

Abstract
In the mid 1990’s, a convergence of discoveries in dendritic cell (DC) biology and tumor antigen identification led investigators to study DCs as adjuvants for cancer vaccines. On the twentieth anniversary of a seminal clinical study by Jacques Banchereau and colleagues, we revisit the key events that prompted the initial wave of DC vaccine clinical studies and lessons learned that, in our opinion, helped forge the path for the field that we now call immuno-oncology. It is essential to recall that prior to the discovery of immune checkpoint therapy and chimeric antigen receptor (CAR) T-cell therapy, skepticism prevailed regarding the potential therapeutic benefit of immunotherapies. In hindsight, we can now appreciate how the early DC cancer vaccine trials helped investigators sustain their attention on adaptive immunity specific for malignant cells. These vaccines demonstrated clear evidence for induction of antigen-specific T cells and were well tolerated despite low rates of objective clinical response. In the context of the current era some 20 years later, harnessing DC vaccines has been shown to increase the breadth and diversity of tumor-specific T cells, and by trafficking to sites of metastases promote an inflamed tumor microenvironment.
Dendritic Cells
Initially described by Ralph Steinman and Zanvil Cohn in 1973, dendritic cells (DC) are antigen-presenting cells notable for their sparse distribution in tissues. In a remarkable series of studies, Steinman and collaborators demonstrated how DCs link innate to adaptive immunity by sensing danger and pathogen-associated signals, migrate between lymphoid and nonlymphoid tissues, produce cytokines and chemokines, and prime naïve T cells through endogenous pathways and cross-presentation of antigens in the context of major histocompatibility complex molecules (1). By the late 1990’s, there was consensus that DCs are the “professional” antigen-presenting cell. Further studies suggested that DC may serve to maintain immune homeostasis and limit autoimmunity. These distinct functionalities are reflective of the heterogeneity of DC populations as well as indicative of cues received from the tissue microenvironment. Given their exquisite ability to prime and boost immune responses, the potential therapeutic benefit of formulating DCs into cancer vaccines was widely appreciated; however, their scarcity presented a challenge for clinical translation. The identification of a bipotential CD34+ hematopoietic precursor cell (HPC) that could differentiate into DCs when cultured in the presence of GM-CSF and TNFα was a pivotal advance for the field as well as the finding that peripheral blood monocytes could differentiate into DCs in the presence of GM-CSF and IL4 (2, 3). These discoveries, along with protocols to purify CD11c+ DCs from peripheral blood, provided the requisite methodology for the generation of myeloid-derived DCs to sufficient numbers for basic science investigators as well as translational researchers seeking to advance DC vaccines to the clinic. Since then, the field has evolved into an international community of investigators that have unraveled the heterogeneity of DC populations. In its simplest inception, DC populations are represented by 3 major subsets of myeloid origin: conventional DC1 (cDC1, CD141+) and DC2 (cDC2, CD1c+), monocyte-derived (moDC), and one subset of lymphoid origin, plasmacytoid DC (pDC). cDC1, cDC2, and pDC populations are defined by phenotypic markers and transcription factors and are found in steady state, while moDC arise after infection or inflammation (4). More recently, additional heterogeneity has been identified by single-cell technologies, although their physiologic significance is yet to be fully examined (4). In addition, we have gained a greater understanding of how the tumor microenvironment (TME), through production of immunosuppressive factors and chemokines, impairs DC differentiation, maturation, and migration, leading to dysfunction and contributing to tumor progression. Altogether, this wealth of knowledge has informed on the contribution of the various DC populations to antitumor immunity, their potential role in immune checkpoint inhibition therapy, and helped identify actionable molecular targets to optimize their use in therapeutic applications.
Tumor Antigen Discovery
The success of cancer vaccines relies on the ability of elicited T-cell responses to discriminate between tumor and healthy cells. In the 1990’s, we witnessed a series of seminal discoveries related to the identification of novel tumor-associated antigens. Initial studies used cDNA cloning approaches to identify peptides derived from MAGEA1, a cancer-germline–encoded antigen, as a target of cytotoxic T lymphocyte recognition. In fast succession, additional antigens encoded by other cancer-germline genes as well as melanocyte differentiation antigens were identified and in some cases, confirmed in landmark studies using immune-peptidomics (5). Although most of these antigens represented tumor-associated and not tumor-specific targets, their formulation with therapeutic vaccines enhanced the specificity of antitumor immunity. Furthermore, the molecular identification of specific tumor antigens enabled precision immune monitoring using MHC-peptide tetramer binding assays in addition to ELISpot assays as means for a quantitative assessment of antitumor immunity. More recently, the advent of next-generation sequencing has propelled into the spotlight tumor-specific antigens arising from single nucleotide variants and frameshift alterations, known as canonical neoantigens. In addition, using a combination of ribosome profiling, RNA sequencing, and proteomics, cryptic proteins arising in noncoding regions in cancer cells have been proposed to generate additional noncanonical neoantigens. In sum, neoantigen identification by next-generation sequencing methods has revitalized the cancer vaccine field and accelerated the push for personalized cancer vaccines.
DC Vaccine Trials: The Initial Wave
Reported in 1996, the first DC vaccine trial targeted patients with low-grade B-cell follicular lymphoma and employed autologous DCs isolated from peripheral blood (6). Investigators took advantage of the unique immunoglobulin variable region (idiotype) of each patient’s malignant clone as a tumor-specific antigen. This study confirmed that 5 × 106 DCs given for 3 doses was sufficient to generate measurable antitumor T-cell immunity and to promote clinical activity with a complete response in 1 patient. This experience prompted investigators to perform additional DC vaccine trials in prostate cancer that ultimately led to the 2010 regulatory approval for Sipuleucel-T, the first cell therapy for cancer (7). Additional studies with Sipuleucel-T in combination with checkpoint inhibitors and radiation therapy are ongoing.
In rapid succession, a series of DC vaccine trials were initiated, resulting in the first wave of reports from US and EU investigators. Among the initial wave of pilot/phase I clinical trials was the report from Banchereau and colleagues at Baylor Institute for Immunology Research and the Rockefeller University describing their experience treating 18 patients (all HLA-A*02:01+) with metastatic melanoma (8). Patients were treated with peptide-pulsed autologous CD34 HPC-derived DCs obtained after 8-day culture in GM-CSF, FLT3-L, and TNFα. Four doses of CD34-DCs were administered subcutaneously every 14 days in a dose-escalation study. Four melanoma peptide antigens (Melan-A/Mart1, gp100, tyrosinase, and MAGE-3) were used along with an influenza A peptide and keyhole limpet hemocyanin as controls. The vaccine was well tolerated, and no infusion reactions were seen. Most notably, 16 of 18 patients responded to the control antigens and had enhanced responses to one or more melanoma antigens, as measured in vitro by ELISpot assay. In addition, 10 of 14 evaluable patients also displayed delayed-type hypersensitivity upon skin testing using individual immunizing peptides, providing in vivo support for the immunogenicity of the vaccine. The 2 patients that failed to elicit immune responses to any of the antigens demonstrated rapid clinical progression of disease. Of significance, only 7 of 17 evaluable patients experienced tumor progression upon clinical status reevaluation; remarkably, the 3 patients with limited metastatic disease experienced complete resolution of melanoma upon reevaluation.
The key lessons provided by Banchereau and other investigators working in the field during this period were multifold. First, manufacturing autologous cell products from patients with cancer under Current Good Manufacturing Practice is time-consuming, labor-intensive and costly, yet the first wave of trials demonstrated that this process could be successfully carried out in the academic setting. Academic investigators able to secure adequate funding were free to develop investigator-initiated Investigational New Drug applications overseen by regulatory agencies and as a result were unencumbered by industry agreements. As the field moves forward, academic investigator-initiated research continues to serve as a primary model for the development of new cell therapies for various disease indications. Second, DCs isolated from peripheral blood or differentiated from several sources (CD34+ HPC and monocytes) could be formulated into vaccines. Banchereau and colleagues chose to pursue CD34+ HPC, a less committed progenitor, as a source of myeloid DCs with the potential to give rise to interstitial DCs and Langerhans cells. Differentiation of DCs from monocytes also produces sufficient cell numbers that upon maturation yields a population of cells capable of eliciting antitumor immunity. Yet, it is the rare cDC1 (~0.1% in blood) that may constitute the ideal source given its capacity to cross-present antigen to CD8+ T cells. These cells could be obtained either by differentiation from CD34 HPC (9) or direct isolation from blood; the latter presenting a challenge given their scarcity and the lack of a facile method of purification. Important questions remain; for example, are cDC1s genuinely superior antigen-presenting cells for the induction of antitumor immunity in humans? Do human-induced pluripotent stem cell (iPSC)–derived DCs, leukemic pDC cell lines, and engineered moDCs represent alternative suitable sources for clinical investigation? Third, patients that experienced more favorable clinical outcomes had limited disease and were, in many instances, chemotherapy-naive. Banchereau and colleagues suggested their finding “supports the concept of testing DC vaccines earlier, i.e., in a surgical adjuvant setting.” Indeed, increased tumor burden along with immunosuppressive barriers imposed by the TME likely contributed to the lackluster clinical efficacy of DC vaccine monotherapy seen in the initial wave of studies. It is now apparent to many scientists working in the field that combinatorial approaches administered in conjunction with cancer vaccines is the way forward.
Epilogue
Next-generation sequencing technologies coupled with improvements in computational biology have created efficient bioinformatic pipelines to foster the development of personalized cancer vaccines. Pending a breakthrough that can simplify and streamline DC manufacturing, messenger RNA-lipid nanoparticle (mRNA-LNP)–based vaccines may offer the potential to efficiently target tumor-specific neoantigens and reprogram DCs in vivo (10). Hence, mRNA-LNP may emerge as the vaccine platform in the Next Wave. However, significant gaps in our knowledge remain related to the identification of bona fide tumor rejection antigens and whether T-cell (CD4+ and CD8+) immunity elicited by the mRNA-LNP platform results in memory induction as it has been reported for DC vaccines. Clinicians appear ready to embrace the next wave of cancer vaccines and this enthusiasm signifies a genuine opportunity to improve patient outcomes and to broaden the impact of cancer immunotherapies.
Figure 1.
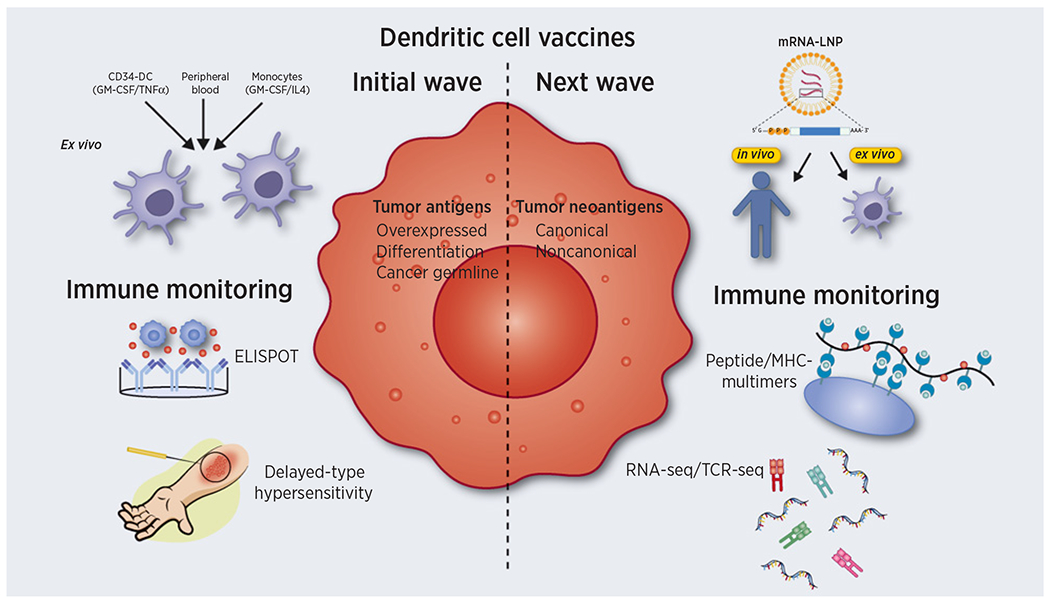
DC vaccines. The initial wave of DC vaccines involved the use of multiple DC sources differentiated ex vivo using a mixture of cytokines or directly isolated from blood. Nonmutated peptides encoded by overexpressed, differentiation or cancer-germline genes were employed as tumor antigens and vaccine responses monitored by ELISPOT and delayed type hypersensitivity (DTH) assays. The next wave may take advantage of mRNA-LNP to target DC either ex vivo or in vivo. Personalized formulations incorporating neoantigens arising from genomic alterations such as frameshift or missense mutations (canonical) or errors in transcription/translation (noncanonical) are now feasible due to advances in next-generation sequencing technologies, immune peptidomics, and bioinformatics. Immune-monitoring at the population and single-cell level can now be performed by peptide/MHC multimers, RNA sequencing (RNA-seq), and T-cell receptor sequencing (TCR-seq).
Acknowledgments
Due to space limitations, the authors were unable to cite the many contributions of investigators working in the immuno-oncology field. This work is supported by NIH R01CA204261.
Authors’ Disclosures
B.M. Carreno reports grants from NIH R01CA204261 during the conduct of the study. No disclosures were reported by the other author.
References
- 1.Steinman RM. Decisions about dendritic cells: past, present, and future. Annu Rev Immunol 2012;30:1–22. [DOI] [PubMed] [Google Scholar]
- 2.Caux C, Vanbervliet B, Massacrier C, Dezutter-Dambuyant C, de Saint-Vis B, Jacquet C, et al. CD34+ hematopoietic progenitors from human cord blood differentiate along two independent dendritic cell pathways in response to GM-CSF+TNFα. J Exp Med 1996;184:695–706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Sallusto F, Lanzavecchia A. Efficient presentation of soluble antigen by cultured human dendritic cells is maintained by granulocyte–macrophage colony-stimulating factor plus interleukin 4 and downregulated by tumor necrosis factor alpha. J Exp Med 1994;179:1109–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Cabeza-Cabrerizo M, Cardoso A, Minutti CM, Pereira da Costa M, Reis ESC. Dendritic cells revisited. Annu Rev Immunol 2021;39:131–66. [DOI] [PubMed] [Google Scholar]
- 5.Coulie PG, Van den Eynde BJ, van der Bruggen P, Boon T. Tumor antigens recognized by T lymphocytes: at the core of cancer immunotherapy. Nat Rev Cancer 2014;14:135–46. [DOI] [PubMed] [Google Scholar]
- 6.Hsu FJ, Benike C, Fagnoni F, Liles TM, Czerwinski D, Taidi B, et al. Vaccination of patients with B-cell lymphoma using autologous antigen-pulsed dendritic cells. Nat Med 1996;2:52–58. [DOI] [PubMed] [Google Scholar]
- 7.Kantoff PW, Higano CS, Shore ND, Berger ER, Small EJ, Penson DF, et al. Sipuleucel-T immunotherapy for castration-resistant prostate cancer. N Engl J Med 2010;363:411–22. [DOI] [PubMed] [Google Scholar]
- 8.Banchereau J, Palucka AK, Dhodapkar M, Burkeholder S, Taquet N, Rolland A, et al. Immune and clinical responses in patients with metastatic melanoma to CD34+ progenitor-derived dendritic cell vaccine. Cancer Res 2001;61:6451–8. [PubMed] [Google Scholar]
- 9.Balan S, Arnold-Schrauf C, Abbas A, Couespel N, Savoret J, Imperatore F, et al. Large-scale human dendritic cell differentiation revealing notch-dependent lineage bifurcation and heterogeneity. Cell Rep 2018;24:1902–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sahin U, Tureci O. Personalized vaccines for cancer immunotherapy. Science 2018;359:1355–60. [DOI] [PubMed] [Google Scholar]