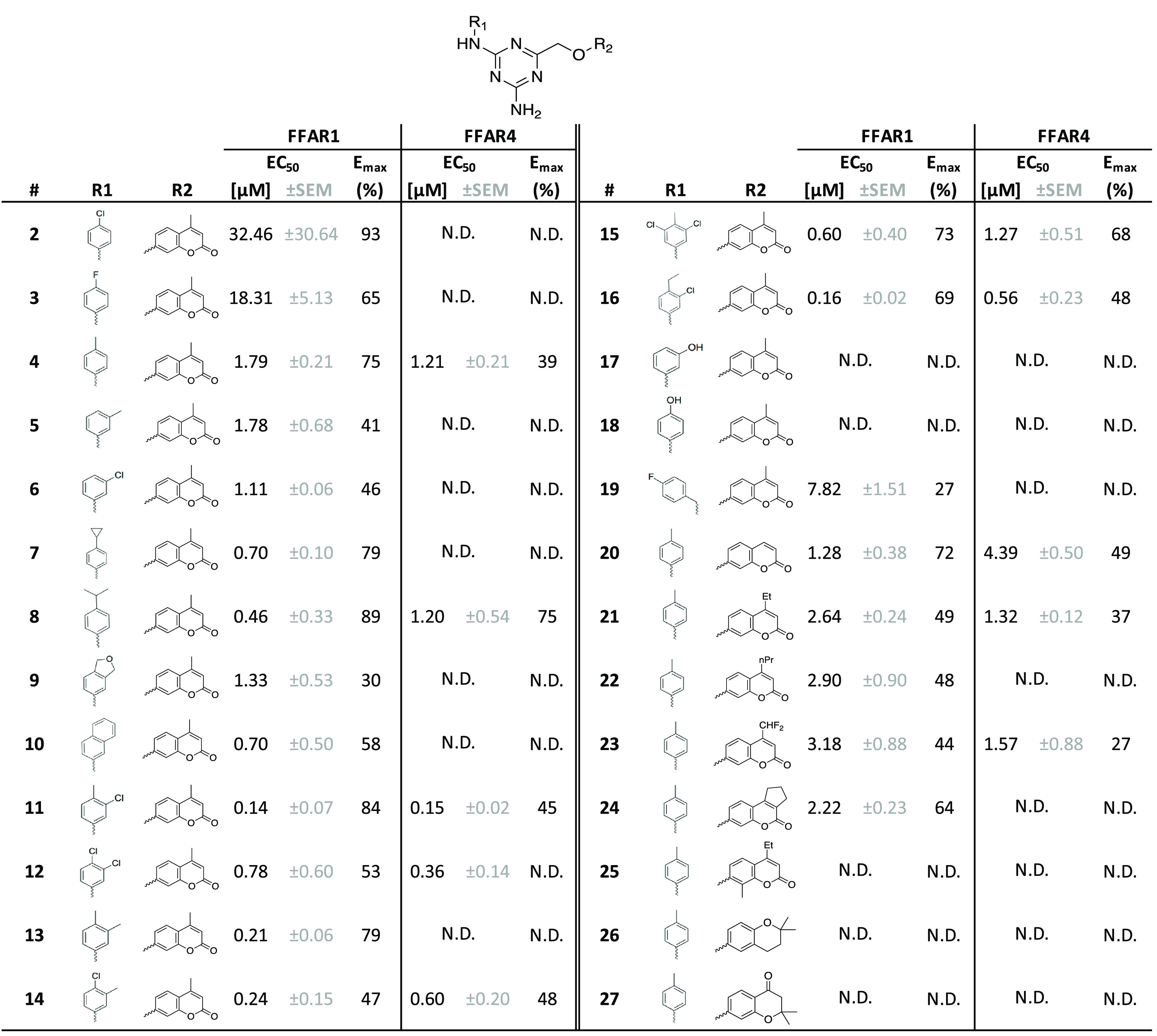
All compounds were tested as duplicates in two independent experiments (n = 2) with the highest concentration being 10 μM. N.D.: non-determinable.
We then moved to probe variations of the right-hand 4-methylchroman-2-one motif (R2-position). Removal of the methyl in the 4-position (20) or replacement by Et (21), nPr (22), CHF2 (23), or fused cyclopentyl in the 3,4 position (24) did not lead to major changes in activity. Substitution with Me in the 8-position (25) was not tolerated, nor were the 2,2-dimethylchroman (26) or 2,2-dimethylchroman-4-one (27) moieties.
Many of the chroman-2-one analogues from Table 1 that were active on FFAR1 also showed activity on FFAR4, albeit often with markedly reduced efficacy (generally <50%). The best overall compound on FFAR1, 11 (EC50 = 0.138 μM), was also the one with the highest potency on FFAR4 (EC50 = 0.155 μM). Several of the compounds that had a decent activity on FFAR1, such as 7 and 13, were entirely inactive on FFAR4.
In the second series of optimization rounds (Tables 2 and 3), we attempted a more thorough exploration of the right-hand 4-methylchroman-2-one motif. Initial data had indicated a solubility below 2 μM in phosphate-buffered saline (PBS) for compound 11, so we aimed to improve overall polarity and solubility while keeping the most favorable left-hand 3-chloro-4-methylphenyl. We observed that it was possible to replace the 4-methylchroman-2-one moiety with a more hydrophilic pyrrolidin-2-one in the 3-position to the ether-linked phenyl with only minor (∼6-fold) loss of potency on FFAR1 (28), leading to a significantly improved solubility (28 μM in PBS). In the same position, we tested other synthetically feasible modifications, such as piperidin-2-one (29), piperidine-2,4-dione (30), or isothiazolidine 1,1-dioxide (31), that did not lead to comparably active compounds. Shifting the pyrrolidin-2-one to the 2-position (32) or substitution with pyrrolidine-2,5-dione in the 4-position (33) was likewise not tolerated.
Table 2. SAR Exploration of the Right-Hand R2-Positiona.
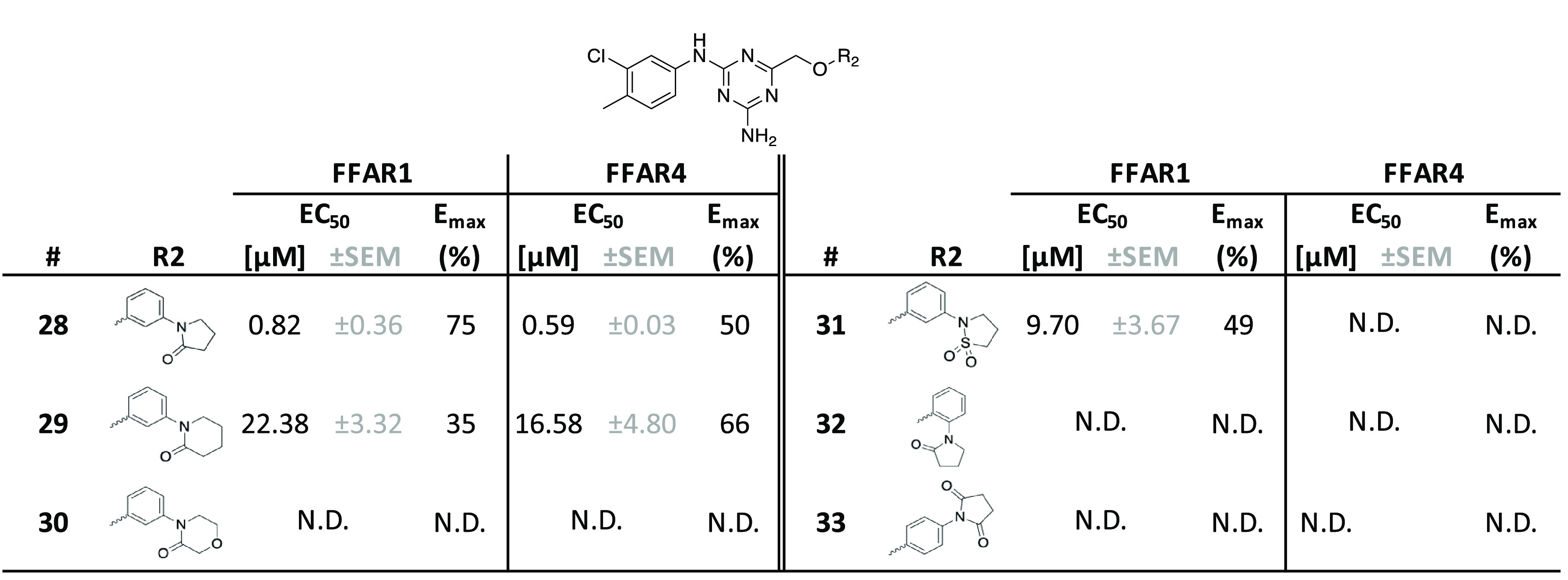
All compounds were tested as duplicates in two independent experiments (n = 2) with the highest concentration being 10 μM. N.D., non-determinable.
Table 3. SAR Exploration of the Right-Hand R3-Positiona.
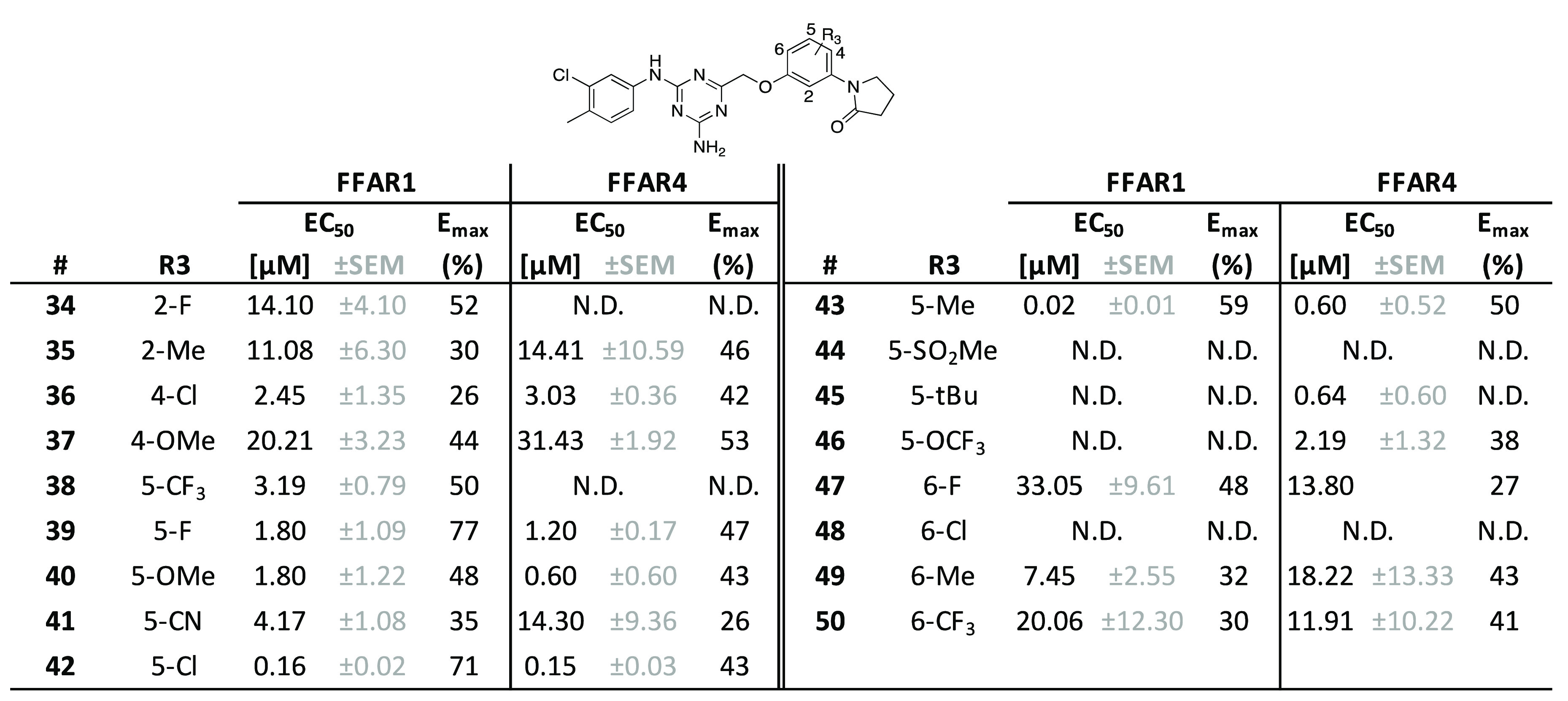
All compounds were tested as duplicates in two independent experiments (n = 2), with the highest concentration being 10 μM. N.D., non-determinable.
We then moved toward systematically substituting the remaining ring positions of the phenyl linker while maintaining the pyrrolidin-2-one as previously described in 28. 2-F (34) and 2-Me (35) substitutions lead to potency and efficacy decreases compared to 28 (Table 3). Probing the 4-position of the ether-linked phenyl with Cl (36) and OMe (37) also yielded less potent compounds. In contrast, we observed several compounds with substitutions in the 5-position of both O-phenyl linker and pyrrolidin-2-one that led to similar or improved potency. While 5-CF3 (38), 5-F (39), 5-OMe (40), and 5-CN (41) were roughly equipotent and mostly less efficacious than 28, 5-Cl (42) showed a 5-fold gain of potency as well as a robust level of efficacy. The overall best compound was the 5-Me (43), with a potency in the double-digit nM range. Other substitutions in this position, such as 5-SO2Me (44), 5-t-Bu (45), or 5-OCF3 (46), were not tolerated. Substituting the 6-position with F (47), Cl (48), Me (49), or CF3 (50) resulted in markedly decreased efficacy or complete loss of activity. Expanding the phenyl-linker into a naphthalenyl was only tolerated for the 4,5-fused naphthalenyl 51 (Supporting Information, Table S1), albeit with a minor loss of activity, whereas the 5,6-fused naphthalenyl 52 (Supporting Information, Table S1) was inactive. Introducing a nitrogen at the 5-position of the O-phenyl linker in 53 (Supporting Information, Table S1) to increase polarity likewise led to an inactive compound.
The activity profile on FFAR4 was similar to the one observed on FFAR1 for the majority of compounds in Table 3. In general, we noticed that substitutions on the phenyl linker were slightly less well tolerated for FFAR4 activity, as can be seen in the cases of compounds 34, 38, 41, and 47 that show decreased potency and/or efficacy. The 5-position of the O-phenyl linker appeared to be most relevant for receptor selectivity: substitution with 5-CF3 (38) or 5-OCF3 (46) led to a selective compound for FFAR1 and FFAR4, respectively.
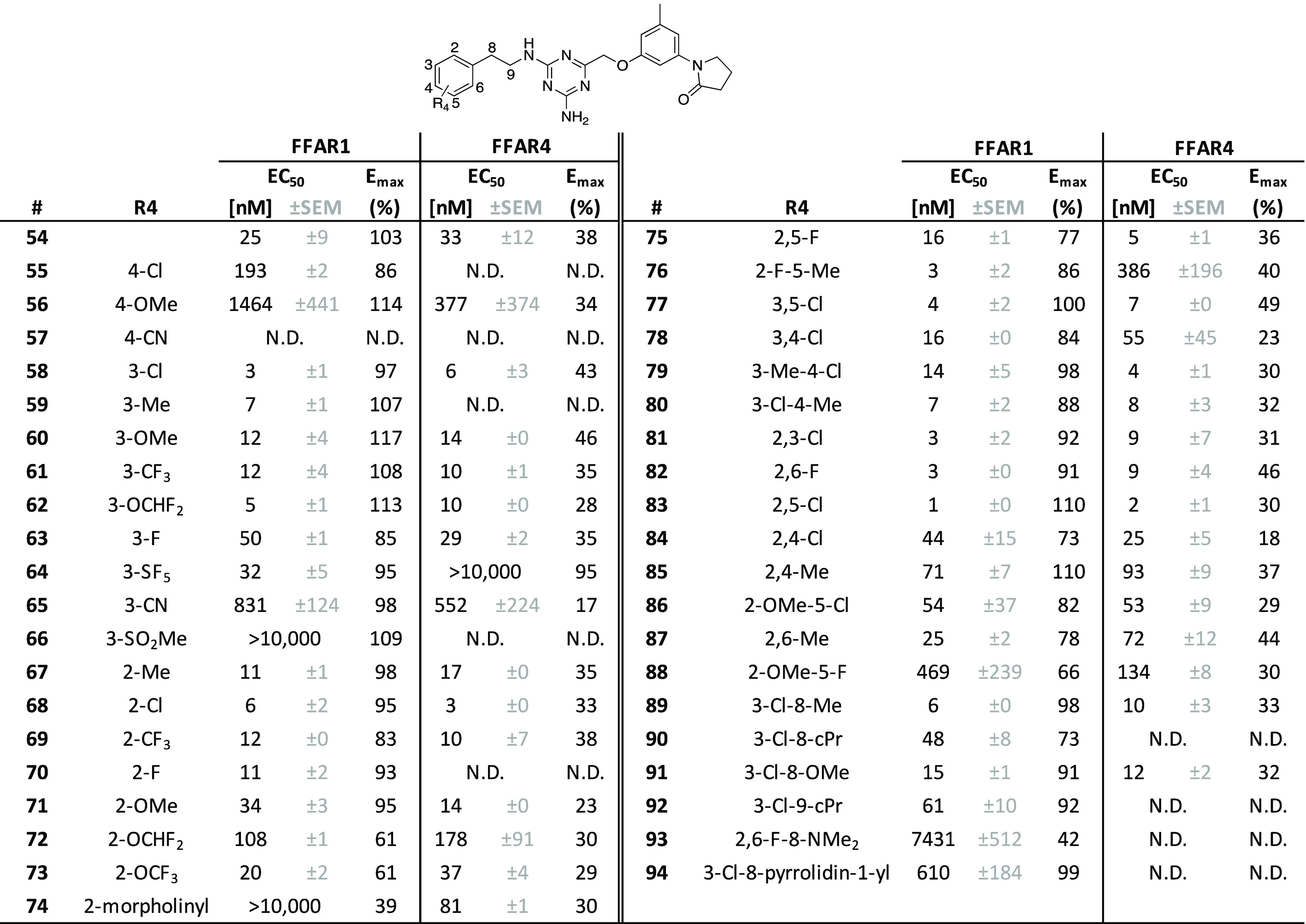
We subsequently shifted focus back to exploring the left-hand phenyl moiety while maintaining the favorable right-hand 5-methylphenyl-pyrrolidin-2-one motif (Table 4). Crucially, we discovered that insertion of an ethylene linker between the amino group and the left-hand phenyl (54) was beneficial for activity on FFAR1, increasing efficacy by 44% as compared to compound 43, while maintaining a similar EC50.
Table 4. SAR Exploration of the Left-Hand R4-Positiona.
All compounds were tested as duplicates in two independent experiments (n = 2), with the highest concentration being 10 μM. N.D., non-determinable.
Following this observation, we attempted to improve activity further by introducing smaller substitutions on the terminal phenyl moiety. At the 4-position, Cl (55), OMe (56), and CN (57) led to a significant decrease or complete loss of activity. In contrast, introducing nonpolar substitutions at the 3-position was very favorable for activity, resulting in analogues 58 (3-Cl), 59 (3-Me), 60 (3-OMe), 61 (3-CF3), and 62 (3-OCHF2) with 2–8-fold improved potency range (single to double-digit nM range) as compared to the unsubstituted phenyl 54 and generally high levels of efficacy, comparable to the prototype compound TAK-875. Minor potency decreases were observed for the moieties in compounds 63 (3-F) and 64 (3-SF5), whereas increasingly polar substituents, such as 3-CN (65) and 3-SO2Me (66), dramatically reduced potency. Generally, a similar trend was observed for the 2-position: most compounds that had improved potency were nonpolar substituents, such as 2-Me (67), 2-Cl (68), and 2-CF3 (69), but also the slightly more polar 2-F (70). Minor loss of potency and/or efficacy was seen for 2-OMe (71), 2-OCHF2 (72), and 2-OCF3 (73), while the hydrophilic 2-morpholinyl (74) was entirely inactive.
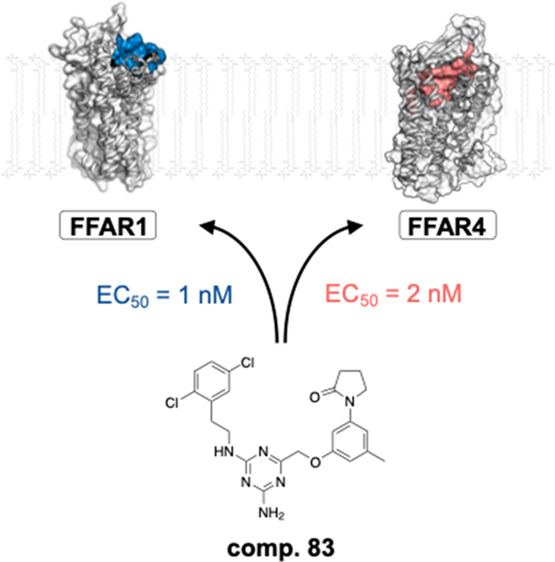
For double substitutions at the left-hand phenyl moiety, our aim was to repeat the successful strategy from the first set of 4-methylchroman-2-one analogues (2–19) and add alkyl, alkoxy, or halogen moieties to improve potency and efficacy on FFAR1. In general, substitutions at the 2- and/or 3-position tended to be more beneficial for activity than those involving the 4-position, consistent with our observations for single substitutions. 2,5-F (75), 2-F-5-Me (76) 3,5-Cl (77), 3,4-Cl (78), 3-Me-4-Cl (79), 3-Cl-4-Me (80), 2,3-Cl (81), and 2,6-F (82) all showed improved potency, albeit with a certain loss of efficacy, which was most pronounced for compound 75 (−26%). The overall most potent and efficacious compound in this analogue set was the 2,5-Cl (83), with an EC50 value of 1 nM and an Emax that superseded the efficacy of the reference compound TAK-875 by 10% (Figure 2). Other combinations of substitutions, such as 2,4-Cl (84), 2,4-Me (85), 2-OMe-5-Cl (86), and 2,6-Me (87), led to a slight (2−3-fold) reduction of potency, which was more pronounced for the 2-OMe-5-F (88).
Figure 2.

Chemical structures and dose–response curves for key compounds in this chemical series in comparison with the reference compound TAK-875.
At this point in the study, initial data from ADME studies indicated, however, metabolic instability in the liver S9 fraction for the most potent compounds from the analogue set containing the left-hand ethylene linker (54–88). Therefore, we tested several modifications of the ethylene linker as well as several heterocyclic and cycloalkylic left-hand substitutions (Table 4, Supporting Information, Table S2) to probe whether these would prevent potential oxidation by liver enzymes. Smaller alkyl or alkoxy groups on the benzyl methylene (89, 90, 91) were tolerated but did not lead to any gains in activity. A similar trend was observed for the cPr addition alpha to the benzyl methylene (92). We also attempted to add basic moieties on the benzyl methylene, such as dimethylamino (93) and pyrrolidin-1-yl (94), to increase polarity and solubility, but these modifications led to a major decrease in potency. Likewise did the majority of ligands with more substantial left-hand modifications, such as replacement of the terminal phenyl ring with cycloalkyls, aromatic heterocycles, or naphthalyl in compounds 95–107 (Supporting Information, Table S2) not lead to any activity improvements.
Most compounds from this set acted as strong agonists on the FFAR4 receptor, often with similar potency compared to FFAR1, but significantly reduced efficacy. We noted a few moieties, often located at the 2-position in the left-hand phenyl group, that seemed to play a role in increasing selectivity for one receptor over the other. Addition of 2-F (70) or 2-F-5-Me (76) rendered the activity as highly selective for FFAR1 whereas 2-morpholinyl (74) led to a FFAR4-selective compound. Many of the ethylene linker modifications (90, 92, 93, 94) led to loss of activity on FFAR4 while maintaining a certain level of FFAR1 activation. Finally, substitution of the left-hand phenyl with 2,2-difluorobenzo[1,3]dioxol-4-yl in 101 (Supporting Information, Table S2) resulted in a highly FFAR1-selective compound.
The key intermediates of this chemical series are highlighted in Figure 2, with the overall most potent (EC50 = 1 nM) compound 83 surpassing both the potency and efficacy of the prototype compound TAK-875.
During optimization of this chemical series, we noted a few general trends. First and foremost, the modifications that led to the overall highest activity tended to be nonpolar alkyl and/or electron-rich halogens, while attempts to increase polarity with, e.g., addition of hydroxyl or nitrile groups (17, 18, 41, 57, 65) or pyridyl moieties (53) in most cases eliminated all biological activity. To a certain extent, this trend was expected due to the highly lipophilic nature of the endogenous and synthetic ligands for both receptors. However, considering the entirely different chemotype and expected binding to a solvent-exposed allosteric binding site,13 we were nevertheless surprised by the steepness of the decrease in activity observed for most hydrophilic substitutions. No clear trend regarding bioactivity could be observed for electronic effects in any of the ring positions that were systematically probed, as exemplified by the roughly equipotent compounds 60 and 61 with strong electron-donating and -withdrawing effects, respectively.
A second trend was the high level of correlation between potency values for FFAR1 and FFAR4 (Figure 3). Depending on the circumstances, this might be considered as an indication of unspecific activity. However, we also observed that key compounds from this chemical series were highly specific for FFAR1 and FFAR4 in a counterscreening involving a broad range of other metabolite GPCRs including the closely related free fatty acid receptors 2 and 3 (Supporting Information, Figure S1). The underlying mechanistic basis of this effect remains unclear, due to the absence of experimental structures of FFAR4. Multiple dual-acting agonists have been described in the literature,7,8 despite the low sequence homology shared between FFAR1 and FFAR4 and lack of conserved key residues in their binding sites,16 pointing toward a mechanism of activation that is potentially independent of both receptors sharing an analogous binding site.
Figure 3.

Comparison between pharmacological activity on FFAR1 and FFAR4. Log EC50 values for FFAR1 and FFAR4 of all compounds tested in Tables 1–3 were plotted against each other, while respective Emax values for FFAR1 (A) and FFAR4 (B) correspond to each data point’s color.
We also noted a markedly lower efficacy of most compounds on FFAR4, which in most cases was below 50%. Efficacy values were normalized based on the reference compounds TAK-875 (FFAR1) and Metabolex-36.17 A similar trend has been observed by others for a set of dual-acting non-esterified fatty acids.8 The most efficacious FFAR4 agonists were among the earlier 4-methylchroman-2-one-based scaffold (Table 1, 8, 15), which during our optimization efforts ultimately was replaced with pyrrolidin-2-one, driven by the observations on FFAR1. Since FFAR4 efficacy and potency appear to be only weakly correlated for this chemical series, a separate optimization drive focusing entirely on improving FFAR4 efficacy would be needed to potentially yield highly efficacious FFAR4 agonists.
Finally, although many compounds were roughly equipotent on both FFAR1 and FFAR4, we were able to identify key positions in the chemical scaffold that appeared to modulate selectivity. Most notably, fused heterocycles and generally substitutions involving the ortho-position on the left-hand phenyl moiety often acted as an important site for selectivity that was able to act in both directions (70, 74, 76) On the other hand, substitutions on the right-hand phenyl linker (34, 38, 39, 41, and 47) or the ethylene linker (90, 92, 93, 94) predominantly increased selectivity for FFAR1.
Several key compounds from this chemical series were subsequently tested for important ADME properties, such as solubility in PBS, log D, and metabolic stability in mouse liver S9 fraction (Table 5). Unfortunately, all tested compounds showed high metabolic instability and were eliminated after a few minutes, despite generally having high chemical stability (data not shown). To identify the potential site of degradation in the chemical scaffold, we tested a diverse set of compounds, consisting of different left-hand halogenated substitutions, 58 (3-Cl), 62 (3-OCHF2), 83 (2,5-Cl), modifications of the ethylene linker (89, 91, 92, 94) as well as replacement of the left-hand phenyl with non-aromatic (98) or heterocyclic moieties (101). To rule out a degradation related to the right-hand pyrrolidin-2-one moiety, we tested the 4-methylchroman-2-one 13, which also showed rapid degradation in mouse liver S9 fraction (t1/2 = 7.8 min). High metabolic instability seemed to be neither related to low solubility, as the data for the polar morpholinyl 74 and the basic pyrrolidin-1-yl 94 shows. A potential degradation mechanism involving the central 1,3,5-triazine-2-amine appears possible but would require further studies.
Table 5. Experimental ADME Parameters for Selected Key Compounds.
| # | solubility [μM in PBS] | log D | metabolic stability (mouse liver S9 fraction) t1/2, [min] |
|---|---|---|---|
| 58 | 11 | 3.77 | 2.3 |
| 63 | 84 | 3.32 | 1.1 |
| 74 | 119 | 2.93 | 0.9 |
| 83 | 19 | 3.46 | 1.9 |
| 89 | 14 | 3.93 | 0.8 |
| 91 | 36 | 3.52 | 2.0 |
| 92 | 23 | 3.82 | 0.7 |
| 94 | 294 | 2.79 | 3.0 |
| 98 | 55 | 3.53 | 0.8 |
| 101 | 13 | 3.42 | 0.8 |
To rationalize some of our SAR observations and understand the mechanistic basis for FFAR1 activity of this set of compounds, we conducted a combined molecular modeling and experimental mutagenesis study. We applied the automated ligand-guided backbone ensemble receptor optimization (ALiBERO), developed by Rueda et al.,18 thereby utilizing the available experimental information on compound activity in the computational protocol. The input FFAR1 receptor structure was generated by running a 600 ns molecular dynamics simulation of FFAR1 in complex with TAK-875 and subsequently extracting a representative frame based on geometric clustering of 48 amino acid residues that form the solvent-exposed extracellular binding site.13 We divided our set of chemical compounds into active (EC50 < 10 nM) and decoy (EC50 > 100 μM) portions that together form the training set used in the iterative ALiBERO optimization protocol. In this protocol, multiple receptor conformations are generated based on elastic network normal mode and then evaluated in a small-scale docking run of the training set. Receptor conformations are assessed based on their ability to discriminate between known actives and decoy molecules, and the best-performing combination of receptor structures is chosen for the next iterative round of optimization. The protocol yielded an ensemble of four highly optimized FFAR1 structures after 10 rounds of optimization that was able to accommodate known actives in consistent, overlapping docking poses and with high discriminatory ability over the decoy molecules in the training set (Supporting Information, Table S1). The docking poses of the active compounds (Figure 4) are characterized by several key interactions. A hydrogen bond between the hydroxyl group of Y12 and the amino group of the ligands is acting as an anchor point that is conserved in all docking poses, together with van der Waals contributions from A66 and L262. The carbonyl oxygen of the pyrrolidin-2-one moiety appears to form a polar network that consists of both intra- and intermolecular interactions with the left-hand NH linker as well as the charged residues K62 and E65 in TM-II. Finally, the terminal left-hand moiety (halogen-substituted phenyl in most cases) is accommodated, among others, by S8 in TM-I and K259 in TM-VII, which potentially could form halogen bonds and/or cation−π interactions.
Figure 4.

Mutagenesis analysis of the solvent-exposed extracellular binding site and outer-leaflet pocket of FFAR1. Dose–response curves for compound 83 from this chemical series are shown together with the reference compounds TAK-875 and AM-5262 for wild-type (WT) and seven FFAR1 mutants. The best-performing FFAR1 structure is displayed with the corresponding docking poses for the active ligands used in the ALiBERO training set and the crystallographic pose (PDB ID 4PHU) of TAK-875. Hydrogen bonds are displayed as yellow spheres.
Based on the computational model of the ligand–receptor interactions, we conducted an experimental mutagenesis studies involving six key side chains in the extracellular binding site located between the extracellular ends of TM-I, -II, -VII, and ECL2. Our results indicated that mutation of five of the six residues (Y12, E65, A66, K259, L262) had a profound effect on the activity of compound 83, with the majority of them eliminating FFAR1 activation entirely (Figure 4), while one mutation (S8A) did affect the potency of compound 83 only slightly (∼10-fold decrease) and led to a 35% decrease in efficacy. As anticipated, the effect on the reference compound AM-5262, that has been shown to bind to a lipid-exposed inner leaflet pocket formed of TM-III, -IV, and -V as well as ICL2, was minimal, except for a minor decrease in efficacy for K259A (−32%) and the steric hindrance mutation L262W (−25%), but there wasa no effect on compound potency. The high level of FFAR1 activity induced by AM-5262 furthermore shows the ability of the mutant receptors to be fully activated via both the Gq and Gs signaling pathways. For comparison, we introduced a steric hindrance mutation (A83F) at the entry of the outer-leaflet pocket, in direct contact with TAK-875, but not with compound 83 in our computational model. While TAK-875’s activity was dramatically reduced (∼500-fold potency decrease), the mutation only had a minimal impact on the activity of compound 83 (∼2-fold potency decrease) and none on AM-5262. We noted that the activity of TAK-875 was affected by most of the mutations in the extracellular site, showing a certain dependence on their proximity to the TAK-875 binding site, with S8A having the smallest effect. We can therefore relate this observation to the close proximity and allosteric connectivity of the two binding pockets and consequential indirect effects on several important residues in TM-VII (R258) and ECL2 (E172) that are in direct contact with TAK-875. It has previously been described how rearrangement of these polar residues is critical in the activation process of FFAR1 by TAK-875,13 and it is conceivable that mutations in the extracellular site are disruptive to this mechanism.
In summary, the chemical series presented in this study represents a unique tool to modulate the activity of FFAR1 and FFAR4 and underlines the interesting pharmacological relation between these two receptors. Exhaustive investigation of the structure–activity relationship and mechanistic basis for activation have highlighted the optimization potential of chemotypes stemming from structure-based screening campaigns targeting previously unexploited binding sites.13 These dual-acting modulators could ultimately lead to new ways to overcome known problems related to the previously described lipophilic chemotypes associated with hepatotoxicity and β-cell toxicity, while potentially maintaining an increased insulin sensitization and control over glucose homeostasis. However, an in-depth analysis of the metabolic vulnerability of our chemical series will be required to translate the observed specific activity into a viable tool that can be used in in vivo studies.
Acknowledgments
The authors would like to thank Anja A. Petersen for performing the experimental compound screening and mutagenesis studies.
Glossary
Abbreviations
- GPCR
G protein-coupled receptor
- FFAR1
free fatty acid receptor 1
- FFAR4
free fatty acid receptor 4
- T2DM
type-2 diabetes mellitus
- TM
transmembrane helix
- PBS
phosphate-buffered saline
- SAR
structure–activity relationship
- ADME
absorption, distribution, metabolism and excretion
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsmedchemlett.2c00160.
Additional bioactivity and counterscreening data for selected compounds, computational parameters for the ALiBERO optimization, material and methods for biological assays, computational protocols, and compound synthesis and characterization (PDF)
Author Contributions
M.L., D.K., and T.M.F. prepared the manuscript. M.L. and T.M.F. designed the strategy for medicinal chemistry optimization and ADME studies. M.L., A.S., J.E.P., T.W.S., and T.M.F. designed the setup for experimental compound screening and mutagenesis. A.S., T.A.D.N., and J.E.P. performed experimental compound screening and mutagenesis. M.L., D.K., and T.M.F. conducted the analysis of the structure–activity relationship. M.L. and T.M.F. designed, conducted, and analyzed computational molecular dynamics simulations and molecular modeling studies.
The Novo Nordisk Foundation Center for Basic Metabolic Research is supported by an unconditional grant (NNF10CC1016515) from the Novo Nordisk Foundation to the University of Copenhagen. This study was further supported by a Proof of Concept (PoC) grant (#0058362) from the Novo Nordisk Foundation.
The authors declare no competing financial interest.
Supplementary Material
References
- Morgan N. G.; Dhayal S. G-Protein Coupled Receptors Mediating Long Chain Fatty Acid Signalling in the Pancreatic Beta-Cell. Biochem. Pharmacol. 2009, 78 (12), 1419–1427. 10.1016/j.bcp.2009.07.020. [DOI] [PubMed] [Google Scholar]
- Kaku K.; Katou M.; Igeta M.; Ohira T.; Sano H. Efficacy and Safety of Pioglitazone Added to Alogliptin in Japanese Patients with Type 2 Diabetes Mellitus: A Multicentre, Randomized, Double-Blind, Parallel-Group, Comparative Study. Diabetes, Obes. Metab. 2015, 17 (12), 1198–1201. 10.1111/dom.12555. [DOI] [PubMed] [Google Scholar]
- Li X.; Zhong K.; Guo Z.; Zhong D.; Chen X. Fasiglifam (TAK-875) Inhibits Hepatobiliary Transporters: A Possible Factor Contributing to Fasiglifam-Induced Liver Injury. Drug Metab. Dispos. 2015, 43 (11), 1751–1759. 10.1124/dmd.115.064121. [DOI] [PubMed] [Google Scholar]
- Li Z.; Qiu Q.; Geng X.; Yang J.; Huang W.; Qian H. Free Fatty Acid Receptor Agonists for the Treatment of Type 2 Diabetes: Drugs in Preclinical to Phase II Clinical Development. Expert Opin. Investig. Drugs 2016, 25 (8), 871–890. 10.1080/13543784.2016.1189530. [DOI] [PubMed] [Google Scholar]
- Li Z.; Zhou Z.; Zhang L. Current Status of GPR40/FFAR1Modulators in Medicinal Chemistry (2016–2019): A Patent Review. Expert Opin. Therapeutic Patents 2020, 30 (1), 27–38. 10.1080/13543776.2020.1698546. [DOI] [PubMed] [Google Scholar]
- Hauge M.; Vestmar M. A.; Husted A. S.; Ekberg J. P.; Wright M. J.; Di Salvo J.; Weinglass A. B.; Engelstoft M. S.; Madsen A. N.; Lückmann M.; Miller M. W.; Trujillo M. E.; Frimurer T. M.; Holst B.; Howard A. D.; Schwartz T. W. GPR40 (FFAR1) - Combined Gs and Gq Signaling in Vitro Is Associated with Robust Incretin Secretagogue Action Ex Vivo and in Vivo. Mol. Metab. 2015, 4 (1), 3–14. 10.1016/j.molmet.2014.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Satapati S.; Qian Y.; Wu M. S.; Petrov A.; Dai G.; Wang S. P.; Zhu Y.; Shen X.; Muise E. S.; Chen Y.; Zycband E.; Weinglass A.; Di Salvo J.; Debenham J. S.; Cox J. M.; Lan P.; Shah V.; Previs S. F.; Erion M.; Kelley D. E.; Wang L.; Howard A. D.; Shang J. GPR120 Suppresses Adipose Tissue Lipolysis and Synergizes with GPR40 in Antidiabetic Efficacy. J. Lipid Res. 2017, 58 (8), 1561–1578. 10.1194/jlr.M075044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christiansen E.; Watterson K. R.; Stocker C. J.; Sokol E.; Jenkins L.; Simon K.; Grundmann M.; Petersen R. K.; Wargent E. T.; Hudson B. D.; Kostenis E.; Ejsing C. S.; Cawthorne M. A.; Milligan G.; Ulven T. Activity of Dietary Fatty Acids on FFA1 and FFA4 and Characterisation of Pinolenic Acid as a Dual FFA1/FFA4 Agonist with Potential Effect against Metabolic Diseases. Br. J. Nutr. 2015, 113 (11), 1677–1688. 10.1017/S000711451500118X. [DOI] [PubMed] [Google Scholar]
- Ichimura A.; Hirasawa A.; Poulain-Godefroy O.; Bonnefond A.; Hara T.; Yengo L.; Kimura I.; Leloire A.; Liu N.; Iida K.; Choquet H.; Besnard P.; Lecoeur C.; Vivequin S.; Ayukawa K.; Takeuchi M.; Ozawa K.; Tauber M.; Maffeis C.; Morandi A.; Buzzetti R.; Elliott P.; Pouta A.; Jarvelin M. R.; Körner A.; Kiess W.; Pigeyre M.; Caiazzo R.; Van Hul W.; Van Gaal L.; Horber F.; Balkau B.; Lévy-Marchal C.; Rouskas K.; Kouvatsi A.; Hebebrand J.; Hinney A.; Scherag A.; Pattou F.; Meyre D.; Koshimizu T. A.; Wolowczuk I.; Tsujimoto G.; Froguel P. Dysfunction of Lipid Sensor GPR120 Leads to Obesity in Both Mouse and Human. Nature 2012, 483 (7389), 350–354. 10.1038/nature10798. [DOI] [PubMed] [Google Scholar]
- Oh D. Y.; Talukdar S.; Bae E. J.; Imamura T.; Morinaga H.; Fan W. Q.; Li P.; Lu W. J.; Watkins S. M.; Olefsky J. M. GPR120 Is an Omega-3 Fatty Acid Receptor Mediating Potent Anti-Inflammatory and Insulin Sensitizing Effects. Cell 2010, 142 (5), 687. 10.1016/j.cell.2010.07.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonnefond A.; Lamri A.; Leloire A.; Vaillant E.; Roussel R.; Lévy-Marchal C.; Weill J.; Galan P.; Hercberg S.; Ragot S.; Hadjadj S.; Charpentier G.; Balkau B.; Marre M.; Fumeron F.; Froguel P. Contribution of the Low-Frequency, Loss-of-Function p.R270H Mutation in FFAR4 (GPR120) to Increased Fasting Plasma Glucose Levels. J. Med. Genet. 2015, 52 (9), 595–598. 10.1136/jmedgenet-2015-103065. [DOI] [PubMed] [Google Scholar]
- Srivastava A.; Yano J.; Hirozane Y.; Kefala G.; Gruswitz F.; Snell G.; Lane W.; Ivetac A.; Aertgeerts K.; Nguyen J.; Jennings A.; Okada K. High-Resolution Structure of the Human GPR40 Receptor Bound to Allosteric Agonist TAK-875. Nature 2014, 513 (7516), 124–127. 10.1038/nature13494. [DOI] [PubMed] [Google Scholar]
- Lückmann M.; Trauelsen M.; Bentsen M. A.; Nissen T. A. D.; Martins J.; Fallah Z.; Nygaard M. M.; Papaleo E.; Lindorff-Larsen K.; Schwartz T. W.; Frimurer T. M. Molecular Dynamics-Guided Discovery of an Ago-Allosteric Modulator for GPR40/FFAR1. Proc. Natl. Acad. Sci. U. S. A. 2019, 116 (14), 7123–7128. 10.1073/pnas.1811066116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lückmann M.; Trauelsen M.; Frimurer T. M.; Schwartz T. W. Structural Basis for GPCR Signaling by Small Polar versus Large Lipid Metabolites-Discovery of Non-Metabolite Ligands. Curr. Opin. Cell Biol. 2020, 63, 38–48. 10.1016/j.ceb.2019.12.005. [DOI] [PubMed] [Google Scholar]
- Milligan G.; Shimpukade B.; Ulven T.; Hudson B. D. Complex Pharmacology of Free Fatty Acid Receptors. Chem. Rev. 2017, 117 (1), 67–110. 10.1021/acs.chemrev.6b00056. [DOI] [PubMed] [Google Scholar]
- Sundström L.; Myhre S.; Sundqvist M.; Ahnmark A.; McCoull W.; Raubo P.; Groombridge S. D.; Polla M.; Nyström A. C.; Kristensson L.; Någård M.; Winzell M. S. The Acute Glucose Lowering Effect of Specific GPR120 Activation in Mice Is Mainly Driven by Glucagon-like Peptide 1. PLoS One 2017, 12 (12), e0189060. 10.1371/journal.pone.0189060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rueda M.; Totrov M.; Abagyan R. ALiBERO: Evolving a Team of Complementary Pocket Conformations Rather than a Single Leader. J. Chem. Inf. Model. 2012, 52 (10), 2705–2714. 10.1021/ci3001088. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.