

Abstract
Purpose:
A lack of sufficient information exists for appropriately classifying a large number of myocilin (MYOC) variants, and their involvement in primary open angle glaucoma, hindering their definitive classification. Most glaucoma-causing MYOC mutations result in protein non-secretion and intracellular insoluble aggregate formation in cultured cells. Herein, we generated a Gaussia luciferase-based MYOC fusion protein to quickly and sensitively track the secretion of MYOC variants and compared these results to the better-established MYOC assay of western blotting.
Methods:
Fourteen clinically-derived MYOC variants with varying degrees of predicted pathogenicity were transfected into HEK-293A cells and analyzed by either a luciferase assay or western blotting.
Results:
Eight of the variants (G12R, V53A, T204T, P254L, T325T, D380H, D395_E396insDP, and P481S) had not been biochemically assessed previously. Off these, P254L and D395_E396insDP demonstrated significant secretion defects from reminiscent of glaucoma-causing mutations. The luciferase assay results agreed with western blotting for thirteen of the fourteen variants (93%), suggesting a strong concordance.
Conclusions:
These results suggest that the Gaussia luciferase assay may be used as a complementary or standalone assay for quickly assessing MYOC variant behavior and we anticipate that these results will be useful in MYOC variant curation and reclassification.
Keywords: myocilin, secretion, protein misfolding, glaucoma, luciferase assay
INTRODUCTION
Glaucoma is the leading cause of irreversible blindness worldwide, accounting for 11% of the total cases of vision loss1. Primary open angle glaucoma (POAG), the most common subset of glaucoma, is a chronic ocular hypertensive disease that triggers the death of retinal ganglion cells. The myocilin (MYOC) gene was the first gene to be associated with POAG, and its mutation is also the most common genetic cause of glaucoma2. Little is known regarding the normal function of the MYOC protein. Wild-type MYOC is a secreted, extracellular matrix protein that is expressed in the trabecular meshwork, which is a key tissue involved in regulating aqueous outflow and governance of intraocular pressure (IOP)3. Glaucoma-causing autosomal dominant mutations in MYOC lead to protein aggregation4–6, are poorly secreted6–9, linked to IOP elevation10,11, and trabecular meshwork death/dysfunction12,13. Yet, many times it is difficult to assess whether an indicated MYOC variant is truly pathogenic, and therefore a possible therapeutic target. Accordingly, the American College of Medical Genetics (ACMG) and the Association for Molecular Pathology (AMP) have developed recommendations for improving the consistency of clinical variant interpretation14, one aspect of which is to use a “well-established” functional assay to provide support for either a benign or pathogenic effect15. This evidence can then be potentially used to reclassify certain variants16.
Previously, we utilized the Gaussia luciferase (eGLuc2)17 to conveniently monitor the secretion of fibulin-3 variants18–20, and evaluate the potential effects of mutations on MYOC secretion7. In the latter publication, we compared the characteristics of WT and Y437H MYOC as FLAG and eGLuc2 fusions but did not extend a comparison to additional mutations. The eGLuc2 tag has no effect on MYOC secretion and the assay is inexpensive, easy to use, and convenient21. Yet, it has not been thoroughly vetted as a true functional assay for additional MYOC mutations through direct comparison to conventional MYOC biochemical assays (protein non-secretion/intracellular insoluble aggregation determined by western blotting)4–6,8,9,13,22. Herein, we sought to characterize fourteen mutations in MYOC that had previously not been tested as MYOC eGLuc2 fusion proteins and compare/contrast these results with the equivalent mutations generated as FLAG-tagged MYOC proteins and quantified by western blotting.
METHODS
General.
Additional, in-depth methods are included in the supplemental information due to space limitations.
Plasmid Generation.
The Q5 Site-Directed Mutagenesis Kit (New England Biolabs (NEB), Ipswich, MA, USA) was used to generate FLAG-tagged (FT) mutant constructs in the pcDNA3 vector. Gaussia luciferase FT MYOC variants were generated by DNA assembly (NEBuilder HiFi DNA Assembly Cloning Kit, NEB). Constructs were verified through full Sanger sequencing.
Cell Culture.
Human embryonic kidney cells (HEK-293A, Life Technologies) were cultured in Dulbecco’s modified Eagle’s Medium (DMEM; Corning, Corning, NY, USA) supplemented with fetal bovine serum (FBS, Omega Scientific, Tarzana, CA, USA) and penicillin/streptomycin/L-glutamine (Corning). Cells were grown at 37°C and 5% CO2.
Transfection.
HEK-239A cells were transfected using Lipofectamine 3000 (Life Technologies, Carlsbad, CA, USA). Twenty-four hours after transfection, media was then changed to low serum media to minimize FBS-related assay disruptions. Cells and media were then collected 24 h later used for either western blotting or a luciferase assay.
Gaussia luciferase assay.
Fifty μL of media was assayed using the NanoFuel® GLOW Assay (NanoLight Technology, Prolume, Pinetop, AZ) in a black 96 well plate (Corning). After a brief incubation, luminescence was measured with a BioTek Synergy 2 plate reader (BioTek, Winooski, VT, USA).
Preparation of Insoluble MYOC.
Transfected cells were lysed using PBS (Corning) supplemented with 0.1% Triton X-100 (Fisher, Waltham, MA, USA) protease inhibitor (Pierce Thermo Fisher, Rockford, IL, USA). The insoluble fraction was pelleted and washed. The soluble protein concentration (as determined via bicinchoninic acid assay (Pierce Thermo Fisher)) was used to determine the amount of 8 M urea (dissolved in PBS, Fisher) used to resuspend the insoluble protein pellet.
Western Blotting.
Media and insoluble samples and were denatured and loaded onto a 4–20%Tris-Gly SDS-PAGE gel (Life Technologies) and transferred to a nitrocellulose membrane. Blots were probed for total protein using Ponceau S (Fig. S1, Sigma), then blocked and probed with either a goat anti-MYOC antibody (Santa Cruz, Dallas, TX), or a rabbit anti-FLAG antibody (Thermo Fisher Scientific) followed by a near-infrared secondary antibody (LI-COR). Similar results were obtained with both primary antibodies. A LI-COR Odyssey CLx infrared scanner was used to image blots and its software was used to quantify protein bands.
RESULTS
Glaucoma-related mutations in MYOC cause protein misfolding, non-secretion/inefficient secretion, and an increase in Triton-X insoluble protein4–7,9,13,22. Many pathogenic MYOC mutations increase levels of endoplasmic reticulum (ER) stress10,11,23–25. Yet, the exact mechanism by which mutations cause higher IOP and glaucoma is unclear. Nonetheless pathogenic MYOC variants can be distinguished from benign mutants through biochemical characteristics4–6,8,9,13,22. In this work, we asked whether a simple Gaussia luciferase-based MYOC secretion assay could be used to complement or replace the more cumbersome western blotting approach for MYOC variants.
HEK-293A cells were transfected with sixteen constructs (Fig. 1A), including a WT MYOC eGLuc2 FT control and an established pathogenic control, Y437H MYOC eGLuc2 FT. The fourteen remaining variants were selected because they range in predicted behavior from ‘benign’ to ‘variant of uncertain significance (VUS)’ to ‘pathogenic’ (ClinVar (https://www.ncbi.nlm.nih.gov/clinvar/), MYOC database (www.myocilin.com)). To be more encompassing of the variants identified in the MYOC gene, we included two synonymous mutations, T204T (c.855G>T, identified in one POAG patient) and T325T (c.975G>A, identified in two controls)26. Importantly, we purposely included five mutants (Q48H, R76K, G367R, E396dup, and A445V) that had been characterized previously. Each of these MYOC eGLuc2 FT ‘calibrators’ displayed secretion profiles in accordance with past results (Fig. 1B), namely WT-like secretion of Q48H, R76K, and A445V and poor secretion of G367R and E396dup (Fig. 1B). Of the remaining previously uncharacterized MYOC variants (G12R, V53A, T204T, P254L, T325T, D380H, D395_E396insDP, and P481S), only P254L and D395_E396insDP demonstrated significant secretion defects using the Gaussia luciferase assay (Fig. 1B), hinting that these mutations are potentially pathogenic.
Figure 1. Testing new and existing luciferase-tagged MYOC variants.
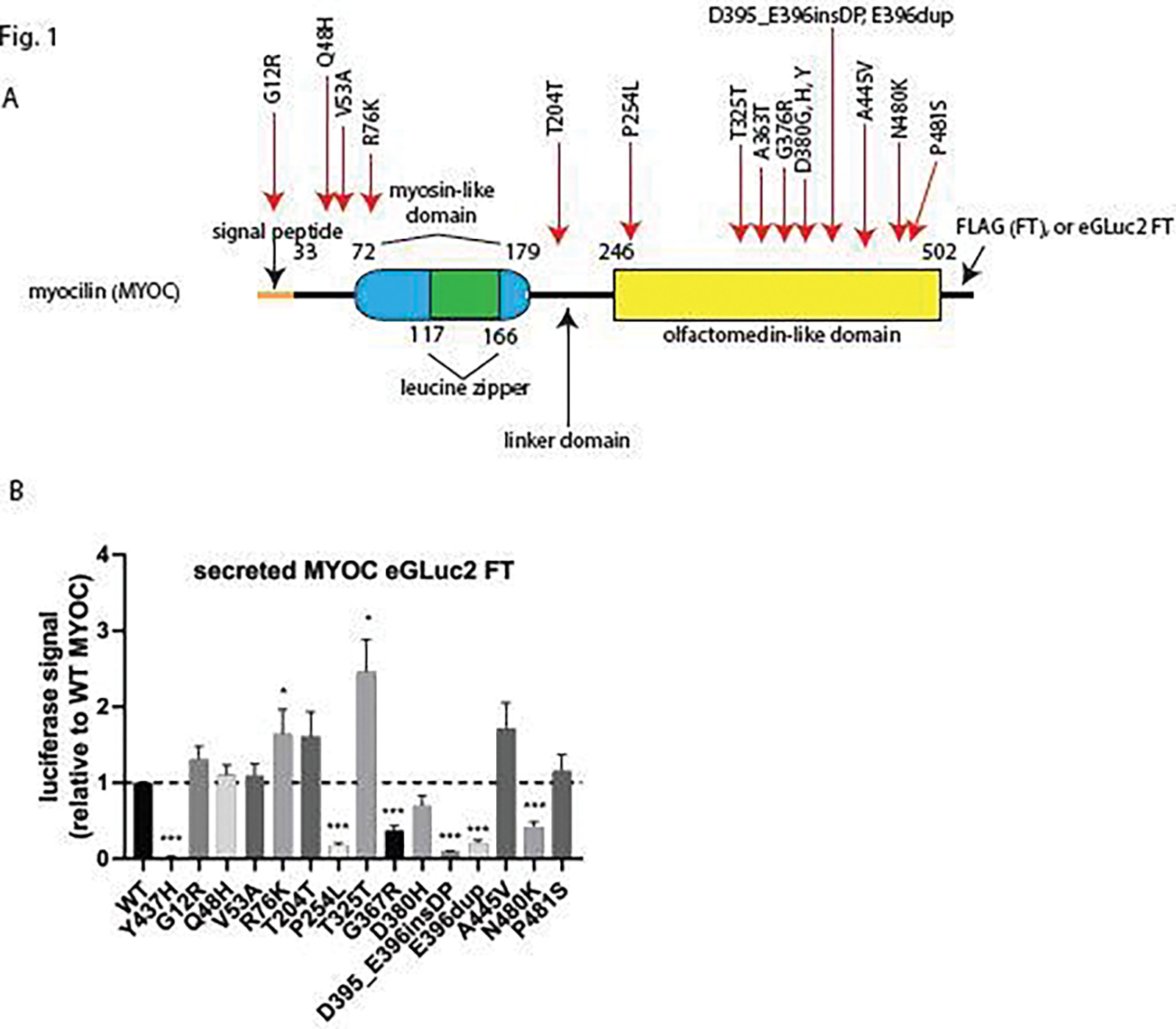
Screening of fourteen MYOC eGLuc2 FT variants demonstrates a range of secretion characteristics. (A) Cartoon schematic of MYOC and the generated mutations. (B) Secreted Gaussia luciferase (eGLuc2) luminescence results after transfection of the indicated constructs in HEK-293A. * = p < 0.05, ** = p < 0.01, *** = p < 0.001 using a 1 sample t-test against a hypothetical value of 1 (i.e., unchanged relative to WT). n ≥ 5 independent experiments.
In parallel, we performed western blotting on secreted (Fig. 2A, B) and insoluble (Fig. 2A, C) MYOC FT variants by conventional western blotting4,5,8,9,13,27. The Gaussia luciferase assay agreed with western blotting for thirteen of the fourteen variants tested (93%), excluding D380H (cf. Fig. 1B to Fig. 2B). MYOC FT variants that demonstrated significant secretion defects (P254L, G367R, D380H, D395_E396insDP, E396dup, and N480K) all had significantly higher insoluble intracellular levels (Fig. 2A, C). While the secreted data for nearly all variants were consistent across assays (and demonstrated a reasonable inter-assay linear correlation [Fig. S2A, B]), the P481S variant seemed to have a unique behavior of being secreted efficiently, but also having significantly higher insoluble levels as detected by western blotting (Fig. 2A–C). This behavior of partial solubility has also been demonstrated with other MYOC variants including G364V, T377M, D380A, and R422C4. Such an observation will likely complicate applying functional rules to determine whether its behavior supports benign or pathogenic classification.
Figure 2. Evaluation and comparison of MYOC variants by western blotting.
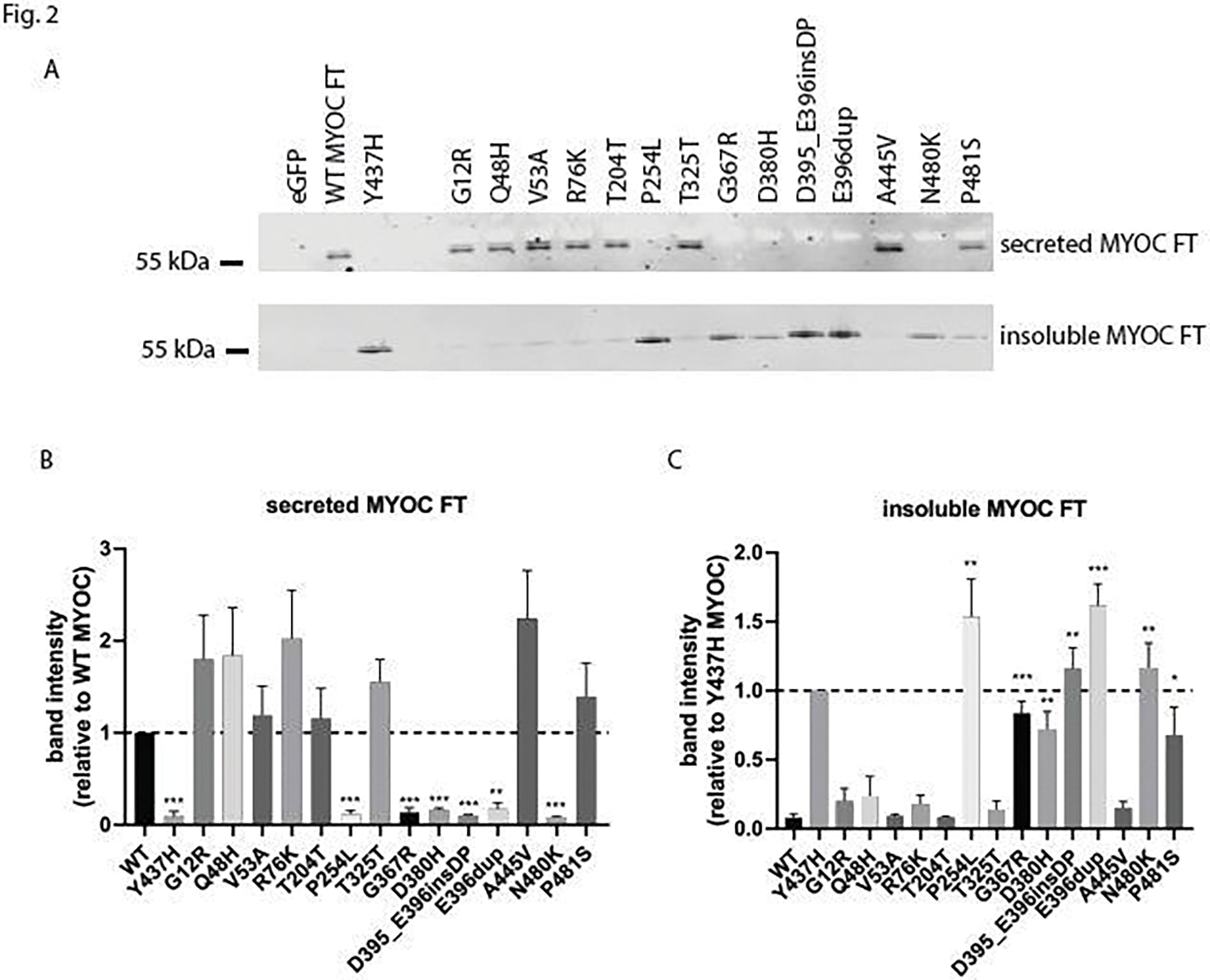
Parallel analysis of MYOC variants by western blotting demonstrates a strong concordance with the MYOC eGLuc2 FT results. (A) Western blotting results of secreted and insoluble intracellular levels of transfected MYOC FT constructs using a goat anti-MYOC antibody. (B, C) Quantification of secreted (B) and insoluble (C) MYOC FT levels from western blotting performed in (A). Because of the more reliable signal obtained from insoluble Y437H MYOC FT, insoluble levels of new variants were first compared to those of Y437H MYOC FT, and then we performed a significance test asking whether this obtained value was significantly different from WT MYOC FT insoluble levels. * = p < 0.05, ** = p < 0.01, *** = p < 0.001 using a 1 sample t-test against a hypothetical value of 1 (i.e., unchanged relative to WT MYOC for secreted) or a 2 sample t-test compared to WT MYOC for insoluble protein. n ≥ 4 independent experiments.
CONCLUSIONS
In summary, the MYOC Gaussia luciferase assay results agreed with secretion data from western blotting for thirteen of the fourteen variants (93%), suggesting that this assay could be used as a surrogate, or in addition to, conventional western blotting for MYOC. For example, the luciferase assay may be used as an initial screening method for large set of mutant MYOCs followed by confirmation of the behavior of select variants via western blotting. These studies also reveal some notable points. The first point highlights the importance of functional evidence for variant reclassification15, which is especially needed since almost half of all MYOC variants in ClinVar are classified as VUS. The second point is that we have demonstrated for the first time that the P254L28 and D395_E396insDP29 mutations appear to behave in a manner consistent with pathogenicity. The third point is that our results would suggest that the V53A, Q48H, and R76K variants are likely benign since they are readily secreted and soluble. These observations could clarify their ClinVar classification of “uncertain significance: and “conflicting interpretations of pathogenicity”, respectively. Fourthly, the discrepancy between the Gaussia luciferase fusion and FT version of D380H highlights the difficulty with applying one single criteria or assay to help in reclassification strategies based on function. This residue appears to be a hot-spot mutation involved in calcium binding since additional D380 mutations have been identified in patients30,31. Intriguingly, a recent study has shown that mutation of D380 can lower MYOC thermal stability, cause protein non-secretion and result in a stable, non-native structure32. However, previous studies indicate that the D380A mutant was partially soluble, suggesting a unique intermediate behavior4,5. Overall, these observations indicate the potential challenges in classification of certain MYOC variants that do not seem to abide by more common MYOC behaviors. Moving forward, co-transfection of an orthogonal reporter, such as a Cypridina luciferase33, could improve the rigor of Gaussia luciferase-based MYOC studies, helping discriminate between subtle secretion defects and possible slight alterations in transfection efficiency. Yet, in conclusion, our results suggest that the Gaussia luciferase assay maybe used as a complementary or standalone assay for quickly assessing MYOC variant behavior, and we predict that these observations will be useful in MYOC variant curation and reclassification.
Supplementary Material
ACKNOWLEDGEMENTS
The work described herein was supported by an endowment from the Roger and Dorothy Hirl Research Fund and a National Eye Institute Visual Science Core Grant (P30 EY030413).
Footnotes
Declarations of interest: none
DATA AVAILABILITY STATEMENT
The data that support the findings of this study are available from the corresponding author, JDH, upon reasonable request.
REFERENCES
- 1.Blindness GBD, Vision Impairment C, Vision Loss Expert Group of the Global Burden of Disease S. Trends in prevalence of blindness and distance and near vision impairment over 30 years: an analysis for the Global Burden of Disease Study. Lancet Glob Health 2021;9(2):e130–e143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Fingert JH. Primary open-angle glaucoma genes. Eye (Lond) 2011;25(5):587–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Tamm ER. The trabecular meshwork outflow pathways: structural and functional aspects. Exp Eye Res 2009;88(4):648–55. [DOI] [PubMed] [Google Scholar]
- 4.Zhou Z, Vollrath D. A cellular assay distinguishes normal and mutant TIGR/myocilin protein. Hum Mol Genet 1999;8(12):2221–8. [DOI] [PubMed] [Google Scholar]
- 5.Vollrath D, Liu Y. Temperature sensitive secretion of mutant myocilins. Exp Eye Res 2006;82(6):1030–6. [DOI] [PubMed] [Google Scholar]
- 6.Shimizu S, Lichter PR, Johnson AT, Zhou Z, Higashi M, Gottfredsdottir M, Othman M, Moroi SE, Rozsa FW, Schertzer RM and others. Age-dependent prevalence of mutations at the GLC1A locus in primary open-angle glaucoma. Am J Ophthalmol 2000;130(2):165–77. [DOI] [PubMed] [Google Scholar]
- 7.Zadoo S, Nguyen A, Zode G, Hulleman JD. A Novel Luciferase Assay For Sensitively Monitoring Myocilin Variants in Cell Culture. Invest Ophthalmol Vis Sci 2016;57(4):1939–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Jacobson N, Andrews M, Shepard AR, Nishimura D, Searby C, Fingert JH, Hageman G, Mullins R, Davidson BL, Kwon YH and others. Non-secretion of mutant proteins of the glaucoma gene myocilin in cultured trabecular meshwork cells and in aqueous humor. Hum Mol Genet 2001;10(2):117–25. [DOI] [PubMed] [Google Scholar]
- 9.Gobeil S, Letartre L, Raymond V. Functional analysis of the glaucoma-causing TIGR/myocilin protein: integrity of amino-terminal coiled-coil regions and olfactomedin homology domain is essential for extracellular adhesion and secretion. Exp Eye Res 2006;82(6):1017–29. [DOI] [PubMed] [Google Scholar]
- 10.Zode GS, Bugge KE, Mohan K, Grozdanic SD, Peters JC, Koehn DR, Anderson MG, Kardon RH, Stone EM, Sheffield VC. Topical ocular sodium 4-phenylbutyrate rescues glaucoma in a myocilin mouse model of primary open-angle glaucoma. Invest Ophthalmol Vis Sci 2012;53(3):1557–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zode GS, Kuehn MH, Nishimura DY, Searby CC, Mohan K, Grozdanic SD, Bugge K, Anderson MG, Clark AF, Stone EM and others. Reduction of ER stress via a chemical chaperone prevents disease phenotypes in a mouse model of primary open angle glaucoma. J Clin Invest 2011;121(9):3542–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kwon YH, Fingert JH, Kuehn MH, Alward WL. Primary open-angle glaucoma. N Engl J Med 2009;360(11):1113–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Liu Y, Vollrath D. Reversal of mutant myocilin non-secretion and cell killing: implications for glaucoma. Hum Mol Genet 2004;13(11):1193–204. [DOI] [PubMed] [Google Scholar]
- 14.Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, Grody WW, Hegde M, Lyon E, Spector E and others. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med 2015;17(5):405–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Brnich SE, Abou Tayoun AN, Couch FJ, Cutting GR, Greenblatt MS, Heinen CD, Kanavy DM, Luo X, McNulty SM, Starita LM and others. Recommendations for application of the functional evidence PS3/BS3 criterion using the ACMG/AMP sequence variant interpretation framework. Genome Med 2019;12(1):3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Brnich SE, Rivera-Munoz EA, Berg JS. Quantifying the potential of functional evidence to reclassify variants of uncertain significance in the categorical and Bayesian interpretation frameworks. Hum Mutat 2018;39(11):1531–1541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Tannous BA. Gaussia luciferase reporter assay for monitoring biological processes in culture and in vivo. Nat Protoc 2009;4(4):582–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hulleman JD, Balch WE, Kelly JW. Translational attenuation differentially alters the fate of disease-associated fibulin proteins. FASEB J 2012;26(11):4548–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hulleman JD, Kaushal S, Balch WE, Kelly JW. Compromised mutant EFEMP1 secretion associated with macular dystrophy remedied by proteostasis network alteration. Mol Biol Cell 2011;22(24):4765–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hulleman JD, Brown SJ, Rosen H, Kelly JW. A high-throughput cell-based Gaussia luciferase reporter assay for identifying modulators of fibulin-3 secretion. J Biomol Screen 2013;18(6):647–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Maguire CA, Deliolanis NC, Pike L, Niers JM, Tjon-Kon-Fat LA, Sena-Esteves M, Tannous BA. Gaussia luciferase variant for high-throughput functional screening applications. Anal Chem 2009;81(16):7102–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Gobeil S, Rodrigue MA, Moisan S, Nguyen TD, Polansky JR, Morissette J, Raymond V. Intracellular sequestration of hetero-oligomers formed by wild-type and glaucoma-causing myocilin mutants. Invest Ophthalmol Vis Sci 2004;45(10):3560–7. [DOI] [PubMed] [Google Scholar]
- 23.Stothert AR, Fontaine SN, Sabbagh JJ, Dickey CA. Targeting the ER-autophagy system in the trabecular meshwork to treat glaucoma. Exp Eye Res 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Suntharalingam A, Abisambra JF, O’Leary JC, 3rd, Koren J, 3rd, Zhang B, Joe MK, Blair LJ, Hill SE, Jinwal UK, Cockman M and others. Glucose-regulated protein 94 triage of mutant myocilin through endoplasmic reticulum-associated degradation subverts a more efficient autophagic clearance mechanism. J Biol Chem 2012;287(48):40661–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Yam GH, Gaplovska-Kysela K, Zuber C, Roth J. Sodium 4-phenylbutyrate acts as a chemical chaperone on misfolded myocilin to rescue cells from endoplasmic reticulum stress and apoptosis. Invest Ophthalmol Vis Sci 2007;48(4):1683–90. [DOI] [PubMed] [Google Scholar]
- 26.Melki R, Belmouden A, Brezin A, Garchon HJ. Myocilin analysis by DHPLC in French POAG patients: increased prevalence of Q368X mutation. Hum Mutat 2003;22(2):179. [DOI] [PubMed] [Google Scholar]
- 27.Izumi K, Mashima Y, Obazawa M, Ohtake Y, Tanino T, Miyata H, Zhang Q, Oguchi Y, Tanaka Y, Iwata T. Variants of the myocilin gene in Japanese patients with normal-tension glaucoma. Ophthalmic Res 2003;35(6):345–50. [DOI] [PubMed] [Google Scholar]
- 28.Souzeau E, Burdon KP, Ridge B, Dubowsky A, Ruddle JB, Craig JE. A novel de novo Myocilin variant in a patient with sporadic juvenile open angle glaucoma. BMC Med Genet 2016;17:30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Braghini CA, Neshich IA, Neshich G, Soardi FC, de Mello MP, Costa VP, de Vasconcellos JP, de Melo MB. New mutation in the myocilin gene segregates with juvenile-onset open-angle glaucoma in a Brazilian family. Gene 2013;523(1):50–7. [DOI] [PubMed] [Google Scholar]
- 30.Campos-Mollo E, Sanchez-Sanchez F, Lopez-Garrido MP, Lopez-Sanchez E, Lopez-Martinez F, Escribano J. MYOC gene mutations in Spanish patients with autosomal dominant primary open-angle glaucoma: a founder effect in southeast Spain. Mol Vis 2007;13:1666–73. [PubMed] [Google Scholar]
- 31.Wirtz MK, Samples JR, Choi D, Gaudette ND. Clinical features associated with an Asp380His Myocilin mutation in a US family with primary open-angle glaucoma. Am J Ophthalmol 2007;144(1):75–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hill SE, Kwon MS, Martin MD, Suntharalingam A, Hazel A, Dickey CA, Gumbart JC, Lieberman RL. Stable calcium-free myocilin olfactomedin domain variants reveal challenges in differentiating between benign and glaucoma-causing mutations. J Biol Chem 2019;294(34):12717–12728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hulleman JD, Kaushal S, Balch WE, Kelly JW. Compromised mutant EFEMP1 secretion associated with macular dystrophy remedied by proteostasis network alteration. Molecular biology of the cell 2011;22(24):4765–4775. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data that support the findings of this study are available from the corresponding author, JDH, upon reasonable request.