

Keywords: idiopathic pulmonary fibrosis, lung fibrosis, Pim kinase, secretome, senescence
Abstract
Cellular senescence is emerging as a driver of idiopathic pulmonary fibrosis (IPF), a progressive and fatal disease with limited effective therapies. The senescence-associated secretory phenotype (SASP), involving the release of inflammatory cytokines and profibrotic growth factors by senescent cells, is thought to be a product of multiple cell types in IPF, including lung fibroblasts. NF-κB is a master regulator of the SASP, and its activity depends on the phosphorylation of p65/RelA. The purpose of this study was to assess the role of Pim-1 kinase as a driver of NF-κB-induced production of inflammatory cytokines from low-passage IPF fibroblast cultures displaying markers of senescence. Our results demonstrate that Pim-1 kinase phosphorylates p65/RelA, activating NF-κB activity and enhancing IL-6 production, which in turn amplifies the expression of PIM1, generating a positive feedback loop. In addition, targeting Pim-1 kinase with a small molecule inhibitor dramatically inhibited the expression of a broad array of cytokines and chemokines in IPF-derived fibroblasts. Furthermore, we provide evidence that Pim-1 overexpression in low-passage human lung fibroblasts is sufficient to drive premature senescence, in vitro. These findings highlight the therapeutic potential of targeting Pim-1 kinase to reprogram the secretome of senescent fibroblasts and halt IPF progression.
INTRODUCTION
Idiopathic pulmonary fibrosis (IPF) is the most common interstitial lung disease (1), with an annual incidence in North America and Europe of ∼3–9/100,000 (2) and a median survival rate of 2–4 years (3, 4). There are currently two FDA-approved therapies for IPF: nintedanib and pirfenidone (5). Although these therapies slow down IPF progression, they have minimal impact on patient mortality (6, 7). Therefore, it remains a significant priority to elucidate the signaling pathways and cell populations regulating IPF progression, to design targeted and effective therapies.
The expansion of single-cell RNA sequencing has helped identify spatially, functionally, and transcriptionally distinct populations of lung fibroblasts in IPF (8–11). These findings have been further validated by examining low passage explanted fibroblast cultures derived from IPF lung tissue. Compared with age-matched controls, lung fibroblasts derived from patients with IPF display multiple features of cellular senescence, including increased expression of cyclin-dependent kinase inhibitors CDKN2A (p16INK4A) and CDKN1A (p21Cip1), and prominent senescence-associated β-galactosidase (SA-β-gal) staining (12–14). In addition, fibroblasts derived from patients with IPF exhibit a senescence-associated secretory phenotype (SASP), producing heightened inflammatory cytokines including IL-6 (IL6), IL-1β (IL1B), and monocyte chemoattractant protein-1 (MCP-1) (CCL2), which contribute to fibrosis progression (13, 15, 16). Nuclear factor κ-light-chain-enhancer of activated B cells (NF-κB) is a master regulator of SASP component transcription (17, 18). NF-κB is an inducible transcription factor complex made up of five different DNA binding proteins (NF-κB1, NF-κB2, p65/RelA, RelB, and c-Rel) that can form diverse homodimers and heterodimers. In brief, a canonical NF-κB heterodimer complex, composed of NF-κB1 and p65/RelA, is sequestered in the cytosol by the inhibitory proteins IκB. Multiple upstream kinases can phosphorylate IκB, releasing the NF-κB complex to translocate into the nucleus and induce expression of target genes (18). In addition, posttranslational modifications in the transactivation domain of p65/RelA are essential for NF-κB complex activity, including phosphorylation of serine 276 (S276) (19). Inhibiting NF-κB phosphorylation may block the transcription of SASP-associated genes and prevent the aberrant secretome of senescent IPF fibroblasts.
Proviral integration site for Moloney murine leukemia virus (Pim) kinases are serine/threonine kinases involved in the regulation of cellular proliferation and survival (20, 21). There are three isoforms of Pim kinases: Pim-1, Pim-2, and Pim-3, with Pim-1 being the most extensively studied, largely due to its overexpression in multiple cancers and its oncogenic potential (22–24). Pim-1 kinase has also been shown to play a role in lung fibrosis. In a recent publication, a Pim-1 kinase inhibitor or intratracheal delivery of adenoviral short hairpin RNA targeting PIM1 prevented collagen accumulation and aberrant alveolar histology following a pulmonary injury in the bleomycin mouse model of lung fibrosis (25). This study emphasized the impact of Pim-1 kinase in regulating epithelial-to-mesenchymal transition but did not explore the role of Pim-1 kinase in lung fibroblasts. To this end, we recently identified Pim-1 kinase is a central driver in age-associated chronic fibroblast activation in mouse lungs after bleomycin injury (26). In addition to its well-defined roles in proliferation, evidence also suggests that Pim-1 plays a role in the initiation of cellular senescence by upregulating CDKN2A (p16INK4A) through multiple mechanisms (27, 28). Furthermore, previous work with cervical cancer cells has shown that Pim-1 kinase has the capacity to directly phosphorylate serine 276 of p65/RelA (29), and studies in pancreatic cancer cells suggest that the IL-6/JAK/STAT axis regulates Pim-1 kinase expression (30).
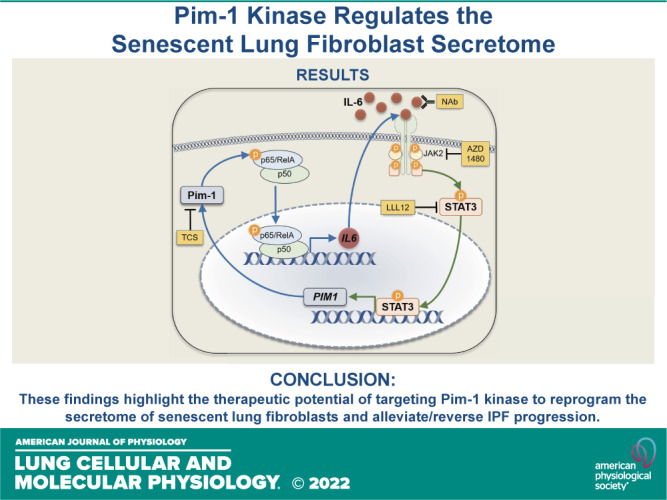
In this study, we aimed to assess the role of Pim-1 kinase in modulating NF-κB activity and downstream SASP presentation in lung fibroblasts derived from patients with IPF. Our results demonstrate that Pim-1 kinase promotes inflammatory cytokine production by activating NF-κB transcription and enhancing IL-6 production, which in turn elevates PIM1 expression, generating a positive feedback loop in cultured lung fibroblasts derived from patients with IPF. These findings highlight the therapeutic potential of targeting Pim-1 kinase to reprogram the secretome of senescent fibroblasts and mitigate IPF progression.
METHODS
Cell Culture
Primary human lung fibroblasts (non-IPF) or fibroblasts derived from patients with IPF (IPF) were purchased from ATCC or Lonza or provided by Peter Bitterman and Craig Henke from the University of Minnesota (Minneapolis, MN); fibroblasts were isolated by explant culture from IPF donors or non-IPF donors whose lungs were rejected for transplantation, under a protocol approved by the University of Minnesota Institutional Review Board. Fibroblasts were cultured in a humidified 37°C, 5% CO2 incubator with Eagle’s minimal essential medium (EMEM) (ATCC) containing 10% fetal bovine serum (FBS) and antibiotic-antimycotic (Thermo Fisher Scientific) unless otherwise noted. Cells were routinely characterized by PCR or Western blotting using specific fibroblast markers including type I collagen and vimentin. Mycoplasma contamination was monitored routinely by PCR. Experiments were performed with cells between passages 3 and 6. The donor age and sex of all cells used in this are provided in Supplemental Table S1 (all Supplemental Material is available at https://doi.org/10.6084/m9.figshare.18737336).
Chemicals and Reagents
Dimethyl sulfoxide (DMSO) was purchased from Fisher Scientific. 2-Mercaptoethanol was purchased from Bio-Rad Laboratories and used for RNA isolation, per the RNeasy Plus Mini Kit (Qiagen) protocol. TCS Pim-1 1 (TCS) and AZD 1480 were purchased from Cayman Chemical. LLL12 was purchased from BioVision. Endotoxin/azide-free IL-6 neutralizing antibody (IL-6 NAb) was purchased from Biolegend (501110). Recombinant TNFα was purchased from R&D systems (10291-TA).
RNA Interference
Cells were transfected in six-well plates using Lipofectamine RNAiMAX (Life Technologies) with siGENOME siRNA SMARTpool (Dharmacon) targeting PIM1 (L-003923-00-0005) or a nontargeting SMARTpool (D-001810-10-05) according to the manufacturers’ suggested protocols. Cells were cultured for 72 h before collecting RNA or protein.
Western Blot
Fibroblasts were plated in six-well plates and treated as specified for each experiment. Total cellular protein was collected using Radioimmunoprecipitation assay buffer (Thermo) containing Pierce Phosphatase Inhibitor and Halt Protease Inhibitor Cocktail (Thermo). BCA Protein Assay Kit (Thermo) was used to measure the total protein concentration of the lysate samples, which were then run on gradient polyacrylamide gels. After transfer and blocking with 5% nonfat dry milk for 60 min, the membranes were incubated with the following primary antibodies overnight: GAPDH (Cell Signaling, 14C10), Pim-1 (Santa Cruz, sc-13513), p65 (Cell Signaling, 8242), and pS276-p65 (Novus, NB100-82086). Blocking buffer was used to dilute all primary antibodies 1:1,000. Tris-buffered saline (TBS)-Tween was used to wash the blots, which were then incubated with horseradish peroxidase (HRP)-conjugated secondary antibodies for 1 h. Secondary antibodies were diluted 1:10,000 in the blocking buffer. Membranes were imaged via a ChemiDoc Imaging System (Bio-Rad) and protein quantification was performed via densitometry using Image Lab v6.0 (Bio-Rad).
PIM1 Overexpression
The control lentiviral vector (Lenti-Con) (pLV-EGFP-CMV-mCherry) and lentiviral vector carrying the human PIM1 gene (Lenti-PIM1) (pLV-EGFP-CMV-hPIM1) were bought packaged in viral particles from VectorBuilder (Chicago, IL). Normal human lung fibroblasts at passage 3 were transduced at a multiplicity of infection (MOI) of 3 with Lenti-Con or Lenti-PIM1 in MEM containing 10% FBS, 100 U/mL of penicillin, 100 µg/mL of streptomycin, and 5 µg/mL of polybrene for 48 h, after which transduced cells were expanded and FACS sorted for GFP+ cells. After sorting, cells were expanded and used for experiments.
RNA Isolation/qPCR
Fibroblasts were treated as indicated for each experiment. The RNeasy Plus Mini Kit (Qiagen) was used to isolate RNA, following Qiagen’s protocol. The SuperScript VILO cDNA Synthesis Kit (Invitrogen) and PTC-200 Peltier Thermal Cycler (MJ Research) were used to synthesize cDNA. The FastStart Essential DNA Green Master (Roche) and LightCycler 96 (Roche) were used to perform quantitative PCR (qPCR) on the cDNA. Data are expressed as a fold change compared with control-treated cells, by ΔΔCt relative to GAPDH. Primers were designed using the IDT RealTime PCR tool: GAPDH (F: ACATCGCTCAGACACCATG, R: TGTAGTTGAGGTCAATGAAGGG), PIM1 (F: CGACATCAAGGACGAAAACATC, R: ACTCTGGAGGGCTATACACTC), IL6 (F: CCACTCACCTCTTCAGAACG, R: CATCTTTGGAAGGTTCAGGTTG), CDKN2A (F: GATGTCGCACGGTACCTG, R: TCTCTGGTTCTTTCAATCGGG), CDKN1A (F: TGTCCGTCAGAACCCATGC, R: AAAGTCGAAGTTCCATCGCTC), CCL7 (F: GAGAGCTACAGAAGGACCAC, R: GTTTTCTTGTCCAGGTGCTTC), CXCL10 (F: CCTTATCTTTCTGACTCTAAGTGGC, R: ACGTGGACAAAATTGGCTTG), CCL2 (F: TGTCCCAAAGAAGCTGTGATC, R: ATTCTTGGGTTGTGGAGTGAG), CXCL8 (F: ATACTCCAAACCTTTCCACCC, R: TCTGCACCCAGTTTTCCTTG), IL1B (F: ATGCACCTGTACGATCACTG, R: ACAAAGGACATGGAGAACACC).
Cellular Proliferation
Fibroblasts were plated into 96-well plates (3,000 cells/well) and cultured for 4 days. Cells were then fixed in 4% paraformaldehyde, then permeabilized with 0.25% Triton X-100, and DAPI stained. Images were then taken using a Cytation5 imaging reader (BioTek) and the number of DAPI objects within each field of view was quantified by automated Gen5 software (BioTek).
Senescence-Associated β-Galactosidase Staining
Fibroblasts were plated into 12-well plates and SA-β-galactosidase staining was performed per the manufacturer’s protocol (Senescence β-Galactosidase Staining Kit No. 9860, Cell Signaling Technology). Briefly, cells were washed with 1× PBS and then 1× fixative solution for 15 min at room temperature. The plates were then washed two times with 1× PBS. β-Galactosidase staining solution was made by diluting 100× solution A and solution B and 20 mg/mL of X-gal solution in DMSO to 1× staining solution, diluted from 10× solution provided with distilled water; then, the pH was adjusted to 6.0. β-Galactosidase staining solution was added to each well. The plate was sealed with parafilm to prevent evaporation and crystal formation. The plate was incubated at 37°C overnight in a dry incubator (no CO2). The β-galactosidase staining solution was removed and rinsed with 1× PBS. The cells were imaged with an optical microscope using a ×10 objective. For quantification of SA-β-galactosidase staining (Fig. 6), phase contrast images were first taken of the β-galactosidase staining followed by DAPI staining, imaging, and quantification using a Cytation5 imager with on-board automated software- Gen5 (BioTek) as previously performed (31–33). Data are plotted as the percentage of SA-β-galactosidase positive cells/total number of cells in each field of view.
Figure 6.

Proviral Integration site for Moloney murine leukemia virus 1 (Pim-1) promotes lung fibroblast premature senescence. A: non-idiopathic pulmonary fibrosis (IPF) adult lung fibroblasts were infected with control lentiviral vector or lentiviral vector carrying the human PIM1 gene. Total protein was isolated in three independent Western blot experiments. Representative images are shown and band density was quantified (**P < 0.01 vs. the Lenti-Control group). B: Lenti-Control and Lenti-PIM1 expressing cells were treated for 3 h or 24 h with 3 µM TCS before total protein isolation and Western blot analysis. Representative images are shown and band density for three independent experiments was quantified (**P < 0.01, ***P < 0.001 vs. the indicated group). C and E: qPCR analysis from three independent experiments comparing gene expression between Lenti-Control and Lenti-PIM1 fibroblasts (*P < 0.05, **P < 0.01, ***P < 0.001 vs. the Lenti-Control group). D: Lenti-Control and Lenti-PIM1 expressing fibroblasts were cultured for 0 and 4 days and then stained for DAPI and quantified using automated imaging software. Representative images are shown for the number of DAPI objects (nuclei) on day 4. Quantification of three independent experiments plots the number of cells in each field of view, for each cell line, relative to the number of cells in each field of view immediately after the cells attached (day 0) (*P < 0.05 vs. the Lenti-Control group). The scale bar represents 200 µm. F: senescence-associated β-galactosidase (SA-β-gal) staining comparing Lenti-Control vs. Lenti-PIM1 lung fibroblasts as they progress through cell culture passages 4–8. Representative images comparing fibroblasts at passage 8. Data are automated quantification of SA-β-gal positive cells relative to the total number of cells in each field of view from three independent experiments (*P < 0.05, ****P < 0.0001 vs. the Lenti-Control group). Scale bar represents 100 µm.
Cytokine and Chemokine PCR Array
Fibroblasts were plated into six-well plates and treated with 3 µM TCS in EMEM containing 0.1% FBS. Total RNA was isolated using RNeasy Plus Mini Kit (Qiagen), following the manufacturer’s protocol. RNA (1 μg) was then used for the synthesis of cDNA using SuperScript VILO (Invitrogen). qPCR was performed using RT2 SYBR Green qPCR Mastermix (Qiagen) and run on a QuantStudio 5 (Applied Biosystems) 384-well real-time PCR thermocycler. Data are expressed as a fold change compared with control-treated cells, by ΔΔCt relative to GAPDH.
Cytokine Protein Array
Fibroblasts were plated into six-well plates (Thermo Fischer Scientific) in EMEM containing 10% FBS and allowed to attach for 24 h. Media was then exchanged with EMEM containing 0.1% FBS and cells were treated for 24 h with 3 µM TCS. All wells were treated with a final concentration of 0.1% DMSO. Conditioned media was harvested for the detection of cytokines, using the Human Cytokine Antibody Array (Abcam, ab133997) according to the manufacturer’s instructions. All reagents and conditioned media samples were thawed on ice and prepared before use, and all membrane incubation periods were performed under gentle rocking. Briefly, array membranes were incubated with a blocking buffer for 30 min at room temperature. Subsequently, conditioned media samples were incubated with array membranes overnight at 4°C. Membranes were washed and incubated with biotin-conjugated anti-cytokines for 2 h at room temperature. Afterward, membranes were washed and incubated with HRP-conjugated streptavidin for 2 h at room temperature. Membranes were then washed and incubated with chemiluminescent detection reagents and imaged via ChemiDoc Imaging System (Bio-Rad). Spot density was quantified via densitometry analysis using Image Lab v6.0 (Bio-Rad). Data are presented as raw signal intensity-background (Fig. 5B) and fold change relative to the control treated well for each donor sample (Fig. 5C).
Figure 5.

Proviral Integration site for Moloney murine leukemia virus 1 (Pim-1) regulates secretion of GRO, IL-6, MCP-1, and MCP-3. Lung fibroblasts derived from patients with idiopathic pulmonary fibrosis (IPF) were treated for 24 h with the Pim-1 inhibitor (TCS, 3 µM) in media without FBS. After incubation, conditioned media was collected and assessed by a cytokine immuno array. A: representative images of the cytokine array from control and TCS-treated samples. +/− controls labeled along with the six cytokines exhibiting the strongest signal intensity. B: raw signal intensity—background quantified for all cytokines included in the array. n = 3 biological and experimentally independent experiments. C: signal intensity for each cytokine was normalized relative to the same DMSO-treated, representative sample. n = 3 biologically and experimentally independent experiments (*P < 0.05, **P < 0.01 vs. DMSO treated groups).
LIVE/DEAD Assay
Fibroblasts were plated into 96-well plates. Cells were treated with the indicated compounds for 24 h. Cells were incubated with each component of the LIVE/DEAD kit (Thermo Fisher Scientific) for 30 min. A Cytation5 microplate fluorescence microscope (BioTek) was used to obtain fluorescent images of each well and cell counts were determined using Gen5 software (BioTek).
Statistics
Data analysis and plotting were performed using Prism 9.0 (GraphPad). Statistical comparison between two groups was performed by paired or unpaired t test, or Mann–Whitney test, according to the data characteristics and the need for parametric or nonparametric testing, respectively. A statistical analysis of three or more groups was performed by repeated-measures (RM) one-way ANOVA with Dunnett’s multiple comparison test, where the mean value of each group was compared against a control group. Results are expressed as the means ± SE. The sample number (n) indicates the number of independent samples in each experiment.
RESULTS
Pim-1 Phosphorylates and Activates p65/RelA
To evaluate the role of Pim-1 kinase in lung fibroblast NF-κB activity, we performed a Western blot to measure S276 phosphorylation on p65/RelA, relative to total p65 in non-IPF lung fibroblasts treated with or without TNF-α after knocking down PIM1 using siRNA. Consistent with previous findings, fibroblast stimulation with TNF-α promoted phosphorylation of p65/RelA on S276 (pS276-p65) (34), and this response was dramatically reduced by the knockdown of PIM1 (Fig. 1A). PIM1 targeting siRNA also reduced total p65 expression (Fig. 1A), which is consistent with a prior report demonstrating S276 phosphorylation of p65 by Pim-1 also prevents ubiquitin-mediated proteolysis (34). To confirm that Pim-1 kinase regulates NF-κB signaling, we measured transcript expression of well-defined NF-κB target genes by qPCR. As shown in Fig. 1B, TNFα enhanced IL6, CCL2, and IL1B gene expression, which was prevented by the knockdown of PIM1 (Fig. 1B). We also performed siRNA targeting of PIM1 in non-IPF lung fibroblasts in the absence of TNFα stimulation and did not observe changes in IL6, CCL2, or IL1B expression, suggesting this pathway is inactive in these cells in the absence of exogenous stimuli (Supplemental Fig. S1). To further confirm that Pim-1 kinase is necessary for the phosphorylation of p65 on S276, we treated non-IPF lung fibroblasts with or without TNFα in the presence of a previously identified (35), selective Pim-1 kinase inhibitor: TCS Pim-1 1 (hereinafter referred to as: TCS). As shown in Fig. 1C, consistent with PIM1 siRNA experiments, TCS blocked p65/RelA phosphorylation induced by TNFα and promoted a loss in p65 total protein. Taken together, our results demonstrate that Pim-1 kinase promotes phosphorylation of p65/RelA on S275 and modulates the activity of NF-κB in cultured human lung fibroblasts stimulated with TNF-α.
Figure 1.

Proviral integration site for Moloney murine leukemia virus 1 (Pim-1) regulates phosphorylation and activity of p65/RelA. A: non-idiopathic pulmonary fibrosis (IPF) adult lung fibroblasts were transfected with nontargeting (NT) siRNA or siRNA targeting PIM1 for 72 h. Fifteen minutes before collecting total cell protein for Western blot analysis, the indicated wells were stimulated with 50 ng/mL TNF-α. Representative image is shown and band density for three independent experiments was quantified (*P < 0.05, **P < 0.01 vs. the indicated group). B: non-IPF adult lung fibroblasts were transfected with nontargeting (NT) siRNA or siRNA targeting PIM1 for 72 h. Six hours before collecting RNA for qPCR analysis, the indicated wells were stimulated with 50 ng/mL TNF-α. n = 3 independent experiments (**P < 0.01, ***P < 0.001, ****P < 0.0001 vs. the indicated group). C: non-IPF adult lung fibroblasts were treated for 3 h with TCS Pim-1 1 (TCS) (3 µM) and 15 min before collecting total cell protein for Western blot analysis, the indicated wells were stimulated with 50 ng/mL TNF-α. Representative image is shown and band density for three independent experiments was quantified (**P < 0.01, ***P < 0.001 vs. the indicated group).
Senescent Lung Fibroblasts Derived from Patient with IPF Express Elevated Pim-1 and p65/RelA Phosphorylation
Pim-1 transcript and protein levels are upregulated in mouse lung tissue after intratracheal exposure to bleomycin (25). To determine if this is also observed in lung fibroblasts derived from patients with IPF, we compared protein expression of Pim-1 and pS276-p65 in low passage fibroblasts derived from patient with IPF to age-matched, non-IPF control fibroblasts. Both Pim-1 protein expression and phosphorylation of p65/RelA (S276) were increased in IPF fibroblasts compared with non-IPF fibroblasts (Fig. 2A, Supplemental Table S1). To begin investigating the mechanism of Pim-1 overexpression, we compared transcript levels of PIM1 and IL6 along with markers of senescence CDKN2A (p16INK4A) and CDKN1A (p21Cip1). Consistent with previous findings, lung fibroblasts derived from patients with IPF expressed elevated levels of senescence-associated cyclin-dependent kinase inhibitors and IL6 (Fig. 2B) (16). We plotted correlation comparisons for each gene and found PIM1 expression in cultured lung fibroblasts to align better with CDKN2A than either IL6 or CDKN1A, though the sample sizes were limited (Supplemental Fig. S2). We performed senescence associated β-galactosidase (SA-β-Gal) staining to confirm the senescence phenotype in IPF-derived fibroblasts (Fig. 2C). Consistent with previous findings (12, 14), fibroblasts derived from patients with IPF were positive for β-galactosidase staining.
Figure 2.

Senescent idiopathic pulmonary fibrosis (IPF) patient-derived lung fibroblasts express spontaneously elevated Pim-1 and p65/RelA phosphorylation. A: lung fibroblasts from non-IPF or IPF donors were cultured for 24 h in the absence of serum before total cell protein isolation and Western Blot analysis. n = 3 non-IPF and 4 IPF biologically independent samples (**P < 0.01 vs. non-IPF). B: lung fibroblasts from non-IPF or IPF donors were cultured for 24 h in the absence of serum before RNA isolation and qPCR analysis. RNA transcript levels are quantified relative to the same representative non-IPF sample, using GAPDH as a housekeeping gene. n = 4 non-IPF and 7 IPF biologically independent samples (*P < 0.05, **P < 0.01 vs. non-IPF). C: lung fibroblasts from non-IPF or IPF donors were stained for senescence-associated β-galactosidase (SA-β-gal) staining as a marker of cellular senescence. Representative examples for non-IPF and IPF are shown.
Pim-1/NF-κB and IL-6/JAK2/STAT3 Form a Positive Feedback Loop
IL-6 binds its cognate receptor IL6R, which promotes the formation of and signaling via a heterohexamer complex of IL-6, IL6R, and gp130. This complex leads to the activation of downstream mediators, including JAK2 and STAT3 (36, 37). Previous studies in pancreatic cancer cells found Pim-1 kinase expression to be regulated by STAT3 transcriptional activity, and STAT3 has been shown to play an important role in pulmonary fibrosis (30, 38). Here, we measured the expression of PIM1 in IPF lung fibroblasts treated with a neutralizing antibody against IL-6 (IL-6 NAb), a JAK2 kinase inhibitor (AZD 1480) (39), and a STAT3 kinase inhibitor (LLL12) (38). In all scenarios, blockade of the IL-6/JAK/STAT axis resulted in decreased PIM1 expression (Fig. 3A), confirming the involvement of STAT3 pathways in maintaining/regulating PIM1 expression in IPF fibroblasts. Next, to confirm that Pim-1 kinase is essential for phosphorylation and activation of NF-κB in IPF fibroblasts, which display high Pim-1 and phosphorylation of p65/RelA, we knocked down PIM1 using siRNA and observed that Pim-1 inhibition reduced S276 phosphorylation and p65 total protein expression in these cells (Fig. 3B). Consistent with these findings, we also detected reduced expression of IL6 transcript in fibroblasts derived from patients with IPF treated with PIM1-targeted siRNA (Fig. 3C). These findings suggest that the IL-6/STAT3 axis promotes expression of Pim-1 kinase, which in turn, stimulates the expression of IL-6, demonstrating a positive feedback loop existing within senescent IPF lung fibroblasts (Fig. 3D).
Figure 3.

Proviral Integration site for Moloney murine leukemia virus 1 (Pim-1)/nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB) and IL-6/JAK2/STAT3 form a positive feedback loop. A: lung fibroblasts derived from patients with idiopathic pulmonary fibrosis (IPF) were cultured for 24 h in the presence of an IL-6 neutralizing antibody (IL-6 NAb, 10 μg/mL), a JAK2 inhibitor (AZD 1480, 10 µM), or a STAT3 inhibitor (LLL12, 1 µM) before RNA isolation and qPCR analysis. RNA transcript levels are quantified relative to the same representative nontargeting (NT) sample, using GAPDH as a housekeeping gene. n = 3 biologically independent and experimentally independent experiments (**P < 0.01 vs. the control treated groups). B: lung fibroblasts derived from patients with IPF were transfected with nontargeting (NT) siRNA or siRNA targeting PIM1 for 72 h before collecting whole cell protein for Western blot analysis. Representative images are shown and band density for three independent experiments was quantified (*P < 0.05, **P < 0.01 vs. the NT control group). C: lung fibroblasts derived from patients with IPF were transfected with nontargeting (NT) siRNA or siRNA targeting PIM1 and cultured for 72 h before collecting RNA for qPCR analysis. RNA transcript levels are quantified relative to the same representative NT sample, using GAPDH as a housekeeping gene. n = 3 independent experiments (***P < 0.001 vs. the indicated group). D: positive feedback model. Pim-1 phosphorylates p65/RelA, supporting NF-κB transcriptional activity that stimulates IL-6 synthesis. IL-6 signals through its receptor system and JAK2/STAT3 to transcribe PIM1. Tools used throughout our studies are highlighted in yellow. Schematic was prepared in part using Motifolio Scientific Illustration Toolkits.
Pim-1 is Essential for the Expression of a Broad Range of Cytokines and Chemokines
Having initially focused analyses on only a few cytokines, we next evaluated the broader role of Pim-1 kinase in regulating the secretion of other inflammatory mediators in IPF fibroblasts. We treated fibroblasts derived from patients with IPF for 24 h with the Pim-1 kinase inhibitor TCS followed by gene expression analysis using a profiler PCR array. We amplified the transcript of 84 genes encoding for cytokines and chemokines and found that among them, 49 were expressed. Intriguingly, in response to Pim-1 inhibition, numerous genes encoding chemokines and NF-κB responders were repressed (Fig. 4A). We designed primers for the three most repressed genes (CCL7, CXCL10, and CCL2) and further confirmed robust repression of these genes after exposure of IPF fibroblasts to TCS (Fig. 4B). To validate that this effect was not the result of cytotoxic effects of the compound, we performed a LIVE/DEAD assay in IPF lung fibroblasts treated for the same duration with TCS. Our findings confirmed that 3 µM TCS was not cytotoxic to our cells (Supplemental Fig. S3). We also compared the effects of TCS to two additional established Pim kinase inhibitors (Supplemental Fig. S4A). Similar to TCS, SMI-4a is thought to be selective for Pim-1 (40), whereas AZD1208 is a pan-Pim kinase inhibitor (41). All three compounds blocked the expression of IL6, CCL2, and IL1B but did not impact the expression of CXCL8, consistent with what we have observed in the cytokine and chemokine array. In looking closely at the literature, the selectivity of TCS and SMI-4a is based on their ability to inhibit Pim-1 over Pim-2 (35, 42). Unfortunately, neither compound has been tested against Pim-3, which shares the greatest homology to Pim-1 (43). This leaves the door open for Pim-3 playing a role in the effects we observe with TCS throughout this manuscript. We performed siRNA targeting PIM3 in IPF lung fibroblasts and found no reduction in IL6, CCL2, and IL1B, further supporting that the effects of TCS are through inhibition of Pim-1 (Supplemental Fig. S4B). These findings suggest that Pim-1 kinase is a broadly influential regulator of inflammatory secretome transcription in cultured lung fibroblasts derived from patients with IPF.
Figure 4.

Proviral Integration site for Moloney murine leukemia virus 1 (Pim-1) is essential for the expression of a broad range of cytokines and chemokines. A: lung fibroblasts derived from patients with idiopathic pulmonary fibrosis (IPF) were treated for 24 h with the Pim-1 inhibitor (TCS, 3 µM). RNA was isolated and a PCR profiler measuring expression of 84 cytokines and chemokines was performed, amplifying the cDNA 40 cycles, detecting 49 genes. RNA transcript levels were calculated and plotted to a heatmap by normalizing all samples relative to the average delta Ct for the two control-treated samples, using GAPDH as a housekeeping gene. Genes were ranked by the average change in the two TCS-treated samples. B: lung fibroblasts derived from patients with IPF were cultured for 24 h with the Pim-1 inhibitor (TCS, 3 µM) before RNA isolation and qPCR analysis. RNA transcript levels in the TCS treated group were quantified relative to their respective control, using GAPDH as a housekeeping gene. n = 3 biologically independent and experimentally independent experiments (**P < 0.01, ***P < 0.001, ****P < 0.0001 vs. the control-treated groups).
Building upon our RNA findings, we next evaluated the expression of inflammatory cytokines at protein levels. We performed a cytokine array measuring the protein abundance of inflammatory cytokines in conditioned media derived from IPF lung fibroblasts, treated with or without TCS. Consistent with what we observed for RNA (Fig. 4), Pim-1 kinase inhibition repressed the secretion of numerous chemokines, including GRO (detected via an antibody recognizing CXCL1/2/3), GRO-α (detected via an antibody that selectively binds to CXCL1), IL-6 (IL6), MCP-1 (CCL2), and MCP-3 (CCL7) (Fig. 5). IL-8 was also strongly secreted by cultured fibroblasts, as previously shown (15). However, it was not repressed by Pim-1 inhibition, demonstrating exclusivity in Pim-1 in mediating IPF lung fibroblast inflammatory signaling. Collectively, these findings further confirm Pim-1 as a major regulator of the inflammatory secretome and warrant further investigations into the therapeutic potential of Pim-1 kinase inhibition for the treatment of IPF (25).
Pim-1 Promotes Premature Senescence of Human Lung Fibroblasts In Vitro
To investigate Pim-1-mediated gain-of-function, we stably overexpressed Pim-1 in low-passage non-IPF human lung fibroblasts by lentiviral infection, which we confirmed by Western blot (Fig. 6A). TCS is an ATP competitive inhibitor of Pim-1 kinase. As such, overexpression of Pim-1, while maintaining the same concentration of TCS, should reduce inhibitor potency. We treated infection control (Lenti-Control) and Pim-1 overexpressing cells (Lenti-PIM1) with 3 µM TCS for 3 and 24 h before collecting total protein. p65 phosphorylation was impacted by TCS only in control cells. Pim-1 overexpressing cells were refractory to the impact of TCS (Fig. 6B). Also consistent with our previous findings, p65 phosphorylation was stably enhanced in the Pim-1 overexpressing cells. These results reinforce the role of Pim-1 in p65 phosphorylation and validate the specificity of TCS. We also found that Pim-1 overexpression enhanced RNA production of inflammatory SASP factors: IL6, CCL2, and IL1B (Fig. 6C). Based on previous studies demonstrating that Pim-1 promotes cell cycle progression in multiple cancers (44–46), we tested whether Pim-1 overexpression causes lung fibroblasts to proliferate more quickly. Conversely, we observed consistent repression of proliferation in Pim-1 overexpressing cells (Fig. 6D). We next explored if the enhanced expression of SASP factors and reduction in cellular proliferation were indicative of senescence. Indeed, Pim-1 overexpression coincided with increased transcription of senescence-associated cyclin-dependent kinase inhibitors CDKN2A (p16INK4a) and CDKN1A (p21waf1) (Fig. 6E). We further confirmed premature senescence in vitro by monitoring SA-β-Gal staining in control or Pim-1 overexpressing lung fibroblasts from culture passage 4–8. Remarkably, by passage 8 nearly 50% of the Pim-1 overexpressing cells stained positively for SA-β-Gal, which was significantly earlier than matched passage control cells (Fig. 6F). Thus, Pim-1 overexpression is sufficient to drive lung fibroblast senescence.
DISCUSSION
Our study indicates that Pim-1 kinase promotes lung fibroblast inflammatory secretome production through a positive feedback loop involving the phosphorylation of NF-κB and the IL-6/JAK/STAT3 pathway. Moreover, NF-κB is known to stimulate the production of the SASP, a feature of senescent cells composed of inflammatory cytokines and profibrotic mediators (18). Pim-1 kinase inhibition has been shown to alleviate fibrosis in an experimental mouse model of pulmonary fibrosis (25) and ex vivo in organotypic cultures of lung slices derived from patients with IPF (26). Therefore, our study builds upon previous findings and reveals a novel role for Pim-1 kinase in regulating the secretome of senescent, lung fibroblasts derived from patients with IPF, and identifies Pim-1 as a driver of lung fibroblast senescence. Based on these findings, Pim-1 kinase shows promise as a regulator of the fibroblast secretome, which may be an important driver of IPF.
Pan-Pim kinase (Pim-1,2,3) inhibitors have entered clinical trials for the treatment of solid tumors, leukemias, and lymphomas, and were found to have minimal efficacy and multiple dose-limiting toxicities (47). In a more recent study, a clinical candidate (TP-3654) with modest selectivity for Pim-1 reduced tumor volume and exhibited no associated toxicity (48). Whether the toxicities associated with pan-Pim inhibitors are on-target and can be mitigated by selective Pim-1 inhibition remains to be determined. Here, we focused on Pim-1 based on previous studies supporting a role for Pim-1 in experimental lung fibrosis (25, 26), and Pim-1 directly phosphorylates p65/RelA at serine 276 (29). Such phosphorylation is essential for the activity of the NF-κB complex, the master regulator of SASP component transcription (17–19).
We present evidence for Pim-1 as a regulator of SASP secretion in lung fibroblasts derived from patients with IPF. The onset of senescence burden as a major target for IPF was very recently supported by the application of single-cell resolution RNA sequencing of patient lungs with IPF (8–11) and earlier by multiple efforts profiling IPF donor samples (49, 50). In addition, clearance of senescent cells improves or resolves experimental lung fibrosis (13, 51). One of the perceived challenges with clearing senescent cells from fibrotic lungs is the potential positive impact they have on lung homeostasis. Therefore, targeting the senescence-associated secretome could serve as an alternative strategy to eliminate their detrimental features while preserving their beneficial properties (52, 53). The transcription factor NF-κB is known to be the primary regulator of genes associated with SASP (17). The data presented here suggest Pim-1 kinase is an upstream regulator of NF-κB activity, and the positive feedback loop supported by our findings offers an opportunity to target this pathway in senescent lung fibroblasts. However, it remains to be investigated if Pim-1 inhibition would also interrupt NF-κB signaling in other resident lung cell populations that rely on NF-κB activation to support host defense (54, 55). Multiple kinases, in addition to Pim-1, phosphorylate p65/RelA on serine 276, including PKA/C, MSK1/2, and RSK (19). Here, we show phosphorylation of p65 (S276) to be tightly dependent on Pim-1 in both TNF-α stimulated cells and senescent IPF lung fibroblasts with elevated S276 phosphorylation. A complete exploration of the specific kinases relevant to NF-κB activation in specific lung cell populations could elucidate selective targeting strategies, potentially necessary to develop a safe and effective therapy for IPF.
We also show in low passage lung fibroblasts derived from non-IPF patients that overexpression of Pim-1 leads to premature senescence. Literature focused on the role of Pim-1 in cell cycle progression appears to be controversial. The bulk of these investigations suggest that Pim-1 is overexpressed in multiple cancer cells (including solid tumor and hematologic malignancies), where it promotes cell cycle progression, proliferation, and apoptosis resistance (43, 44, 46). Conversely, previous reports have also shown Pim-1 can drive premature expression of CDKN2A (p16INK4a) and cellular senescence (27, 28, 56). In our recent publication, we found PIM1, along with a deluge of inflammatory cytokines and chemokines, to be upregulated in freshly sorted lung fibroblasts derived from 18-mo-old mice compared with 2-mo-old mice (26), potentially indicating that the positive feedback loop identified here also occurs during normal aging. This implores the question of whether Pim-1 kinase could be a hub and/or target for multiple age-related diseases, including IPF.
In conclusion, our findings demonstrate a critical role for Pim-1 kinase in regulating the inflammatory secretome of senescence lung fibroblasts and warrant further investigations into the utility of targeting Pim-1 as a therapy for IPF.
DATA AVAILABILITY
Data will be made available upon reasonable request.
SUPPLEMENTAL DATA
Supplemental Table S1 and Supplemental Figs. S1–S4: https://doi.org/10.6084/m9.figshare.18737336.
GRANTS
This work was supported in part by the American Lung Association (to A. J. Haak), Boehringer Ingelheim (BI) (to A. J. Haak), National Heart, Lung, and Blood Institute Grants HL092961 and 1R61HL161804-01 (to D. J. Tschumperlin and A. J. Haak), Mayo Clinic (to A. J. Haak), Pulmonary Fibrosis Foundation (PFF) (to A. J. Haak), and U.S. Department of Defense (DOD) Grant PR181132 (to D. J. Tschumperlin).
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
A.J.H. conceived and designed research; A.Y.G., A.M.D.E., F.G., T.X.P., C.M.C., A.A., N.C., and A.J.H. performed experiments; A.J.H. analyzed data; A.J.H. interpreted results of experiments; C.M.B. and A.J.H. prepared figures; A.Y.G., A.M.D., T.X.P., and A.J.H. drafted manuscript; A.Y.G., A.M.D.E., F.G., T.X.P., G.L., M.J.S., and A.J.H. edited and revised manuscript; A.Y.G., A.M.D.E., F.G., T.X.P., C.M.C., A.A., C.M.B., G.L., N.C., M.J.S., and A.J.H. approved final version of manuscript.
REFERENCES
- 1. King TE Jr, Pardo A, Selman M. Idiopathic pulmonary fibrosis. Lancet 378: 1949–1961, 2011. doi: 10.1016/S0140-6736(11)60052-4. [DOI] [PubMed] [Google Scholar]
- 2. Hutchinson J, Fogarty A, Hubbard R, McKeever T. Global incidence and mortality of idiopathic pulmonary fibrosis: a systematic review. Eur Respir J 46: 795–806, 2015. doi: 10.1183/09031936.00185114. [DOI] [PubMed] [Google Scholar]
- 3. Khor YH, Ng Y, Barnes H, Goh NSL, McDonald CF, Holland AE. Prognosis of idiopathic pulmonary fibrosis without anti-fibrotic therapy: a systematic review. Eur Respir Rev 29: 190158, 2020. doi: 10.1183/16000617.0158-2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Ley B, Collard HR, King TE Jr.. Clinical course and prediction of survival in idiopathic pulmonary fibrosis. Am J Respir Crit Care Med 183: 431–440, 2011. doi: 10.1164/rccm.201006-0894CI. [DOI] [PubMed] [Google Scholar]
- 5. Glassberg MK. Overview of idiopathic pulmonary fibrosis, evidence-based guidelines, and recent developments in the treatment landscape. Am J Manag Care 25: S195–S203, 2019. [PubMed] [Google Scholar]
- 6. Richeldi L, Collard HR, Jones MG. Idiopathic pulmonary fibrosis. Lancet 389: 1941–1952, 2017. doi: 10.1016/S0140-6736(17)30866-8. [DOI] [PubMed] [Google Scholar]
- 7. Valenzuela C, Torrisi SE, Kahn N, Quaresma M, Stowasser S, Kreuter M. Ongoing challenges in pulmonary fibrosis and insights from the nintedanib clinical programme. Respir Res 21: 7, 2020. doi: 10.1186/s12931-019-1269-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Adams TS, Schupp JC, Poli S, Ayaub EA, Neumark N, Ahangari F, Chu SG, Raby BA, DeIuliis G, Januszyk M, Duan Q, Arnett HA, Siddiqui A, Washko GR, Homer R, Yan X, Rosas IO, Kaminski N. Single-cell RNA-seq reveals ectopic and aberrant lung-resident cell populations in idiopathic pulmonary fibrosis. Sci Adv 6: eaba1983, 2020. doi: 10.1126/sciadv.aba1983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Habermann AC, Gutierrez AJ, Bui LT, Yahn SL, Winters NI, Calvi CL, Peter L, Chung MI, Taylor CJ, Jetter C, Raju L, Roberson J, Ding GX, Wood L, Sucre JMS, Richmond BW, Serezani AP, McDonnell WJ, Mallal SB, Bacchetta MJ, Loyd JE, Shaver CM, Ware LB, Bremner R, Walia R, Blackwell TS, Banovich NE, Kropski JA. Single-cell RNA sequencing reveals profibrotic roles of distinct epithelial and mesenchymal lineages in pulmonary fibrosis. Sci Adv 6: eaba1972, 2020. doi: 10.1126/sciadv.aba1972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Nemeth J, Schundner A, Frick M. Insights into development and progression of idiopathic pulmonary fibrosis from single cell RNA studies. Front Med (Lausanne) 7: 611728, 2020. doi: 10.3389/fmed.2020.611728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Tsukui T, Sun KH, Wetter JB, Wilson-Kanamori JR, Hazelwood LA, Henderson NC, Adams TS, Schupp JC, Poli SD, Rosas IO, Kaminski N, Matthay MA, Wolters PJ, Sheppard D. Collagen-producing lung cell atlas identifies multiple subsets with distinct localization and relevance to fibrosis. Nat Commun 11: 1920, 2020. doi: 10.1038/s41467-020-15647-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Alvarez D, Cardenes N, Sellares J, Bueno M, Corey C, Hanumanthu VS, Peng Y, D'Cunha H, Sembrat J, Nouraie M, Shanker S, Caufield C, Shiva S, Armanios M, Mora AL, Rojas M. IPF lung fibroblasts have a senescent phenotype. Am J Physiol Lung Cell Mol Physiol 313: L1164–L1173, 2017. doi: 10.1152/ajplung.00220.2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Schafer MJ, White TA, Iijima K, Haak AJ, Ligresti G, Atkinson EJ, Oberg AL, Birch J, Salmonowicz H, Zhu Y, Mazula DL, Brooks RW, Fuhrmann-Stroissnigg H, Pirtskhalava T, Prakash YS, Tchkonia T, Robbins PD, Aubry MC, Passos JF, Kirkland JL, Tschumperlin DJ, Kita H, LeBrasseur NK. Cellular senescence mediates fibrotic pulmonary disease. Nat Commun 8: 14532, 2017. doi: 10.1038/ncomms14532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Yanai H, Shteinberg A, Porat Z, Budovsky A, Braiman A, Ziesche R, Zeische R, Fraifeld VE. Cellular senescence-like features of lung fibroblasts derived from idiopathic pulmonary fibrosis patients. Aging (Albany NY) 7: 664–672, 2015. doi: 10.18632/aging.100807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Caporarello N, Meridew JA, Jones DL, Tan Q, Haak AJ, Choi KM, Manlove LJ, Prakash YS, Tschumperlin DJ, Ligresti G. PGC1α repression in IPF fibroblasts drives a pathologic metabolic, secretory and fibrogenic state. Thorax 74: 749–760, 2019. doi: 10.1136/thoraxjnl-2019-213064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Lin Y, Xu Z. Fibroblast senescence in idiopathic pulmonary fibrosis. Front Cell Dev Biol 8: 593283, 2020. doi: 10.3389/fcell.2020.593283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Chien Y, Scuoppo C, Wang X, Fang X, Balgley B, Bolden JE, Premsrirut P, Luo W, Chicas A, Lee CS, Kogan SC, Lowe SW. Control of the senescence-associated secretory phenotype by NF-kappaB promotes senescence and enhances chemosensitivity. Genes Dev 25: 2125–2136, 2011. doi: 10.1101/gad.17276711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Salminen A, Kauppinen A, Kaarniranta K. Emerging role of NF-kappaB signaling in the induction of senescence-associated secretory phenotype (SASP). Cell Signal 24: 835–845, 2012. doi: 10.1016/j.cellsig.2011.12.006. [DOI] [PubMed] [Google Scholar]
- 19. Christian F, Smith EL, Carmody RJ. The regulation of NF-kappaB subunits by phosphorylation. Cells-Basel 5: 12, 2016. doi: 10.3390/cells5010012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Warfel NA, Kraft AS. PIM kinase (and Akt) biology and signaling in tumors. Pharmacol Ther 151: 41–49, 2015. doi: 10.1016/j.pharmthera.2015.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Zhang XN, Song MQ, Kundu JK, Lee MH, Liu ZZ. PIM kinase as an executional target in cancer. J Cancer Prev 23: 109–116, 2018. doi: 10.15430/JCP.2018.23.3.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Cao LJ, Wang F, Li SY, Wang XY, Huang DZ, Jiang RC. PIM1 kinase promotes cell proliferation, metastasis and tumor growth of lung adenocarcinoma by potentiating the c-MET signaling pathway. Cancer Lett 444: 116–126, 2019. doi: 10.1016/j.canlet.2018.12.015. [DOI] [PubMed] [Google Scholar]
- 23. Merkel AL, Meggers E, Ocker M. PIM1 kinase as a target for cancer therapy. Expert Opin Investig Drugs 21: 425–436, 2012. doi: 10.1517/13543784.2012.668527. [DOI] [PubMed] [Google Scholar]
- 24. Santio NM, Koskinen PJ. PIM kinases: from survival factors to regulators of cell motility. Int J Biochem Cell Biol 93: 74–85, 2017. doi: 10.1016/j.biocel.2017.10.016. [DOI] [PubMed] [Google Scholar]
- 25. Zhang XY, Zou Y, Liu YQ, Cao YM, Zhu JL, Zhang JH, Chen X, Zhang R, Li JB. Inhibition of PIM1 kinase attenuates bleomycin-induced pulmonary fibrosis in mice by modulating the ZEB1/E-cadherin pathway in alveolar epithelial cells. Mol Immunol 125: 15–22, 2020. doi: 10.1016/j.molimm.2020.06.013. [DOI] [PubMed] [Google Scholar]
- 26. Pham TX, Lee J, Guan JZ, Caporarello N, Meridew JA, Jones DL, Tan Q, Huang SK, Tschumperlin DJ, Ligresti G. Transcriptional analysis of lung fibroblasts identifies PIM1 signaling as a driver of aging-associated persistent fibrosis. JCI Insight 7: e153672, 2022. doi: 10.1172/jci.insight.153672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Wang S, Cao Z, Xue J, Li H, Jiang W, Cheng Y, Li G, Zhang X. A positive feedback loop between Pim-1 kinase and HBP1 transcription factor contributes to hydrogen peroxide-induced premature senescence and apoptosis. J Biol Chem 292: 8207–8222, 2017. doi: 10.1074/jbc.M116.768101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Yang J, Liu K, Yang J, Jin B, Chen H, Zhan X, Li Z, Wang L, Shen X, Li M, Yu W, Mao Z. PIM1 induces cellular senescence through phosphorylation of UHRF1 at Ser311. Oncogene 36: 4828–4842, 2017. doi: 10.1038/onc.2017.96. [DOI] [PubMed] [Google Scholar]
- 29. Nihira K, Ando Y, Yamaguchi T, Kagami Y, Miki Y, Yoshida K. Pim-1 controls NF-kappaB signalling by stabilizing RelA/p65. Cell Death Differ 17: 689–698, 2010. doi: 10.1038/cdd.2009.174. [DOI] [PubMed] [Google Scholar]
- 30. Block KM, Hanke NT, Maine EA, Baker AF. IL-6 stimulates STAT3 and Pim-1 kinase in pancreatic cancer cell lines. Pancreas 41: 773–781, 2012. doi: 10.1097/MPA.0b013e31823cdd10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Parikh P, Britt RD Jr, Manlove LJ, Wicher SA, Roesler A, Ravix J, Teske J, Thompson MA, Sieck GC, Kirkland JL, LeBrasseur N, Tschumperlin DJ, Pabelick CM, Prakash YS. Hyperoxia-induced cellular senescence in fetal airway smooth muscle cells. Am J Respir Cell Mol Biol 61: 51–60, 2019. doi: 10.1165/rcmb.2018-0176OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Parikh P, Wicher S, Khandalavala K, Pabelick CM, Britt RD Jr, Prakash YS. Cellular senescence in the lung across the age spectrum. Am J Physiol Lung Cell Mol Physiol 316: L826–L842, 2019. doi: 10.1152/ajplung.00424.2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Wicher SA, Roos BB, Teske JJ, Fang YH, Pabelick C, Prakash YS. Aging increases senescence, calcium signaling, and extracellular matrix deposition in human airway smooth muscle. PLoS One 16: e0254710, 2021. doi: 10.1371/journal.pone.0254710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Okazaki T, Sakon S, Sasazuki T, Sakurai H, Doi T, Yagita H, Okumura K, Nakano H. Phosphorylation of serine 276 is essential for p65 NF-kappa B subunit-dependent cellular responses. Biochem Biophys Res Commun 300: 807–812, 2003. doi: 10.1016/S0006-291X(02)02932-7. [DOI] [PubMed] [Google Scholar]
- 35. Cheney IW, Yan SQ, Appleby T, Walker H, Vo T, Yao NH, Hamatake R, Hong Z, Wu JZ. Identification and structure-activity relationships of substituted pyridones as inhibitors of Pim-1 kinase. Bioorg Med Chem Lett 17: 1679–1683, 2007. doi: 10.1016/j.bmcl.2006.12.086. [DOI] [PubMed] [Google Scholar]
- 36. Milara J, Hernandez G, Ballester B, Morell A, Roger I, Montero P, Escrivaa J, Lloris JM, Molina-Molina M, Morcillo E, Cortijo J. The JAK2 pathway is activated in idiopathic pulmonary fibrosis. Resp Res 19: 24, 2018. doi: 10.1186/s12931-018-0728-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Pullamsetti SS, Seeger W, Savai R. Classical IL-6 signaling: a promising therapeutic target for pulmonary arterial hypertension. J Clin Invest 128: 1720–1723, 2018. doi: 10.1172/JCI120415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Oh RS, Haak AJ, Smith KMJ, Ligresti G, Choi KM, Xie T, Wang S, Walters PR, Thompson MA, Freeman MR, Manlove LJ, Chu VM, Feghali-Bostwick C, Roden AC, Schymeinsky J, Pabelick CM, Prakash YS, Vassallo R, Tschumperlin DJ. RNAi screening identifies a mechanosensitive ROCK-JAK2-STAT3 network central to myofibroblast activation. J Cell Sci 131: jcs209932, 2018. doi: 10.1242/jcs.209932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Hedvat M, Huszar D, Herrmann A, Gozgit JM, Schroeder A, Sheehy A, Buettner R, Proia D, Kowolik CM, Xin H, Armstrong B, Bebernitz G, Weng SB, Wang L, Ye MW, McEachern K, Chen HW, Morosini D, Bell K, Alimzhanov M, Ioannidis S, McCoon P, Cao ZA, Yu H, Jove R, Zinda M. The JAK2 inhibitor AZD1480 potently blocks stat3 signaling and oncogenesis in solid tumors. Cancer Cell 16: 487–497, 2009. doi: 10.1016/j.ccr.2009.10.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Beharry Z, Zemskova M, Mahajan S, Zhang FX, Ma J, Xia ZP, Lilly M, Smith CD, Kraft AS. Novel benzylidene-thiazolidine-2,4-diones inhibit Pim protein kinase activity and induce cell cycle arrest in leukemia and prostate cancer cells. Mol Cancer Ther 8: 1473–1483, 2009. doi: 10.1158/1535-7163.MCT-08-1037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Keeton EK, McEachern K, Dillman KS, Palakurthi S, Cao YC, Grondine MR, Kaur S, Wang SP, Chen YC, Wu A, Shen MH, Gibbons FD, Lamb ML, Zheng XL, Stone RM, DeAngelo DJ, Platanias LC, Dakin LA, Chen HW, Lyne PD, Huszar D. AZD1208, a potent and selective pan-Pim kinase inhibitor, demonstrates efficacy in preclinical models of acute myeloid leukemia. Blood 123: 905–913, 2014. doi: 10.1182/blood-2013-04-495366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Xia ZP, Knaak C, Ma J, Beharry ZM, McInnes C, Wang WX, Kraft AS, Smith CD. Synthesis and evaluation of novel inhibitors of Pim-1 and Pim-2 protein kinases. J Med Chem 52: 74–86, 2009. doi: 10.1021/jm800937p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Saurabh K, Scherzer MT, Shah PP, Mims AS, Lockwood WW, Kraft AS, Beverly LJ. The PIM family of oncoproteins: small kinases with huge implications in myeloid leukemogenesis and as therapeutic targets. Oncotarget 5: 8503–8514, 2014. doi: 10.18632/oncotarget.2330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Bachmann M, Moroy T. The serine/threonine kinase Pim-1. Int J Biochem Cell Biol 37: 726–730, 2005. doi: 10.1016/j.biocel.2004.11.005. [DOI] [PubMed] [Google Scholar]
- 45. Brault L, Gasser C, Bracher F, Huber K, Knapp S, Schwaller J. PIM serine/threonine kinases in the pathogenesis and therapy of hematologic malignancies and solid cancers. Haematologica 95: 1004–1015, 2010. doi: 10.3324/haematol.2009.017079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Morishita D, Katayama R, Sekimizu K, Tsuruo T, Fujita N. Pim kinases promote cell cycle progression by phosphorylating and down-regulating p27(Kip1) at the transcriptional and posttranscriptional levels. Cancer Res 68: 5076–5085, 2008. doi: 10.1158/0008-5472.CAN-08-0634. [DOI] [PubMed] [Google Scholar]
- 47. Luszczak S, Kumar C, Sathyadevan VK, Simpson BS, Gately KA, Whitaker HC, Heavey S. PIM kinase inhibition: co-targeted therapeutic approaches in prostate cancer. Signal Transduct Target Ther 5: 7 2020. doi: 10.1038/s41392-020-0109-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Foulks JM, Carpenter KJ, Luo B, Xu Y, Senina A, Nix R, Chan A, Clifford A, Wilkes M, Vollmer D, Brenning B, Merx S, Lai SP, McCullar MV, Ho KK, Albertson DJ, Call LT, Bearss JJ, Tripp S, Liu T, Stephens BJ, Mollard A, Warner SL, Bearss DJ, Kanner SB. A small-molecule inhibitor of PIM kinases as a potential treatment for urothelial carcinomas. Neoplasia 16: 403–412, 2014. doi: 10.1016/j.neo.2014.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Liu RM, Liu G. Cell senescence and fibrotic lung diseases. Exp Gerontol 132: 110836, 2020. doi: 10.1016/j.exger.2020.110836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Merkt W, Bueno M, Mora AL, Lagares D. Senotherapeutics: targeting senescence in idiopathic pulmonary fibrosis. Semin Cell Dev Biol 101: 104–110, 2020. doi: 10.1016/j.semcdb.2019.12.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Hohmann MS, Habiel DM, Coelho AL, Verri WA Jr, Hogaboam CM. Quercetin enhances ligand-induced apoptosis in senescent idiopathic pulmonary fibrosis fibroblasts and reduces lung fibrosis in vivo. Am J Respir Cell Mol Biol 60: 28–40, 2019. doi: 10.1165/rcmb.2017-0289OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Mailleux AA, Crestani B. Licence to kill senescent cells in idiopathic pulmonary fibrosis? Eur Respir J 50: 1701360, 2017. doi: 10.1183/13993003.01360-2017. [DOI] [PubMed] [Google Scholar]
- 53. Schafer MJ, Haak AJ, Tschumperlin DJ, LeBrasseur NK. Targeting senescent cells in fibrosis: pathology, paradox, and practical considerations. Curr Rheumatol Rep 20: 3, 2018. doi: 10.1007/s11926-018-0712-x. [DOI] [PubMed] [Google Scholar]
- 54. Hess C, Herr C, Beisswenger C, Zakharkina T, Schmid RM, Bals R. Myeloid RelA regulates pulmonary host defense networks. Eur Respir J 35: 343–352, 2010. doi: 10.1183/09031936.00196408. [DOI] [PubMed] [Google Scholar]
- 55. Sadikot RT, Zeng H, Joo M, Everhart MB, Sherrill TP, Li B, Cheng DS, Yull FE, Christman JW, Blackwell TS. Targeted immunomodulation of the NF-kappaB pathway in airway epithelium impacts host defense against Pseudomonas aeruginosa. J Immunol 176: 4923–4930, 2006. doi: 10.4049/jimmunol.176.8.4923. [DOI] [PubMed] [Google Scholar]
- 56. Jin B, Wang Y, Wu CL, Liu KY, Chen H, Mao ZB. PIM-1 modulates cellular senescence and links IL-6 signaling to heterochromatin formation. Aging Cell 13: 879–889, 2014. doi: 10.1111/acel.12249. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental Table S1 and Supplemental Figs. S1–S4: https://doi.org/10.6084/m9.figshare.18737336.
Data Availability Statement
Data will be made available upon reasonable request.