

Graphical abstract
Keywords: Fluoropolymer, mRNA Delivery, Antigen presentation, Personalized mRNA cancer vaccine, Cancer immunotherapy
Abstract
Messenger RNA (mRNA) vaccines, while demonstrating great successes in the fight against COVID-19, have been extensively studied in other areas such as personalized cancer immunotherapy based on tumor neoantigens. In addition to the design of mRNA sequences and modifications, the delivery carriers are also critical in the development of mRNA vaccines. In this work, we synthesized fluoroalkane-grafted polyethylenimine (F-PEI) for mRNA delivery. Such F-PEI could promote intracellular delivery of mRNA and activate the Toll-like receptor 4 (TLR4)-mediated signaling pathway. The nanovaccine formed by self-assembly of F-PEI and the tumor antigen-encoding mRNA, without additional adjuvants, could induce the maturation of dendritic cells (DCs) and trigger efficient antigen presentation, thereby eliciting anti-tumor immune responses. Using the mRNA encoding the model antigen ovalbumin (mRNAOVA), our F-PEI-based mRNAOVA cancer vaccine could delay the growth of established B16-OVA melanoma. When combined with immune checkpoint blockade therapy, the F-PEI-based MC38 neoantigen mRNA cancer vaccine was able to suppress established MC38 colon cancer and prevent tumor reoccurrence. Our work presents a new tool for mRNA delivery, promising not only for personalized cancer vaccines but also for other mRNA-based immunotherapies.
1. Introduction
Cancer vaccines, which trigger tumor-specific cell-mediated immunity to recognize and kill cancer cells, are one of the most interesting approaches in cancer immunotherapy [1], [2], [3]. mRNA-based vaccines are a promising vaccine platform for several reasons [4], [5]. Firstly, mRNA is delivered into the cytosol for rapid production of large quantity of endogenous protein or peptide antigens, which are processed by the proteasome and presented to the cell surface by the major histocompatibility complex class I (MHC I) to activate CD8+ T cells that are thought to play a key role in the antitumor immunity [6], [7], [8], whereas exogenous peptide/protein antigens are mainly been processed in the lysosome and presented via MHC II, inducing humoral immunity [9]. Therefore, compared with peptide/protein vaccines, mRNA vaccines may induce stronger cellular immune responses for antitumor immunity. Secondly, compared with protein antigens, mRNA antigens have stronger immunogenicity and possess intrinsic adjuvant properties, which could further enhance immune responses [10]. Thirdly, compared with DNA vaccines, mRNA vaccines only need to be internalized into the cytoplasm for translation, without raising the risk of gene integration or other safety issues [11].
However, due to the abundance of RNases and the difficulty of mRNA molecules in entering cells, biocompatible delivery carriers that can improve the mRNA stability and transport mRNA into antigen-presenting cells (APCs) are essential in the development of mRNA vaccines [12], [13], [14]. Currently, lipid nanoparticles (LNPs) have demonstrated great successes as the delivery system in the fabrication of mRNA vaccines [15], [16], [17]. However, LNPs usually are composed by a variety of different lipid components with rather complicated compositions. Moreover, state-of-art microfluidic devices are usually required in the fabrication processes to produce LNP-based mRNA vaccines [18]. Developing new mRNA delivery carriers with simple composition and easy preparation process would still be of great interests for mRNA-based biotechnology.
An effective mRNA delivery carrier should be able to pack and protect the mRNA from enzymatic degradation, to transport the mRNA either directly into the cytosol or via escaping from the lysosome, and finally to release the mRNA cargo to the cellular translation machinery. Therefore, both the affinity of the carrier towards the mRNA cargo and the interaction(s) of the carrier with the target cell would contribute to its delivery efficiency. Fluorine-containing amphiphiles have been reported to show promising gene and protein delivery effects [19], [20], [21], [22]. Fluorinated compounds with both hydrophobic and lipophobic features show a high tendency of phase separation in both polar and non-polar environments [23], [24], enabling their penetration across the lipid bilayer of cell membranes as well as endosomal/lysosomal membranes [25], [26]. In our previous work, we reported that a fluoroalkane modified polyethylenimine (PEI) with a molecular weight of 25 kDa (F-PEI25 kDa), while serving as an agonist for the Toll-like receptor 4 (TLR4)-mediated signaling pathway, could self-assemble with protein or peptide antigens to form nanovaccines without the need of additional adjuvants [27]. However, regular PEI with high molecular weight possesses certain cytotoxicity, thus limiting its potential in bio-applications [28], [29].
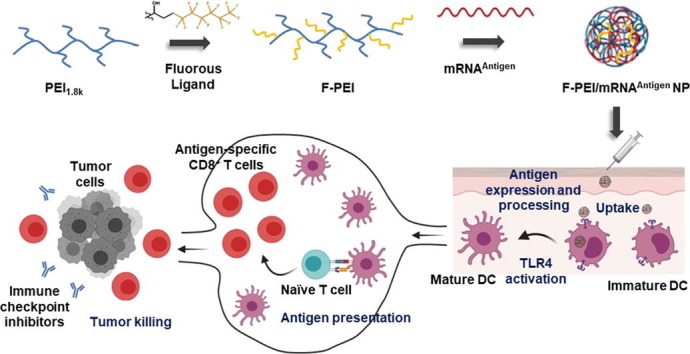
Here, a low molecular weight (1.8 kDa) PEI with low cytotoxicity was chosen for further optimization in this study. Two fluoroalkane-grafted PEI polymers (F-PEI1.8 kDa) were synthesized for efficient mRNA delivery and TLR4 activation. By simply mixing F-PEI with the mRNA encoding tumor antigens (Ag), without additional adjuvants, we obtained F-PEI/mRNAAg nanovaccines which could promote high level of dendritic cells (DCs) activation and MHC I antigen processing and presentation by APCs, subsequently inducing antigen-specific CD8+ T cell immune responses to effectively inhibit the growth of established B16-OVA melanoma tumors. We further demonstrated that using the mRNA encoding neoantigens of MC38 tumors, F-PEI-based personalized nanovaccine, in combination with the immune checkpoint inhibitors, could eradicate established tumors (Fig. 1 ). This work presents a new type of mRNA delivery vector that may be of great interests to the development of mRNA-based personalized cancer vaccines as well as other mRNA-based immunotherapies.
Fig. 1.

Design of the F-PEI/mRNA nanovaccine platform for cancer treatment. (a) Schematic illustration showing the preparation of F-PEI/mRNA nanovaccine. F-PEI was synthesized by grafting fluorine ligands on PEI (MW 1.8 kDa), and then mixed with the mRNA encoding antigen (Ag) to form F-PEI-based mRNA nanovaccine (F-PEI/mRNAAg). (b) After administration, F-PEI/mRNAAg nanovaccine is taken up by dendritic cells (DCs), to promote antigen presentation and maturation of DCs. Then activated DCs would migrate to the draining lymph nodes, triggering robust antigen-specific CD8+ T cell responses. Activated CD8+ T cells would then recognize and kill target cancer cells and exert powerful antitumor efficacy. Combining with immune checkpoint inhibitors would further enhance the efficacy of F-PEI-based mRNA nanovaccine to eliminate established tumors. Ag, antigen. i.d., intradermal.
2. Experimental
2.1. Materials
Branched polyethylenimine (MW 1.8 kDa) was purchased from Sigma-Aldrich (St. Louis, MO). 3-(perfluorohex-1-yl)-1,2-propenoxide was purchased from J&K Scientific (Shanghai, China). Firefly-luciferase and OVA expression mRNAs were obtained from Trilink. MC38 neoantigen expression mRNA was commissioned to construct by Stemirna (Shanghai, China). The firefly-luciferase mRNA labeled with the fluorophore Cy5 was obtained from APExBIO. Lipofectamine MessengerMAX was purchased from Invitrogen.
2.2. Animals and cells
Female C57BL/6 mice (6–8 weeks) were purchased from Nanjing Pengsheng Biological Technology Co., ltd. Female OT-I transgenic mice (6–8 weeks) were a kind gift from Prof. Xuefeng Wang, Soochow University. All animal experiments were performed according to the guidelines for the protection of animal life and protocols approved by Laboratory Animal Ethics Committee in Soochow University. B16-OVA cells (a gift from Prof. Yuhui Huang, Soochow University), RAW264.7 cells (obtained from American Type Culture Collection) were cultured in Dulbecco’s modifed Eagle medium (DMEM) with 10 % fetal bovine serum (FBS) and 1 % penicillin sulfate and streptomycin (PS) at 37 °C in 5 % CO2. DC2.4 cells (a gift from Prof. Chao Wang, Soochow University), MC38 cells (obtained from American Type Culture Collection) were cultured in Roswell Park Memorial Institute (RPMI) 1640 medium 10 % FBS and 1 % PS at 37 °C in 5 % CO2. HEK-Dual mTLR4 (NF/IL8) cells (obtained from InvivoGen) were cultured in DMEM with 10 % FBS and 1 % PS supplemented with 100 μg mL−1 Hygromycin B Gold (InvivoGen) and 50 μg mL−1 Zeocin (InvivoGen) at 37 °C in 5 % CO2.
2.3. Synthesis of F-PEI
Epoxides of fluoroalkanes were added dropwisely into PEI1.8k in methanol at different molar ratios (75:1 or 100:1). The mixture was stirred at room temperature for 48 h, then the products were purified by intensive dialysis against methanol and double distilled water (molecular weight cut off 1000 Da). The products were collected and lyophilized under vacuum to obtain F-PEI.
2.4. In vitro cytotoxicity assessment
DC2.4 cells or RAW264.7 macrophages were seeded into 96-well plates at 5 × 104 cells per well and incubated with different concentrations of F-PEI or PEI for 24 h, and relative cell viability was measured by standard MTT assay.
2.5. Nucleic acid condensation ability assay
Agarose Gel Electrophoresis (AGE) was used to verify the ability of F-PEI to condense mRNA. F-PEI/mRNA complexes at various w/w ratios (from 0.25 to 2) were prepared, added with 2 × RNA loading dye (Solarbio), heat-treated at 65 °C for 8 min, loaded onto a 1 % agarose denaturing gel containing GelRed dye (Beyotime), and then electrophoresed at 125 V for 30 min. The electrophoretic band was imaged by using Amersham Imager 600 UV System.
2.6. Characterization of F-PEI/mRNA
The synthesized F-PEI was mixed with mRNA in deionized water at a mass ratio of 1:1 for 15 min. The size and zeta potential of the formed F-PEI/mRNA NPs were measured using a Zetasizer Nano ZS (Malvern Instruments). The morphology of the NPs was observed with a FEI TF20 transmission electron microscope.
2.7. In vivo mRNA expression assay
The F-PEI/mRNALuc (10 μg mRNALuc per mouse) was intradermally injected at the tail base of each C57BL/6 mouse, followed by intraperitoneal injection of 0.2 mL of d-luciferin (1.5 μg mL−1) after 6 h and 24 h. After 15 min of reaction, the mice were subjected to bioluminescence assays using IVIS Kinetic Imaging System (Perkin Elmer).
2.8. BMDC activation and antigen presentation
For in vitro DC maturation experiments, BMDCs were plated at 106 cells per well in a 24-well plate and incubated with mRNAOVA, F-PEI/mRNAOVA or Lipofectamine MessengerMAX/mRNAOVA (mRNAOVA = 3 μg mL−1, the w/w ratio of material and mRNAOVA was 1:1). After 24 h incubation, BMDCs were harvested and washed with FACS buffer (1 % FBS in PBS), and incubated with anti-CD16/32 at 4 °C, then stained with anti-CD11c-FITC, anti-CD80-APC and anti-CD86-PE for DC maturation analysis, or anti-CD11c-APC and anti-SIINFEKL/H-2Kb-PE for antigen presentation analysis.
2.9. In vitro mRNA cellular uptake
To assess the cellular uptake of mRNA by BMDCs, 106 BMDCs were plated in 24-well plate and incubated with Cy5-labelled mRNALuc (Cy5-mRNALuc), F13-PEI1.8k-1/Cy5-mRNALuc, F13-PEI1.8k-2/Cy5-mRNALuc for 6 h (Cy5-mRNALuc = 1 μg mL−1, The w/w ratio of material and mRNALuc was 1:1). BMDCs were harvested and added with trypan blue to quench the extracellular fluorescence. After washing for three times, BMDCs were incubated with anti-CD16/32 for 15 min at 4 °C before being stained with anti-CD11c-FITC, and then analyzed using flow cytometer (BD Accurit C6 Plus).
2.10. In vitro CD8+ T-cell priming assay
BMDCs were incubated with F-PEI/mRNAOVA NPs. Then the treated BMDCs were washed by PBS containing 0.1 % BSA. CD8+ T lymphocytes were negatively selected from the spleen of OT-I mice by magnetic separation (MACS system, Miltenyi Biotec) according to the manufacturer's instructions and stained with the CellTrace CFSE Cell Proliferation Kit (Invitrogen) according to the experimental protocol. The treated BMDCs (105 mL−1) were then mixed with CFSE-stained splenic CD8+ T cells at a ratio of 1:10, and incubated in round-bottom 96-well plates (Beyotime) for 72 h. Cells were washed by FACS buffer, before being incubated with anti-CD16/32 at 4 °C then stained with anti-CD3-PerCP for flow cytometer measurement.
2.11. Detection of cytokine
The supernatant after incubation in CD8+ T-cell priming assay was collected, and the secretion level of IFN-γ in the cell supernatant was detected by enzyme-linked immunosorbent assay (ELISA). The experimental steps were performed according to the instructions of the ELISA kit (Invitrogen).
2.12. Lymph node analysis
C57BL/6 mice were immunized with PBS, mRNAOVA or F-PEI/mRNAOVA by intradermal injection on Day 0. The injected dose of mRNAOVA per mouse was 10 μg. On Day 3 after mouse immunization, we assessed DC maturation and antigen presentation in mouse inguinal lymph nodes (LNs). LNs from immunized mice were treated by mechanical disruption, then filtered through 300-mesh nylon mesh to obtain single-cell suspensions, which were incubated with anti-CD16/32 for 15 min at 4 °C before being stained with anti-CD11c-FITC, anti-CD86-PE and anti-CD80-APC for DC maturation analysis, or anti-CD11c-APC and anti-SIINFEKL/H-2 Kb-PE for antigen presentation analysis.
2.13. Tetramer analysis and peptide re-stimulation of splenocytes
C57BL/6 mice were intradermally immunized with PBS, mRNAOVA or F-PEI/mRNAOVA on Day 0 and 7. On Day 14, the spleens of the immunized mice were minced and filtered with a 300 mesh cell strainer to obtain splenocytes, and then Red blood cells (RBCs) were lysed with RBC lysis buffer (Beyotime). Splenocytes were stained with PE-labelled SIINFEKL-MHC I tetramer, anti-CD8-APC and anti-CD3-FITC, then analyzed the percentage of OVA-specific CD8+ T cells using the tetramer staining assay following the standard protocol.
For enzyme linked immunospot assay (ELISPOT) analysis of IFN-γ spot-forming cells (BD Biosciences), 5 × 105 splenocytes were plated in each well and incubated with 10 μg mL−1 OVA257–264 peptide (SIINFEKL), Reps1 peptide (AQLANDVVL), Dpagt1 peptide (SIIVFNLL) or Adpgk peptide (ASMTNMELM), respectively. The plates were kept at 37 °C in 5 % CO2 for 24 h. Following the experimental protocol, an automated ELISPOT Plate Reader (AID iSpot) was used to determine the amount of IFN-γ spot-forming cells and the data were presented as spot-forming cells per half million cells.
To demonstrate the cellular responses, 106 immunized mice splenocytes were incubated with 10 μg mL−1 OVA257-264 peptide (SIINFEKL) in the medium containing brefeldin A inhibitor and monensin inhibitor for 6 h. Cells were incubated with anti-CD16/32 antibody at 4 °C for 15 min, and then stained with anti-CD3e-FITC and anti-CD8a-APC at 4 °C for 30 min. Then the cells were stained anti-IFN-γ-PE according to the intracellular staining protocol before the flow cytometry measurement.
2.14. Hematoxylin and eosin (H&E) staining
C57BL/6 mice were intradermally treated two times with F13-PEI1.8k-1/mRNAOVA or F13-PEI1.8k-2/mRNAOVA at 1 week intervals. Mice were euthanized and collected major organs on Day 1, 7 and 21 after two vaccination. The major organs were fixed in 4 % paraformaldehyde solution, and then sectioned for H&E staining.
2.15. Treatment of the B16-OVA tumor model
C57BL/6 mice were subcutaneously inoculated with 5 × 105 B16-OVA cells on Day 0. Mice were intradermally treated with PBS, mRNAOVA or F-PEI/mRNAOVA on Day 4 and 11. The injected dose of mRNAOVA per mouse was 10 μg. The tumor growth were regularly measured and recorded: tumor volume = length × width × width/2. Mice were euthanized when the tumor volume reached 1500 mm3.
2.16. Neoantigen mRNA vaccine combined with immune checkpoint inhibitor for tumor therapy
C57BL/6 mice were subcutaneously injected with 106 MC38 colon cancer cells on Day 0. Tumor-bearing mice were randomly divided into 4 groups and treated with different treatments: (1) PBS, (2) anti-PD-1, (3) F13-PEI1.8k-1/mRNAMC38, (4) F13-PEI1.8k-1/mRNAMC38 + anti-PD-1. Mice were intradermally immunized with F13-PEI1.8k-1/mRNAMC38 on Days 8 and 15. The injected dose of mRNAMC38 per mouse was 10 μg. On Day 9 and 16, mice were intravenously injected with anti-PD-1 (20 μg per mouse). On Day 12 and 19, mice were intravenously injected with anti-PD-1 (10 μg per mouse). Mice were euthanized when the tumor volume reached 1000 mm3.
For tumor rechallenge experiment, tumor-eliminated mice treated with neoantigen mRNA nanovaccine combined with immune checkpoint inhibitors were subcutaneously injected with 5 × 105 MC38 cells on Day 60. The tumor growth was monitored and recorded to evaluate the immune memory effect.
2.17. CD8+ T cell depletion
Each C57BL/6 mouse was subcutaneously injected with 106 MC38 cells on Day 0. The tumor-bearing mice were randomly divided into 4 groups and treated with different methods: (1) mouse-IgG (as Control, Southern Biotech), (2) anti-mouse-CD8a (BioXcell), (3) F13-PEI1.8k-1/mRNAMC38 + anti-PD-1 + anti-mouse-CD8a, (4) F13-PEI1.8k-1/mRNAMC38 + anti-PD-1 + mouse-IgG. On Day 7, 10 and 13, mice were intravenously injected with anti-mouse-CD8a or mouse-IgG antibody (20 μg per mouse). On Day 8, mice were intradermally immunized with F13-PEI1.8k-1/mRNAMC38. On Day 9, mice were intravenously injected with anti-PD-1 (20 μg per mouse). On Day 12, mice were intravenously injected with anti-PD-1 (10 μg per mouse). The peripheral blood of mice was collected on Day 10, and the depletion of CD8+ T cells was detected by flow cytometry.
3. Results and discussion
3.1. Characterization and the mRNA delivery efficiency of fluoropolymers
Branched PEI (MW 1.8 kDa) was grafted with fluoroalkanes (3-(perfluoro-n-hexyl)-1,2-propenoxide, F13) via amine-epoxide reaction [22]. The F13 ligand was mixed with PEI at 2 different feed ratios: 75:1, 100:1, and the obtained materials were named as F13-PEI1.8k-1 and F13-PEI1.8k-2 with the fluorine contents of 35.98 % and 40.36 %, respectively.
Firstly, we verified the ability of F-PEI to condense nucleic acids by agarose gel electrophoresis. The two F-PEIs were simply mixed with mRNA at increasing w/w ratios. Both F13-PEI1.8k-1 and F13-PEI1.8k-2 could successfully encapsulate mRNA at a very low ratio to prevent its leakage by forming particles (Fig. 2 a), demonstrated by the disappearance of the free mRNA band. The cytotoxicity of F-PEIs was also explored. Cell viability was analyzed after incubating RAW264.7 macrophages and DC2.4 cells with the two F-PEIs or bare PEI1.8k. Both F-PEIs showed comparable low cytotoxicity with bare PEI1.8k (Supplementary Fig. S1), demonstrating that the fluorine modification did not affect the biocompatibility. Considering no obvious toxicity could be observed at 10 μg mL−1 (Supplementary Fig. S1), the w/w ratio of F13-PEI1.8k-1/mRNA and F13-PEI1.8k-2/mRNA was set at 1:1 accordingly giving a working concentration of 3–10 μg mL−1. At this ratio, the particle sizes were both about 280 nm in hydrodynamic diameter (Fig. 2b) with spherical shape under TEM (Fig. 2c), and their zeta potentials were approximately −6.5 mV and −12.3 mV, respectively (Fig. 2d).
Fig. 2.

Characterization and mRNA delivery efficiency of F-PEI. (a) Agarose gel electrophoresis of F-PEI/mRNA at different w/w ratios. (b) Dynamic light scattering analysis of F-PEI/mRNA. (c) Transmission electron microscopy imaging shown F13-PEI1.8k-1/mRNA and F13-PEI1.8k-2/mRNA. Scale bar = 200 nm. (d) Zeta potentials of the F13-PEI1.8k-1, F13-PEI1.8k-2, mRNA, F13-PEI1.8k-1/mRNA and F13-PEI1.8k-2/mRNA. (e&f) Representative flow cytometry plots (e) and the mean fluorescence intensity (MFI) of Cy5 (f) in BMDCs incubated with F-PEI/Cy5-mRNALuc for 6 h. (g&h) F-PEI/mRNALuc (10 μg mRNALuc per mouse) was intradermally injected at the tail base of mice to evaluate the in vivo mRNA delivery efficiency, and the bioluminescence images (g) and statistical data (h) were recorded at 6 h and 24 h post injection. d,f, The data show mean ± standard deviation (n = 3). h, The data show mean ± standard error of mean (n = 3 mice per group). f,h, Statistical significance between the indicated groups was determined using two-sided unpaired t-tests. **P < 0.01, ***P < 0.001.
We next investigated the mRNA cellular delivery efficiency of F-PEIs in vitro. mRNA encoding firefly luciferase (mRNALuc) was labeled with Cyanine 5 (Cy5) fluorescent dye (Cy5-mRNALuc) for intracellular tracking. Compared with free Cy5-mRNALuc, both F13-PEI1.8k-1/Cy5-mRNALuc and F13-PEI1.8k-2/Cy5-mRNALuc promoted mRNA uptake by mouse bone marrow-derived dendritic cells (BMDCs) (Fig. 2e & 2f), indicating that F13-PEI1.8k-1 and F13-PEI1.8k-2 could significantly improve the mRNA entry efficiency, and the mRNA delivery efficiency of F13-PEI1.8k-1 was significantly higher than that of F13-PEI1.8k-2.
The mRNA delivery efficacy of F-PEI was also explored in vivo. F-PEI/mRNALuc was intradermally injected at the tail base of mice, and the bioluminescence intensities at the injection site were evaluated after intraperitoneal injection of d-luciferin. The expression of luciferase was recorded at 6 h and 24 h post injection. Based on the bioluminescence intensities, it was found that F13-PEI1.8k-1 showed better in vivo mRNA delivery efficiency than F13-PEI1.8k-2 (Fig. 2g & 2 h), consistent with their abilities of mRNA packing and cellular delivery.
3.2. DC activation and OVA-specific T cell immune responses
The maturation of DCs was essential for antigen presentation and subsequent initiation of T cell immune responses (Fig. 3 a). BMDCs were pulsed with mRNAOVA, F13-PEI1.8k-1/mRNAOVA, F13-PEI1.8k-2/mRNAOVA or Lipofectamine MessengerMAX/mRNAOVA, the latter of which was a commercial mRNA transfection reagent. It was found that F13-PEI1.8k-1/mRNAOVA and F13-PEI1.8k-2/mRNAOVA treated BMDCs showed a high up-regulation in co-stimulatory molecules (CD80 and CD86) compared to the control group and the mRNAOVA group, indicating significantly stimulated maturation of BMDCs (Fig. 3b).
Fig. 3.

Potent DC activation and antigen-specific T cell responses mediated by F-PEI/mRNA. (a) Schematic illustrating the mRNA encoding the specific antigen, the process of DC activation, antigen presentation and subsequent T cell proliferation. UTR, untranslated region. (b&c) Flow-cytometry analysis of CD80+CD86+ (b) and SIINFEKL-H-2Kb (c) in BMDCs treated with mRNAOVA, F-PEI/mRNAOVA, or Lipofectamine MessengerMAX/mRNAOVA. The mRNAOVA concentration was fixed at 3 μg mL−1. (d) HEK-Dual Null (NF/IL8) cells (as control) and HEK-Dual mTLR4 (NF/IL-8) cells were stimulated with 10 μg mL−1 F13-PEI1.8k-1 or F13-PEI1.8k-2, respectively. Lipopolysaccharide (LPS, 2 μg mL−1) was used as the positive control. After 12 h of incubation, activation of the TLR4 signaling pathway was determined by measuring the reporter Lucia luciferase activity. RLU, Relative light unit of treated groups. RLU0, RLU of the blank group. (e&f) Representative flow cytometry plots (e) and statistical data (f) showing proliferation of OT-I CD8+ T cells after co-cultured with BMDCs pre-treated with mRNAOVA or F-PEI/mRNAOVA. (g) Enzyme-linked immunosorbent assay (ELISA) measurement of IFN-γ in cell supernatants from the stimulated OT-I CD8+ T cells in (f). The data show mean ± s.d. from 3 independent experiments (n = 3). b,c,d,f,g, Statistical significance between the indicated groups was determined using two-sided unpaired t-tests. *P < 0.05, **P < 0.01.
We then examined the antigen presentation efficiency via the MHC I pathway in BMDCs treated with F-PEI/mRNAOVA NPs. Compared with the mRNAOVA group, F13-PEI1.8k-1/mRNAOVA, F13-PEI1.8k-2/mRNAOVA and Lipofectamine MessengerMAX/mRNAOVA all could enhance the MHC I-associated SIINFEKL peptide presentation (Fig. 3c). More importantly, the F13-PEI1.8k-1/mRNAOVA could induce a higher level of MHC I antigen presentation, and its efficiency was significantly higher than that of the Lipofectamine MessengerMAX/mRNAOVA (Fig. 3c).
A recent study demonstrated that F-PEI with PEI molecular weight of 25 kDa could activate the TLR4 signaling pathway to stimulate DC activation[27]. TLR4 is an important member of the TLR protein family for pathogen recognition [30]. It has been reported that, by binding to its ligand, TLR4 promotes innate immune activation, and the activation of the TLR4 signaling pathway can enhance antigen-specific adaptive immune response, linking innate and adaptive immune responses [31]. Therefore, we next wanted to verify whether F-PEI with PEI molecular weight of 1.8 kDa could also possess the ability to activate the TLR4-mediated signaling pathway. As shown in Fig. 3d, the activation of TLR4 signaling pathway by F13-PEI1.8k-1 and F13-PEI1.8k-2 was confirmed using murine TLR4 (NF-κB-SEAP/KI-[IL-8]Lucia) dual-reporter HEK293 cells with stably transfected mouse TLR4 (mTLR4) MD-2/CD14 genes. Our results also showed that F13-PEI1.8k-1 could trigger TLR4 activation to a level significantly higher than F13-PEI1.8k-2 (Fig. 3d), suggested that F13-PEI1.8k-1, as an mRNA delivery vehicle, could be the better one for both cellular uptake enhancement and immune stimulation.
The OT-I CD8+ T cell contain transgenic T cell receptor designed to study the response of CD8+ T cells to specific OVA antigen. Using in vitro OT-I CD8+ T cell priming assay, BMDCs pre-treated with F13-PEI1.8k-1/mRNAOVA or F13-PEI1.8k-2/mRNAOVA could induce significant proliferation of OT-I CD8+ T cells, while the BMDCs pre-treated with F13-PEI1.8k-1/mRNAOVA induced higher levels of OT-I CD8+ T cell proliferation compared with those pre-treated with F13-PEI1.8k-2/mRNAOVA (Fig. 3e & 3f). Furthermore, for those CD8+ T cells activated by BMDCs pre-treated with F-PEI/mRNAOVA, a significant increase in the secretion of interferon-γ (IFN-γ) was also observed (Fig. 3g). Again, the above data indicated that F13-PEI1.8k-1/mRNAOVA NPs exhibited a better ability to activate BMDCs and trigger subsequent OVA-specific T cell responses.
3.3. In vivo stimulation of robust immune responses
Next, the immunization efficacy of the F-PEI/mRNAOVA vaccine was evaluated in vivo. C57BL/6 mice were intradermally injected with different formulations, the activation and antigen presentation of DCs in the inguinal lymph nodes of the mice were detected on Day 3 (Fig. 4 a). Compared with the control group, in the lymph nodes of mice immunized with F13-PEI1.8k-1/mRNAOVA, the proportion of DCs with up-regulated expression of the co-stimulatory molecules CD80/CD86 was significantly increased, and the MHC I antigen presentation of the model antigen OVA was also significantly increased (Fig. 4b & 4c).
Fig. 4.

In vivo immune stimulation by the F-PEI/mRNAOVA nanovaccine. (a) Timeline of the experimental design to evaluate the in vivo immune responses triggered by the indicated formulations (mRNAOVA or F-PEI/mRNAOVA, 10 μg mRNAOVA per mouse). (b&c) Proportions of CD80+CD86+ DCs (b) and SIINFEKL-H2Kb+ (MHC I bound SIINFEKL+) DCs (c) among DCs in LNs on Day 3 post one immunization. (d&e) Representative flow dot plots (d) and statistical data (e) of SIINFEKL-specific CD8+ T cells in the spleen on Day 14 post two immunization by flow-cytometry analysis of MHC I/SIINFEKL tetramer+CD8+ T cells. (f&g) ELISPOT analysis of IFN-γ spot-forming cells (f) and statistical data (g) among splenocytes after ex vivo restimulation on Day 14 post two immunization. (h) The percentages of IFN-γ expression T cells in CD8+ T cells from restimulated splenocytes on Day 14 post two immunization. b,c,e,g,h, The data show mean ± s.d. (b,c, n = 4–5 mice per group; e,g,h, n = 6 mice per group). Statistical significance between the indicated groups was determined using two-sided unpaired t-tests. *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001. LNs, lymph nodes. i.d., intradermal.
After C57BL/6 mice were immunized twice with indicated formulations at 1 week intervals, the frequency of OVA-specific CD8+ T cells in the splenocytes was analyzed on Day 14 (Fig. 4a). Notably, compared with the mRNAOVA group and the F13-PEI1.8k-2/mRNAOVA group, the frequency of SIINFEKL-MHC-I tetramer+CD8+ T cells in the F13-PEI1.8k-1/mRNAOVA group increased by 6.7-fold and 3.2-fold, respectively (Fig. 4d & 4e), suggesting that F13-PEI1.8k-1/mRNAOVA vaccination could trigger the most robust OVA-specific immune responses.
Splenocytes from the twice-immunized mice were then restimulated with OVA antigen peptide (SIINFEKL), and OVA-specific T cell responses were examined. Compared with the mRNAOVA group and the F13-PEI1.8k-2/mRNAOVA group, the F13-PEI1.8k-1/mRNAOVA group triggered the strongest IFN-γ responses, as evidenced by the highest level of IFN-γ producing cells from the enzyme-linked immunospot assay (ELISPOT) (Fig. 4f & 4 g) and the flow cytometry analysis (Fig. 4h). The results again indicated that the F13-PEI1.8k-1/mRNAOVA vaccine triggered a strong OVA-specific T cell immune response in vivo.
Based on the strong antigen-specific immune responses induced by F-PEI/mRNA, we further assessed its antitumor efficacy. C57BL/6 mice were subcutaneously inoculated with B16-OVA cells to establish the tumor-bearing mouse model. The mice were then treated two times with either phosphate buffered saline (PBS), mRNAOVA, F13-PEI1.8k-1/mRNAOVA or F13-PEI1.8k-2/mRNAOVA at 1 week interval after 4 days (Fig. 5 a). It could be observed that the F13-PEI1.8k-1/mRNAOVA cancer vaccine showed a strong tumor suppressive effect and effectively prolonged the survival time of mice (Fig. 5b & 5c), suggesting that the F13-PEI1.8k-1/mRNAAg had the potential to be a therapeutic mRNA cancer vaccine.
Fig. 5.

F-PEI/mRNAOVA nanovaccine inhibits tumor growth and prolongs survival in tumor-bearing mice. (a) A scheme of tumor challenge experiment design (n = 6). (b) Tumor growth curves for B16-OVA on mice after the various treatments. The data show mean ± s.e.m. (c) Survival curves of mice bearing B16-OVA tumor in different treatment groups. (d) Timeline of the vaccination and H&E staining experiment of mice major organs. (e) H&E staining of major organs collected from vaccinated mice on Day 8. Major organs from untreated healthy mice of the same age were collected on Day 28 as control. Scale bar = 200 μm. s.c., subcutaneous; i.d., intradermal.
Hematoxylin and eosin (H&E) staining of major organs of the F13-PEI1.8k-1/mRNAOVA or F13-PEI1.8k-2/mRNAOVA vaccinated mice showed no obvious sign of any acute organ damage or inflammatory lesions in mice on Day 8, Day 14, and Day 28 (Fig. 5d, 5e and Supplementary Fig. S2), suggesting the good biosafety of our treatment in mice.
3.4. F-PEI-based neoantigen mRNA vaccination combined with immune checkpoint blockade for personalized cancer therapy
Tumor neoantigens are non-self antigens produced by somatic cell mutations[32]. They only exist in cancer cells, not in normal cells[33]. Tumor neoantigens are highly immunogenic and can drive effective antitumor immune responses [34], [35], and thus are showing strong prospects in the design of cancer vaccines [36], [37], [38]. In order to prove the applicability of our cancer vaccine platform, we constructed F-PEI-based neoantigen vaccines to treat MC38 colon cancer model. It has been reported that the MC38 tumor cell line-specific mutant peptides Reps1, Dpagt1 and Adpgk could be used as neoepitopes for T cells [39], and these mutant peptides could trigger specific CD8+ T cell responses [40], [41]. The encoding sequences of Reps1, Dpagt1 and Adpgk peptides were included in the open reading frame (ORF) of the mRNA to construct the MC38 neoantigen mRNA (mRNAMC38) (Fig. 6 a).
Fig. 6.

F-PEI-based neoantigen mRNA vaccine for personalized immunotherapy. (a) Design of the MC38 tumor neoantigen mRNA vaccine. (b) C57BL/6 mice were immunized with the indicated formulations (PBS, mRNAMC38, and F13-PEI1.8k-1/mRNAMC38, 10 μg mRNAMC38 per mouse). (c) Statistical data of IFN-γ producing cells among splenocytes after ex vivo restimulation with peptide antigens on Day 14 post immunization (6 mice for each group, data shown as mean ± s.d.). (d) The timeline of F13-PEI1.8k-1/mRNAMC38 nanovaccine treatment combined with anti-PD-1 therapy. (e&f) Tumor growth curves (e) and survival curves (f) of mice in different treatment groups (data shown as mean ± s.e.m.) (g) Survival curves of the 4 cured mice (from 6f, the F13-PEI1.8k-1/mRNAMC38 vaccine + anti-PD-1 combination treatment group) rechallenged with MC38 cells on Day 60. Untreated: healthy mice of the same age challenged with MC38 cells. (h) The timeline of CD8+ T cell depletion experiment. (i) Flow cytometry analysis of CD8+ T cells in the mouse blood on the third day after treatment with anti-CD8a or an isotype mouse monoclonal antibody (IgG). (j) Tumor growth curves of the tumor-bearing mice pre-treated with either anti-CD8a or IgG followed by the combined F13-PEI1.8k-1/mRNAMC38 + anti-PD-1 therapy (data shown as mean ± s.d.). e,f,j, 7–8 mice for each group. Statistical significance between the indicated groups was determined using two-sided unpaired t-tests. ***P < 0.001, ****P < 0.0001. s.c., subcutaneous; i.d., intradermal; i.v., intravenous.
Next, we selected F13-PEI1.8k-1, which performed better in the previous experiments, as the neoantigen mRNA delivery vehicle. We simply mixed mRNAMC38 with F13-PEI1.8k-1 to synthesize the F13-PEI1.8k-1/mRNAMC38 nanovaccine with an average size of ∼250 nm and a zeta potential of −6.8 mV (Supplementary Fig. S3). Compared with the control group and the mRNAMC38 group, the splenocytes from mice immunized with F13-PEI1.8k-1/mRNAMC38 showed significantly increased IFN-γ producing cells after restimulation with Reps1, Dpagt1 and Adpgk peptides, respectively, indicating that F13-PEI1.8k-1/mRNAMC38 could induce potent antigen specific CD8+ T cell responses for each displayed antigen (Fig. 6b & 6c).
The F13-PEI1.8k-1/mRNAMC38 cancer vaccine was then combined with the immune checkpoint inhibitor anti-PD-1 to evaluate its antitumor effect (Fig. 6d). It could be seen that the tumor growth in F13-PEI1.8k-1/mRNAMC38 immunized mice was significantly delayed compared with the mice in the control group and the anti-PD-1 group. Moreover, the combined treatment with anti-PD-1 could further improve the antitumor therapeutic effect of F13-PEI1.8k-1/mRNAMC38 vaccine (Fig. 6e). In fact, F13-PEI1.8k-1/mRNAMC38 combined with anti-PD-1 treatment significantly prolonged the survival time of mice (Fig. 6f), and 50 % of mice showed complete tumor regression. Notably, when mice that survived from the combinational treatment were rechallenged with MC38 cells on Day 60, compared to healthy mice challenged with MC38 cells (the untreated group), 100 % of the cured mice survived (Fig. 6g), indicating effective immunological memory effect triggered by the F13-PEI1.8k-1/mRNAMC38 combined with anti-PD-1 treatment against tumor recurrence.
In order to verify the importance of CD8+ T cells in the combination therapy, anti-CD8a was used to perform the CD8+ T cell depletion experiment (Fig. 6h). The depletion of CD8+ T cells in the peripheral blood of mice was analyzed by flow cytometry on the third day after intravenous injection of anti-CD8a or mouse IgG (as control). The results showed complete depletion of CD8+ T cells from the peripheral blood of mice treated with anti-CD8a, while CD8+ T cells in the mice treated with mouse IgG remain unaffected (Fig. 6i). As expected, after CD8+ T cell depletion, the inhibitory effect of the combination therapy on tumor growth was dramatically impaired (Fig. 6j), suggesting that CD8+ T cells played a key role in the tumor suppression by the combination therapy.
4. Conclusions
In this work, we synthesized fluoroalkane modified cationic polymers for mRNA vaccine delivery and immune stimulation. We established a fluorinated polymer-based mRNA nanovaccine platform, by simple blending of F-PEI with mRNA encoding antigen(s). Our fluorinated polymer-based mRNA vaccine could effectively promote the uptake of mRNA by DCs, activate the TLR4 signaling pathway, trigger the DC activation and antigen presentation by DCs, therefore induce T cell priming, stimulate the tumor antigen-specific cellular immunity, and significantly delay the tumor growth after therapeutic vaccination. We further combined the fluorinated polymer-based MC38 neoantigen mRNA vaccine with the immune checkpoint inhibitor, and eradicated tumors in 50 % of the MC38 tumor-bearing mice and successfully prevented tumor reoccurrence. In conclusion, we have developed a novel cancer vaccine platform suitable for the delivery of mRNA vaccines, and demonstrated synergistic therapeutic effects of such personalized neoantigen vaccines in combination with immune checkpoint inhibitors. In the future, the mRNA delivery polymer developed in this work might be extended to applications in other mRNA-based therapeutics where high level of mRNA delivery is desired, or both robust cellular and humoral immune responses are crucial, e.g., in the fight against viral infections, besides neutralizing antibody induction, strong cellular immune responses are required to eliminate the infected cells.
Declaration of Competing Interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Acknowledgements
This work is dedicated to the memory of Dr. Jun Xu, who passed away in a tragic accident. Jun took part in the original design, the functionalization & optimization of the F-PEI polymers, and several efficacy evaluation assays. This work was partially supported by the National Key Research and Development (R&D) Program of China (2022YFB3804604 and 2021YFF0701800), the National Natural Science Foundation of China (32071382, 52032008, and 21927803), the Natural Science Foundation of Jiangsu Higher Education Institutions of China (19KJA310008), Suzhou Science and Technology Development Project-Science and Technology Innovation in Medicine and Health Care (SKY2021033), the Collaborative Innovation Center of Suzhou Nano Science and Technology (Nano-CIC), the 111 Project, the National Center of Technology Innovation for Biopharmaceuticals (NCTIB2022HS01011), and the Suzhou Key Laboratory of Nanotechnology and Biomedicine.
Footnotes
Supplementary data to this article can be found online at https://doi.org/10.1016/j.cej.2022.140930.
Appendix A. Supplementary data
The following are the Supplementary data to this article:
Data availability
No data was used for the research described in the article.
References
- 1.Vermaelen K. Vaccine strategies to improve anti-cancer cellular immune responses. Front. Immunol. 2019;10:00008. doi: 10.3389/fimmu.2019.00008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kim C.G., Sang Y.B., Lee J.H., Chon H.J. combining cancer vaccines with immunotherapy: establishing a new immunological approach. Int. J. Mol. Sci. 2021;22:8035. doi: 10.3390/ijms22158035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Zhang Q.L., Hong S., Dong X., Zheng D.W., Liang J.L., Bai X.F., Wang X.N., Han Z.Y., Zhang X.Z. Bioinspired nano-vaccine construction by antigen pre-degradation for boosting cancer personalized immunotherapy. Biomaterials. 2022;287 doi: 10.1016/j.biomaterials.2022.121628. [DOI] [PubMed] [Google Scholar]
- 4.Deng Z., Tian Y., Song J., An G., Yang P. mRNA vaccines: the dawn of a new era of cancer immunotherapy. Front. Immunol. 2022;13 doi: 10.3389/fimmu.2022.887125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Shi J., Huang M.W., Lu Z.D., Du X.J., Shen S., Xu C.F., Wang J. Delivery of mRNA for regulating functions of immune cells. J. Control. Release. 2022;345:494–511. doi: 10.1016/j.jconrel.2022.03.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Moser M., Leo O. Key concepts in immunology. Vaccine. 2010;28:C2–C13. doi: 10.1016/j.vaccine.2010.07.022. [DOI] [PubMed] [Google Scholar]
- 7.Appay V., Douek D.C., Price D.A. CD8(+) T cell efficacy in vaccination and disease. Nat. Med. 2008;14:623–628. doi: 10.1038/nm.f.1774. [DOI] [PubMed] [Google Scholar]
- 8.Ji X., Guo D., Ma J., Yin M., Yu Y., Liu C., Zhou Y., Sun J., Li Q., Chen N., Fan C., Song H. Epigenetic remodeling hydrogel patches for multidrug-resistant triple-negative breast cancer. Adv. Mater. 2021;33(18):2100949. doi: 10.1002/adma.202100949. [DOI] [PubMed] [Google Scholar]
- 9.Eggenhuizen P.J., Ng B.H., Ooi J.D. Antigen-driven CD4(+) T-cell anergy: a pathway to peripheral T regulatory cells. Immunol. Cell Biol. 2021;99:252–254. doi: 10.1111/imcb.12424. [DOI] [PubMed] [Google Scholar]
- 10.Linares-Fernandez S., Lacroix C., Exposito J.Y., Verrier B. Tailoring mRNA vaccine to balance innate/adaptive immune response. Trends Mol. Med. 2020;26:311–323. doi: 10.1016/j.molmed.2019.10.002. [DOI] [PubMed] [Google Scholar]
- 11.Pardi N., Hogan M.J., Porter F.W., Weissman D. mRNA vaccines - a new era in vaccinology. Nat. Rev. Drug Discov. 2018;17:261–279. doi: 10.1038/nrd.2017.243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Shuai Q., Zhu F.T., Zhao M.D., Yan Y.F. mRNA delivery via non-viral carriers for biomedical applications. Int J Pharmaceut. 2021;607 doi: 10.1016/j.ijpharm.2021.121020. [DOI] [PubMed] [Google Scholar]
- 13.Pardi N., Hogan M.J., Weissman D. Recent advances in mRNA vaccine technology. Curr. Opin. Immunol. 2020;65:14–20. doi: 10.1016/j.coi.2020.01.008. [DOI] [PubMed] [Google Scholar]
- 14.Yin Y., Li X., Ma H., Zhang J., Yu D., Zhao R., Yu S., Nie G., Wang H. In situ transforming RNA nanovaccines from polyethylenimine functionalized graphene oxide hydrogel for durable cancer immunotherapy. Nano Lett. 2021;21:2224–2231. doi: 10.1021/acs.nanolett.0c05039. [DOI] [PubMed] [Google Scholar]
- 15.Polack F.P., Thomas S.J., Kitchin N., Absalon J., Gurtman A., Lockhart S., Perez J.L., Marc G.P., Moreira E.D., Zerbini C., Bailey R., Swanson K.A., Roychoudhury S., Koury K., Li P., Kalina W.V., Cooper D., Frenck R.W., Hammitt L.L., Tureci O., Nell H., Schaefer A., Unal S., Tresnan D.B., Mather S., Dormitzer P.R., Sahin U., Jansen K.U., Gruber W.C., Grp C.C.T. Safety and efficacy of the BNT162b2 mRNA covid-19 vaccine. New Engl J Med. 2020;383:2603–2615. doi: 10.1056/NEJMoa2034577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Baden L.R., El Sahly H.M., Essink B., Kotloff K., Frey S., Novak R., Diemert D., Spector S.A., Rouphael N., Creech C.B., McGettigan J., Khetan S., Segall N., Solis J., Brosz A., Fierro C., Schwartz H., Neuzil K., Corey L., Gilbert P., Janes H., Follmann D., Marovich M., Mascola J., Polakowski L., Ledgerwood J., Graham B.S., Bennett H., Pajon R., Knightly C., Leav B., Deng W.P., Zhou H.H., Han S., Ivarsson M., Miller J., Zaks T., Grp C.S. Efficacy and safety of the mRNA-1273 SARS-CoV-2 vaccine. New Engl J Med. 2021;384:403–416. doi: 10.1056/NEJMoa2035389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hou X.C., Zaks T., Langer R., Dong Y.Z. Lipid nanoparticles for mRNA delivery. Nat. Rev. Mater. 2022;7:65. doi: 10.1038/s41578-021-00358-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Evers M.J.W., Kulkarni J.A., van der Meel R., Cullis P.R., Vader P., Schiffelers R.M. State-of-the-art design and rapid-mixing production techniques of lipid nanoparticles for nucleic acid delivery. Small Methods. 2018;2(9) [Google Scholar]
- 19.Xiong S.-D., Li L., Jiang J., Tong L.-P., Wu S., Xu Z.-S., Chu P.K. Cationic fluorine-containing amphiphilic graft copolymers as DNA carriers. Biomaterials. 2010;31(9):2673–2685. doi: 10.1016/j.biomaterials.2009.12.014. [DOI] [PubMed] [Google Scholar]
- 20.Wang M.M., Cheng Y.Y. Structure-activity relationships of fluorinated dendrimers in DNA and siRNA delivery. Acta Biomater. 2016;46:204–210. doi: 10.1016/j.actbio.2016.09.023. [DOI] [PubMed] [Google Scholar]
- 21.Zhang J.H., Wang W.J., Zhang J., Xiao Y.P., Liu Y.H., Yu X.Q. ROS-responsive fluorinated polycations as non-viral gene vectors. Eur. J. Med. Chem. 2019;182 doi: 10.1016/j.ejmech.2019.111666. [DOI] [PubMed] [Google Scholar]
- 22.Zhang Z.J., Shen W.W., Ling J., Yan Y., Hu J.J., Cheng Y.Y. The fluorination effect of fluoroamphiphiles in cytosolic protein delivery. Nat. Commun. 2018;9:1377. doi: 10.1038/s41467-018-03779-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Horvath I.T., Rabai J. Facile catalyst separation without water - fluorous biphase hydroformylation of olefins. Science. 1994;266:72–75. doi: 10.1126/science.266.5182.72. [DOI] [PubMed] [Google Scholar]
- 24.Cametti M., Crousse B., Metrangolo P., Milani R., Resnati G. The fluorous effect in biomolecular applications. Chem. Soc. Rev. 2012;41:31–42. doi: 10.1039/c1cs15084g. [DOI] [PubMed] [Google Scholar]
- 25.Kasuya M.C.Z., Nakano S., Katayama R., Hatanaka K. Evaluation of the hydrophobicity of perfluoroalkyl chains in amphiphilic compounds that are incorporated into cell membrane. J. Fluor. Chem. 2011;132:202–206. [Google Scholar]
- 26.Wang M.M., Liu H.M., Li L., Cheng Y.Y. A fluorinated dendrimer achieves excellent gene transfection efficacy at extremely low nitrogen to phosphorus ratios. Nat. Commun. 2014;5:3053. doi: 10.1038/ncomms4053. [DOI] [PubMed] [Google Scholar]
- 27.Xu J., Lv J., Zhuang Q., Yang Z.J., Cao Z.Q., Xu L.G., Pei P., Wang C.Y., Wu H.F., Dong Z.L., Chao Y., Wang C., Yang K., Peng R., Cheng Y.Y., Liu Z. A general strategy towards personalized nanovaccines based on fluoropolymers for post-surgical cancer immunotherapy. Nat. Nanotechnol. 2020;15:1043–1052. doi: 10.1038/s41565-020-00781-4. [DOI] [PubMed] [Google Scholar]
- 28.Hall A., Lachelt U., Bartek J., Wagner E., Moghimi S.M. Polyplex evolution: understanding biology, optimizing performance. Mol. Ther. 2017;25:1476–1490. doi: 10.1016/j.ymthe.2017.01.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Pandey A.P., Sawant K.K. Polyethylenimine: a versatile, multifunctional non-viral vector for nucleic acid delivery. Mater. Sci. Eng. C, Mater. Biol. Appl. 2016;68:904–918. doi: 10.1016/j.msec.2016.07.066. [DOI] [PubMed] [Google Scholar]
- 30.Steinhagen F., Kinjo T., Bode C., Klinman D.M. TLR-based immune adjuvants. Vaccine. 2011;29:3341–3355. doi: 10.1016/j.vaccine.2010.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Shetab Boushehri M.A., Lamprecht A. TLR4-based immunotherapeutics in cancer: a review of the achievements and shortcomings. Mol. Pharm. 2018;15:4777–4800. doi: 10.1021/acs.molpharmaceut.8b00691. [DOI] [PubMed] [Google Scholar]
- 32.Xu P., Luo H., Kong Y., Lai W.-F., Cui L., Zhu X. Cancer neoantigen: boosting immunotherapy. Biomed. Pharmacother. 2020;131:110640. doi: 10.1016/j.biopha.2020.110640. [DOI] [PubMed] [Google Scholar]
- 33.Sahin U., Tureci O. Personalized vaccines for cancer immunotherapy. Science. 2018;359:1355–1360. doi: 10.1126/science.aar7112. [DOI] [PubMed] [Google Scholar]
- 34.Perumal D., Imai N., Lagana A., Finnigan J., Melnekoff D., Leshchenko V.V., Solovyov A., Madduri D., Chari A., Cho H.J., Dudley J.T., Brody J.D., Jagannath S., Greenbaum B., Gnjatic S., Bhardwaj N., Parekh S. Mutation-derived Neoantigen-specific T-cell Responses in Multiple Myeloma. Clin. Cancer Res. 2020;26:450–464. doi: 10.1158/1078-0432.CCR-19-2309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Yarchoan M., Johnson B.A., Lutz E.R., Laheru D.A., Jaffee E.M. Targeting neoantigens to augment antitumour immunity (vol 17, pg 209, 2017) Nat. Rev. Cancer. 2017;17:569. doi: 10.1038/nrc.2017.74. [DOI] [PubMed] [Google Scholar]
- 36.Blass E., Ott P.A. Advances in the development of personalized neoantigen-based therapeutic cancer vaccines. Nat. Rev. Clin. Oncol. 2021;18:215–229. doi: 10.1038/s41571-020-00460-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ott P.A., Hu Z.T., Keskin D.B., Shukla S.A., Sun J., Bozym D.J., Zhang W.D., Luoma A., Giobbie-Hurder A., Peter L., Chen C., Olive O., Carter T.A., Li S.Q., Lieb D.J., Eisenhaure T., Gjini E., Stevens J., Lane W.J., Javeri I., Nellaiappan K., Salazar A.M., Daley H., Seaman M., Buchbinder E.I., Yoon C.H., Harden M., Lennon N., Gabriel S., Rodig S.J., Barouch D.H., Aster J.C., Getz G., Wucherpfennig K., Neuberg D., Ritz J., Lander E.S., Fritsch E.F., Hacohen N., Wu C.J. An immunogenic personal neoantigen vaccine for patients with melanoma. Nature. 2017;547:217–221. doi: 10.1038/nature22991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Cafri G., Gartner J.J., Zaks T., Hopson K., Levin N., Paria B.C., Parkhurst M.R., Yossef R., Lowery F.J., Jafferji M.S., Prickett T.D., Goff S.L., McGowan C.T., Seitter S., Shindorf M.L., Parikh A., Chatani P.D., Robbins P.F., Rosenberg S.A. mRNA vaccine-induced neoantigen-specific T cell immunity in patients with gastrointestinal cancer. J. Clin. Invest. 2020;130:5976–5988. doi: 10.1172/JCI134915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Yadav M., Jhunjhunwala S., Phung Q.T., Lupardus P., Tanguay J., Bumbaca S., Franci C., Cheung T.K., Fritsche J., Weinschenk T., Modrusan Z., Mellman I., Lill J.R., Delamarre L. Predicting immunogenic tumour mutations by combining mass spectrometry and exome sequencing. Nature. 2014;515:572–576. doi: 10.1038/nature14001. [DOI] [PubMed] [Google Scholar]
- 40.Kuai R., Ochyl L.J., Bahjat K.S., Schwendeman A., Moon J.J. Designer vaccine nanodiscs for personalized cancer immunotherapy. Nat. Mater. 2017;16:489–496. doi: 10.1038/nmat4822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wang W.J., Liu Z.D., Zhou X.X., Guo Z.Q., Zhang J., Zhu P., Yao S., Zhu M.Z. Ferritin nanoparticle-based SpyTag/SpyCatcher-enabled click vaccine for tumor immunotherapy. Nanomed-Nanotechnol. 2019;16:69–78. doi: 10.1016/j.nano.2018.11.009. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
No data was used for the research described in the article.