

Abstract
Cancer cells show increases in protein degradation pathways, including autophagy, during progression to meet the increased protein degradation demand and support cell survival. On the other hand, reduced autophagy activity during aging is associated with a reduced DNA damage response and increased genomic instability. Therefore, it is a puzzling how DNA repair can be increased in cancer cells that are resistant to chemotherapies or during progression when autophagy activity is intact or increased. We discovered that tripartite motif containing 44 (TRIM44) is a pivotal element regulating the DNA damage response in cancer cells with intact autophagy. TRIM44 deubiquitinates p62, an autophagy substrate, which leads to its oligomerization. This prevents p62 localization to the nucleus upon irradiation. Increased cytoplasmic retention of p62 by TRIM44 prevents the degradation of FLNA and 53BP1, which increases DNA damage repair. Together, our data support TRIM44 a potential therapeutic target for therapy-resistant tumor cells with intact autophagy.
Keywords: DNA damage response, multiple myeloma, oligomerization, p62, TRIM44
Introduction
Maintaining genomic integrity is critical for organismal survival. Organisms have developed a global signaling network known as the DNA-damage response (DDR) to control genomic integrity, a system that senses DNA damage and recruits repair factors [1]. The failure to ameliorate DNA damage results in genomic instability or cancer cell survival after therapeutic intervention. Thus, targeting DNA damage repair mechanisms may improve current cancer therapeutic outcomes and improve cancer patient survival.
Multiple myeloma (MM) is an incurable malignancy of plasma cells characterized by hypercalcemia, renal insufficiency, anemia, and increased bone lesions [2]. Genetic instability of plasma B cells is associated with disease progression from monoclonal gammopathy of undetermined significance (MGUS) to MM to plasma cell leukemia [3, 4]. Despite progress in therapeutic intervention, significant numbers of MM patients still relapse [2, 5]. Therefore, it is important to have new therapeutic targets to design therapies after MM relapse. Upon the application of chemo- and radiotherapies, one mechanism determining MM cell survival is an efficient DDR. Ataxia-telangiectasia-mutated (ATM), ATM and Rad3-related (ATR), and DNA-dependent protein kinases (DNA-PKs) are examples of components of the DNA damage-sensing system. DNA damage initiates phosphorylation events, followed by DNA repair and cell cycle arrest [7]. In G0/G1 phase, DNA double strand breaks are repaired by the nonhomologous end-joining (NHEJ) pathway, an error-prone repair mechanism; in S and G2/M phases, the homologous recombination (HR) pathway is activated to repair DSBs, an error-free repair mechanism [7]. The dysregulation of HR or NHEJ in MM contributes to genomic instability, chromosomal translocation, disease onset and progression [8], [9]. However, the detailed mechanisms of DSB repair pathways affecting MM survival after DNA damage remain unclear. In particular, how DNA damage repair differs among subpopulations of MM in anatomically distinguishable locations in bone marrow (BM) is unknown. Therefore, understanding MM repair mechanisms after chemotherapy-induced DNA damage may improve patient outcome and may help to design a new therapy.
TRIM44 is a deubiquitinase containing a zinc finger ubiquitin protease (ZF-UBP) domain. TRIM family proteins are involved in various biological functions from the virus-mediated immune response to tumor suppression or promotion [10, 11]. TRIM44 is upregulated in several cancers, including head and neck squamous cell carcinoma, lung cancer, hepatocellular carcinoma, prostate cancer, gastric cancer, testicular germ cell tumors, and breast cancer [12–18]. Our lab firstly reported that TRIM44 is highly expressed in quiescent MM cells residing in the osteoblastic niche of BM. Decreased TRIM44 level reduces MM cell localization to BM and decreases bone lytic lesions in xenograft models. On the other hand, increased TRIM44 expression promotes bone lysis, similar to what is observed in MM patients [19]. Quiescent MM cells from the osteoblastic niche are more protected against chemotherapies, and TRIM44 silencing in quiescent MM cells decreases resistance to chemotherapies [19].
Herein, we report novel functions of TRIM44 in the irradiation (IR)-induced DNA damage response and repair in cancer cells. TRIM44 expression protects MM cells from IR-induced cell death, but TRIM44-silencing increases MM cell death. TRIM44 silencing reduces both the NHEJ- and HR-mediated DDR. TRIM44 expression enables MM cells to efficiently reduce IR-induced γH2AX foci and induce DDR repair activity. The upregulation of DNA repair mechanisms induced by TRIM44 and p62 (SQSTM1, sequestosome 1), an autophagy receptor. Upon the induction of DNA damage, p62 interacts with FLNA (filament A), which recruits the DNA repair protein RAD51 (RAD51 recombinase) to double-stranded breaks. p62 promotes the proteasomal degradation of FLNA and RAD51 within the nucleus, resulting in reduced levels of nuclear RAD51 and slower DNA repair [20]. We found that TRIM44 oligomerizes p62, which prevents its translocation to the nucleus during DDR responses. As a result, FLNA and RAD51 are not degraded in the nucleus, which results in increased DNA repair. p62 accumulates in many human age-related diseases, and its activities are correlated with reduced DDR repair and increased genome instability. The only known mechanism resulting in the accumulation of p62 is decreased autophagy activity during aging. Our report is the first to show that without decreasing autophagy, an increase in TRIM44 prevents p62 nuclear translocation in response to irradiation and that TRIM44-mediated p62 oligomerization promotes the DDR repair process and supports cancer cell survival in response to DNA damage.
Results
TRIM44 promotes radioresistance and protects MM cells from IR-induced cell death
Since TRIM44 silencing via the CRISPR-CAS9 method in cancer cells caused cell death (Fig. S1A), we generated MM cells that in which TRIM44 was overexpressed (TRIM44OE) or knocked down (TRIM44KD) via a lentivirus (Fig. S1B) to study the role of TRIM44 in IR-induced DNA damage responses and repair. Control cells were infected with relevant control vectors corresponding to the infecting virus vector (TRIM44OE-CON or TRIM44KD-CON) (Fig. S1B). Compared to TRIM44 control cells, TRIM44OE cells were resistant to irradiation-induced cell death (Fig. 1A). On the other hand, silencing increased sensitivity to irradiation-induced cell death (Fig. 1B). Irradiation elicited prolonged effects on the colony-forming ability of MM cells, and the control cells exhibited a markedly reduced colony-forming ability after 2 weeks. TRIM44 expression reduced the inhibitory effects of irradiation (Fig. 1C), while TRIM44 knockdown cells showed further reduced the number of colonies formed after irradiation (Fig. 1D).
Figure 1. TRIM44 promotes ionizing radiation (IR) resistance and protects MM cells from IR-induced cell death.

(A) TRIM44OE MM cells show decreased IR-induced apoptosis, which was measured using flow cytometry based on annexin V/7-AAD staining. TRIM44OE-CON and TRIM44OE MM cells were treated with 2 Gy IR.
(B) TRIM44KD MM cells show increased IR-induced apoptosis, which was measured using flow cytometry based on annexin V/7-AAD staining. TRIM44KD-CON and TRIM4KD MM cells were treated with 2 Gy IR.
(C) TRIM44OE MM cells are resistant to the IR-mediated inhibition of colony formation. TRIM44OE-CON and TRIM44OE RPMI MM cells (103) were irradiated with 2 Gy IR and seeded in semisolid methylcellulose medium. Colonies containing >40 cells were counted after 10 – 14 days. Colony formation was calculated as the IR-treated colony number/untreated colony number × 100%. Scale bars, 100 μm.
(D) Colony formation after IR was greatly decreased in TRIM44KD MM cells. TRIM44KD-CON and TRIM44KD RPMI MM cells (103) were irradiated with 2 Gy IR and seeded in semisolid methylcellulose medium. Colony formation was calculated as described in (C).
(E) TRIM44OE MM cells display fewer IR-induced γH2AX foci after irradiation (2 Gy). After 1 hour, TRIM44OE-CON and TRIM44OE MM cells were immunostained with anti-γH2AX (red) and DRAQ5 (blue) and analyzed using confocal microscopy. Scale bars, 20 μm.
(F) Graphic presentation of the percentage of γH2AX+ TRIM44OE-CON and TRIM44OE MM cells with or without irradiation. The cells containing > 5 γH2AX foci were recorded as γH2AX+ cells, whose percentage was averaged from at least 100 cells. The data are displayed as mean plus SD of three counts, and statistical significance was calculated and represented as the P value. *, P < 0.05; **, P < 0.01; ***, P < 0.001.
(G) TRIM44KD MM cells show enhanced IR-induced γH2AX foci in MM cells, which indicates more susceptibility to irradiation. γH2AX+ cells were determined as described in (E).
(H) Graphic presentation of the percentage of γH2AX+ TRIM44KD-CON and TRIM44KD MM cells with or without irradiation. The percentage of γH2AX+ cells were calculated as described in (F).
After irradiation, the ATM/ATR-dependent phosphorylation of the histone variant H2AX produces γH2AX as the initial signal for the subsequent accumulation of repair proteins at DNA lesions [21, 22]. The numbers of γH2AX-positive foci (γH2AX+) counted 5 minutes and 1 hour after irradiation were significantly lower in TRIM44OE MM cells than in control cells (Fig. S1C, Fig. 1E and F), whereas TRIM44 silencing led to increased numbers of γH2AX+ cells after irradiation (Fig. S1D, Fig. 1G and H). Hydroxyurea, a DNA replication inhibitor, causes DNA double-strand breaks during S phase [23]. Treatment with hydroxyurea led to similar results; TRIM44 expression in MM cells led to reduced numbers of γH2AX+ cells after hydroxyurea treatment (Fig. S2A), whereas TRIM44 silencing sensitized MM cells to DNA-damaging agents (Fig. S2B).
TRIM44 expression enhances DNA damage repair
To explore whether radioresistance in MM cells after TRIM44 expression is accompanied by enhanced recovery after IR-induced DNA damage, the time-course of the change in γH2AX foci in IR-treated MM cells was analyzed using immunofluorescence. γH2AX foci were counted at 1, 8 and 24 hours after irradiation. TRIM44OE MM cells showed the accelerated elimination of γH2AX foci at 24 h in RPMI cells and at 8 h in U266 cells (Fig. 2A and B, Fig. S3A and B). The number of γH2AX foci at 1 hour were normalized to 100%. On the other hand, TRIM44 silencing in MM cells resulted in significantly increased numbers of γH2AX+ cells after normalization compared to the controls (Fig. 2C and D, Fig. S3C and D), indicating that TRIM44 silencing decreased the DNA damage repair ability in MM cells. This trend continues after hydroxyurea treatment. TRIM44OE decreased the number of γH2AX foci with faster kinetics in MM cells, whereas TRIM44KD did not (Fig. S3E). IR treatment normally induces G2/M arrest [24]. In agreement with faster DNA damage repair kinetics, TRIM44 expression induced escape from IR-induced G2/M arrest at 24 h post-IR (Fig. S3F).
Figure 2. TRIM44 enhances DNA damage repair in MM cells.

(A) TRIM44OE MM cells show a decrease in IR-induced γH2AX foci with faster kinetics compared to control cells (OE-CON). At 1, 8 and 24 hours after irradiation (2 Gy), cells were immunostained with anti-γH2AX (red) and DRAQ5 (blue) and analyzed using confocal microscopy. Scale bars, 20 μm.
(B) Graphic presentation of the relative percentage of γH2AX+ TRIM44OE-CON and TRIM44OE MM cells. The cells containing > 5 γH2AX foci were recorded as γH2AX+ cells, whose percentage was averaged from at least 100 cells. The number of γH2AX foci at 1 hour were normalized to 100%. The data are displayed as the mean plus SD of three counts, and statistical significance was calculated and represented as the P value. *, P < 0.05; **, P < 0.01; ***, P < 0.001.
(C) TRIM44KD MM cells show delayed DNA damage repair after irradiation (2 Gy). γH2AX+ cells were determined as described in (A).
(D) Graphic presentation of the relative percentage of γH2AX+ TRIM44KD-CON and TRIM44KD MM cells. The relative percentage of γH2AX+ cells were calculated as described in (B). NS, non-significant.
(E) The COMET assay confirmed that TRIM44 enhances DNA damage repair in MM cells. Cells were treated with 2 Gy IR, and after 8 hours, COMET images were captured using fluorescence microscopy. Scale bars, 100 μm.
(F) Graphic presentation of the tail moment of TRIM44OE-CON and TRIM44OE MM cells. The tail moment was calculated as the tail length multiplied by the tail DNA percentage in at least 100 cells. The data are displayed as the mean plus SD of three counts, and statistical significance was calculated and represented as the P value. *, P < 0.05; **, P < 0.01; ***, P < 0.001.
(G) The COMET assay confirmed that TRIM44KD MM cells exhibit delayed DNA repair after irradiation. The tail moment was calculated as described in (E).
(H) Graphic presentation of the tail moment of TRIM44KD-CON and TRIM44KD MM cells. The tail moment were calculated as described in (F).
To determine whether the decrease in γH2AX foci was due to ongoing DNA repair or reflected cell loss, we performed neutral COMET assays [25]. Normalization between irradiation doses was accomplished by dividing the COMET tail DNA content score for each group by the total number of visual fields counted [26]. To further verify that the elimination of IR-induced γH2AX foci induced by TRIM44 reflected repaired DNA, a COMET assay was performed at 1h and 8 h post-IR, and the tail moment was calculated as the tail length × tail DNA % to evaluate the levels of DNA breaks. TRIM44OE reduced the tail moment at 1h and 8 h post-IR in MM cells (Fig. 2E and F, Fig. S4A–F), whereas TRIM44 silencing impaired the recovery of the cells from DNA damage, suggesting that TRIM44 supports the repair of IR-induced DNA breaks in MM cells (Fig. 2G and H, Fig. S4G–L). Furthermore, the relative percentage of tail moment were also calculated as in Figure S4E and S4J. As tail moment at 1 hr was set to 100%, tail moment at 8 hr showed an accelerated DNA damage repair in TRIM44 overexpressing cells compared to control cells. Consistently, tail moment at 8 hr of TRIM44 silencing cells were even higher than 1 hr time point (Fig. S4M and N). Together, our data support the involvement of TRIM44 in protecting cancer cells from initial DNA damage as well as the DNA damage repair step by regulating repair kinetics and promoting exit from cell cycle arrest.
TRIM44 expression enhances both NHEJ- and HR-mediated DNA repair
Deregulated DSB repair is one of the mechanisms underlying genomic instability in MM [27]. The resolution of double-strand DNA breaks requires DNA repair pathways, including nonhomologous end joining (NHEJ) and homologous recombination (HR). Both NHEJ and HR are reported to be increased in MM [9].
Since TRIM44 decreases DSBs post-IR in MM cells, we analyzed the roles of TRIM44 in NHEJ or HR. We used mCherry reporter assays to detect NHEJ activity. The NHEJ repair substrate contains a BamHI restriction site, allowing the introduction of a DSB. The transduction of HEK 293T cells was performed to overexpress or silence TRIM44, and the cells were transfected with a digested plasmid carrying a fluorescence gene, where the interruption of fluorescence gene expression mimicked a DNA double-strand break. The fluorescence intensity expressed in the TRIM44-modulated cells reflected NHEJ activity repairing DSBs via direct end ligation. In TRIM44OE cells, NHEJ activity was higher than that in TRIM44OE-CON cells (Fig. 3A, Fig. S4O), whereas the depletion of TRIM44 completely abrogated NHEJ activity (Fig. 3B, Fig. S4O). The DNA-PKcs regulatory subunits Ku70 and Ku80 are core NHEJ factors that are required for NHEJ-directed DSB repair [28]. We further assessed the expression of XRCC6, XRCC5 and PRKDC, which are genes encoding Ku70, Ku80 and DNA-PKcs. TRIM44OE significantly increased XRCC6 and XRCC5 expression (Fig. 3C). On the other hand, TRIM44 silencing resulted in the downregulation of the XRCC6, XRCC5 and PRKDC genes (Fig. 3D). 53BP1, a positive regulator that promotes NHEJ repair and protects DSB ends from processing by the DNA end-resection machinery during G1 phase of the cell cycle [29], was immunostained in IR-treated MM cells in a time-course experiment. TRIM44 expression rapidly increased 53BP1 levels (green) in the early stage of the DNA damage response (post-IR 1 h) and maintained 53BP1 levels for a longer time (24 h post-IR, Fig. 3E–G). Conversely, 53BP1 was undetectable in TRIM44KD cells until the late stage of the DNA damage response (24 h post-IR, Fig. 3H–J). We also measured the efficiency of HR in MM cells using an HR reporter construct to exclusively detect the HR pathway. The HR reporter was based on the recombination substrate DR-GFP [30]. HEK 293T TRIM44OE and TRIM44KD cells were cotransfected with the HR reporter plasmid and I-SceI, and the number of GFP-positive cells counted by flow cytometry provided a quantitative measure of HR efficiency. In TRIM44OE cells, HR activity was higher than that in TRIM44OE-CON cells, whereas the depletion of TRIM44 significantly reduced HR activity (Fig. 4A and B, Fig. S4O). We also investigated RAD51, a recombinase protein that plays an exclusive role in HR repair, in MM cells treated with IR. RAD51 was upregulated in IR-treated MM cells and colocalized with the DNA damage marker γH2AX [31]. TRIM44 expression led to a significant increase in RAD51, which also colocalized with γH2AX (Fig. 4C–E). We further tested RAD51 levels in IR-treated MM cells in a time-course experiment. TRIM44 expression rapidly increased RAD51 levels (red) in the early stage of the DNA damage response (post-IR 1 h) and maintained RAD51 levels for a longer time (24 h post-IR, Fig. 4F–H). Conversely, there RAD51 was undetectable in TRIM44KD cells until the late stage of the DNA damage response (24 h post-IR, Fig. 4I–K). These data support important roles of TRIM44 in enhancing NHEJ and HR activity and coordinating the DNA damage response in MM cells.
Figure 3. TRIM44 promotes NHEJ-mediated DNA repair and genome instability in MM.

(A, B) TRIM44 increases NHEJ activity. The BamHI-digested mCherry2-N1 and pDsRed2 plasmids, which was used as internal control (Fig. S4O), were co-transfected into TRIM44OE-CON, TRIM44OE, TRIM44KD-CON and TRIM44KD HEK 293T cells. After 48 h, the cells were harvested, and the intensity of mCherry fluorescence was measured using flow cytometry.
(C, D) XRCC6, XRCC5, and PRKDC expression was increased in TRIM44OE MM cells (C) but decreased in TRIM44KD MM cells (D), as measured using qRT-PCR.
(E-G) TRIM44OE MM cells showed an increase in IR-induced 53BP1 focus expression. Cells were collected at 1 h post-IR and immunostained with 53BP1 (green) and DRAQ5 (blue) (E). Scale bars, 20 μm. The cells containing > 5 53BP1 foci were recorded as 53BP1+ cells under confocal microscopy. The percentages of 53BP1+ cells were displayed as the mean plus SD of three counts, and statistical significance was performed (F). Quantification of fluorescence intensity was performed from randomized districts on slides from each treatment group. Results were expressed as average fluorescence intensity of nuclear 53BP1 per cell and statistical analysis was performed. Results are shown as means ± SD of 100 randomized cells obtained from three independent experiments (G). Statistical significance was calculated and represented as the P value. *, P < 0.05; **, P < 0.01; ***, P < 0.001.
(H-J) TRIM44KD MM cells showed fewer IR-induced 53BP1 foci compared to the control. 53BP1+ cells were determined as described in (F). Quantification of fluorescence intensity was calculated as described in (G).
Figure 4. TRIM44 upregulates HR-mediated DNA repair mechanisms and genome instability in MM.

(A, B) TRIM44OE cells exhibit increased HR-mediated DNA repair activity, but TRIM44KD cells exhibit decreased HR-mediated DNA repair activity. The I-SceI, pDRGFP, and pDsRed2 plasmids, which was used as internal control (Fig. S4O), were cotransfected into TRIM44OE-CON, TRIM44OE, TRIM44KD-CON and TRIM44KD HEK 293T cells. After 48 h, the cells were harvested, and the intensity of GFP fluorescence was measured using flow cytometry. The data are displayed as the mean plus SD of three counts, and statistical significance was calculated and represented as the P value. *, P < 0.05; **, P < 0.01; ***, P < 0.001.
(C-E) TRIM44 expression in MM cells greatly increased the expression of RAD51 after irradiation (2 Gy). Cells were collected at 8 h post-IR and immunostained with RAD51 (green), γH2AX (red) and DRAQ5 (blue) (C). Scale bars, 20 μm. The cells containing > 5 RAD51 foci were recorded as RAD51+ cells under confocal microscopy. The percentages of RAD51+ cells were displayed as the mean plus SD of three counts, and statistical significance was performed (D). Quantification of fluorescence intensity was performed from randomized districts on slides from each treatment group. Results were expressed as average fluorescence intensity of nuclear RAD51 colocalized with γH2AX per cell and statistical analysis was performed. Results are shown as means ± SD of 100 randomized cells obtained from three independent experiments (E). Statistical significance was calculated and represented as the P value. *, P < 0.05; **, P < 0.01; ***, P < 0.001.
(F-H) TRIM44OE MM cells show an increase in RAD51 foci after irradiation (2 Gy). After 1, 8 and 24 hours, cells were immunostained with anti-RAD51 (red, Alexa-594) and DRAQ5 (blue) and analyzed under confocal microscopy (F). Scale bars, 20 μm. The cells containing > 5 RAD51 foci were recorded as RAD51+ cells. The percentage was averaged from at least 200 cells. The data are displayed as the mean plus SD of three counts, and statistical significance was calculated and represented was performed (G). Quantification of fluorescence intensity was performed from randomized districts on slides from each treatment group. Results were expressed as average fluorescence intensity of nuclear RAD51 per cell and statistical analysis was performed. Results are shown as means ± SD of 100 randomized cells obtained from three independent experiments (H). Statistical significance was calculated and represented as the P value. *, P < 0.05; **, P < 0.01; ***, P < 0.001.
(I-K) TRIM44KD MM cells show fewer RAD51 foci after irradiation (2 Gy) compared to controls (KD-CON). The percentage was averaged from at least 200 cells. RAD51+ cells were determined as described in (G). Quantification of fluorescence intensity was calculated as described in (H)
p62 is a binding partner for TRIM44
To determine the mechanisms of the TRIM44-mediated DNA response and repair, we conducted mass spectrometry analyses. Mass spectrometry analyses revealed the interaction of TRIM44 with the sequestosome protein (p62, also known as SQSTM1). We confirmed the interaction in immunoblots (Fig. 5A). Confocal analyses further confirmed their binding, which demonstrated colocalization between TRIM44 and p62 in MM cells (Fig. 5B). We then generated various truncated forms of p62 to identify TRIM44 interaction domains (Fig. S5A). Quantification of the florescence using confocal microscopy showed that most p62 deletional mutants decreased interaction with TRIM44 compared to full-length of p62 (Fig. S5B).
Figure 5. TRIM44 interacts with p62.

(A) TRIM44OE MM cell lysates were immunoprecipitated with anti-TRIM44 and immunoblotted using TRIM44 or p62 antibodies.
(B) Confocal analyses show colocalization between TRIM44 and p62. U266 cells were treated with MG132 (5 μM) for 1 hour, and the results were visualized with antibodies against p62 and TRIM44. Scale bars, 10 μm.
(C) Illustrations of different deletional TRIM44 constructs. GFP-tagged wild-type (WT) TRIM44; full-length (FL) TRIM44 (aa 1–344); ZF domain (aa 13–48); BB domain (aa 174–215); CC domain (aa 290 to 325); dCC domain (aa 1 to 290), a construct lacking the CC domain; and dZF domain (aa 48 to 344), a construct lacking the ZF domain.
(D) Different deletional GFP-TRIM44 mutants were transfected into HeLa cells with full-length HA-p62 to identify the TRIM44 domains responsible for p62 binding. Overlapping immunofluorescence in the images is shown in yellow. All experiments were replicated three independent times with similar results. Scale bars, 10 μm.
TRIM44 has several known functional domains (Fig. 5C). We also generated TRIM44 mutant protein constructs to identify the TRIM44 domains responsible for colocalization and binding with p62. We generated constructs of (a) full-length TRIM44 (FL, 1–344); (b) only the zinc finger domain (ZF, 13–48); (c) the B-box domain (BB, 174–215); (d) the coiled-coil domain (CC, 290–325); (e) TRIM44 with a deleted CC domain (dCC, 1–290); (f) and TRIM44 with a deleted ZF domain (dZF, 48–344). Transfection with BB domain construct led to the complete loss of the interaction with p62 (Fig. 5D). The deletion of other binding sites resulted in a reduced interaction with p62 to varying degrees. Only the ZF domain construct maintained the same degree of interaction with p62 as the full-length protein.
p62 is oligomerized by TRIM44
Under confocal microscopy, we noted that the transfection of full-length TRIM44 with p62 resulted in distinguishable p62 dots, which were not observed in association with the other truncated forms of TRIM44 or GFP controls (Fig. 5D). p62 can occur as a monomer or dimer, but p62 oligomerization by another protein has not been reported. This could indicate important biological functions of p62 in various diseases since p62 oligomerizes and sequesters target proteins in inclusions, including Keap1, a negative regulator of the antioxidant response [32].
Due to its high molecular weight, oligomerized p62 does cannot be resolved in a standard nonreducing SDS gel. Treatment with reducing agents such as βME or DTT disrupts disulfide bonds and helps to detect monomeric p62. To analyze the effects of TRIM44 on p62 oligomerization, mCherry-p62 was transfected into TRIM44OE, TRIM44KD, and control MM (lentivirus-mediated) cells and HEK 293T cells. We generated mCherry-p62 clones from single HEK 293T or HeLa cell sorting to ensure that all cells contained the same amount of p62. Confocal analyses showed that the transfection of TRIM44 resulted in its colocalization with p62 and increased the polymerization of p62 (Fig. 6A). These cells were cross-linked with DSP after MG132 treatment, and the lysates were run under reducing or nonreducing conditions. The data showed that TRIM44 expression increased the amount of oligomerized p62 (Fig. 6B, Fig. S6A). The ratios of oligomerized and monomeric p62 under nonreducing versus reducing conditions were quantified by immunoblotting. Since TRIM44 promotes p62 oligomerization, we examined whether TRIM44 interacts with oligomerized p62. We used the reversible crosslinking agent DSP to treat cell lysates and subjected them to co-IP, which showed that TRIM44 interacts with both the p62 monomer and polymer (Fig. 6C). We then cotransfected HA-p62 and the TRIM44 mutants with deleted domains into HeLa cells to determine the domains responsible for p62 oligomerization. The percentage of cells containing p62 aggregates was then quantified. The number of aggregates in the cells transfected with full-length TRIM44 was 60% higher than in the cells transfected with GFP alone, indicating that TRIM44 is essential for p62 aggregation (Fig. 6D). In contrast, the BB and dZF mutants significantly decreased the level of p62 aggregation (Fig. 6D). Taken together, these findings indicate that TRIM44 promotes p62 oligomerization via the ZF domain.
Figure 6. TRIM44 promotes p62 deubiquitination.

(A) GFP-TRIM44 and HA-p62 plasmids were transfected into HeLa cells and immunofluorescence imaging were analyzed using confocal microscopy (Left). Graphic presentation of the average volume of p62 oligomerization 48h after transfection. The volume of p62 oligomerization in 50 cells was counted for each data point shown (each bar) (Right). Student’s t test was used for statistical analysis. *, P < 0.05; **, P < 0.01; ***, P < 0.001. Scale bars, 10 μm.
(B) HA-p62 plasmids were transfected into HEK 293T cells expressing different levels of TRIM44 (TRIM44KD-CON, TRIM44KD, TRIM44OE-CON, or TRIM44OE). The cells were treated with or without DSP and lysed in RIPA buffer 2 hours after irradiation. The lysates were subjected to immunoblotting.
(C) TRIM44 interacts with monomer as well as oligomerized p62. 293T TRIM44OE cells are transfect with mCherry-p62 and then treated with MG132 (5 μM, 6 hours) and cross-linked using DSP. For reduced condition, we used 0.1%SDS. For non-Reduced condition, normal lysis buffer was used. Immunoprecipitation were performed using anti-TRIM44 followed by immunoblots using anti-p62.
(D) ZF domain of TRIM44 is responsible for p62 oligomerization. HeLa cells were co-transfected with HA-p62 and full-length or different GFP-TRIM44 truncations. Shown the increased percentage of cells with p62 oligomerization compared to the cells transfected with GFP and HA-p62 (means + SD, n=3). The data are displayed as the mean plus SD of three counts, and statistical significance was calculated and represented as the P value. *, P < 0.05; **, P < 0.01; ***, P < 0.001. NS, non-significant.
(E) Deubiquitination assay using HEK293T cells transfected with HA empty vector or HA-p62, His-Ubiquitin and GFP-TRIM44 plasmids and treated with proteasome inhibitor MG132 (5 μM, 6 hours). Cells were collected and IP against HA antibody. Ubiquitinated HA-p62 was immunoblotted with anti-Ub antibody.
(F) HA-Ub (K48R, and K63R) were expressed with or without GFP-TRIM44 in HEK293T cells. Cells were subjected to HA IP and IB.
(G) Identification of the TRIM44 functional domain for p62 deubiquitination. HEK293T cells were transfected with a combination of HA-p62, His-Ub, and various truncated forms of GFP-TRIM44 (ZF UBP domain (ZF), B-box domain (BB), or coiled-coil domain (CC)). The cells were treated with MG132 (5 μM) for 6 hrs before collection. Ubiquitinated p62 was measures by immunoblotting.
It has been reported that the ubiquitylation of p62 prevents its dimerization and sequestration [32]. Since TRIM44 functions as a deubiquitinase, we performed deubiquitination assays with exogenous p62. TRIM44 reduced the levels of ubiquitinated p62 (Fig. 6E). These data suggest that TRIM44 promotes p62 oligomerization through its deubiquitination. A deubiquitination assay further revealed that TRIM44 deubquitylates p62 via the K48 and K63 linkage (Fig. 6F). To identify the functional domains responsible for p62 deubiquitination by TRIM44, we cotransfected p62 and Ub with the ZF, BB, or CC domains of TRIM44 into HEK 293T cells. Compared with the BB or CC domain, full-length TRIM44 and the ZF domain significantly inhibited p62 polyubiquitination, indicating that the ZF domain is critical for p62 deubiquitination (Fig. 6G). TRIM21 can prevent p62 oligomerization via p62 ubiquitination [32], therefore, we analyzed whether TRIM44 can reverse the effect of TRIM21 in p62 oligomerization. We co-transfected HA-p62 with TRIM21 and TRIM44 or GFP as controls and quantified p62 oligomerization using confocal. The numbers and size of aggregates in the cells co-transfected with TRIM21 and TRIM44 are 3-fold higher than the cells transfected with TRIM21 alone (Fig. S6B–D). Our data support TRIM44 promotes oligomerization of p62 and reverses p62 monomerization mediated by TRIM21.
TRIM44 prevents p62 from localizing to the nucleus in response to irradiation
p62 shuttles between the cytoplasm and the nucleus, mediated by a nuclear export signal (NES) and two nuclear localization signals (NLS) [33]. To investigate the mechanisms by which TRIM44-p62 contributes to DNA damage repair, we first detected the nuclear transportation of p62 after irradiation in TRIM44-expressing cells. HeLa cells were stably transfected with mCherry-p62 together with GFP or GFP-TRIM44, pretreated with LMB to inhibit the export of nuclear proteins, and then subjected to irradiation and fixed at different time points. A time course experiment after irradiation showed that p62 gradually translocated to the nucleus after irradiation, but TRIM44 transfection prevented p62 translocation to the nucleus (Fig. 7A).
Figure 7. TRIM44 increases FLNA, RAD51 and 53BP1 expression by inhibiting p62 nuclear translocation after irradiation.

(A) TRIM44 expression inhibits p62 nuclear translocation after irradiation. TRIM44OE-CON and TRIM44OE, TRIM44KD-CON and TRIM44KD HeLa cells were transfected with mCherry-p62. Images were captured at the indicated times.
(B) TRIM44 increases nuclear FLNA expression. TRIM44OE-CON and TRIM44OE, TRIM44KD-CON and TRIM44KD U266 cells were treated with or without IR. Cells were collected 2 hours after irradiation.
(C) TRIM44 increases nuclear RAD51 expression in MM cells, which decreases in TRIM44KD MM cells. Cells were treated with or without IR and collected 2 hours after irradiation.
(D) TRIM44 increases nuclear 53BP1 expression in U266 cells, which decreases in TRIM44KD MM cells.
(E) TRIM44 increases the protein half-life of FLNA, 53BP1 and RAD51. TRIM44OE-CON and TRIM44OE MM cells were treated with IR, and cells were collected at the indicated times after CHX treatment. The protein levels of FLNA, 53BP1 and RAD51 were analyzed using immunoblots.
(F) Down-regulation of TRIM44 decreases the protein half-life of FLNA, 53BP1 and RAD51. TRIM44KD-CON and TRIM44KD MM cells were treated with or without IR, and cells were collected at the indicated times after CHX treatment. The protein levels of FLNA, 53BP1 and RAD51 were analyzed using immunoblots.
Upon irradiation, p62 translocates to the nucleus. Nuclear p62 facilitates the proteasomal degradation of FLNA and RAD51, two essential DNA repair machinery components [20]. Since TRIM44 prevents p62 translocation to the nucleus via polymerization, we tested whether TRIM44 prevents the proteasomal degradation of FLNA and RAD51, which promote DNA repair after irradiation.
To test our hypothesis, we analyzed RAD51 and FLNA levels in the nuclear fraction of TRIM44OE and TRIM44KD cells after irradiation. FLNA interacts with BRCA1/2 and recruits RAD51 for the HR repair pathway [34]. TRIM44 overexpression led to increased FLNA in the nucleus, whereas TRIM44 silencing decreased nuclear FLNA (Fig. 7B, Fig. S7A and B). RAD51 levels were also reduced upon TRIM44 silencing (Fig. 7C, Fig. S7A and B).
53BP1 is an enigmatic DNA damage response factor that functions as a DSB escort that guards against illegitimate and potentially tumorigenic recombination [35]. Nuclear 53BP1 levels were increased in TRIM44OE cells, and the silencing of TRIM44 decreased nuclear 53BP1 (Fig. 7D, Fig. S7A and B). We also analyzed the half-life of FLNA, RAD51 and 53BP1 in TRIM44-expressing and TRIM44-silenced cells. The expression of TRIM44 stabilized FLNA, RAD51 and 53BP1 and extended their half-life (Fig. 7E, Fig. S7C). TRIM44 silencing decreased FLNA, RAD51 and 53BP1 protein stability (Fig. 7F, Fig. S7D).
To further confirm our hypothesis that the TRIM44 mediated 53BP1 and FLNA overexpression could increase IR resistance in MM cells, we silenced 53BP1 or FLNA in TRIM44OE MM cells. Both 53BP1 and FLNA knockdown markedly increased apoptosis in TRIM44OE MM cells at 24 h post-IR (Fig. S8A and B). Moreover, 53BP1 knockdown significantly decreased the NHEJ activity in TRIM44OE MM cells (Fig. S8C), and FLNA knockdown significantly decreased the HR activity (Fig. S8D). Together, these data suggest that TRIM44 prevents p62 localization to the nucleus via oligomerization, resulting in the prevention of the degradation of FLNA, RAD51, and 53BP1, and increased protein stability upon irradiation.
TRIM44 knock down induced sensitization to IR is p62 dependent
To further validate our hypothesis that TRIM44KD-induced sensitivity is due to increased p62 nuclear translocation, we silenced p62 in TRIM44KD MM cells. p62 knockdown markedly decreased apoptosis in TRIM44KD MM cells at 24 h post-IR (Fig. 8A, Fig. S9A–C). Furthermore, p62 silencing significantly decreased the formation of IR-induced γH2AX foci at 1 h post-IR (Fig. 8B and C, Fig. S9D and E). Accordingly, faster elimination of γH2AX foci was observed in p62-depleted TRIM44KD MM cells (Fig. 8D and E, Fig. S9F and G). Additionally, stronger 53BP1 staining was observed in p62-depleted TRIM44KD MM cells than in p62-proficient TRIM44KD MM cells in the early stage of DNA damage. Moreover, an increased 53BP1 half-life was observed in p62-depleted TRIM44KD MM cells (Fig. 8F and G, Fig. S9H and I). Concomitantly, increased 53BP1, RAD51 and FLNA protein levels were confirmed using immunoblotting in p62 downregulated TRIM44KD MM cells after IR (Fig. 8H, Fig. S9J–L). These data indicate that TRIM44 increases the DNA damage response and repair via p62.
Figure 8. Knockdown of p62 rescues TRIM44KD-mediated IR sensitivity.
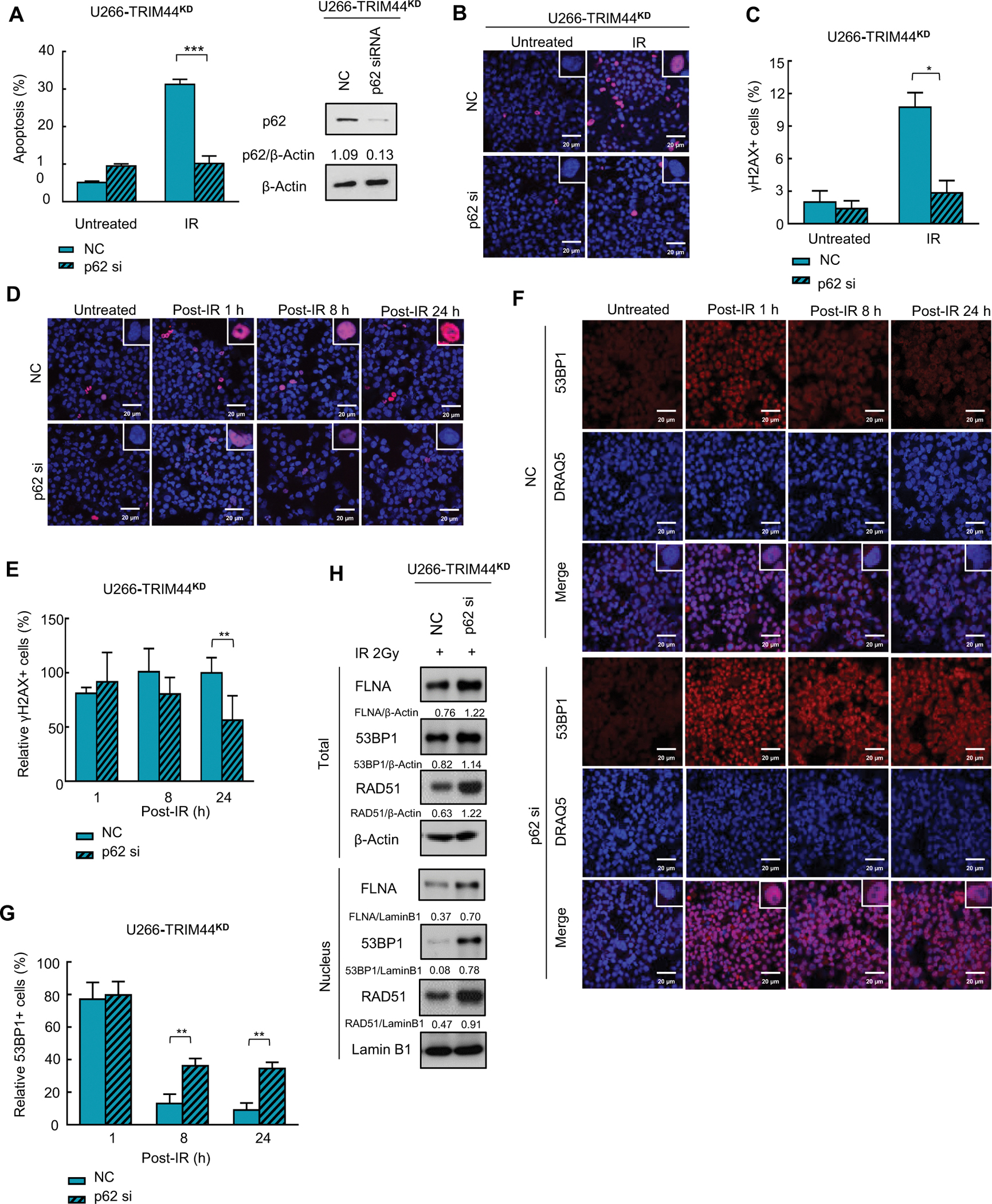
(A) Downregulation of p62 decreases IR-induced apoptosis in TRIM44KD MM cells. TRIM44KD MM cells were transfected with NC (sc-37007, Santa Cruz, CA, USA) or p62 siRNA (sc-29679, Santa Cruz, CA, USA), and the cells were then treated with 2 Gy IR. Apoptosis was evaluated by using annexin V/7-AAD staining and flow cytometry.
(B) Downregulation of p62 decreases IR-induced γH2AX foci in irradiated TRIM44KD MM cells. TRIM44KD U266 cells were transfected with NC or p62 siRNA, and the cells were then treated with 2 Gy IR. After 1 hour, the cells were immunostained with anti-γH2AX (red) and DRAQ5 (blue) and observed by confocal microscopy. Scale bars, 20 μm.
(C) Graphic presentation of the percentage of γH2AX+ MM cells with or without irradiation. The cells containing > 5 γH2AX foci were recorded as γH2AX+ cells, whose percentage was averaged from at least 100 cells. The data are displayed as the mean plus SD of three counts, and statistical significance was calculated and represented as the P value. *, P < 0.05; **, P < 0.01; ***, P < 0.001.
(D) p62 knockdown accelerated the decrease in IR-induced γH2AX foci in TRIM44KD MM cells. TRIM44KD U266 cells were transfected with NC or p62 siRNA, and the cells were then treated with 2 Gy IR. The cells were collected at indicated time after IR, then the cells were immunostained with anti-γH2AX (red) and DRAQ5 (blue) and observed by confocal microscopy. Scale bars, 20 μm.
(E) Graphic presentation of the relative percentage of γH2AX+ MM cells after irradiation. The cells containing > 5 γH2AX foci were recorded as γH2AX+ cells, whose percentage was averaged from at least 100 cells. The data are displayed as the mean plus SD of three counts, and statistical significance was calculated and represented as the P value. *, P < 0.05; **, P < 0.01; ***, P < 0.001.
(F) p62 knockdown preserves IR-induced 53BP1 foci in TRIM44KD MM cells. TRIM44KD U266 cells were transfected with NC or p62 siRNA, and the cells were then treated with 2 Gy IR. Cells were collected at 2 h post-IR and immunostained with 53BP1 (green) and DRAQ5 (blue) and observed by confocal microscopy. Scale bars, 20 μm.
(G) Graphic presentation of the relative percentage of 53BP1+ MM cells after irradiation. The cells containing > 5 53BP1 foci were recorded as 53BP1+ cells under confocal microscopy, whose percentage was averaged from at least 100 cells. The data are displayed were displayed as the mean plus SD of three counts, and statistical significance was calculated and represented as the P value. *, P < 0.05; **, P < 0.01; ***, P < 0.001.
(H) Downregulation of p62 stabilizes nuclear FLNA, 53BP1 and RAD51 in TRIM44 knockdown cells. TRIM44KD U266 cells were transfected with NC or p62 siRNA, and the cells were collected 2 hours after irradiation. The protein levels of FLNA, 53BP1 and RAD51 were analyzed using immunoblots. LaminB1-Nucleus, β-Actin-Total.
Discussion
TRIM44 is highly expressed in different cancers, and its expression is associated with drug resistance and progression in many cancer types [13, 14, 36]. However, the detailed mechanisms by which TRIM44 regulates these processes in cancers still need to be elucidated.
Our lab first discovered that TRIM44 is highly upregulated in drug-resistant quiescent MM stem-like cells in the osteoblastic niche [37].As a deubiquitinase, TRIM44 can stabilize HIF-1α to promote MM survival in a hypoxic niche [19]. In this study, we delineated novel roles of TRIM44 in DNA damage responses and repair. Extrinsic and intrinsic factors such as chemical drugs, oxidative stress and proliferative stress result in DNA damage. Unrepaired DNA damage leads to cell cycle arrest, cell senescence or even cell death. By manipulating the TRIM44 gene in MM cells, we discovered that TRIM44 expression protects MM cells from IR-induced DNA damage, decreases apoptosis, and increases colony formation in MM cells. We also discovered that TRIM44 accelerates DNA repair kinetics at IR-mediated DSBs and promotes HR- and NHEJ-mediated DNA damage repair in MM cells.
Error-free DNA repair is essential for cells to survive DNA damage induced by extrinsic attack and intrinsic stress [38]. However, erroneous DNA repair causes genetic mutations, chromosomal abnormalities, and genomic instability [39]. Two common DNA repair pathways, HR and NHEJ, are responsible for the repair of DSBs [40]. Although HR is considered an error-free DSB repair pathway, genomic rearrangement can occur when the HR pathway is activated [41]. The NHEJ repair pathway tends to generate small insertions or deletions at the junction site and to be highly mutagenic for chromosomal translocation [40]. In a study by Herrero et al., both HR and NHEJ were found to be upregulated in MM, and increases in DNA deletions and microhomology usage suggested that the NHEJ repair pathway is highly activated in MM.[9]
It has been reported that p62 may regulate DNA repair by interacting with RNF168, result in downregulating RNF169’s activity and decreasing chromatin ubiquitination which ultimately decreases BRCA1 and Rad51 recruitment to the damage sites and decreases DNA repair [42]. We found that p62/sequestosome 1 (SQSTM1) is a novel binding partner of TRIM44. p62 constitutively shuttles between the nucleus and the cytoplasm [33]. Because of the small size of nuclear pores (~50 nm), with a functional diameter of opening of 9 nm, only small water-soluble molecules or small proteins can passively diffuse through nuclear pores. Proteins larger than ~40 kDa have to be actively transported by karyopherins [43]. p62 contains one leucine-rich nuclear export signal motif between amino acids 303–320 and two basic monopartite nuclear localization signals at amino acids 186–189 (NLS1) and amino acids 264–267 (NLS2). Several mechanisms are known to regulate the nucleocytosolic distribution of p62, including polymerization, the phosphorylation of serine and threonine residues in NLS2 and binding to ubiquitin substrates. The polymerization of p62 is essential for the recognition of ubiquitin cargos [44] and for the autophagic degradation of p62 [45]. p62 polymerization can delay nuclear localization and reduce the rate of nuclear-cytoplasmic shuttling. We discovered that TRIM44 deubiquitinates p62 and promotes its polymerization. Polymerized p62 is not able to shuttle to the nucleus in response to irradiation, which prevents the proteasomal degradation of FLNA and RAD51 in the nucleus. Increased FLNA and RAD51 levels within the nucleus increase DNA repair upon DNA damage. In summary, our data indicate TRIM44 as a potential therapeutic target for MM cells in which the DNA damage response and repair are active and suggest a new paradigm of p62 regulation and DNA damage responses and repair in cancers.
Methods
Cells
HeLa, HEK293T, and MM cell lines (U266 and RPMI8226) were purchased from the ATCC (Rockland, MD). We verified these cell lines via STR profiling (short tandem repeat analysis of DNA). Cell lines that contained knockout or overexpression constructs were not passaged beyond ~8 generations (1–1.5 months).
HeLa and HEK293T cells were cultured in in Dulbecco’s Modified Eagle’s medium (Invitrogen, Carlsbad, CA, USA) with 10% FBS (Gibco). RPMI8226 and U266 cells were cultured in RPMI medium (Invitrogen, Carlsbad, CA, USA) with 10% FBS (Gibco). Both RPMI8226 and U266 cells are B lymphocyte isolated from peripheral blood. U266 cells have been reported to produce human IL-6, and RPMI8226 cells have been reported to produce immunoglobulin light chain. Moreover, RPMI8226 cells have also been tested for KRAS mutation (p.G12A c.35G>C).
We generated cells that in which TRIM44 was overexpressed (TRIM44OE) or knocked down (TRIM44KD) via a lentivirus. Control cells were infected with relevant control vectors corresponding to the infecting virus vector (TRIM44OE-CON or TRIM44KD-CON). Lentivirus were packaged using transfer vector (), viral packaging (psPAX2), and viral envelope (pMD2G) in 293FT cells.
Antibodies and reagents
An anti-TRIM44 polyclonal (11511-1-AP) antibody was purchased from the Proteintech Group (Chicago, IL, USA). An anti-Ub (646301) antibody was purchased from Biolegend (San Diego, CA, USA). Anti-mCherry (PA5-34974) antibody was purchased from ThermoFisher (Rockford, IL, USA). Anti-GFP (sc-9996), anti-β-Actin (sc-47778), and anti-HA (sc-805) antibodies were purchased from Santa Cruz Biotechnology Biotech (Santa Cruz, CA, USA). Anti-γH2AX was purchased from Millipore (Temecula, CA, USA). An anti-RAD51, anti-53BP1, anti-FLNA and anti-p62 (88588) monoclonal antibody was purchased from CST (Cell Signaling Technology, MA, USA). MG132 and cycloheximide (CHX) were purchased from Selleck Chemicals (Houston, TX, USA). Hydroxyurea was purchased from Sigma-Aldrich (Burlington, MA, USA).
Plasmids
To construct p62 truncates, a series of deletions of p62 were amplified by PCR using appropriate primers and cloned into the HA empty vector. GFP-TRIM44, and its deletion mutant expression plasmids have been described previously [19].
Apoptosis assay
MM cells were irradiated with 2 Gy irradiation and harvested after 24 h. The cells were stained with PE-Annexin V and 7-AAD (BD Bioscience) and examined with an LSR-II flow cytometer. The percentage of apoptosis was analyzed using FACS Diva software (BD Bioscience).
Cell cycle analysis
MM cells were treated with 2 Gy irradiation and harvested at the indicated times for cell cycle analysis. The cells were fixed with ice-cold 70% ethanol for 1 h, treated with 100 μg/ml RNase A and stained with 50 μg/ml propidium iodide for 20 min. The distribution of cell cycle stages was assessed using an LSR-II flow cytometer.
Immunofluorescence and confocal microscopy
MM cells were treated with 2 Gy irradiation, collected at the indicated times and cytospun onto slides at 800 rpm for 4 min. The cells were fixed with formalin at RT for 15 min, blocked with Animal-free Blocker (Vector), and incubated with the primary antibody Alexa Fluor® 555-conjugated-anti-γH2AX (EMD Millipore), anti-RAD51 (Santa Cruz) or Alexa Fluor® 594-conjugated-anti-53BP1 (Bioss) at 4°C overnight. After washing with PBS, the slides were incubated with the nuclear dye Draq5 (Abcam) at RT for 2 h. Immunofluorescence imaging was performed using a Leica TSC SP5 confocal microscope. Nuclei with five or more γH2AX foci were recorded as γH2AX+. In each experiment, a minimum of 100 cells were analyzed.
Colony formation assay
MM cells treated with/without 2 Gy irradiation were plated at 5000 cells per 35 mm petri dish. After 10 – 14 days, colonies were counted by using an Olympus IX70 microscope, and images were analyzed by using CellSens software (Olympus). A colony composed of over 40 cells was scored.
RNA extraction and real-time PCR
Total RNA was isolated using the Direct-zolTM RNA miniPrep kit (Zymo Research), and cDNA was synthesized using random hexamers and the ImProm-IITM Reverse Transcription System (Promega). Gene expression was determined using an ABI 7900 system (Applied Biosystems) with SYBR Green MasterMix Plus (Thermo Scientific) and normalized to ACTB expression. Relative expression was calculated as 2−(Ct Target - Ct Control).
COMET assay
The COMET assay was described in detail previously.[26] Briefly, MM cells were treated with/without 2 Gy irradiation, collected at 8 h postirradiation and embedded in 1% low-gelling-temperature agarose. The embedded cells were lysed overnight, followed by electrophoresis under alkaline conditions. The slides were stained with propidium iodide and observed using an Olympus IX70 microscope. Images were analyzed using the ImageJ plug-in in OpenComet software, and the tail moment was calculated as the tail length × tail DNA %. At least 100 cells were examined on each slide, and the results were averaged.
Immunoprecipitation and immunoblotting
Cells were lysed in RIPA buffer containing a protease inhibitor (Sigma) and incubated with indicated antibodies (anti-TRIM44 (Proteintech, 11511-1-AP), anti-HA (Santa Cruz, sc-805, and anti-mCherry (ThermoFisher, PA5-34974) antibodies) together with the protein A/G plus-agarose immunoprecipitation reagent (Santa Cruz) at 4°C overnight. Immunoprecipitates were eluted with Laemmli sample buffer (Bio-Rad) and analyzed using the indicated antibodies.
Cell transfection
Cells were transfected by Lipofectamine® 3000 (Invitrogen, L3000015) following the protocol.
Nuclear fraction extraction
Nuclear fractions were extracted by kit ().
CHX chase assay
MM cells were seeded into 6-well plates at a density of 3 × 105 cells/well and incubated overnight at 37 °C in a CO2 incubator. Cells were treated with 50 μg/ml of cycloheximide (CHX) dissolved in absolute ethanol, and harvested in ice cold phosphate buffered saline (PBS, pH 7.4) at varying chase points by centrifugation at 2500 × g for 10 min at 4 °C. Cell pellets were lysed in a lysis buffer. Samples were heated at 95 °C for 10 min.
NHEJ activity assay
HEK 293T cells were transfected with the BamHI-digested mCherry2-N1 plasmid (Addgene). After 48 h, the cells were detached, and the mCherry signal was determined by flow cytometry.
HR activity assay
HEK 293T cells were cotransfected with pDRGFP and I-SceI plasmids (Addgene). After 48 h, the cells were detached, and the GFP signal was determined by flow cytometry.
Statistical analysis
Data were analyzed by using the unpaired two-tailed Student’s t test, and P<0.05 was considered statistically significant. The results are expressed as the mean ± SD from at least 3–5 independent experiments.
Supplementary Material
Acknowledgements
This work is supported by NIH grant (R01CA181319) and CPRIT grant (RP20093) given to NM.
Footnotes
Competing Interests statement
All authors declare no conflict of interest.
Reference
- 1.Harper JW, Elledge SJ. The DNA damage response: ten years after. Molecular cell 2007; 28: 739–745. [DOI] [PubMed] [Google Scholar]
- 2.Anderson KC. Multiple myeloma: a clinical overview. Oncology (Williston Park) 2011; 25 Suppl 2: 3–9. [PubMed] [Google Scholar]
- 3.Boehrer S, Ades L, Tajeddine N, Hofmann WK, Kriener S, Bug G et al. Suppression of the DNA damage response in acute myeloid leukemia versus myelodysplastic syndrome. Oncogene 2009; 28: 2205–2218. [DOI] [PubMed] [Google Scholar]
- 4.Walters DK, Wu X, Tschumper RC, Arendt BK, Huddleston PM, Henderson KJ et al. Evidence for ongoing DNA damage in multiple myeloma cells as revealed by constitutive phosphorylation of H2AX. Leukemia 2011; 25: 1344–1353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Munshi NC, Anderson KC. New strategies in the treatment of multiple myeloma. Clinical cancer research : an official journal of the American Association for Cancer Research 2013; 19: 3337–3344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Michels TC, Petersen KE. Multiple Myeloma: Diagnosis and Treatment. Am Fam Physician 2017; 95: 373–383. [PubMed] [Google Scholar]
- 7.Blanpain C, Mohrin M, Sotiropoulou PA, Passegue E. DNA-damage response in tissue-specific and cancer stem cells. Cell stem cell 2011; 8: 16–29. [DOI] [PubMed] [Google Scholar]
- 8.Shammas MA, Shmookler Reis RJ, Koley H, Batchu RB, Li C, Munshi NC. Dysfunctional homologous recombination mediates genomic instability and progression in myeloma. Blood 2009; 113: 2290–2297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Herrero AB, San Miguel J, Gutierrez NC. Deregulation of DNA double-strand break repair in multiple myeloma: implications for genome stability. PloS one 2015; 10: e0121581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Nisole S, Stoye JP, Saib A. TRIM family proteins: retroviral restriction and antiviral defence. Nature reviews Microbiology 2005; 3: 799–808. [DOI] [PubMed] [Google Scholar]
- 11.Cambiaghi V, Giuliani V, Lombardi S, Marinelli C, Toffalorio F, Pelicci PG. TRIM proteins in cancer. Advances in experimental medicine and biology 2012; 770: 77–91. [DOI] [PubMed] [Google Scholar]
- 12.Jarvinen AK, Autio R, Kilpinen S, Saarela M, Leivo I, Grenman R et al. High-resolution copy number and gene expression microarray analyses of head and neck squamous cell carcinoma cell lines of tongue and larynx. Genes, chromosomes & cancer 2008; 47: 500–509. [DOI] [PubMed] [Google Scholar]
- 13.Luo Q, Lin H, Ye X, Huang J, Lu S, Xu L. Trim44 facilitates the migration and invasion of human lung cancer cells via the NF-kappaB signaling pathway. International journal of clinical oncology 2015; 20: 508–517. [DOI] [PubMed] [Google Scholar]
- 14.Kawaguchi T, Komatsu S, Ichikawa D, Hirajima S, Nishimura Y, Konishi H et al. Overexpression of TRIM44 is related to invasive potential and malignant outcomes in esophageal squamous cell carcinoma. Tumour biology : the journal of the International Society for Oncodevelopmental Biology and Medicine 2017; 39: 1010428317700409. [DOI] [PubMed] [Google Scholar]
- 15.Tan Y, Yao H, Hu J, Liu L. Knockdown of TRIM44 Inhibits the Proliferation and Invasion in Prostate Cancer Cells. Oncology research 2017; 25: 1253–1259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kashimoto K, Komatsu S, Ichikawa D, Arita T, Konishi H, Nagata H et al. Overexpression of TRIM44 contributes to malignant outcome in gastric carcinoma. Cancer science 2012; 103: 2021–2026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Yamada Y, Takayama KI, Fujimura T, Ashikari D, Obinata D, Takahashi S et al. A novel prognostic factor TRIM44 promotes cell proliferation and migration, and inhibits apoptosis in testicular germ cell tumor. Cancer science 2017; 108: 32–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kawabata H, Azuma K, Ikeda K, Sugitani I, Kinowaki K, Fujii T et al. TRIM44 Is a Poor Prognostic Factor for Breast Cancer Patients as a Modulator of NF-kappaB Signaling. International journal of molecular sciences 2017; 18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Chen Z, Lin TC, Bi X, Lu G, Dawson BC, Miranda R et al. TRIM44 promotes quiescent multiple myeloma cell occupancy and survival in the osteoblastic niche via HIF-1alpha stabilization. Leukemia 2019; 33: 469–486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hewitt G, Carroll B, Sarallah R, Correia-Melo C, Ogrodnik M, Nelson G et al. SQSTM1/p62 mediates crosstalk between autophagy and the UPS in DNA repair. Autophagy 2016; 12: 1917–1930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Huen MS, Grant R, Manke I, Minn K, Yu X, Yaffe MB et al. RNF8 transduces the DNA-damage signal via histone ubiquitylation and checkpoint protein assembly. Cell 2007; 131: 901–914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kolas NK, Chapman JR, Nakada S, Ylanko J, Chahwan R, Sweeney FD et al. Orchestration of the DNA-damage response by the RNF8 ubiquitin ligase. Science 2007; 318: 1637–1640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Petermann E, Orta ML, Issaeva N, Schultz N, Helleday T. Hydroxyurea-stalled replication forks become progressively inactivated and require two different RAD51-mediated pathways for restart and repair. Mol Cell 2010; 37: 492–502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Gogineni VR, Nalla AK, Gupta R, Dinh DH, Klopfenstein JD, Rao JS. Chk2-mediated G2/M cell cycle arrest maintains radiation resistance in malignant meningioma cells. Cancer Lett 2011; 313: 64–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Klaude M, Eriksson S, Nygren J, Ahnstrom G. The comet assay: mechanisms and technical considerations. Mutation research 1996; 363: 89–96. [DOI] [PubMed] [Google Scholar]
- 26.Olive PL, Banath JP. The comet assay: a method to measure DNA damage in individual cells. Nature protocols 2006; 1: 23–29. [DOI] [PubMed] [Google Scholar]
- 27.Gourzones-Dmitriev C, Kassambara A, Sahota S, Reme T, Moreaux J, Bourquard P et al. DNA repair pathways in human multiple myeloma: role in oncogenesis and potential targets for treatment. Cell Cycle 2013; 12: 2760–2773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Nubel U, Dordel J, Kurt K, Strommenger B, Westh H, Shukla SK et al. A timescale for evolution, population expansion, and spatial spread of an emerging clone of methicillin-resistant Staphylococcus aureus. PLoS Pathog 2010; 6: e1000855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Panier S, Boulton SJ. Double-strand break repair: 53BP1 comes into focus. Nat Rev Mol Cell Biol 2014; 15: 7–18. [DOI] [PubMed] [Google Scholar]
- 30.Pierce AJ, Johnson RD, Thompson LH, Jasin M. XRCC3 promotes homology-directed repair of DNA damage in mammalian cells. Genes Dev 1999; 13: 2633–2638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ahmed KM, Pandita RK, Singh DK, Hunt CR, Pandita TK. beta1-Integrin Impacts Rad51 Stability and DNA Double-Strand Break Repair by Homologous Recombination. Mol Cell Biol 2018; 38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Pan JA, Sun Y, Jiang YP, Bott AJ, Jaber N, Dou Z et al. TRIM21 Ubiquitylates SQSTM1/p62 and Suppresses Protein Sequestration to Regulate Redox Homeostasis. Mol Cell 2016; 62: 149–151. [DOI] [PubMed] [Google Scholar]
- 33.Pankiv S, Lamark T, Bruun JA, Overvatn A, Bjorkoy G, Johansen T. Nucleocytoplasmic shuttling of p62/SQSTM1 and its role in recruitment of nuclear polyubiquitinated proteins to promyelocytic leukemia bodies. J Biol Chem 2010; 285: 5941–5953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Velkova A, Carvalho MA, Johnson JO, Tavtigian SV, Monteiro AN. Identification of Filamin A as a BRCA1-interacting protein required for efficient DNA repair. Cell Cycle 2010; 9: 1421–1433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Mirman Z, de Lange T. 53BP1: a DSB escort. Genes Dev 2020; 34: 7–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ong CA, Shannon NB, Ross-Innes CS, O’Donovan M, Rueda OM, Hu DE et al. Amplification of TRIM44: pairing a prognostic target with potential therapeutic strategy. Journal of the National Cancer Institute 2014; 106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Chen Z, Orlowski RZ, Wang M, Kwak L, McCarty N. Osteoblastic niche supports the growth of quiescent multiple myeloma cells. Blood 2014; 123: 2204–2208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lindahl T Molecular biology: ensuring error-free DNA repair. Nature 2004; 427: 598. [DOI] [PubMed] [Google Scholar]
- 39.Torgovnick A, Schumacher B. DNA repair mechanisms in cancer development and therapy. Front Genet 2015; 6: 157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Dietlein F, Thelen L, Reinhardt HC. Cancer-specific defects in DNA repair pathways as targets for personalized therapeutic approaches. Trends Genet 2014; 30: 326–339. [DOI] [PubMed] [Google Scholar]
- 41.Rodgers K, McVey M. Error-Prone Repair of DNA Double-Strand Breaks. J Cell Physiol 2016; 231: 15–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Wang Y, Zhang N, Zhang L, Li R, Fu W, Ma K et al. Autophagy Regulates Chromatin Ubiquitination in DNA Damage Response through Elimination of SQSTM1/p62. Mol Cell 2016; 63: 34–48. [DOI] [PubMed] [Google Scholar]
- 43.Terry LJ, Shows EB, Wente SR. Crossing the nuclear envelope: hierarchical regulation of nucleocytoplasmic transport. Science 2007; 318: 1412–1416. [DOI] [PubMed] [Google Scholar]
- 44.Kirkin V, McEwan DG, Novak I, Dikic I. A role for ubiquitin in selective autophagy. Mol Cell 2009; 34: 259–269. [DOI] [PubMed] [Google Scholar]
- 45.Ichimura Y, Kumanomidou T, Sou YS, Mizushima T, Ezaki J, Ueno T et al. Structural basis for sorting mechanism of p62 in selective autophagy. J Biol Chem 2008; 283: 22847–22857. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.