

Abstract
Advanced age is associated with accumulation of damage and other deleterious changes and a consequential systemic decline of function. This decline affects all organs and systems in an organism, leading to their inadaptability to the environment, and therefore is thought to be inevitable for humans and most animal species. However, in vitro and in vivo application of reprogramming strategies, which convert somatic cells to induced pluripotent stem cells, has demonstrated that the aged cells can be rejuvenated. Moreover, the data and theoretical considerations suggest that reversing the biological age of somatic cells (from old to young) and de‐differentiating somatic cells into stem cells represent two distinct processes that take place during rejuvenation, and thus they may be differently targeted. We advance a stemness‐function model to explain these data and discuss a possibility of rejuvenation from the perspective of damage accumulation. In turn, this suggests approaches to achieve rejuvenation of cells in vitro and in vivo.
Keywords: aging, biomarkers of aging, partial reprogramming, regeneration, rejuvenation
1. CURRENT UNDERSTANDING OF AGING
1.1. Aging as a combination of systematic transitions and random events
Aging is associated with an inevitable decline of organ and system functions, the cause of which remains a matter of debate. Although the possibility of lifespan extension by dietary, genetic and pharmacological interventions has been demonstrated for all common model organisms, indicating an association with slowing down aging, it has been less clear whether under certain circumstances aged organisms can be rejuvenated. Some researchers posit that aging is a programmed process, that the reverse‐program can be achieved, and some others believe that aging is mainly associated with random events, which cannot be reversed. 1 Yet, these different views appear to point to particular features of aging, without emphasizing its multi‐dimensional nature. In fact, as we discuss below, emerging evidence suggests that aging is a combination of systematic and random changes (Figure 1).
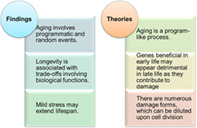
FIGURE 1.
Facts and theories of aging. There are important experimental observations (facts) in the field of aging as well as many proposed theories to explain the aging process. They offer insights into particular aspects of the aging process, but require integration to better describe the complexity of aging
One of the most obvious features of aging, besides its inevitability, is that aging is determined by numerous gradual changes that together relate to functional decline, including some that manifest as aging hallmarks. 2 These changes follow certain age‐related trajectories, and although their origin is not well understood, they can be integrated into a set of biomarkers that track and characterize the process of aging. For example, on the phenotypic level, human facial phenotypes are known to gradually shift with age, which led to the development of biomarkers based on photographic images. 3 This is also reflected in phenotypes such as cognitive deficiency. 4 On the molecular level, the most precise biomarker of aging thus far is based on DNA methylation profiling and is known as the epigenetic clock. 5 , 6 , 7 Different versions of this clock can estimate the biological or chronological age of humans and mice, thus distinguishing the rate of aging among different organs, and reporting the effects of longevity interventions. 5 , 8 , 9 Importantly, interventions such as caloric restriction, rapamycin treatment, and certain gene perturbations can extend lifespan, regardless of differences between animals or even species. 10 , 11 , 12 Moreover, age‐related events happen with surprising predictability across organisms and species, suggesting program‐like (programmatic, quasi‐programmed) features of aging. 13 , 14 , 15 , 16 However, aging is not a program like the developmental program of an organism, and it has no purpose. It is characterized by increasing randomness with time. For example, it has been observed through single‐cell RNA‐seq analyses that cell‐to‐cell heterogeneity increases with age, along with the perturbation of T‐cell activation that is associated with a decrease in T cell transcriptional variability. 17 Variance in other biological parameters also changes with age, for example, DNA methylation and chromatin modifications. 18 , 19 Overall, aging can be described as a combination of predictable (program‐like) transitions and random events.
1.2. Longevity is associated with functional trade‐offs
It is important to distinguish the “rate of aging” and aging in general from longevity. First, aging is a process, whereas longevity is a quantitative feature of an aging organism. Secondly, aging may be understood by considering whether it applies to a living organism because not all organisms age, and why it happens. In contrast, longevity is best understood by asking questions about how long an organism lives and how this variable may be modified. Thirdly, longevity or lifespan is not only determined by the rate of aging, but also by lethal diseases and even conditions unrelated to aging but which may happen often in the old age. Such diseases, for example, lymphoma in lab mice may be targeted by certain interventions without affecting the rate of aging. Likewise, some diseases causing early death change mortality in early life without affecting aging. Aging is not necessarily accelerated in individuals with a markedly shorter lifespan or always happen proportionally to lifespan, although some short‐lived cases called progeroid syndromes do display certain features of accelerated aging. Although the nature of aging is not well understood, or at least there is still no consensus on its understanding, it is clear that it is possible to manipulate lifespan. Many interventions are now known that increase the lifespan of model organisms. 10 , 11 , 12 , 20 , 21 , 22 , 23 , 24 , 25 , 26 , 27 In fact, increasing the lifespan of any model species is not at all unusual, as numerous conditions have been identified in large‐scale screens that lead to longevity.
As a rule, increased lifespan is associated with certain trade‐offs. It has been shown that there is a relationship between lifespan and factors that represent fitness and reproductive capacity. For example, inhibition of mTOR signaling promotes longevity, but suppresses the immune function. 28 Growth hormone receptor (GHR) knockout mice have an extended lifespan, but the trade‐off is dwarfism. 10 , 24 Overexpression of telomerase can extend lifespan, but it is also associated with tumorigenesis. 29 , 30 The trade‐offs associated with longevity are also observed in the wild, as long‐lived species are often characterized by lower fecundity than short‐lived species 1 , 31 . In other words, an intraspecific and interspecific extension of lifespan is usually accompanied by trade‐offs in “functions.” It is important to note that these functions usually benefit organisms when they are young. This relationship between longevity and fitness is also associated with the decline in the force of selection with age, which is widespread in nature with a few exceptions. 32 , 33 , 34
Consideration of this aspect of aging leads to the concept of antagonistic pleiotropy (AP). 35 This aging theory posits that there are genes and alleles with two‐sided effects that manifest differently with age, specifically being beneficial in the young (at or soon after the onset of reproduction) but deleterious in the older population. Because the benefits offered by such genes in early life can affect fitness, these genes are more likely to be selected, regardless of their negative effects in old age. This model explains the decline in the force of natural selection for aging population and is supported for example, by the effect of p53 gene on aging—it can protect against cancer‐related mortality, but can also impair normal tissue homeostasis and accelerate aging. 36 On the other hand, the AP theory is less clear in addressing the “reverse trade‐off” in aging—hormesis 37 , 38 . Hormesis refers to the observations that mildly harmful interventions often extend the lifespan of organisms. This effect is often explained by the fact that these manipulations activate stress response genes, benefiting animals throughout their life. However, if such genes follow the AP model, they should harm old animals and shorten lifespan, as is the normal case in high‐stress conditions when organisms exhibit distinct gene expression profiles. 26 , 39 Therefore, the AP concept, at least in its original form, is not an exhaustive description of aging, but rather a description of one aspect of aging. Although many theoretical models, such as AP, offer important insights into aging, they are incomplete in describing the complexity of aging. It is therefore important to unite these concepts into a single model, which not only defines the origin of aging and the effects of interventions on lifespan, but also explains approaches to rejuvenation.
2. AN INTEGRATIVE MODEL IN WHICH DAMAGE DRIVES AGING
2.1. Aging and entropy increase
Increased variation of different biological parameters during aging, as well as elevated Shannon information entropy (the degree of uncertainty or the amount of “surprise” in information such as those observed in DNA methylation profiles or gene expression patterns), suggests an analogy to the famous statement of the second law of thermodynamics: The total entropy or degree of disorder of an isolated system never decreases over time (Figure 2). This law stipulates that the order of energy flow favor the process that increases the degree of chaos. This analogy is consistent with the fact that Shannon entropy increases with age 24 , 40 , 41 . It is also interesting that the Horvath clock CpG sites exhibit higher Shannon entropy, providing insights into the relationship between entropy and systemic transitions during aging. 42 Of course, what is different between this age‐related entropy increase and the law of thermodynamics is that living organisms including humans are not an isolated system. It is an open system that exchanges substances and energy with the environment.
FIGURE 2.
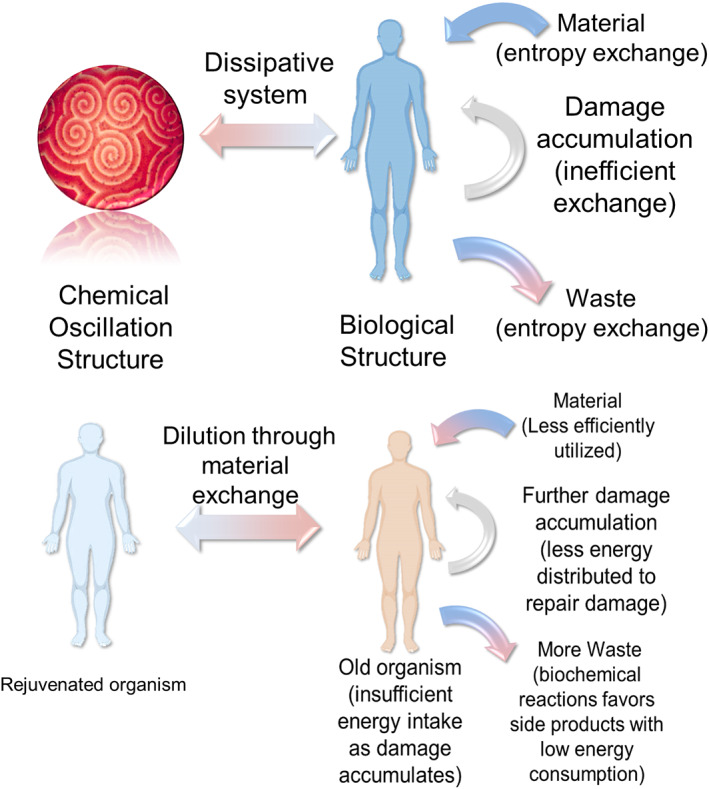
Aging from an entropy perspective. Upper: Analogy of aging to a chemical oscillation structure. Humans continuously exchange entropy with the environment by exchanging materials and energy in order to maintain the ordered biological structure. A restricted exchange of substances leads to aging. Lower: Further damage is caused by limits to obtain energy. The inability to take in substances disrupts functions and limits the ability to consume energy and repair damage, and the existing damage then makes biological systems produce more damage
The aging organism may also be viewed as a system with chemical fluctuations caused by biological reactions in the organism. This notion takes the whole organism as collected microscopically ordered patterns resembling oscillating chemical reactions. In such reactions, the chemicals involved form a dissipative structure far from the equilibrium, which allowed the raw materials to be transformed to maintain as a certain self‐assembled biological structure that is ordered both spatially and temporally. 43 , 44 , 45 , 46 With aging, program‐like changes reflect the temporal order of this structure, while the spatial order is damped over time as fewer and fewer materials effectively join the reaction. Like oscillating chemical reactions, this chemical fluctuation model suggests that the biological system gets materials (a negative entropy flow) from the environment to maintain the spontaneous self‐assembly of biological structures. 47 However, it is harder to maintain such structures when the biological organization of an organism is constrained by its own biology and the genome evolution needed to maximize fitness, and is further impacted by age‐related changes, diminishing the ability to effectively exchange and distribute substances and energy. Therefore, these spatially ordered biological structures collapse as the negative entropy flow cannot be effectively transferred from the environment into the organism. Because the entropy of the system tends to increase, the organism must exchange substances with the environment to battle the increasing degree of disorder of the biological system over time, and it is increasingly unable to do so.
2.2. Aging is caused by systematic damage
The increased entropy problem arises from the fact that molecular damage and other age‐related deleterious changes are not efficiently removed from the organism. Organisms are composed of systems with all sorts of biochemical reactions. From an organic chemistry perspective, no perfect reactions exist that generate only products and no side products 48 , 49 . The same idea applies to living organisms: biochemical reactions generate deleterious side products resulting in abnormal functions, such as somatic DNA mutations, extraneous RNA splicing variants, protein misfolding, and various other types. Taking the glycolytic pathway as an example, its side‐products are pervasively produced by essentially every participating gene product, and therefore glycolysis requires a specialized maintenance system composed of additional enzymes that target major by‐products of the 10 glycolytic reactions. 50 Even for such a highly conserved pathway, there might be a certain degree of randomness that may generate the molecular damage; however, the specific side products of glycolysis are always produced, suggesting that much of this metabolic damage is not random. Organisms have evolved numerous mechanisms to deal with this damage; however, the protection mechanisms seem to be always inferior to the variety of damage forms generated; in addition, these mechanisms may also be imperfect themselves and thus introduce other forms of damage. Moreover, damage to energy‐producing systems (eg, mitochondrial dysfunction) limits energy production by organisms, compromising the damage control processes that require energy, for example, DNA damage repair. In turn, the accumulation of damage impairs biological functions, causing the increasingly frequent breakdown of biological systems. 51 It should be stressed that the diversity of damage is not limited to molecular damage but involves many levels of age‐related deleterious changes. For this reason, it is convenient to designate the sum of this damage as the deleteriome. 48 , 49
The diverse types of damage in the aging cells suggest that an isolated non‐dividing cell or an organism with such nonrenewable cells cannot deal with them all, so the damage accumulates. These damages affect their normal cellular function and may trigger immune responses, which can in turn cause malfunction at many levels. For example, a mutation will lead to the respective error in the transcript and then in the synthesized protein or impair its regulatory function or gene expression. This type of damage may be random. A similarly damaged protein form may also occur due to a random error during transcription, translation, or protein modification. Nevertheless, most of such damages are systematic because they reflect particular gene expression processes involving multiple intermediate products, or are being part of functional molecular complexes, thus having a genetic or a program‐like nature. This damage is not entirely random, as it is generated by particular processes, involving genes that are encoded in the genome. This hierarchical reaction of damage explains why certain age‐related changes are program‐like, while others emerge in a random manner.
Since genes are not perfectly precise in performing their functions, they will make mistakes that may cause damage as a result of these functions. In addition, a protein can perform different functions temporally and spatially, depending on the specific protein complex involving it. 52 Therefore, all genes are AP‐like genes, or more broadly speaking, all biological products, and functions are antagonistically pleiotropic with regard to their functions and contribution to cumulative damage. Yet, judging by the normal function, not all genes are equal in the contribution of their antagonistic features. We may expect at least four distinct cases (described in Table 1): first, the AP genes that increase fitness significantly, whereas their resulting deleterious contribution is relatively small. These genes are highly beneficial in early life, but do not produce much damage over the lifetime, or organisms may die before the occurrence of the negative action of these genes. Certain genes related to age‐related diseases may be characterized into this category, as most organisms bear high mortality in the wild and cannot live up to the age when the negative effect of these genes takes place. Second, the AP genes that provide little benefit in early life, but significantly contribute to cumulative damage (Figure 3 ) corresponding to the restricted case represented by another aging theory—mutation accumulation. 70 Third, we predict inducible genes that cope with damage most effectively in later life when the damage they target most strongly affects function, thereby increasing lifespan. Such genes function in the maintenance and protection, via tumor suppression, apoptosis and senescent cell removal, and in some cases, autophagy. 55 , 71 , 72 The fourth type of the AP genes comprises those that are essential during a certain stage of development but are damaging during other developmental stages and even after completion of development when the organism is supposed to have maximal fitness. Overall, it is clear that AP properties penetrate all of the biology but come in a variety of mechanistically distinct forms. In a way, the beneficial aspects of AP are the essence of life, whereas each deleterious property of AP is a side product or a consequence of life, and together these properties define the essence of aging.
TABLE 1.
Examples of antagonistically pleiotropic features of genes and processes
| Examples | Beneficial features | Deleterious features | Reference |
|---|---|---|---|
| Oncogene MYC WNT1 | Cell proliferation stem cell maintenance | Tumorigenesis | 53, 54 |
| Tumor suppressor gene TP53 | Inhibition cell proliferation of damaged cells | Apoptotic cell death necrotic cell death accelerated aging | 55, 56, 57, 58, 59, 60 |
| Cellular senescence | Prevents cell proliferation of damaged cells | Loss of cell function inflammation promotes tumorigenesis | 61, 62, 63, 64 |
| Immune response | Remove pathogens kill damaged cells kill neoplastic cells | Inflammation (inflammaging) | 65, 66 |
| Mild stress | Lifespan extension | Death frailty | 38 |
| Glyceraldehyde‐3‐phosphate dehydrogenase | Conversion of glyceraldehyde‐3‐phosphate to 1,3‐bisphosphoglycerate (glycolysis) | Side products: 1,4‐bisphospho‐erythronate, 4‐phospho‐erythronate, NADPHX, NADHX | 50 |
| Molecular oxygen | Aerobic respiration energy derivation | Intrinsic apoptotic signaling pathway in response to oxidative stress | 67, 68, 69 |
FIGURE 3.
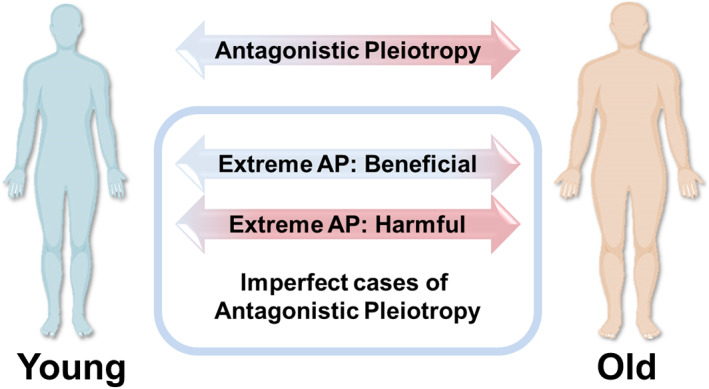
Antagonistic pleiotropy. The antagonistic pleiotropy (AP) theory states that certain alleles and genes that are beneficial in early life can be detrimental in later life, causing aging. However, the great majority of genes exhibit AP features, and this happens regardless of whether the organism containing these genes ages or not. We point out that the beneficial features of AP genes are represented by their functions, whereas the deleterious features are represented by damage generated because of these functions. This damage accumulates over time, leading to the appearance of the damaging effects of genes in late life. However, genes are not equal in their AP properties since in the extreme cases their beneficial effects may span most of the lifespan or be confined to a certain stage of development, and their deleterious effects may significantly contribute to the cumulative damage even in early life or contribute little over the entire life
2.3. Explaining rejuvenation from the damage dilution perspective in a stemness‐function model
Aging is an irreversible process, and most organisms can never escape the diversity and accumulation of damage that their own functions generate. To reduce damage, species with a simple organization may opt to discard some damage with a part of the cytoplasm, but this mechanism needs to be investigated in more complex species. 73 interventions such as parabiosis may partially restore aged organ functions through transfusion of young blood to an old organism. 74 This may be considered as a damage dilution process, where the old blood is diluted by the less damaged young blood. It was shown that, following hematopoietic stem cell transfer, the blood of the recipient follows the epigenomic age of the donor, suggesting a possibility to consistently generate younger blood than the actual age of the organism, if the source of hematopoietic stem cells is a young donor. 75 It is important to emphasize that the transition to a younger age, based on one or more tissues being younger than the rest and younger than the chronological age, does not necessarily mean a longer lifespan for the subject, particularly if the lifespan is limited by a particular dysfunction or disorder that causes death. Recent studies suggested a possibility that certain drugs may slow down, and even reverse the epigenomic age, as defined by epigenetic clock biomarkers. 76 These and other developments brought to light the possibility of “rejuvenation,” and even the prospect of reversing the biological age of an organism from an old to a young state.
Although somatic aging appears at first sight irreversible, we cannot bypass the fact that it is successfully reset to zero from generation to generation, suggesting that, during germline development, embryonic development, or some other phases of life there is a process that rewinds the aging clock. Somatic cell nuclear transfer shows that this rewinding process can be also induced in differentiated cell nuclei, although the mechanism is unknown. 77 As discussed above, aging is caused by the accumulation of damage. However, this damage does not typically pass to the next generation to accelerate aging in the following generations, ultimately leading to species extinction. Therefore, some mechanisms must operate in the process of germline production, development, and maintenance that reverses aging and might provide the clue for selection against, diluting or even erasing of such damage. These mechanisms of dilution are currently unclear, although evidence suggests that they may involve a combination of cell division, cell selection, epigenetic remodeling, and global activation of genes, especially those genes for controlling DNA damage 78 , 79 . These mechanisms allow cells to dilute even the scarcest molecular species such as functionally abnormal RNA, proteins, harmful metabolites, and those that would not be sensed by a cell. Thus, a combination of cell growth, selection, and proliferation dilutes mild damage, in addition to the removal of damage through specialized detoxification, repair, excretion, preemption, and other approaches. These mechanisms together allow the cells to keep the damage in control. 80
It should be noted that division and dilution are not necessarily related in the context of proliferation of differentiated somatic cells, as, unlike germ cells or stem cells, these cells may undergo senescence or tumor transformation when proliferating in culture 81 , 82 . This suggests that there is a particular relationship between cell division and damage dilution, whose mechanism is not yet understood. We think that this relationship is reflected, for instance, in the differences between early embryonic and aged cells, partially due to their different differentiation states. The former may stay in quiescent stage to avoid further damage or proliferate to select the cells with less damage. Compared to adult cells, embryonic cells specifically experience two waves of global demethylation and re‐methylation, establishing the same DNA methylation pattern for every generation. 83 These differences suggest a possibility that certain embryonic cells and somatic cells have different modes and rate of damage accumulation and dilution through proliferation. From the damage perspective, the proliferation of cells with more specialized functions bears higher damage, as more specialized molecules are produced, allowing more side‐products to be generated. Furthermore, adult stem cells may overcome the proliferation limit when exposed to a mixed pro‐stemness signal. 84 This shows that the combined effect of niche pathways that promote the stemness of the adult stem cells may act similarly to reprogramming. Thus, the difference in the damage accumulation between somatic cells and stem cells may lie, at least in part, in the cell matrix environment in which cells reside. Moreover, the environment may undergo a transition to sacrifice stemness for specific biological functions.
To visualize this stage‐shifting concept, we advance a weight‐scale metaphor, which we call a “stemness‐function” model (Figure 4 and Table 2). We designate the two states as “pro‐stemness” and “pro‐function” based on the balance between damage production and its removal by proliferation and apoptosis. During early life, organisms remain in a “pro‐stemness” state, encouraging cells to proliferate and grow so that the damage is unchecked and does not cause cell cycle arrest. In that state, although stem cells exhibit a limited intrinsic immune function, the function to recognize “self” and “nonself” is not yet fully developed, allowing a lower level of inflammation and an increased potential for regeneration. 97 In contrast, in somatic cells, the damage generation can be sensed easier, triggering the reactions such as the DNA damage repair process, growth arrest, apoptosis, and immune responses. Therefore, organisms must undergo a transition from the “pro‐stemness” to “pro‐function” states, wherein differentiation and specification of cells are supported. Following this transition, the cells enhance their function in reproduction, damage sensing and apoptosis pathway, complete the immune function, and increase fitness by generating specific biological products related to their functions, while adult stem cells at this stage undergo gradual exhaustion 2 , 98 , 99 . At this stage, damage accumulation is spontaneous while damage dilution via proliferation is not supported in most cell types. During the process of fertilization or before/after it, this damage gets thoroughly checked, cleared and diluted by the transition to the “pro‐stemness” state 78 , 79 , 80 , 100 . An example of such a “reset” function exists at fertilization in C. elegans where lysosomal functions in oocytes are enhanced by sperm‐secreted hormones, allowing degradation of protein aggregates and protein homeostasis. 101 Such transitions are then followed by cell replication, allowing cells to enter the “pro‐stemness” state. Some cells may not enter this state successfully due to damage they bear, and these cells will be competed out by apoptosis, contributing to mortality in early life 102 , 103 .
FIGURE 4.
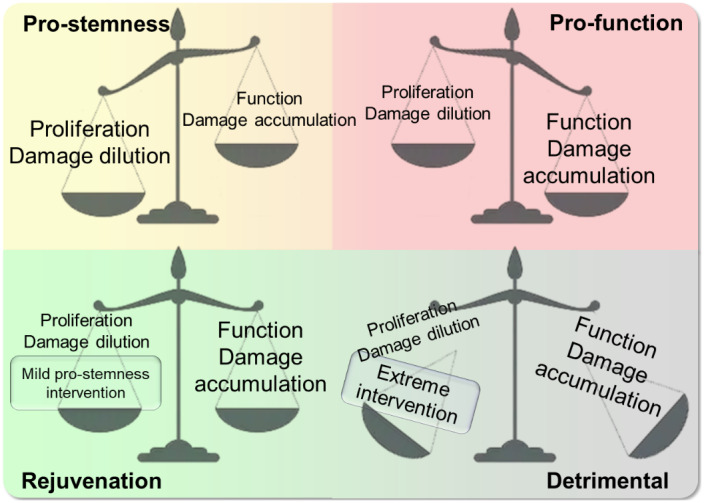
The stemness‐function model. This model posits the existence of two types of environment in an organism: the pro‐stemness state in the early life and regenerating cells, and probiological function in most tissues in the adult stage. Mild global activation of pro‐stemness genes in the pro‐function state may extend lifespan, whereas global overexpression of pro‐stemness genes may result in a detrimental effect
TABLE 2.
Examples of age‐related manipulations following the stemness‐function model
| Type | Manipulation | Outcome | Reference |
|---|---|---|---|
| Generation of iPSCs from somatic cells | Dilution of damage in pro‐stemness state | Age reset | 5, 7, 8, 85, 86 |
| Proliferation of somatic cells | Dilution of damage in pro‐functional state | Age acceleration, cellular senescence | 6, 81, 82 |
| Partial reprogramming | Mild pro‐stemness intervention | Potential age reset longer lifespan | 87, 88, 89 |
| Rapamycin | Potential mild pro‐stemness intervention | Potential age reset longer lifespan | 90, 91 |
| Hormesis mild activation of autophagy protein quality control tumor suppressor gene | Mild pro‐functional intervention | Potential longer lifespan | 37, 38 |
| Senolytics | Mild pro‐functional intervention | Potential longer lifespan | 27, 72 |
| Potent overexpression of iPSC reprogramming factors | Extreme pro‐stemness intervention | Loss of cell function tumorigenesis potential age reset | 92, 93, 94 |
| Forced proliferation of adult stem cells | Extreme pro‐functional intervention | Stem cell exhaustion age acceleration | 95, 96 |
What perturbations might then be expected to delay or reverse aging? If a mild “pro‐function” feature is induced in the cells with the “pro‐stemness” state, it may extend lifespan as we learn from mild overexpression of certain tumor suppressors 104 , 105 . Similarly, the weakened immune system upon rapamycin treatment provides an example that the opposite may also work 28 , 90 , 106 . On the other hand, if a specific function (supported by a certain gene) that shifts the system toward the “pro‐function” state is introduced, it may lead to death or premature aging, caused by a sudden increase in function and damage. This might be the case when tumor‐suppressor Tp53 is overexpressed in mice, and the animals show a significantly shorter lifespan. 56 It should be noted, however, that similar cases of Tp53 overexpression in mouse models show an indistinguishable lifespan. 57 Nevertheless, considering that cancer‐related deaths are more common in lab mice than in humans and that these risks are limited in these cancer‐resistant mouse models, there is still a possibility that the overexpression accelerates aging 57 , 107 . Conversely, if a “pro‐stemness” signal introduced to cells in the “pro‐function” state, it may also cause deleterious effects, resulting in cell death or aberrant immortality. For instance, forcing cell proliferation by expressing oncogenes in fibroblasts promotes tumor transformation. 108 A further support for this model is offered by the finding that human aging and cancer transformation exhibit transcriptional changes essentially in the opposite direction, along with the observation that cancer incidence in the elderly increases with age, together supporting the idea that cancer can be initiated by spontaneous processes that introduce aberrant “pro‐stemness” stimuli into the “pro‐function state”. 109
3. APPROACHES TO REJUVENATION AND THEIR THEORETICAL BASIS
3.1. Rejuvenation through reprogramming
In our proposed stemness‐function model, the cell environment in the pro‐stemness state is different from that in the pro‐function state. In contrast to the rejuvenation of a whole organism, it should be easier to rejuvenate specific cell types in culture, because cultured cells are more homogeneous and less affected by extrinsic signaling crosstalk, hence more prone to go back to the pro‐stemness state. Supporting this notion is the induction of pluripotent stem (iPS) cells by Yamanaka and colleagues 85 , 86 . Reprogramming adult somatic cells into iPS cells through the expression of four transcription factors (OSKM) achieves a cell fate like that of embryonic stem cells with the potential to develop as any part of the embryo. This can be interpreted as an example of damage dilution since the cells first undergo rapid proliferation along with the reduction of the size of each cell at the early state to become iPS cells, and iPS cells have a close‐to‐zero biological age as judged by the epigenetic clock biomarker 5 , 8 , 9 . These cells essentially revert from the “pro‐function” state to the “pro‐stemness” state and gradually reverse their age‐related damage, of which the aging biomarkers are the readouts.
However, as described before, forcing the expression of the stemness genes to overdo their effect on the “pro‐function” state may result in a detrimental consequence, such as weakened biological functions or tumorigenesis. One example is that promoting hematopoietic stem cell proliferation can lead to the accelerated aging potentially through exhausting stem cell. 95 However, this may be different from treatments, such as young blood transfusion, which brings both the dilution of damage and a “pro‐stemness” general environment.
It is important to note that even in cell culture, only a small portion of cells eventually become iPS cells. Reprogramming in vitro generates cell heterogeneity and most of the cells that undergo reprogramming differentiate into specific cell lineages, or transition through senescence, apoptosis, or premature differentiation. 110 In addition, the generated iPS cells are more prone to tumor transition. 111 This brings a major problem when researchers attempt rejuvenation using this approach in vivo. Such reprogramming in vivo in mice results in the loss of cell function and tumorigenesis 92 , 93 . Therefore, mice constantly expressing OSKM die early rather than live longer. Later, researchers applied reprogramming with the premature withdrawal of the expression of OSKM to prevent tumor formation and loss of cell identity (Figure 5). 87 Interestingly, an increased lifespan was observed in a fast‐aging mouse model, which may be explained by the partial transition from “pro‐function” to “pro‐stemness” states. In addition, a clear sign was found that the regenerative capacity increases upon injury, suggesting a transition to a more youthful state. Although an increase in lifespan has not been observed in normally aging mice, research suggests that this could eventually be achieved. A human in vitro reprogramming study based on the methylation clock biomarker found that there is a steady decrease in the methylation age upon reprogramming, as well as subsequent de‐differentiation characterized by the loss of molecular markers of cell commitment. 112 We suggest an interpretation that the dilution of damage during the reprogramming results in the selection of a cell subpopulation that erases the age‐related methylation signature in the genome.
FIGURE 5.
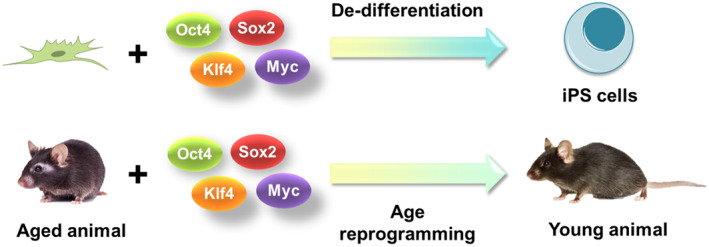
Two effects of in vivo reprogramming by Yamanaka (OSKM) factors are de‐differentiation and age reversal. De‐differentiation causes cells to go back to the stem cell lineage, whereas age reversal may lead to a younger biological age without de‐differentiation
To move from partial reprogramming to true rejuvenation requires a mechanism to shift cells to a decreased age state without forcing them into a non‐physiological state of gene expression, or “confused” cell identity. This process might involve restoring original stem cell function through reprogramming differentiated cells to stem cells or the provision of specific signaling molecules that support stem cell functions by reprogramming differentiated cells in the stem cell niche. Recently, a study involving human muscle cells revealed a possibility that, by transiently expressing reprogramming factor (OSKM plus Lin28 and Nanog) mRNAs, one can restore young regenerative capacity and methylation age in human muscle stem cells without changing their identity, yet restore muscle physiological function of these muscle stem cells. 88 This study, along with the previous work on partial reprogramming, suggests a possibility that there are indeed different states that represent age‐related transitions from those of developmental differentiation. If the gene sets contributing to age reversal can be distinguished from those involved in the reversal of differentiation state, rejuvenation may be accomplished more effectively in vivo. Considering that OSKM is not the only combination to induce reprogramming, and that some of them are predicted via omics methods and applied to trans‐differentiation, an interesting future approach may seek to identify the factors orthogonal to/downstream of OSKM that are only responsible for the age reversal and isolate their effects from the original reprogramming factors. 113 As trans‐differentiation among adult cell types can also be achieved by inducing the expression of combinations of transcription factors, a similar strategy could be employed to find the genes of age reversal, skipping the requirement for pluripotency reprogramming but still erasing the cellular aging signatures. 114
3.2. Rejuvenation by regeneration
The process of fibroblast reprogramming shares several interesting features with the wound healing process. Both involve massive cell senescence and cell death; both are related to cell de‐differentiation generating the cells that can grow into multiple cell types. 61 Further, it has been shown that senescent cells promote both reprogramming and wound healing processes and that in vivo reprogramming promotes the regenerative capacity of animals, suggesting an intrinsic relationship between reprogramming and regeneration‐mediated wound healing. 61 , 62 , 115 Thus, the possibility that regeneration can be harnessed to rejuvenate an entire organism should be taken into consideration and the differences between regeneration and rejuvenation should also be informative.
Animals have multiple ways to repair their wounds. The two main approaches are wound healing through regeneration and wound healing with scar tissues. 116 When organisms employ the regeneration method, they de‐differentiate their tissues next to the wound to form a blastema. An almost identical tissue is then grown and differentiated from the blastema tissue. This process does not generate scars. Organisms such as axolotls, zebrafish, spiny mice, and even neonatal humans and mice can use this way to repair their wounded tissues. 117 , 118 , 119 , 120 Axolotls can repair their amputated limb and regenerate a completely new limb. They can also regenerate the cryo‐injured heart. Zebrafish can regenerate the tail if the wound is not severe. Neonatal mice can repair their skin without scar 3 to 5 days before birth, and they have the same heart regeneration capacity prior to being 7 days old. 121 In contrast, adult humans and mice repair the majority of wounds with scar tissue. 117 This process is triggered by growth factors produced by macrophages, which cause fibroblasts to proliferate and generate scar tissue to repair wounds. 122 Although multiple biocompatible scaffolds have been applied as artificial and extracellular matrices to improve wound healing, there has not yet been any dramatic difference found through such biomaterials. 123 Compared to the normal tissues, the scar tissues have collagen I aggregation and a different collagen structure. 124 Notably, this scar‐inducing process does not restore the function of the originally wounded tissue. Therefore, the shift between regeneration and scar tissue formation suggests that the regenerative capacity of mammals decreases with age. Yet, it is unknown whether such a shift is merely due to the process of development or aging itself, as the question when the aging begins in early life is still not fully understood.
A recent study of deleterious somatic mutations and biomarkers of aging coupled with demographic analyses revealed that aging starts very early in life, whereas mortality is initially high and decreases in early life. 125 Therefore, this shift in regenerative capacity may be due to the state of aging or the declining capacity to adapt to the changed environment. Weakened regenerative capacity in aged animals leads to impaired wound healing, especially in the skin and muscle 2 , 126 , 127 , 128 , 129 . It was suggested that this decline is closely related to the lowered immune function and inflammatory response during aging. Consistently, embryonic wounds have a low level of inflammatory cells and TGF‐beta 1 and 2 proteins, which might promote the regeneration process. 97
This association between inflammatory responses and regeneration suggests that recognition and removal of non‐self and dying cells and cell debris might play a role in regeneration. A part of hydra can generate a new hydra if being pressed against the same part of another hydra, indicating that they do not distinguish self from non‐self. 130 On the other hand, mammalian transplanted organs need treatment with immunosuppressors to keep their function. Depleting macrophages in humans and axolotls, or treating mice with the immunosuppressant, rapamycin can lead to impaired wound healing. 131 Interestingly, besides inhibiting growth via mTOR function, rapamycin also downregulates TGF beta 1 and leads to a weaker immune function. 132 This is consistent with our stemness‐function model: humans lose the regenerative capacity due to the transition to a “functional” state, and after this transition, the damage‐accumulating cells lead to aging and dying cells that increase inflammatory responses, exhaust stem cells, and accelerate aging. Rapamycin may partially reverse the organism to the “pro‐stemness” state by impairing certain functions including the immune response, thereby extending lifespan.
An important question is then unavoidable: If an organism has an unlimited regenerative capacity, will it have an unlimited lifespan? Hydra, planarians and some other species exhibit an almost unlimited capacity to regenerate with an exceptional lifespan 130 , 133 . Axolotls show the ability to regenerate their limbs and heart, although it is not unlimited as axolotls fail to generate their limbs after multiple amputations. 134 Humans and mice lost most of their regenerative capacity already during embryonic development, trading it for specific functions such as a tumor suppression. 95 The application of partial reprogramming can now address this question; for example, the neurons responsible for the retina function are regenerated in the partially reprogrammed mice, challenging the dogma that neurons cannot be regenerated. 89 This suggests another possibility, namely that partial reprogramming achieves its effect through a global increase of regenerative capacity. Such reprogramming can be accessed in vivo at several levels: by directing gene expression, changing the extracellular environment, and the exchange of signaling molecules, providing animals with a “pro‐stemness” environment.
The main question in the context of regeneration and aging is then whether the newly regenerated tissue is younger than the previously amputated tissue (Figure 6). Because regenerated tissues arise from de‐differentiated cells, in vitro cell reprogramming may be analogous to this process. It was shown that iPSC reprogramming erases epigenomic or transcriptomic features of aging from primary fibroblast cells and differentiate into the neurons with the same features, while directly converted neuron cells from primary fibroblast cells still retain age‐related transcriptome phenotypes. 135 From this, we hypothesize that the regenerated tissues may be younger than the tissues they arise from. However, a more precise type of aging biomarker will be needed in the future to test this hypothesis. If the unlimited capacity to regenerate leads to a totally self‐renewable organism, a possible future approach for rejuvenation may be to identify the transition that allows humans to temporarily reverse to the “pro‐stemness” stage. With the transition to a state supporting unlimited regenerative capacity, one may be able to achieve an infinite self‐renewal of tissues with minimal loss of developmental identity or neoplastic transformation.
FIGURE 6.
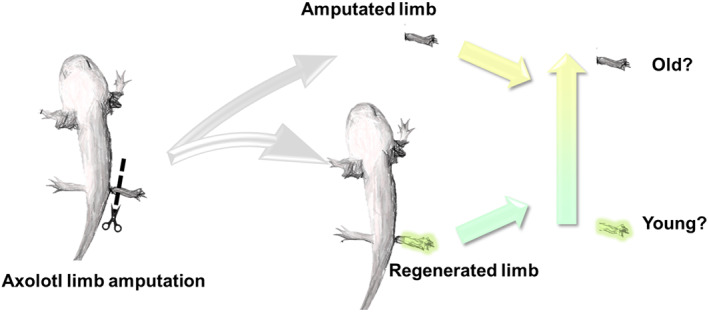
Assessing the biological age of regenerated tissues. Tissues undergo spontaneous de‐differentiation and re‐differentiation during wound healing by regeneration. Many features are shared between this process and reprogramming. It is possible that the regenerated tissues are younger than the original tissues based on their biological age. An example of axolotl limb regeneration is shown
AUTHOR CONTRIBUTIONS
Bohan Zhang: Conceptualization; visualization; writing‐original draft; writing‐review and editing. Vadim Gladyshev: Conceptualization; funding acquisition; supervision; writing‐review and editing.
CONFLICT OF INTEREST
The authors declare they have no conflicts of interest.
[Correction added on 1 December 2020, after first online publication: Peer review history is not available for this article, so the peer review history statement has been removed.]
Supporting information
Transparent‐Peer‐Review‐Record
ACKNOWLEDGMENTS
Supported by grants NIH AG021518, AG047200, and GM065204.
Zhang B, Gladyshev VN. How can aging be reversed? Exploring rejuvenation from a damage‐based perspective. Advanced Genetics. 2020;1:e10025. 10.1002/ggn2.10025
Funding information National Institutes of Health, Grant/Award Numbers: GM065204, AG047200, AG021518
REFERENCES
- 1. Kirkwood TB, Melov S. On the programmed/non‐programmed nature of ageing within the life history. Curr Biol. 2011;21(18):R701‐R707. [DOI] [PubMed] [Google Scholar]
- 2. Lopez‐Otin C, Blasco MA, Partridge L, Serrano M, Kroemer G. The hallmarks of aging. Cell. 2013;153(6):1194‐1217. 10.1016/j.cell.2013.05.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Chen W, Qian W, Wu G, et al. Three‐dimensional human facial morphologies as robust aging markers. Cell Res. 2015;25(5):574‐587. 10.1038/cr.2015.36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Harada CN, Natelson Love MC, Triebel KL. Normal cognitive aging. Clin Geriatr Med. 2013;29(4):737‐752. 10.1016/j.cger.2013.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Horvath S. DNA methylation age of human tissues and cell types. Genome Biol. 2013;14(10):R115. 10.1186/gb-2013-14-10-r115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Lowe D, Horvath S, Raj K. Epigenetic clock analyses of cellular senescence and ageing. Oncotarget. 2016;7(8):8524‐8531. 10.18632/oncotarget.7383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Horvath S, Raj K. DNA methylation‐based biomarkers and the epigenetic clock theory of ageing. Nat Rev Genet. 2018;19(6):371‐384. 10.1038/s41576-018-0004-3. [DOI] [PubMed] [Google Scholar]
- 8. Petkovich DA, Podolskiy DI, Lobanov AV, Lee SG, Miller RA, Gladyshev VN. Using DNA methylation profiling to evaluate biological age and longevity interventions. Cell Metab. 2017;25(4):954‐960 e956. 10.1016/j.cmet.2017.03.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Meer MV, Podolskiy DI, Tyshkovskiy A, Gladyshev VN. A whole lifespan mouse multi‐tissue DNA methylation clock. elife. 2018;7:e40675. 10.7554/eLife.40675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Bartke A, Brown‐Borg H. Life extension in the dwarf mouse. Curr Top Dev Biol. 2004;63:189‐225. 10.1016/S0070-2153(04)63006-7. [DOI] [PubMed] [Google Scholar]
- 11. Colman RJ, Anderson RM, Johnson SC, et al. Caloric restriction delays disease onset and mortality in rhesus monkeys. Science. 2009;325(5937):201‐204. 10.1126/science.1173635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Swindell WR. Dietary restriction in rats and mice: a meta‐analysis and review of the evidence for genotype‐dependent effects on lifespan. Ageing Res Rev. 2012;11(2):254‐270. 10.1016/j.arr.2011.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. de Magalhaes JP. Programmatic features of aging originating in development: aging mechanisms beyond molecular damage? FASEB J. 2012;26(12):4821‐4826. 10.1096/fj.12-210872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Longo VD, Mitteldorf J, Skulachev VP. Programmed and altruistic ageing. Nat Rev Genet. 2005;6(11):866‐872. 10.1038/nrg1706. [DOI] [PubMed] [Google Scholar]
- 15. Prinzinger R. Programmed ageing: the theory of maximal metabolic scope. How does the biological clock tick? EMBO Rep. 2005;6(19):S14. 10.1038/sj.embor.7400425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Blagosklonny MV. Aging and immortality: quasi‐programmed senescence and its pharmacologic inhibition. Cell Cycle. 2006;5(18):2087‐2102. 10.4161/cc.5.18.3288. [DOI] [PubMed] [Google Scholar]
- 17. Martinez‐Jimenez CP, Eling N, Chen HC, et al. Aging increases cell‐to‐cell transcriptional variability upon immune stimulation. Science. 2017;355(6332):1433‐1436. 10.1126/science.aah4115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Cheung P, Vallania F, Warsinske HC, et al. Single‐cell chromatin modification profiling reveals increased epigenetic variations with aging. Cell. 2018;173(6):1385‐1397 e1314. 10.1016/j.cell.2018.03.079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Ashapkin VV, Kutueva LI, Vanyushin BF. Aging as an epigenetic phenomenon. Curr Genomics. 2017;18(5):385‐407. 10.2174/1389202918666170412112130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Friedman DB, Johnson TE. A mutation in the age‐1 gene in Caenorhabditis elegans lengthens life and reduces hermaphrodite fertility. Genetics. 1988;118(1):75‐86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Anisimov VN. Metformin: do we finally have an anti‐aging drug? Cell Cycle. 2013;12(22):3483‐3489. 10.4161/cc.26928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Hine C, Mitchell JR. Calorie restriction and methionine restriction in control of endogenous hydrogen sulfide production by the transsulfuration pathway. Exp Gerontol. 2015;68:26‐32. 10.1016/j.exger.2014.12.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Melo DS, Costa‐Pereira LV, Santos CS, et al. Severe calorie restriction reduces Cardiometabolic risk factors and protects rat hearts from ischemia/reperfusion injury. Front Physiol. 2016;7:106. 10.3389/fphys.2016.00106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Wang T, Tsui B, Kreisberg JF, et al. Epigenetic aging signatures in mice livers are slowed by dwarfism, calorie restriction and rapamycin treatment. Genome Biol. 2017;18(1):57. 10.1186/s13059-017-1186-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Kaeberlein M. Longevity and aging. F1000Prime Rep. 2013;5(5):5. 10.12703/P5-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Mitchell SJ, Madrigal‐Matute J, Scheibye‐Knudsen M, et al. Effects of sex, strain, and energy intake on hallmarks of aging in mice. Cell Metab. 2016;23(6):1093‐1112. 10.1016/j.cmet.2016.05.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Xu M, Pirtskhalava T, Farr JN, et al. Senolytics improve physical function and increase lifespan in old age. Nat Med. 2018;24(8):1246‐1256. 10.1038/s41591-018-0092-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Li J, Kim SG, Blenis J. Rapamycin: one drug, many effects. Cell Metab. 2014;19(3):373‐379. 10.1016/j.cmet.2014.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Artandi SE, Alson S, Tietze MK, et al. Constitutive telomerase expression promotes mammary carcinomas in aging mice. Proc Natl Acad Sci U S A. 2002;99(12):8191‐8196. 10.1073/pnas.112515399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Gonzalez‐Suarez E, Samper E, Ramirez A, et al. Increased epidermal tumors and increased skin wound healing in transgenic mice overexpressing the catalytic subunit of telomerase, mTERT, in basal keratinocytes. EMBO J. 2001;20(11):2619‐2630. 10.1093/emboj/20.11.2619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Kirkwood TB. Evolution of ageing. Mech Ageing Dev. 2002;123(7):737‐745. [DOI] [PubMed] [Google Scholar]
- 32. Chen HY, Maklakov AA. Longer life span evolves under high rates of condition‐dependent mortality. Curr Biol. 2012;22(22):2140‐2143. 10.1016/j.cub.2012.09.021. [DOI] [PubMed] [Google Scholar]
- 33. Maklakov AA, Rowe L, Friberg U. Why organisms age: evolution of senescence under positive pleiotropy? BioEssays. 2015;37(7):802‐807. 10.1002/bies.201500025. [DOI] [PubMed] [Google Scholar]
- 34. Williams SA, Shattuck MR. Ecology, longevity and naked mole‐rats: confounding effects of sociality? Proc Biol Sci. 2015;282(1802):20141664. 10.1098/rspb.2014.1664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Williams GC. Pleiotropy, natural selection, and the evolution of senescence. Evolution. 1957;11(4):398‐411. 10.1111/j.1558-5646.1957.tb02911.x. [DOI] [Google Scholar]
- 36. Ungewitter E, Scrable H. Antagonistic pleiotropy and p53. Mech Ageing Dev. 2009;130(1–2):10‐17. 10.1016/j.mad.2008.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Rattan SI. Aging, anti‐aging, and hormesis. Mech Ageing Dev. 2004;125(4):285‐289. 10.1016/j.mad.2004.01.006. [DOI] [PubMed] [Google Scholar]
- 38. Rattan SI. Hormesis in aging. Ageing Res Rev. 2008;7(1):63‐78. 10.1016/j.arr.2007.03.002. [DOI] [PubMed] [Google Scholar]
- 39. Sullivan J, Mirbahai L, Lord JM. Major trauma and acceleration of the ageing process. Ageing Res Rev. 2018;48:32‐39. 10.1016/j.arr.2018.10.001. [DOI] [PubMed] [Google Scholar]
- 40. Slieker RC, van Iterson M, Luijk R, et al. Age‐related accrual of methylomic variability is linked to fundamental ageing mechanisms. Genome Biol. 2016;17(1):191. 10.1186/s13059-016-1053-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Sziraki A, Tyshkovskiy A, Gladyshev VN. Global remodeling of the mouse DNA methylome during aging and in response to calorie restriction. Aging Cell. 2018;17(3):e12738. 10.1111/acel.12738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Martin‐Herranz DE, Aref‐Eshghi E, Bonder MJ, et al. Screening for genes that accelerate the epigenetic aging clock in humans reveals a role for the H3K36 methyltransferase NSD1. Genome Biol. 2019;20(1):146. 10.1186/s13059-019-1753-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Zhabotinskii AM. Periodic course of the oxidation of malonic acid in a solution (studies on the kinetics of Beolusov's reaction). Biofizika. 1964;9:306‐311. [PubMed] [Google Scholar]
- 44. Zhabotinsky AM. A history of chemical oscillations and waves. Chaos. 1991;1(4):379‐386. 10.1063/1.165848. [DOI] [PubMed] [Google Scholar]
- 45. Goldbeter A. Dissipative structures and biological rhythms. Chaos. 2017;27(10):104612. 10.1063/1.4990783. [DOI] [PubMed] [Google Scholar]
- 46. Lefever R, Nicolis G, Prigogine I. On the occurrence of oscillations around the steady state in systems of chemical reactions far from equilibrium. J Chem Phys. 1967;47(3):1045‐1047. [Google Scholar]
- 47. Schrödinger E. What Is Life? The physical aspect of the living cell and mind. Cambridge: Cambridge University Press; 1944. [Google Scholar]
- 48. Gladyshev VN. The origin of aging: imperfectness‐driven non‐random damage defines the aging process and control of lifespan. Trends Genet. 2013;29(9):506‐512. 10.1016/j.tig.2013.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Gladyshev VN. Aging: progressive decline in fitness due to the rising deleteriome adjusted by genetic, environmental, and stochastic processes. Aging Cell. 2016;15(4):594‐602. 10.1111/acel.12480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Bommer GT, Van Schaftingen E, Veiga‐da‐Cunha M. Metabolite repair enzymes control metabolic damage in glycolysis. Trends Biochem Sci. 2019;45:228‐243. 10.1016/j.tibs.2019.07.004. [DOI] [PubMed] [Google Scholar]
- 51. Gavrilov LA, Gavrilova NS. The reliability theory of aging and longevity. J Theor Biol. 2001;213(4):527‐545. 10.1006/jtbi.2001.2430. [DOI] [PubMed] [Google Scholar]
- 52. Jeffery CJ. Moonlighting proteins. Trends Biochem Sci. 1999;24(1):8‐11. 10.1016/s0968-0004(98)01335-8. [DOI] [PubMed] [Google Scholar]
- 53. Blum B, Benvenisty N. The tumorigenicity of human embryonic stem cells. Adv Cancer Res. 2008;100:133‐158. 10.1016/S0065-230X(08)00005-5. [DOI] [PubMed] [Google Scholar]
- 54. Ben‐David U, Benvenisty N. The tumorigenicity of human embryonic and induced pluripotent stem cells. Nat Rev Cancer. 2011;11(4):268‐277. 10.1038/nrc3034. [DOI] [PubMed] [Google Scholar]
- 55. Krizhanovsky V, Lowe SW. Stem cells: the promises and perils of p53. Nature. 2009;460(7259):1085‐1086. 10.1038/4601085a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Tyner SD, Venkatachalam S, Choi J, et al. p53 mutant mice that display early ageing‐associated phenotypes. Nature. 2002;415(6867):45‐53. 10.1038/415045a. [DOI] [PubMed] [Google Scholar]
- 57. Garcia‐Cao I, Garcia‐Cao M, Martin‐Caballero J, et al. "super p53" mice exhibit enhanced DNA damage response, are tumor resistant and age normally. EMBO J. 2002;21(22):6225‐6235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Rasmussen MA, Holst B, Tumer Z, et al. Transient p53 suppression increases reprogramming of human fibroblasts without affecting apoptosis and DNA damage. Stem Cell Reports. 2014;3(3):404‐413. 10.1016/j.stemcr.2014.07.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Christophorou MA, Martin‐Zanca D, Soucek L, et al. Temporal dissection of p53 function in vitro and in vivo. Nat Genet. 2005;37(7):718‐726. 10.1038/ng1572. [DOI] [PubMed] [Google Scholar]
- 60. Aloni‐Grinstein R, Shetzer Y, Kaufman T, Rotter V. p53: the barrier to cancer stem cell formation. FEBS Lett. 2014;588(16):2580‐2589. 10.1016/j.febslet.2014.02.011. [DOI] [PubMed] [Google Scholar]
- 61. Mosteiro L, Pantoja C, Alcazar N, et al. Tissue damage and senescence provide critical signals for cellular reprogramming in vivo. Science. 2016;354(6315):aaf4445. 10.1126/science.aaf4445. [DOI] [PubMed] [Google Scholar]
- 62. Demaria M, Ohtani N, Youssef SA, et al. An essential role for senescent cells in optimal wound healing through secretion of PDGF‐AA. Dev Cell. 2014;31(6):722‐733. 10.1016/j.devcel.2014.11.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. van Deursen JM. The role of senescent cells in ageing. Nature. 2014;509(7501):439‐446. 10.1038/nature13193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Banito A, Rashid ST, Acosta JC, et al. Senescence impairs successful reprogramming to pluripotent stem cells. Genes Dev. 2009;23(18):2134‐2139. 10.1101/gad.1811609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Franceschi C, Garagnani P, Parini P, Giuliani C, Santoro A. Inflammaging: a new immune‐metabolic viewpoint for age‐related diseases. Nat Rev Endocrinol. 2018;14(10):576‐590. 10.1038/s41574-018-0059-4. [DOI] [PubMed] [Google Scholar]
- 66. Ferrucci L, Fabbri E. Inflammageing: chronic inflammation in ageing, cardiovascular disease, and frailty. Nat Rev Cardiol. 2018;15(9):505‐522. 10.1038/s41569-018-0064-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Velarde MC, Demaria M, Melov S, Campisi J. Pleiotropic age‐dependent effects of mitochondrial dysfunction on epidermal stem cells. Proc Natl Acad Sci U S A. 2015;112(33):10407‐10412. 10.1073/pnas.1505675112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Brace LE, Vose SC, Stanya K, et al. Increased oxidative phosphorylation in response to acute and chronic DNA damage. NPJ Aging Mech Dis. 2016;2:16022. 10.1038/npjamd.2016.22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Harman D. Aging: a theory based on free radical and radiation chemistry. J Gerontol. 1956;11(3):298‐300. 10.1093/geronj/11.3.298. [DOI] [PubMed] [Google Scholar]
- 70. Medawar, P. (1951). An unsolved problem in biology: An inaugural lecture delivered at University College. London: HK Lewis and Company, 6. [Google Scholar]
- 71. Shintani T, Klionsky DJ. Autophagy in health and disease: a double‐edged sword. Science. 2004;306(5698):990‐995. 10.1126/science.1099993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Zhu Y, Tchkonia T, Pirtskhalava T, et al. The Achilles' heel of senescent cells: from transcriptome to senolytic drugs. Aging Cell. 2015;14(4):644‐658. 10.1111/acel.12344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Melentijevic I, Toth ML, Arnold ML, et al. C. elegans neurons jettison protein aggregates and mitochondria under neurotoxic stress. Nature. 2017;542(7641):367‐371. 10.1038/nature21362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Conboy IM, Conboy MJ, Wagers AJ, Girma ER, Weissman IL, Rando TA. Rejuvenation of aged progenitor cells by exposure to a young systemic environment. Nature. 2005;433(7027):760‐764. 10.1038/nature03260. [DOI] [PubMed] [Google Scholar]
- 75. Soraas A, Matsuyama M, de Lima M, et al. Epigenetic age is a cell‐intrinsic property in transplanted human hematopoietic cells. Aging Cell. 2019;18(2):e12897. 10.1111/acel.12897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Fahy GM, Brooke RT, Watson JP, et al. Reversal of epigenetic aging and immunosenescent trends in humans. Aging Cell. 2019;18:e13028. 10.1111/acel.13028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Gurdon JB, Elsdale TR, Fischberg M. Sexually mature individuals of Xenopus laevis from the transplantation of single somatic nuclei. Nature. 1958;182(4627):64‐65. [DOI] [PubMed] [Google Scholar]
- 78. Stergiou L, Hengartner MO. Death and more: DNA damage response pathways in the nematode C. elegans . Cell Death Differ. 2003;11(1):21‐28. [DOI] [PubMed] [Google Scholar]
- 79. Gumienny TL, Lambie E, Hartwieg E, Horvitz HR, Hengartner MO. Genetic control of programmed cell death in the Caenorhabditis elegans hermaphrodite germline. Development. 1999;126(5):1011‐1022. [DOI] [PubMed] [Google Scholar]
- 80. Xia B, Yan Y, Baron M, et al. Widespread transcriptional scanning in the testis modulates gene evolution rates. Cell. 2020;180(2):248‐262 e221. 10.1016/j.cell.2019.12.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Hayflick L, Moorhead PS. The serial cultivation of human diploid cell strains. Exp Cell Res. 1961;25(3):585‐621. 10.1016/0014-4827(61)90192-6. [DOI] [PubMed] [Google Scholar]
- 82. Hayflick L. The limited in vitro lifetime of human diploid cell strains. Exp Cell Res. 1965;37(3):614‐636. 10.1016/0014-4827(65)90211-9. [DOI] [PubMed] [Google Scholar]
- 83. Zeng Y, Chen T. DNA methylation reprogramming during mammalian development. Genes (Basel). 2019;10(4):257. 10.3390/genes10040257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Sato T, Clevers H. Growing self‐organizing mini‐guts from a single intestinal stem cell: mechanism and applications. Science. 2013;340(6137):1190‐1194. 10.1126/science.1234852. [DOI] [PubMed] [Google Scholar]
- 85. Takahashi K, Yamanaka S. Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell. 2006;126(4):663‐676. 10.1016/j.cell.2006.07.024. [DOI] [PubMed] [Google Scholar]
- 86. Takahashi K, Tanabe K, Ohnuki M, et al. Induction of pluripotent stem cells from adult human fibroblasts by defined factors. Cell. 2007;131(5):861‐872. 10.1016/j.cell.2007.11.019. [DOI] [PubMed] [Google Scholar]
- 87. Ocampo A, Reddy P, Martinez‐Redondo P, et al. In vivo amelioration of age‐associated hallmarks by partial reprogramming. Cell. 2016;167(7):1719‐1733 e1712. 10.1016/j.cell.2016.11.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Sarkar TJ, Quarta M, Mukherjee S, et al. Transient non‐integrative nuclear reprogramming promotes multifaceted reversal of aging in human cells. bioRxiv. 2019;573386. 10.1101/573386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Lu Y, Krishnan A, Brommer B, et al. Reversal of ageing‐ and injury‐induced vision loss by tet‐dependent epigenetic reprogramming. bioRxiv. 2019;710210. 10.1101/710210. [DOI] [Google Scholar]
- 90. Chen C, Liu Y, Liu Y, Zheng P. mTOR regulation and therapeutic rejuvenation of aging hematopoietic stem cells. Sci Signal. 2009;2(98):ra75. 10.1126/scisignal.2000559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Antikainen H, Driscoll M, Haspel G, Dobrowolski R. TOR‐mediated regulation of metabolism in aging. Aging Cell. 2017;16(6):1219‐1233. 10.1111/acel.12689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Abad M, Mosteiro L, Pantoja C, et al. Reprogramming in vivo produces teratomas and iPS cells with totipotency features. Nature. 2013;502(7471):340‐345. 10.1038/nature12586. [DOI] [PubMed] [Google Scholar]
- 93. Ohnishi K, Semi K, Yamamoto T, et al. Premature termination of reprogramming in vivo leads to cancer development through altered epigenetic regulation. Cell. 2014;156(4):663‐677. 10.1016/j.cell.2014.01.005. [DOI] [PubMed] [Google Scholar]
- 94. Marion RM, Lopez de Silanes I, Mosteiro L, et al. Common telomere changes during in vivo reprogramming and early stages of tumorigenesis. Stem Cell Reports. 2017;8(2):460‐475. 10.1016/j.stemcr.2017.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Kirschner K, Chandra T, Kiselev V, et al. Proliferation drives aging‐related functional decline in a subpopulation of the hematopoietic stem cell compartment. Cell Rep. 2017;19(8):1503‐1511. 10.1016/j.celrep.2017.04.074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Zhang B, Ma S, Rachmin I, et al. Hyperactivation of sympathetic nerves drives depletion of melanocyte stem cells. Nature. 2020;577:676‐681. 10.1038/s41586-020-1935-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Ferguson MW, O'Kane S. Scar‐free healing: from embryonic mechanisms to adult therapeutic intervention. Philos Trans R Soc Lond Ser B Biol Sci. 2004;359(1445):839‐850. 10.1098/rstb.2004.1475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Ahmed AS, Sheng MH, Wasnik S, Baylink DJ, Lau KW. Effect of aging on stem cells. World J Experiment Med. 2017;7(1):1‐10. 10.5493/wjem.v7.i1.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Rossi DJ, Bryder D, Seita J, Nussenzweig A, Hoeijmakers J, Weissman IL. Deficiencies in DNA damage repair limit the function of haematopoietic stem cells with age. Nature. 2007;447(7145):725‐729. 10.1038/nature05862. [DOI] [PubMed] [Google Scholar]
- 100. Vermezovic J, Stergiou L, Hengartner MO, d'Adda di Fagagna F. Differential regulation of DNA damage response activation between somatic and germline cells in Caenorhabditis elegans . Cell Death Differ. 2012;19(11):1847‐1855. 10.1038/cdd.2012.69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. Bohnert KA, Kenyon C. A lysosomal switch triggers proteostasis renewal in the immortal C. elegans germ lineage. Nature. 2017;551(7682):629‐633. 10.1038/nature24620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Clavería C, Giovinazzo G, Sierra R, Torres M. Myc‐driven endogenous cell competition in the early mammalian embryo. Nature. 2013;500:39‐44. 10.1038/nature12389. [DOI] [PubMed] [Google Scholar]
- 103. Kermi C, Aze A, Maiorano D. Preserving genome integrity during the early embryonic DNA replication cycles. Genes (Basel). 2019;10(5):398. 10.3390/genes10050398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104. Matheu A, Maraver A, Klatt P, et al. Delayed ageing through damage protection by the Arf/p53 pathway. Nature. 2007;448(7151):375‐379. 10.1038/nature05949. [DOI] [PubMed] [Google Scholar]
- 105. Carrasco‐Garcia E, Arrizabalaga O, Serrano M, Lovell‐Badge R, Matheu A. Increased gene dosage of Ink4/Arf and p53 delays age‐associated central nervous system functional decline. Aging Cell. 2015;14(4):710‐714. 10.1111/acel.12343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106. Schmelzle T, Hall MN. TOR, a central controller of cell growth. Cell. 2000;103(2):253‐262. [DOI] [PubMed] [Google Scholar]
- 107. Mendrysa SM, O'Leary KA, McElwee MK, et al. Tumor suppression and normal aging in mice with constitutively high p53 activity. Genes Dev. 2006;20(1):16‐21. 10.1101/gad.1378506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108. Hubbard K, Ozer HL. Mechanism of immortalization. Age (Omaha). 1999;22(2):65‐69. 10.1007/s11357-999-0008-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109. Chatsirisupachai K, Palmer D, Ferreira S, de Magalhaes JP. A human tissue‐specific transcriptomic analysis reveals a complex relationship between aging, cancer, and cellular senescence. Aging Cell. 2019;e13041. 10.1111/acel.13041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110. Schiebinger, G. , Shu, J. , Tabaka, M , et al. (2017). Reconstruction of developmental landscapes by optimal‐transport analysis of single‐cell gene expression sheds light on cellular reprogramming. bioRxiv . doi: 10.1101/191056 [DOI]
- 111. Yamanaka S. A fresh look at iPS cells. Cell. 2009;137(1):13‐17. 10.1016/j.cell.2009.03.034. [DOI] [PubMed] [Google Scholar]
- 112. Olova N, Simpson DJ, Marioni RE, Chandra T. Partial reprogramming induces a steady decline in epigenetic age before loss of somatic identity. Aging Cell. 2019;18(1):e12877. 10.1111/acel.12877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113. Buganim Y, Markoulaki S, van Wietmarschen N, et al. The developmental potential of iPSCs is greatly influenced by reprogramming factor selection. Cell Stem Cell. 2014;15(3):295‐309. 10.1016/j.stem.2014.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114. Rackham OJ, Firas J, Fang H, et al. A predictive computational framework for direct reprogramming between human cell types. Nat Genet. 2016;48(3):331‐335. 10.1038/ng.3487. [DOI] [PubMed] [Google Scholar]
- 115. Mosteiro L, Pantoja C, de Martino A, Serrano M. Senescence promotes in vivo reprogramming through p16(INK)(4a) and IL‐6. Aging Cell. 2017;17:e12711. 10.1111/acel.12711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116. Atala A, Irvine DJ, Moses M, Shaunak S. Wound healing versus regeneration: role of the tissue environment in regenerative medicine. MRS Bull. 2010;35(8):597‐606. 10.1557/mrs2010.528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117. Olutoye OO, Cohen IK. Fetal wound healing: an overview. Wound Repair Regen. 1996;4(1):66‐74. 10.1046/j.1524-475X.1996.40112.x. [DOI] [PubMed] [Google Scholar]
- 118. Haas BJ, Whited JL. Advances in decoding axolotl limb regeneration. Trends Genet. 2017;33(8):553‐565. 10.1016/j.tig.2017.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119. Pfefferli C, Jazwinska A. The art of fin regeneration in zebrafish. Regeneration (Oxford, England). 2015;2(2):72‐83. 10.1002/reg2.33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120. Seifert AW, Kiama SG, Seifert MG, Goheen JR, Palmer TM, Maden M. Skin shedding and tissue regeneration in African spiny mice (Acomys). Nature. 2012;489(7417):561‐565. 10.1038/nature11499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121. Porrello ER, Mahmoud AI, Simpson E, et al. Transient regenerative potential of the neonatal mouse heart. Science. 2011;331(6020):1078‐1080. 10.1126/science.1200708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122. Simpson DM, Ross R. The neutrophilic leukocyte in wound repair a study with antineutrophil serum. J Clin Invest. 1972;51(8):2009‐2023. 10.1172/JCI107007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123. Moore AL, Marshall CD, Longaker MT. Minimizing skin scarring through biomaterial design. J Funct Biomater. 2017;8(1):3. 10.3390/jfb8010003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124. Linares HA. From wound to scar. Burns. 1996;22(5):339‐352. 10.1016/0305-4179(95)00164-6. [DOI] [PubMed] [Google Scholar]
- 125. Kinzina ED, Podolskiy DI, Dmitriev SE, Gladyshev VN. Patterns of aging biomarkers, mortality, and damaging mutations illuminate the beginning of aging and causes of early‐life mortality. Cell Rep. 2019;29(13):4276‐4284 e4273. 10.1016/j.celrep.2019.11.091. [DOI] [PubMed] [Google Scholar]
- 126. Kennedy BK, Berger SL, Brunet A, et al. Geroscience: linking aging to chronic disease. Cell. 2014;159(4):709‐713. 10.1016/j.cell.2014.10.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127. Oh J, Lee YD, Wagers AJ. Stem cell aging: mechanisms, regulators and therapeutic opportunities. Nat Med. 2014;20(8):870‐880. 10.1038/nm.3651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128. Keyes BE, Liu S, Asare A, et al. Impaired epidermal to dendritic T cell signaling slows wound repair in aged skin. Cell. 2016;167(5):1323‐1338 e1314. 10.1016/j.cell.2016.10.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129. Ashcroft GS, Mills SJ, Ashworth JJ. Ageing and wound healing. Biogerontology. 2002;3(6):337‐345. [DOI] [PubMed] [Google Scholar]
- 130. Bode HR. Head regeneration in hydra. Dev Dyn. 2003;226(2):225‐236. 10.1002/dvdy.10225. [DOI] [PubMed] [Google Scholar]
- 131. Weinreich J, Lob S, Loffler M, et al. Rapamycin‐induced impaired wound healing is associated with compromised tissue lactate accumulation and extracellular matrix remodeling. Eur Surg Res. 2011;47(1):39‐44. 10.1159/000327972. [DOI] [PubMed] [Google Scholar]
- 132. Cheng KY, Hao M. Mammalian target of rapamycin (mTOR) regulates transforming growth factor‐beta1 (TGF‐beta1)‐induced epithelial‐mesenchymal transition via decreased pyruvate kinase M2 (PKM2) expression in cervical cancer cells. Med Sci Monit. 2017;23:2017‐2028. 10.12659/msm.901542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133. Reddien PW, Sanchez Alvarado A. Fundamentals of planarian regeneration. Annu Rev Cell Dev Biol. 2004;20:725‐757. 10.1146/annurev.cellbio.20.010403.095114. [DOI] [PubMed] [Google Scholar]
- 134. Bryant DM, Sousounis K, Payzin‐Dogru D, et al. Identification of regenerative roadblocks via repeat deployment of limb regeneration in axolotls. NPJ Regen Med. 2017;2(1):30. 10.1038/s41536-017-0034-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135. Mertens J, Paquola AC, Ku M, et al. Directly reprogrammed human neurons retain aging‐associated transcriptomic signatures and reveal age‐related nucleocytoplasmic defects. Cell Stem Cell. 2015;17(6):705‐718. 10.1016/j.stem.2015.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Transparent‐Peer‐Review‐Record