

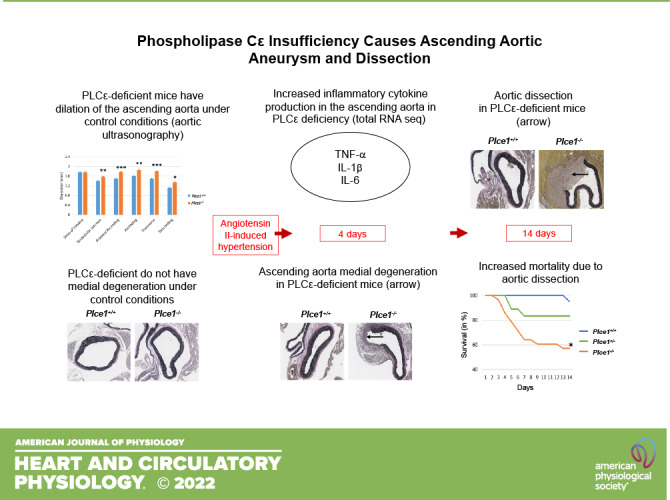
Keywords: angiotensin II, aortic dissection, aortic insufficiency, signaling pathway analysis
Abstract
Phospholipase Cε (PLCε) is a phospholipase C isoform with a wide range of physiological functions. It has been implicated in aortic valve disorders, but its role in frequently associated aortic disease remains unclear. To determine the role of PLCε in thoracic aortic aneurysm and dissection (TAAD) we used PLCε-deficient mice, which develop aortic valve insufficiency and exhibit aortic dilation of the ascending thoracic aorta and arch without histopathological evidence of injury. Fourteen days of infusion of Plce1+/+ and Plce1−/− mice with angiotensin II (ANG II), which induces aortic dilation and dissection, led to sudden death secondary to ascending aortic dissection in 43% of Plce1−/− versus 5% of Plce1+/+ mice (P < 0.05). Medial degeneration and TAAD were detected in 80% of Plce1−/− compared with 10% of Plce1+/+ mice (P < 0.05) after 4 days of ANG II. Treatment with ANG II markedly increased PLCε expression within the ascending aortic adventitia. Total RNA sequencing demonstrated marked upregulation of inflammatory and fibrotic pathways mediated by interleukin-1β, interleukin-6, and tumor necrosis factor-α. In silico analysis of whole exome sequences of 258 patients with type A dissection identified 5 patients with nonsynonymous PLCE1 variants. Our data suggest that PLCε deficiency plays a role in the development of TAAD and aortic insufficiency.
NEW & NOTEWORTHY We describe a novel phenotype by which PLCε deficiency predisposes to aortic valve insufficiency and ascending aortic aneurysm, dissection, and sudden death in the setting of ANG II-mediated hypertension. We demonstrate PLCE1 variants in patients with type A aortic dissection and aortic insufficiency, suggesting that PLCE1 may also play a role in human aortic disease. This finding is of very high significance because it has not been previously demonstrated that PLCε directly mediates aortic dissection.
INTRODUCTION
Phospholipase Cε (mouse gene ID, Plce1; human gene ID, PLCE1; protein, PLCε) is a novel phospholipase C isoform involved in canonical diacylglycerol (DAG) and protein kinase C (PKC) signaling pathways and as nexus for rho, rap1, ras, and G protein-coupled receptor (GPCR) signaling (1). PLCε is known to regulate the response of ANG II and the β-adrenergic receptor and has previously been implicated in the pathogenesis of various cardiovascular disorders (2, 3). Global deletion of PLCε results in left ventricular hypertrophy and cardiac fibrosis in mice (4). Additionally, global deletion of PLCε results in pathological thickening of the aortic and pulmonic valve during development, leading to semilunar valve regurgitation (5). Although PLCε has been implicated in aortic valve disorders in murine models, PLCε has not been previously implicated in aortic aneurysm or dissection.
Thoracic aortic dissection (TAD) is a highly morbid condition that can result in sudden death from aortic rupture. Numerous risk factors exist for the development of TAD, including underlying aortic diameter and/or aneurysm, hypertension, and genetic predisposition (6). Many cases of thoracic aortic aneurysm and dissection are comorbid with aortic valve disorders, typically bicuspid aortic valves or aortic insufficiency (6). Aortic dilation is one of the major underlying risk factors for eventual aortic dissection and rupture (7, 8). Within the ascending aorta, medial degeneration is an important contributor to development of aortic aneurysms and progressive aortic dilation (6). Histological changes that occur during this process include abnormal mucopolysaccharide extracellular matrix accumulation (MEMA), loss of vascular smooth muscle nuclei, and degeneration of the aortic elastin fibers (6, 9). In those with genetic predisposition to aortic aneurysm and dissection, highly penetrant conditions are thought to contribute to ∼20% of TAD cases (10). Highly penetrant mutations known to contribute to TAD include fibrillin-1 (FBN1, Marfan syndrome), smooth muscle actin-2 (ACTA-2, smooth muscle dysfunction syndrome), collagen 3a1 (COL3A1, vascular Ehlers–Danlos syndrome), and various mutations in the transforming growth factor-β ligand, receptor, or signaling family (Loeys–Dietz syndrome 1–5) (10). However, in many cases, additional environmental insults are necessary to express the full severity of an aortopathy (11–13).
Multiple inflammatory signaling cascades have been implicated in the development of aortic aneurysms and dissection. Interleukin-1β (IL-1β), interleukin-6 (IL-6), and tumor necrosis factor-α (TNF-α) have been implicated in aortic dissection. Specifically, pharmacological blockade of these inflammatory cytokines has been shown to decrease the aneurysmal growth rate and aortic dissection. IL-6 production in the tunica adventitia is thought to increase vascular macrophage infiltration and aortic dissection (14–17). Whether any of these various mediators of aortic aneurysm and dissection interact with PLCε in the context of aortic dissection has not been previously explored.
Angiotensin II (ANG II) is the major hypertensive effector molecule of the renin-angiotensin system. ANG II is known to cause variably penetrant ascending aortic aneurysms and aortic dissection in murine models within 7 days of treatment initiation (18, 19). The mechanisms of this are complex and include effects mediated by the AT1 receptor (AT1R) in adventitial fibroblasts as opposed to the AT1R expressed in either the vascular smooth muscle or endothelium (20, 21).
Since PLCε deficiency in murine models leads to aortic valve insufficiency (5), we tested the hypothesis that PLCε deficiency will predispose to ascending thoracic aortic aneurysm and dissection as well. We characterize a novel phenotype by which PLCε deficiency causes ascending aortic dilation and aortic valve insufficiency and dissection and sudden death in the setting of ANG II-mediated hypertension. We determined that PLCε expression is strongly increased in the aortic adventitia in response to ANG II, with a pattern that is most consistent with expression in myofibroblasts. We determined that multiple dysregulated signaling cascades, including TNF-α, IL-1β, and IL-6, may contribute to this propensity for aortic dissection. Finally, we characterized five patients with PLCE1 variants who were found to have ascending aortic aneurysm and dissection associated with aortic regurgitation.
METHODS
Mice
Global Plce1−/− mice were developed as previously described and had been crossed for at least 10 generations on a C57BL/6J background (3, 4). F1 Plce1+/− mice were bred to generate F2 mice used in all studies. Male and female mice were used for experiments except where indicated. Experiments were performed on 10- to 12-wk-old mice. Hemagglutinin-tagged PLCε (Plce1-HA) mice were generated as previously described (3). All mice were genotyped as previously described (3).
Surgical Procedures and Euthanasia
Osmotic minipumps were implanted as previously described (3). Mice were anesthetized with 2–5% inhaled isoflurane. Perioperative analgesia was provided with 0.1 mg/kg subcutaneous buprenorphine before surgery and every 12 h for 72 h postoperatively as needed. Ophthalmic ointment was applied. Body temperature was maintained with a heating pad for the duration of the surgery. The nape was shaved and cleaned with 70% ethanol and Betadine. The minipump was implanted in the subcutaneous tissue through a small incision under sterile conditions. The wound was closed with silk suture (Ethicon, Somerville, NJ). Mice were monitored for recovery and then returned to their cages. Osmotic minipumps (models 1002 and 1004) were purchased from Alzet (Durect, Cupertino, CA) for 2- and 4-wk infusions, respectively, and primed overnight in sterile PBS (pH 7.4) at 37°C before implantation. Euthanasia was performed via intraperitoneal injection of 160 mg/kg pentobarbital sodium (Fatal Plus, Vortech Pharmaceuticals, Dearborn, MI). At the cessation of spontaneous breathing, the thoracic cavity was opened and the heart was perfused via a left ventricular puncture with 10 mL of cold PBS.
Histological Processing, Immunohistochemistry, and Analysis
Aortas were dissected out after perfusion and submerged overnight in 10% neutral buffered formalin at room temperature before switching to 70% ethanol. Samples were processed and infiltrated with paraffin on an automated histology processor (Tissue Tek VIP; Sakura, Torrance, CA) The aorta was trimmed after processing into segments 4 to 5 mm in length, encompassing the entire span from the aortic sinus to the descending thoracic aorta. The segments were embedded in cross section into paraffin blocks, and replicate serial sections of 4-μm thickness were taken at multiple levels spaced 100 μm apart for the aortic root and 250 µm apart for the ascending and descending thoracic aorta. At each level, replicate sections were mounted on glass slides and stained with hematoxylin and eosin (H&E) and Verhoeff–Van Gieson stain. Additional replicates were left unstained and subsequently used for immunohistochemistry.
Slides were evaluated by a board-certified veterinary pathologist (I.L.B.) without knowledge of the experimental groups using a BX45 Olympus light microscope. Criteria for lesion identification were as follows: aneurysm was defined as thinning of the media and dilation of the aorta to ≥1.5 times the normal aortic diameter. Dissection was defined as hemorrhage within the aortic media. Rupture was defined as periaortic hemorrhage with the presence of fibrin. Medial degeneration was defined as loss of smooth muscle, elastin fragmentation/loss/disorganization, or mucopolysaccharide extracellular matrix accumulation per standard histological criteria (9).
Immunohistochemistry was performed on 4-μm sections as previously described (3). Primary antibodies were incubated overnight at 4°C and were then developed with the Vectastain anti-rabbit HRP kit (Vector, Burlingame, CA) with diaminobenzidine (Sigma, St. Louis, MO) as per the manufacturer’s instructions. Slides for histological analysis were digitally scanned by the University of Michigan In Vivo Animal Core and were visualized for images with Aperio ImageScope (Leica Biosystems, Buffalo Grove, IL). Quantitation of tissue areas and cell counts for immunohistochemistry were performed with QuPath v0.2.3 (Edinburgh, UK) (22). Quantification of histological parameters was performed on a minimum of three separate aortic sections from each animal to develop a mean value for the parameter, which is reported.
Aortic Ultrasonography
Aortic ultrasonography was performed by the University of Michigan Cardiovascular Phenotyping Core with a VisualSonics Vevo 2100s system. The ultrasonographer was blinded to genotype and treatment. Measurements were obtained in B mode or M mode. Aortic diameter measurements were obtained in diastole. Proximal ascending aorta diameter was obtained 1 mm distal to the sinotubular junction. Ascending aortic diameter was obtained 1 mm distal to the proximal ascending aortic measurement point. All other measurement points were taken where described in results. Measurements of aortic ejection velocity were obtained in Doppler mode.
Drugs and Antibodies
Angiotensin II was procured from Sigma (St. Louis, MO) and was delivered at 1.5 mg/kg/day in sterile PBS (pH = 7.4; Thermo-Fisher Scientific, Waltham, MA). This is a standard dose for studying aortic dissection (11, 18, 19). The rabbit anti-hemagglutinin (C29F4) and -F4/80 (D2S9R) monoclonal antibodies were obtained from Cell Signaling Technology (Danvers, MA) at 1:500 dilution. Rabbit polyclonal antibodies to periostin (Ab92460) were obtained from Abcam (Cambridge, UK) and were used at 1:200 dilutions.
Blood Pressure Measurement
Systolic blood pressure was measured noninvasively with the CODA tail-cuff system (Kent Scientific, Torrington, CT) as previously described (3). Mice were placed on a heating pad (38°C) in the restrainer for 3 min before 2 blood pressure measurements were taken, which were not recorded, followed by 10 measurements, which were used to calculate the mean. All measurements were completed in one session.
Total RNA Sequencing and Analysis
Ascending aortic tissue and arch, proximal to the left subclavian artery, was obtained and trimmed of fat and lymphatic tissue and stored in RNAlater (Ambion, Carlsbad, CA) at −80°C until further processing. Tissue was obtained from four and five control-treated Plce1+/+ and Plce1−/− mice, respectively, and three and five Plce1+/+ and Plce1−/− mice treated with 4 days of ANG II, respectively. As above, all mice were male. The rationale for this approach was that male mice had a more penetrant phenotype, and as such we would be more likely to find pathologically meaningful differences in transcripts contributing to the severe aortic dissection phenotype. The tissue was minced and sonicated in TRIzol, and RNA was extracted with chloroform and an RNeasy Kit (Qiagen, Hilden, Germany) as per the manufacturer’s instructions. At least 75 ng of RNA per sample was sent to the University of Michigan Advanced Genomics Core for total RNA preparation and library creation. Library construction and 150-bp paired-end sequencing were performed on the Illumina NovaSeq S4300 platform by the Advanced Genomics Core at the University of Michigan. Sequencing was performed with 47 million reads per sample. Reads were trimmed with Cutadapt v2.3 and quality assessed with FastQC v0.11.8 (23, 24). Mapping to the ENSEMBL GRCm38 reference genome was done with STAR v2.7.8a, and read counts were obtained with RSEM v1.3.3 (25, 26). Quality control metrics were integrated by multiQC v1.7 and found to be acceptable for all samples (27).
Count data were further evaluated by principal component analysis (PCA) using the prcomp function (R v4.0.4) with no indication of extreme outliers. Differential gene expression (DGE) was then estimated in R with DESeq2 v1.30.1 (28). To assess the significance of DGE, false discovery rate (FDR) calculations were performed with the method of Benjamini and Hochberg (29). For additional analyses, count data were normalized and log-transformed with the voom function in limma v3.46.0 (30).
In Silico Analysis and Variant Calling
Sequenced exomes (n = 258) with aortic dissection were obtained from the University of Michigan Cardiovascular Health Improvement Project (CHIP) (30, 31). Variant annotation was performed as previously described (32), with the exception that nonsynonymous variants were predicted to be deleterious if their CADD Phred score was ≥20 and their non-Finnish European and All Population frequency (gnomAD) was ≤0.1%.
Statistics
Comparisons of the means between two groups were performed with a two-sided Student’s t test. Paired comparisons within the same mouse under two different treatment conditions were performed with a paired Student’s t test. Comparisons of the mean between three or more groups were performed with one-way ANOVA and Tukey’s post hoc test. Categorical data comparing two groups were evaluated with Fisher’s exact test. Categorical data comparing more than two groups were evaluated with a χ2 test. Survival curve analysis was performed with log-rank test with a Bonferroni correction for multiple comparisons. A P value of <0.05 was considered significant, except for the survival curve analyses, in which a P value of <0.0167 was considered significant in controlling for multiple comparisons between three groups. All figures present means ± SE unless otherwise specified. Individual data points are displayed within each figure.
Research Assurances
All procedures were approved by the University of Michigan Institutional Animal Care and Use Committee and adhered to the guiding principles in the care and use of experimental animals in accordance with the National Institute of Health Guidelines. The University of Michigan operates an AALAC-certified animal facility. All studies derived from the CHIP cohort were approved by the University of Michigan Institutional Review Board. Written informed consent was obtained from patients during their initial enrollment into the CHIP cohort. Studies adhere to the requirements of the US Federal Policy for the Protection of Human Subjects (45 CFR, Part 46).
RESULTS
Untreated PLCε-Deficient Mice Have Ascending Aortic Dilation and Aortic Valve Insufficiency
To provide experimental evidence that PLCε deficiency contributes to ascending aortic aneurysm and dissection, we evaluated globally deficient Plce1−/− mice for evidence of aortic aneurysms. Plce1−/− mice showed evidence of ascending aortic dilation (Fig. 1). This ascending aortic dilation spared the aortic root and was most pronounced in the ascending aorta and arch. We next performed a histological assessment of the ascending aortas and aortic valves. There was no significant difference in the number of elastin breaks or medial degeneration in untreated Plce1−/− mice (Fig. 2 and Table 1), and no Plce1−/− mice had evidence of aortic dissection. Consistent with previous reports (5), 75% of Plce1−/− mice showed echocardiographic evidence of aortic insufficiency as well (Supplemental Fig. S1A; see https://doi.org/10.6084/m9.figshare.19787476), and the aortic valves of Plce1−/− mice were markedly thickened (Supplemental Fig. S1, B and C; see https://doi.org/10.6084/m9.figshare.19787476). All valves from Plce1−/− mice were noted to be tricuspid.
Figure 1.

PLCε deficiency causes dilation primarily of the ascending aorta and arch under control conditions. This dilation of the ascending aorta and arch worsens after 4 days of ANG II treatment. A: aortic ultrasonography demonstrating baseline dilation under control conditions and increasing expansion of the ascending aorta and arch in Plce1−/− mice treated with ANG II for 4 days. ○, Male; △, female. B: representative ultrasonographic images from the root and arch of Plce1+/+ and Plce1−/− mice. Samples were chosen from N = 10–12 per group. Paired comparisons of aortic size in the same mouse under control and ANG II-treated conditions were analyzed with a paired Student’s t test. Single intergroup comparisons were evaluated with an unpaired Student’s t test. *P < 0.05 vs. Plce1+/+, **P < 0.01 vs. Plce1+/+, ***P < 0.001 vs. Plce1+/+, †P < 0.05 vs. same genotype + control, ††P < 0.01 vs. same genotype + control, †††P < 0.001 vs. same genotype + control. N, number of mice per group.
Figure 2.

Plce1−/− mice show no evidence of medial degeneration under control conditions but rapidly develop medial degeneration of the ascending aorta after 4 days of ANG II infusion. Arrow points to the area of elastolysis with medial fibrosis and associated adventitial inflammation in the ANG II-treated Plce1−/− mouse. All slides stained with Verhoeff–Van Gieson stain. Images acquired at ×20 magnification. All representative pictures taken from male mice from 6–10 samples per group.
Table 1.
ANG II leads to exaggerated elastin degradation and medial degeneration in PLCε deficiency after 4 days of ANG II infusion
| Plce1+/+ + Control | Plce1−/− + Control | Plce1+/+ + ANG II | Plce1−/− + ANG II | |
|---|---|---|---|---|
| Elastin breaks, number/aortic section | 0.81 ± 0.26 | 1.44 ± 0.60 | 1.56 ± 0.46 | 6.17 ± 1.34**## |
| Medial degeneration, n mice | 0/6 | 1/6 | 1/10 | 9/10††† |
| Ascending aortic aneurysm, n mice | 0/6 | 0/6 | 1/10 | 7/10†† |
| Descending aortic aneurysm, n mice | 0/6 | 0/6 | 0/10 | 2/10 |
| Aortic dissection, n mice | 0/6 | 0/6 | 1/10 | 7/10†† |
| Sudden death from aortic rupture, n mice | 0/6 | 0/6 | 1/10 | 2/10 |
Values are means ± SE and number of mice. All experiments were performed in male mice. This leads preferentially to ascending aortic aneurysm and dilation, predisposing to aortic dissection. There is no difference in mortality after 4 days of ANG II infusion. N = 6 in both the Plce1+/+ + control and Plce1−/− + control groups for elastin breaks analysis and N = 9 and 8 for ANG II-treated Plce1+/+ and Plce1−/− groups, respectively. Data analysis performed with 1-way ANOVA with Tukey post hoc test and χ2 test. **P < 0.01 vs. Plce1+/+ + ANG II, ##P < 0.01 vs. Plce1−/− + control. ††P < 0.01 for χ2, †††P < 0.001 for χ2. N, number of mice per group.
ANG II Induces Sudden Death by Ascending Aortic Dilation, Dissection, and Rupture in PLCε Deficiency
Since increased aortic diameter is a risk factor for aortic dissection (7), we infused Plce1+/+, Plce1+/−, and Plce1−/− mice with ANG II, which is known to promote aortic aneurysm and dissections within the ascending aorta in murine models (19). All Plce1+/− and Plce1−/− mice found dead had either hemothorax and/or hemopericardium, with necropsy showing ascending aortic dissection and rupture as the cause of death (Fig. 3). No mice died of a ruptured abdominal aortic aneurysm. Some 42% of Plce1−/− mice died (12/28 mice) during the 14-day infusion, compared with only 5% (1/20 mice) of Plce1+/+ mice (P < 0.017; Fig. 4). Some 17% of Plce1+/− mice died during the ANG II infusion as well, which was not significantly different compared with Plce1−/− mice. In Plce1+/− mice PLCe1 transcript expression in kidney tissue was detected at a significantly lower level than in Plce1+/+ mice and undetected in Plce1−/− mice (Supplemental Fig. S2; see https://doi.org/10.6084/m9.figshare.19787437). Male Plce1−/− mice were more susceptible to ANG II, with 60% (9/15 mice) suddenly dying (P < 0.017 vs. Plce1+/+ males; Fig. 4), whereas only 23% of female Plce1−/− mice (3/13 mice) succumbed to ANG II [P = nonsignificant (NS) vs. Plce1+/+ females; Fig. 4).
Figure 3.

ANG II causes sudden death by ascending thoracic aortic dissection and rupture in PLCε deficiency. Ascending aortic dissection and rupture in Plce1−/− mouse with Verhoeff–Van Gieson (left) and hematoxylin and eosin (H&E; right) stains. Arrows points to area of dissection and rupture. Images obtained with ×10 magnification. Representative pictures were obtained from 12 representative male mice.
Figure 4.

Sudden death from aortic dissection is more penetrant in male as opposed to female PLCε-deficient mice. Left: higher mortality rate in Plce1−/− mice treated with ANG II compared with Plce1+/+ mice (P = 0.0056). Mortality rate for Plce1−/− mice is not significantly greater compared with Plce1+/− mice (P = 0.0689) (Plce1+/+, N = 20; Plce1+/−, N = 18; and Plce1−/−, N = 28). Center: mortality was significantly greater in male Plce1−/− mice treated with ANG II compared with Plce1+/+ mice (P = 0.011) (Plce1+/+, N = 10; Plce1+/−, N = 13; and Plce1−/−, N = 15). Right: mortality was not significantly elevated in female Plce1−/− mice treated with ANG II compared with other genotypes (P = 0.0629) (Plce1+/+, N = 10; Plce1+/−, N = 5; and Plce1−/−, N = 13). Log-rank test with Bonferroni correction was performed. *P < 0.0167. N, number of mice per group.
We next characterized the changes in the ascending aorta in PLCε deficiency before ascending aortic rupture and sudden death. We evaluated mice euthanized after only 4 days of ANG II infusion, based on our survival analysis suggesting that spontaneous death from aortic dissection was unlikely during this time frame (Fig. 4). Given the increased susceptibility of male mice to sudden death (Fig. 4), we used them exclusively for this study. ANG II caused significantly more elastin breaks in the ascending aorta Plce1−/− mice (Table 1) and a higher incidence of medial degeneration, evident as elastin fragmentation, mucoid extracellular matrix accumulation (MEMA), and smooth muscle cell loss, after 4 days of infusion. Medial degeneration of the ascending aorta and ascending aortic aneurysms with dissecting hemorrhage were evident, respectively, in 90% and 70% of Plce1−/− mice (Table 1 and Fig. 2), whereas both findings were present in only 10% of Plce1+/+ mice (P < 0.05), which were affected. Histological findings corresponded with progressive dilation, which mostly affected the ascending aorta and arch in Plce1−/− mice (Fig. 1A). Descending aortic aneurysms were also noted in 20% of Plce1−/− mice, although the incidence in the descending aorta did not achieve statistical significance (Table 1). Despite the high incidence of ascending aortic aneurysm and dissecting hemorrhage, only 20% of Plce1−/− mice died of spontaneous aneurysmal rupture during the 4-day ANG II infusion period (Table 1). These findings support that medial degeneration was the antecedent event leading to aneurysmal changes and caused the aortic dissection and rupture in Plce1−/− mice, as some mice were already showing evidence of aortic dissection and early aortic rupture. Additionally, these changes were not due to an enhanced pressor response to ANG II in the Plce1−/− mice, as blood pressure rose equally in Plce1+/+ and Plce1−/− mice treated with ANG II (Table 2) over 4 days.
Table 2.
Effect of ANG II on systolic blood pressure in PLCε deficiency
| Plce1+/+ + Control | Plce1−/− + Control | Plce1+/+ + ANG II | Plce1−/− + ANG II | |
|---|---|---|---|---|
| Systolic BP, mmHg | 135 ± 4 | 127 ± 4 | 173 ± 7** | 164 ± 3## |
Values are means ± SE; N, number of mice per group. Data were obtained from male mice. BP, blood pressure. Data analysis performed with 1-way ANOVA with Tukey post hoc test. N per group: Plce1+/+ + control = 8, Plce1−/− + control = 9, Plce1+/+ + ANG II = 13, Plce1−/− + ANG II = 13. **P < 0.01 vs. Plce1+/+ + control, ##P < 0.01 vs. Plce1−/− + control.
PLCε Is Markedly Upregulated in the Ascending Aortic Adventitia in Response to ANG II
To determine how PLCε deficiency contributes to the development of ascending aortic aneurysms and dissections, we evaluated the expression of PLCε within the ascending aorta at baseline and in response to ANG II, using a mouse model that expresses a hemagglutinin-tagged PLCε (Plce1-HA) (3). Surprisingly, PLCε was minimally expressed in the ascending aorta at baseline (Fig. 5A). As demonstrated in Fig. 5 and Fig. 6, ANG II significantly increased the percentage of PLCε-expressing cells within the aortic adventitia. As such, PLCε is minimally expressed within the aorta at baseline but is robustly increased in the adventitia in response to ANG II. To determine which cells express increased levels of PLCε, we stained additional aortic sections for periostin or F4/80, which are markers of myofibroblasts and macrophages, respectively. Predominantly, the cells in the adventitia appear to be periostin expressing (Supplemental Fig. S3; see https://doi.org/10.6084/m9.figshare.19787494), suggesting that most PLCε-expressing cells are myofibroblasts.
Figure 5.

PLCε is markedly upregulated in the ascending aortic adventitia after ANG II-mediated injury. A: ascending aortas from wild-type (WT) and hemagglutinin-tagged PLCε (Plce1-HA) mice stained with anti-hemagglutinin antibody before or after treatment with ANG II for 7 days. All mice were male except for the ANG II-treated wild-type mouse, which was female, and were taken from samples of 2–3 mice per group. Note the aortic dissection and medial necrosis at the arrows; ×5 magnification. B, top: Plce1-HA ascending aorta under control conditions (male mouse). B, bottom: Plce1-HA ascending aorta treated with ANG II (male mouse). *Area of significant Plce1-HA positivity within the adventitia. Arrow points to an area of aortic dissection. ×20 magnification. L, lumen; M, media; A, adventitia.
Figure 6.

Quantification of number of PLCε-expressing cells in the media or adventitia. All experiments were performed in male mice. N, number of mice per group.
ANG II-Induced Hypertension Increases TNF-α, Il-1β, and IL-6 Signaling in PLCε Deficiency
To determine the mechanisms by which ANG II caused aortic dissection in PLCε deficiency, we performed total RNA sequencing of the ascending aorta and arch in Plce1+/+ and Plce1−/− mice under control conditions and with 4 days of ANG II treatment. As demonstrated in Fig. 7, only 11 of 22,246 genes were differentially expressed in Plce1+/+ and Plce1−/− mice under control conditions. ANG II treatment resulted in the differential expression of a significant number of genes (Fig. 7). A count of 1,180 genes were differentially expressed with ANG II treatment in Plce1+/+ mice and 8,194 genes in Plce1−/− mice (Fig. 7). A count of 3,778 genes were differentially expressed between ANG II-treated Plce1+/+ and Plce1−/− mice, respectively (Supplemental Fig. S4; see https://doi.org/10.6084/m9.figshare.19787497). Upstream analysis of ingenuity pathways in ANG II-treated Plce1−/− mice identified multiple effector molecules known to affect aortic aneurysm or dissection formation such as TNF-α, IL-1β, and IL-6 (Fig. 8A). Consistent with this, the relative differential gene expression of TNF-α, IL-1β, and IL-6 was increased 3.8-, 34-, and 17-fold in Plce1−/− mice treated with ANG II compared with Plce1+/+ mice (Fig. 8B). There was no difference in the expression of the Agtr1a gene between the two different genotypes (Supplemental Fig. S5; see https://doi.org/10.6084/m9.figshare.21187738). Downstream ingenuity pathway analysis of differentially expressed genes in ANG II-treated Plce1−/− mice compared with Plce1+/+ mice demonstrated marked activation Z scores in signaling pathways related to inflammation, activation of fibrotic pathways, and IL-6 and IL-17 signaling (Supplemental Fig. S6A; see https://doi.org/10.6084/m9.figshare.21187804). Select PCR analysis confirmed increased transcript signaling through fibrotic and inflammatory pathways (Supplemental Fig. S6B; see https://doi.org/10.6084/m9.figshare.21187804). Network analysis of signaling was consistent with our upstream signaling analysis; IL-1β was central to all these responses (Supplemental Fig. S7; see https://doi.org/10.6084/m9.figshare.21187822). Raw data for RNA sequencing are available as a supplemental file (http://doi.org/10.6084/m9.figshare.19787626).
Figure 7.

Volcano plots demonstrating differentially expressed genes in Plce1−/− mice compared with Plce1+/+ mice under control conditions (left), Plce1+/+ mice under control conditions and with 4 days of ANG II treatment (center), and Plce1−/− mice under control conditions and with 4 days of ANG II treatment (right). Negative log fold change (fc) represents decreased expression in Plce1−/− mice relative to Plce1+/+ mice and deceased expression in ANG II-treated compared with control conditions. All experiments were performed in male mice.
Figure 8.

A: top 10 predicted upstream regulators of ANG II signaling in PLCε deficiency by activation z score (denoted by color scheme). B: relative differential gene expression of relevant upstream regulators. WT.ANG II, predicted upstream activators in control and ANG II-treated Plce1+/+ mice; KO.ANG IIb, predicted upstream activators in and ANG II-treated Plce1−/− mice; ANG IIKO_WT, differentially expressed genes between ANG II-treated Plce1−/− and Plce1+/+ mice. All experiments were performed in male mice.
Human PLCE1 Genetic Variants Are Present in Type A Aortic Dissection
To determine whether variants in PLCE1 could contribute to human aortic disease, we queried exome sequencing data from the Cardiovascular Health Improvement Project. We discovered six nonsynonymous single-nucleotide variants in five different patients with type A aortic aneurysms and dissections (Table 3). None of the variants was a missense or nonsense variant. However, in silico analysis with UMD, CADD_phred, SIFT, and Polyphen2_HVAR score suggested the potential for pathogenicity for most variants (Table 3).
Table 3.
PLCE1 variants discovered in CHIP cohort and annotation
| Gene | Exon | Nucleotide Change (HGVS) | gnomAD | CADD | Sift | Polyphen2 |
|---|---|---|---|---|---|---|
| PLCE1 | 10 | c.3281G>T, (p.Gly1094Val) | 3.0 × 10−4 | 27.3 | D | D |
| PLCE1 | 14 | c.4000G>A, (p.Val1334Met) | 3.3 × 10−5 | 29.5 | T | D |
| PLCE1 | 18 | c.4484G>A, (p.Arg1495Gln) | 2.8 × 10−5 | 29.6 | T | D |
| PLCE1 | 19 | c.4640C>T, (p.Thr1547Met) | 6.5 × 10−5 | 34 | D | D |
| PLCE1 | 26 | c.5756T>C, (p.Met1919Thr) | 6.1 × 10−5 | 24.1 | T | D |
| PLCE1 | 26 | c.5809C>T, (p.Arg1937Cys) | 35 | D | D |
Annotation was based on NM_016341. gnomAD frequency is inferred from gnomAD exome (v2.1). CADD score is CADD phred score. SIFT: D, deleterious; T, tolerant. Polyphen2 is Polyphen2 HVAR score: D, deleterious. CHIP, Cardiovascular Health Improvement Project.
We obtained select clinical data to better characterize the patients with PLCE1 variants (Table 4). Two patients possessed other potential primary causes of their aortic dissection (bicuspid aortic valve and pathogenic FBN1 variant). Aside from the patient with the FBN1 variant, all other patients showed maximal aneurysmal diameter of the aorta in the ascending segment as opposed to the aortic root (Table 5). All patients suffered from aortic regurgitation at the time of diagnosis of their aortic dissection. One patient developed progressive aortic regurgitation after their initial valve-sparing type A dissection repair, requiring surgical intervention 5 yr after their initial surgery (c.5809C>T). As such, patients with PLCE1 variants share many of the pathological aortic characteristics of Plce1-deficient mice, suggesting that PLCE1 variants may play a role in human type A aortic dissection.
Table 4.
Characteristics of patients with PLCE1 variants in the CHIP cohort
| Nucleotide Change | Age at First Dissection, yr | Sex | HTN | Smoking | Family History | Other Factors | Genetic Findings |
|---|---|---|---|---|---|---|---|
| c.3281G>T | 42 | F | Yes | Yes | No | Lupus, prednisone use | None |
| c.4000G>A and c.4484G>A | 34 | M | Yes | No | Yes | Bicuspid aortic valve | None |
| c.4640C>T | 31 | M | Yes | No | No | Chronic kidney disease | Pathogenic FBN1 variant |
| c.5756T>C | 61 | M | No | Yes | No | No | None |
| c.5809C>T | 42 | M | No | No | No | No | None |
CHIP, Cardiovascular Health Improvement Project; HTN, hypertension; F, female; M, male.
Table 5.
Characteristics of ascending aorta and aortic valve of patients with PLCE1 variants from CHIP cohort
| Nucleotide Change | Aortic Valve/Valve Repair | Root Diameter/Ascending Diameter, cm | Aortic Pathology |
|---|---|---|---|
| c.3281G>T | Tricuspid, regurgitant, no repair | 4.6/5.7 | Aortic dissection and mild atherosclerosis |
| c.4000G>A and c.4484G>A | Bicuspid, regurgitant, valve repaired during initial surgery | 5.0/7.8 | Myxoid degeneration |
| c.4640C>T | Tricuspid, regurgitant, no repair | 5.0/4.6 | Acute aortic dissection, cystic medial changes, ulcerated atheromas |
| c.5756T>C | Tricuspid, regurgitant, no repair | 4.5/5.0 | Cystic medial degeneration, atherosclerosis |
| c.5809C>T | Tricuspid, regurgitant, valve repaired 5 yr after initial surgery | 5.9/6.1 | Mild atherosclerosis, myxoid degeneration of aortic valve |
CHIP, Cardiovascular Health Improvement Project.
DISCUSSION
PLCε deficiency has been previously shown to cause aortic insufficiency in a murine model (5). This is thought to be due to a developmental defect secondary to activation of Smad 1/5/8 on embryonic day (E)14.5 leading to pathological thickening in the developing semilunar valves. Since aortic valve insufficiency is frequently comorbid with aortic aneurysm and dissection, we tested whether global PLCε-deficient mice have aortic aneurysms as well. We found that Plce1−/− mice have dilation of the ascending aorta under unchallenged conditions and that this dilation spares the aortic root and is most pronounced in the ascending aortic segment and arch. The magnitude of aortic dilation we noted is similar to that which is seen in other aortopathies (33).
Since there was no evidence of medial degeneration, aneurysm, or dissection in untreated Plce1−/− mice, we challenged them with ANG II to determine whether an environmental stressor could produce ascending aortic aneurysmal growth and dissection. ANG II is known to cause progressive dilation of the ascending aorta leading to aortic dissection in 12–25% of wild-type mice within 7 days of treatment initiation (19). ANG II caused sudden death by aortic dissection and rupture in 42% of Plce1−/− mice, significantly greater than the 5% of ANG II-treated Plce1+/+ mice. We also treated Plce1+/− mice with ANG II as well to determine whether there was a dose-response effect of Plce1 deficiency in the development of aortic dissections. Interestingly, 17% of Plce1+/− mice died from ascending aortic dissection in response to ANG II as well. This was likely explained by the significantly reduced Plce1 transcript expression seen in the Plce1+/− mice. The fact that PLCε deficiency requires an environmental stressor to develop the full dissection phenotype is consistent with other models of aortopathy as well. Both pregnancy and calcium channel blocker use increase the risk of aortic dissection in Marfan mice (12, 13). Additionally, ACTA2 variants result in human aortic disease (34). However, Acta2-null mice require ANG II-induced hypertension to develop significant aortic pathology (11). We also noted that male Plce1−/− mice had a higher propensity for dissection compared with female mice. This is consistent with previous data from the ANG II model (35) but is also generally seen in human aortic aneurysm as well (36, 37).
Elastin degradation after 4 days of treatment was more severe in PLCε-deficient mice compared with wild-type mice. Histologically, there was evidence of significant medial degeneration in Plce1−/− mice treated with ANG II as well. We surmise that these degenerative changes caused progressive aneurysmal dilation of the aorta, as this was noted on echocardiography as well as histologically in Plce1−/− mice treated with ANG II. We suspect that this progressive dilation of the aorta is what led to the eventual aortic dissection and rupture, as there were early signs of aortic dissection in some Plce1−/− mice antemortem. Additionally, hypertension is a known risk factor for the development of aortic dissection (37). We measured blood pressure in the mice to determine whether a differential blood pressure response to ANG II could account for the different dissection rate between wild-type and PLCε-deficient mice. Plce1−/− mice have attenuated responses to longer infusions of hypertensive substances including ANG II, deoxycorticosterone acetate, and norepinephrine over 2–4 wk (3). However, within the 4 days of ANG II infusion in the present study, there was no significant difference in the hypertensive response between genotypes, suggesting that the aortic dissection phenotype in PLCε deficiency was not due to an exaggerated hypertensive response to ANG II. The blood pressure values determined by tail-cuff method under control conditions and with ANG II are similar to those reported by other groups (38).
PLCε was minimally expressed within the ascending aorta under control conditions but increased markedly in the ascending aortic adventitia in response to ANG II-mediated injury. This is consistent with previous data showing minimal expression of PLCε in large vessels (main renal artery) under untreated conditions (3). Periostin is also strongly expressed in the aortic adventitia after treatment with ANG II, supporting that the majority of PLCε-expressing cells are likely myofibroblasts. Myofibroblasts have not been well studied in aortic pathology but have been well described in cardiac pathology (39, 40). There, myofibroblasts are integral in the fibrotic response to injury. Loss of periostin expression impairs myofibroblast function and increases the risk of left ventricular wall rupture in response to myocardial infarction in mice (41). We speculate that loss of PLCε function may impair myofibroblast function within the aorta and that this may contribute to the aortic dissection phenotype seen in Plce1−/− mice. Further experiments are needed to confirm whether this hypothesis is correct.
Upstream ingenuity pathway analysis indicated that upregulation of TNF-α, IL-1β, and IL-6 signaling contributed to the transcriptomic signature in PLCε deficiency. TNF-α, IL-1β, and IL-6 have all been implicated in the pathogenesis of aortic aneurysm and dissection. Genetic deletion of TNF-α or pharmacological inhibition with infliximab impairs aortic dilation and abdominal aortic aneurysm formation (16). IL-1β is known to promote the development of aortic aneurysms and ruptures, as pharmacological blockade of IL-1β and genetic deletion of the IL-1β receptor ameliorate the rate of aneurysmal growth and dissection (14, 15). IL-6 is known to contribute to the development of aortic dissections as well. Specifically, IL-6 release from the aortic adventitia is known to increase MCP-1 secretion leading to increased aortic macrophage accumulation, leading to aortic destabilization, dissection, and rupture (17). Our studies did not experimentally investigate the specific mechanism through which PLCε deficiency leads to increased levels of these inflammatory mediators. It remains to be determined whether this is due to PLCε-mediated upregulation of the expression of these inflammatory mediators identified in the gene expression profiling or whether the altered expression is secondary to the aortic dissection process. Even though PLCε has previously been shown to directly and positively regulate the production of TNF-α, IL-1β, and IL-6 (42–44), additional experiments are needed to clarify the role of PLCε in regulating the expression of these inflammatory mediators in the aorta.
In addition to its role in aortic dissection in mice, we provide a preliminary proof-of-concept association for PLCε contributing to development of aortic dissection in humans. We identified five patients with six different PLCE1 variants in the CHIP cohort with aortic aneurysms and dissections. Although none of these variants was either a frameshift or stop codon variant, the UMD, CADD Phred, SIFT, and Polyphen2 HVAR scores were all suggestive of pathogenicity. All five patients were noted to have ascending aortic aneurysms and/or type A aortic dissections. Except for one patient with a concomitant FBN1 variant, all patients had maximal aneurysmal dilation of the aorta in the ascending segment, and not the aortic root. This is similar to the pattern of aortic dilation and dissection we saw in PLCε-deficient mice. Another characteristic the patients shared with our mouse model is that all of them suffered from aortic regurgitation. Although this may have been secondary to the acute effects of aortic dissection, one patient developed progressive worsening of his aortic insufficiency after his type A dissection repair and required eventual aortic valve replacement, suggesting that the valve defect may have been separate from the aortic dissection. Our initial proof-of-concept analysis does not provide a conclusive analysis of the association of PLCE1 variants with aortic disease and does not clarify a precise mechanism by which PLCE1 variants contribute to human aortic disease. However, within these limitations, additional studies have implicated genetic variants of PLCE1 in aortic disease. A recent study using genomewide association has identified a PLCE1 variant that is associated with expanding ascending and descending aortic diameter (45).
In conclusion, we characterize a novel phenotype by which PLCε deficiency predisposes to aortic valve insufficiency and ascending aortic aneurysm, dissection, and sudden death in the setting of ANG II-mediated hypertension. PLCε expression is markedly increased in aortic adventitial myofibroblasts in response to ANG II, suggesting that PLCε may play an integral role in the inflammatory and/or reparative response of the adventitia during injury. This is associated with increased inflammatory signaling via known mediators of aortic dissection such as TNF-α, IL-1β, and IL-6. Finally, we demonstrate six PLCE1 variants in five patients with type A aortic dissection and aortic insufficiency, suggesting that PLCE1 may contribute to human aortic disease.
DATA AVAILABILITY
Raw data for RNA sequencing are available as a supplemental file (http://doi.org/10.6084/m9.figshare.19787626).
SUPPLEMENTAL DATA
Supplemental Fig. S1: https://doi.org/10.6084/m9.figshare.19787476.
Supplemental Fig. S2: https://doi.org/10.6084/m9.figshare.19787437.
Supplemental Fig. S3: https://doi.org/10.6084/m9.figshare.19787494.
Supplemental Fig. S4: https://doi.org/10.6084/m9.figshare.19787497.
Supplemental Fig. S5: https://doi.org/10.6084/m9.figshare.21187738.
Supplemental Fig. S6: https://doi.org/10.6084/m9.figshare.21187804.
Supplemental Fig. S7: https://doi.org/10.6084/m9.figshare.21187822.
GRANTS
This work was supported by National Institutes of Health (NIH) Grant 5T32DK007378 (to D.K.A.); George C. O’Brien Pilot Funding Program NIH Grant P30DK081943 (to D.K.A. and J.R.H.); and NIH Grants R01DK100449 (to M.B.); R35GM127303 (to A.V.S.); T32-HL007853 (to Y.W.); and R01HL139672, R01HL139672, and R01HL086694 (to S.K.G.) and the A. Alfred Taubman Institute (to S.K.G.).
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
A.V.S., S.K.G., and M.B. conceived and designed research; D.K.A., C.L.O., K.C.-B., I.L.B., and H.Z. performed experiments; D.K.A., K.C.-B., I.L.B., Y.W., J.R.H., and W.J. analyzed data; D.K.A., I.L.B., Y.W., W.J., A.V.S., S.K.G., and M.B. interpreted results of experiments; D.K.A. and J.R.H. prepared figures; D.K.A. drafted manuscript; A.V.S., S.K.G., and M.B. edited and revised manuscript; D.K.A., C.L.O., K.C.-B., I.L.B., H.Z., Y.W., J.R.H., W.J., A.V.S., S.K.G., and M.B. approved final version of manuscript.
ACKNOWLEDGMENTS
We thank the Frankel Cardiovascular Center Physiological Phenotyping Core at the University of Michigan for work with the aortic ultrasonography and the In Vivo Animal Core in the Unit for Laboratory Animal Medicine at the University of Michigan for assistance with tissue processing. We thank all participants and the collection team of the Cardiovascular Health Improvement Project (CHIP) at the University of Michigan. The collection of samples for Michigan Medicine–Cardiovascular Health Improvement Project was supported by the Frankel Cardiovascular Center. We appreciate the Aikens Fund for Aortic Research and McKay research award for supporting this project. We acknowledge the support of the George C. O’Brien Applied Systems Biology Core.
REFERENCES
- 1. Smrcka AV, Brown JH, Holz GG. Role of phospholipase Cε in physiological phosphoinositide signaling networks. Cell Signal 24: 1333–1343, 2012. doi: 10.1016/j.cellsig.2012.01.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Nash CA, Wei W, Irannejad R, Smrcka AV. Golgi localized β1-adrenergic receptors stimulate Golgi PI4P hydrolysis by PLCε to regulate cardiac hypertrophy. Elife 8: e48167, 2019. doi: 10.7554/eLife.48167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Atchison DK, O'Connor CL, Menon R, Otto EA, Ganesh SK, Wiggins RC, Smrcka AV, Bitzer M. Hypertension induces glomerulosclerosis in phospholipase C-ε1 deficiency. Am J Physiol Renal Physiol 318: F1177–F1187, 2020. doi: 10.1152/ajprenal.00541.2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Wang H, Oestreich EA, Maekawa N, Bullard TA, Vikstrom KL, Dirksen RT, Kelley GG, Blaxall BC, Smrcka AV. Phospholipase C epsilon modulates beta-adrenergic receptor-dependent cardiac contraction and inhibits cardiac hypertrophy. Circ Res 97: 1305–1313, 2005. doi: 10.1161/01.RES.0000196578.15385.bb. [DOI] [PubMed] [Google Scholar]
- 5. Tadano M, Edamatsu H, Minamisawa S, Yokoyama U, Ishikawa Y, Suzuki N, Saito H, Wu D, Masago-Toda M, Yamawaki-Kataoka Y, Setsu T, Terashima T, Maeda S, Satoh T, Kataoka T. Congenital semilunar valvulogenesis defect in mice deficient in phospholipase C epsilon. Mol Cell Biol 25: 2191–2199, 2005. doi: 10.1128/MCB.25.6.2191-2199.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Isselbacher EM. Thoracic and abdominal aortic aneurysms. Circulation 111: 816–828, 2005. doi: 10.1161/01.CIR.0000154569.08857.7A. [DOI] [PubMed] [Google Scholar]
- 7. Juvonen T, Ergin MA, Galla JD, Lansman SL, Nguyen KH, McCullough JN, Levy D, de Asla RA, Bodian CA, Griepp RB. Prospective study of the natural history of thoracic aortic aneurysms. Ann Thorac Surg 63: 1533–1545, 1997. [Erratum in Ann Thorac Surg 64: 594, 1997]. doi: 10.1016/s0003-4975(97)00414-1. [DOI] [PubMed] [Google Scholar]
- 8. Kuzmik GA, Sang AX, Elefteriades JA. Natural history of thoracic aortic aneurysms. J Vasc Surg 56: 565–571, 2012. doi: 10.1016/j.jvs.2012.04.053. [DOI] [PubMed] [Google Scholar]
- 9. Halushka MK, Angelini A, Bartoloni G, Basso C, Batoroeva L, Bruneval P, Buja LM, Butany J, d’Amati G, Fallon JT, Gallagher PJ, Gittenberger-de Groot AC, Gouveia RH, Kholova I, Kelly KL, Leone O, Litovsky SH, Maleszewski JJ, Miller DV, Mitchell RN, Preston SD, Pucci A, Radio SJ, Rodriguez ER, Sheppard MN, Stone JR, Suvarna SK, Tan CD, Thiene G, Veinot JP, van der Wal AC. Consensus statement on surgical pathology of the aorta from the Society for Cardiovascular Pathology and the Association For European Cardiovascular Pathology: II. Noninflammatory degenerative diseases—nomenclature and diagnostic criteria. Cardiovasc Pathol 25: 247–257, 2016. doi: 10.1016/j.carpath.2016.03.002. [DOI] [PubMed] [Google Scholar]
- 10. Pinard A, Jones GT, Milewicz DM. Genetics of thoracic and abdominal aortic diseases. Circ Res 124: 588–606, 2019. doi: 10.1161/CIRCRESAHA.118.312436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Cheng J, Zhou X, Jiang X, Sun T. Deletion of ACTA2 in mice promotes angiotensin II induced pathogenesis of thoracic aortic aneurysms and dissections. J Thorac Dis 10: 4733–4740, 2018. doi: 10.21037/jtd.2018.07.75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Habashi JP, MacFarlane EG, Bagirzadeh R, Bowen C, Huso N, Chen Y, Bedja D, Creamer TJ, Rykiel G, Manning M, Huso D, Dietz HC. Oxytocin antagonism prevents pregnancy-associated aortic dissection in a mouse model of Marfan syndrome. Sci Transl Med 11: eaat4822, 2019. doi: 10.1126/scitranslmed.aat4822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Doyle JJ, Doyle AJ, Wilson NK, Habashi JP, Bedja D, Whitworth RE, Lindsay ME, Schoenhoff F, Myers L, Huso N, Bachir S, Squires O, Rusholme B, Ehsan H, Huso D, Thomas CJ, Caulfield MJ, Van Eyk JE, Judge DP, Dietz HC; GenTAC Registry Consortium, MIBAVA Leducq Consortium. A deleterious gene-by-environment interaction imposed by calcium channel blockers in Marfan syndrome. Elife 4: e08648, 2015. doi: 10.7554/eLife.08648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Johnston WF, Salmon M, Su G, Lu G, Stone ML, Zhao Y, Owens GK, Upchurch GR Jr, Ailawadi G. Genetic and pharmacologic disruption of interleukin-1beta signaling inhibits experimental aortic aneurysm formation. Arterioscler Thromb Vasc Biol 33: 294–304, 2013. doi: 10.1161/ATVBAHA.112.300432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Da Ros F, Carnevale R, Cifelli G, Bizzotto D, Casaburo M, Perrotta M, Carnevale L, Vinciguerra I, Fardella S, Iacobucci R, Bressan GM, Braghetta P, Lembo G, Carnevale D. Targeting interleukin-1beta protects from aortic aneurysms induced by disrupted transforming growth factor beta signaling. Immunity 47: 959–973.e9, 2017. doi: 10.1016/j.immuni.2017.10.016. [DOI] [PubMed] [Google Scholar]
- 16. Xiong W, MacTaggart J, Knispel R, Worth J, Persidsky Y, Baxter BT. Blocking TNF-alpha attenuates aneurysm formation in a murine model. J Immunol 183: 2741–2746, 2009. doi: 10.4049/jimmunol.0803164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Tieu BC, Lee C, Sun H, Lejeune W, Recinos A 3rd, Ju X, Spratt H, Guo DC, Milewicz D, Tilton RG, Brasier AR. An adventitial IL-6/MCP1 amplification loop accelerates macrophage-mediated vascular inflammation leading to aortic dissection in mice. J Clin Invest 119: 3637–3651, 2009. doi: 10.1172/JCI38308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Daugherty A, Rateri DL, Charo IF, Owens AP, Howatt DA, Cassis LA. Angiotensin II infusion promotes ascending aortic aneurysms: attenuation by CCR2 deficiency in apoE−/− mice. Clin Sci (Lond) 118: 681–689, 2010. doi: 10.1042/CS20090372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Rateri DL, Davis FM, Balakrishnan A, Howatt DA, Moorleghen JJ, O’Connor WN, Charnigo R, Cassis LA, Daugherty A. Angiotensin II induces region-specific medial disruption during evolution of ascending aortic aneurysms. Am J Pathol 184: 2586–2595, 2014. doi: 10.1016/j.ajpath.2014.05.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Poduri A, Rateri DL, Howatt DA, Balakrishnan A, Moorleghen JJ, Cassis LA, Daugherty A. Fibroblast angiotensin II type 1a receptors contribute to angiotensin II-induced medial hyperplasia in the ascending aorta. Arterioscler Thromb Vasc Biol 35: 1995–2002, 2015. doi: 10.1161/ATVBAHA.115.305995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Tieu BC, Ju X, Lee C, Sun H, Lejeune W, Recinos A 3rd, Brasier AR, Tilton RG. Aortic adventitial fibroblasts participate in angiotensin-induced vascular wall inflammation and remodeling. J Vasc Res 48: 261–272, 2011. doi: 10.1159/000320358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Bankhead P, Loughrey MB, Fernández JA, Dombrowski Y, McArt DG, Dunne PD, McQuaid S, Gray RT, Murray LJ, Coleman HG, James JA, Salto-Tellez M, Hamilton PW. QuPath: open source software for digital pathology image analysis. Sci Rep 7: 16878, 2017. doi: 10.1038/s41598-017-17204-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Martin M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet.journal 17: 10–12, 2011. doi: 10.14806/ej.17.1.200. [DOI] [Google Scholar]
- 24. Andrews S. FastQC: quality control tool for high throughput sequence data. 2010. https://www.bioinformatics.babraham.ac.uk/projects/fastqc/.
- 25. Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, Batut P, Chaisson M, Gingeras TR. STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29: 15–21, 2013. doi: 10.1093/bioinformatics/bts635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Li B, Dewey CN. RSEM: accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinformatics 12: 323, 2011. doi: 10.1186/1471-2105-12-323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Ewels P, Magnusson M, Lundin S, Käller M. MultiQC: summarize analysis results for multiple tools and samples in a single report. Bioinformatics 32: 3047–3048, 2016. doi: 10.1093/bioinformatics/btw354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Love MI, Huber W, Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol 15: 550, 2014. doi: 10.1186/s13059-014-0550-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Ritchie ME, Phipson B, Wu D, Hu Y, Law CW, Shi W, Smyth GK. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res 43: e47, 2015. doi: 10.1093/nar/gkv007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Helgadottir A, Thorleifsson G, Gretarsdottir S, Stefansson OA, Tragante V, Thorolfsdottir RB, et al. Genome-wide analysis yields new loci associating with aortic valve stenosis. Nat Commun 9: 987, 2018. doi: 10.1038/s41467-018-03252-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Yang B, Zhou W, Jiao J, Nielsen JB, Mathis MR, Heydarpour M, et al. Protein-altering and regulatory genetic variants near GATA4 implicated in bicuspid aortic valve. Nat Commun 8: 15481, 2017. doi: 10.1038/ncomms15481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Richer J, Hill HL, Wang Y, Yang ML, Hunker KL, Lane J, Blackburn S, Coleman DM, Eliason J, Sillon G, D’Agostino MD, Jetty P, Mongeon FP, Laberge AM, Ryan SE, Fendrikova-Mahlay N, Coutinho T, Mathis MR, Zawistowski M, Hazen SL, Katz AE, Gornik HL, Brummett CM, Abecasis G, Bergin IL, Stanley JC, Li JZ, Ganesh SK. A novel recurrent COL5A1 genetic variant is associated with a dysplasia-associated arterial disease exhibiting dissections and fibromuscular dysplasia. Arterioscler Thromb Vasc Biol 40: 2686–2699, 2020. doi: 10.1161/ATVBAHA.119.313885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Gallo EM, Loch DC, Habashi JP, Calderon JF, Chen Y, Bedja D, van Erp C, Gerber EE, Parker SJ, Sauls K, Judge DP, Cooke SK, Lindsay ME, Rouf R, Myers L, Ap Rhys CM, Kent KC, Norris RA, Huso DL, Dietz HC. Angiotensin II-dependent TGF-β signaling contributes to Loeys-Dietz syndrome vascular pathogenesis. J Clin Invest 124: 448–460, 2014. doi: 10.1172/JCI69666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Guo DC, Pannu H, Tran-Fadulu V, Papke CL, Yu RK, Avidan N, Bourgeois S, Estrera AL, Safi HJ, Sparks E, Amor D, Ades L, McConnell V, Willoughby CE, Abuelo D, Willing M, Lewis RA, Kim DH, Scherer S, Tung PP, Ahn C, Buja LM, Raman CS, Shete SS, Milewicz DM. Mutations in smooth muscle alpha-actin (ACTA2) lead to thoracic aortic aneurysms and dissections. Nat Genet 39: 1488–1493, 2007. [Erratum in Nat Genet 40: 255, 2008]. doi: 10.1038/ng.2007.6. [DOI] [PubMed] [Google Scholar]
- 35. Alsiraj Y, Thatcher SE, Blalock E, Fleenor B, Daugherty A, Cassis LA. Sex chromosome complement defines diffuse versus focal angiotensin II-induced aortic pathology. Arterioscler Thromb Vasc Biol 38: 143–153, 2018. doi: 10.1161/ATVBAHA.117.310035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Holmes KW, Maslen CL, Kindem M, Kroner BL, Song HK, Ravekes W, Dietz HC, Weinsaft JW, Roman MJ, Devereux RB, Pyeritz RE, Bavaria J, Milewski K, Milewicz D, LeMaire SA, Hendershot T, Eagle KA, Tolunay HE, Desvigne-Nickens P, Silberbach M; GenTAC Registry Consortium. GenTAC registry report: gender differences among individuals with genetically triggered thoracic aortic aneurysm and dissection. Am J Med Genet A 161A: 779–786, 2013. doi: 10.1002/ajmg.a.35836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Rylski B, Hoffmann I, Beyersdorf F, Suedkamp M, Siepe M, Nitsch B, Blettner M, Borger MA, Weigang E; Multicenter Prospective Observational Study. Acute aortic dissection type A: age-related management and outcomes reported in the German Registry for Acute Aortic Dissection Type A (GERAADA) of over 2000 patients. Ann Surg 259: 598–604, 2014. doi: 10.1097/SLA.0b013e3182902cca. [DOI] [PubMed] [Google Scholar]
- 38. Wilde E, Aubdool AA, Thakore P, Baldissera L Jr, Alawi KM, Keeble J, Nandi M, Brain SD. Tail-cuff technique and its influence on central blood pressure in the mouse. J Am Heart Assoc 6: e005204, 2017. doi: 10.1161/JAHA.116.005204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Kanisicak O, Khalil H, Ivey MJ, Karch J, Maliken BD, Correll RN, Brody MJ, J Lin SC, Aronow BJ, Tallquist MD, Molkentin JD. Genetic lineage tracing defines myofibroblast origin and function in the injured heart. Nat Commun 7: 12260, 2016. doi: 10.1038/ncomms12260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Fu X, Khalil H, Kanisicak O, Boyer JG, Vagnozzi RJ, Maliken BD, Sargent MA, Prasad V, Valiente-Alandi I, Blaxall BC, Molkentin JD. Specialized fibroblast differentiated states underlie scar formation in the infarcted mouse heart. J Clin Invest 128: 2127–2143, 2018. doi: 10.1172/JCI98215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Oka T, Xu J, Kaiser RA, Melendez J, Hambleton M, Sargent MA, Lorts A, Brunskill EW, Dorn GW 2nd, Conway SJ, Aronow BJ, Robbins J, Molkentin JD. Genetic manipulation of periostin expression reveals a role in cardiac hypertrophy and ventricular remodeling. Circ Res 101: 313–321, 2007. doi: 10.1161/CIRCRESAHA.107.149047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Dusaban SS, Purcell NH, Rockenstein E, Masliah E, Cho MK, Smrcka AV, Brown JH. Phospholipase C epsilon links G protein-coupled receptor activation to inflammatory astrocytic responses. Proc Natl Acad Sci USA 110: 3609–3614, 2013. doi: 10.1073/pnas.1217355110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Dusaban SS, Kunkel MT, Smrcka AV, Brown JH. Thrombin promotes sustained signaling and inflammatory gene expression through the CDC25 and Ras-associating domains of phospholipase Cϵ. J Biol Chem 290: 26776–26783, 2015. doi: 10.1074/jbc.M115.676098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Bijli KM, Fazal F, Slavin SA, Leonard A, Grose V, Alexander WB, Smrcka AV, Rahman A. Phospholipase C-ε signaling mediates endothelial cell inflammation and barrier disruption in acute lung injury. Am J Physiol Lung Cell Mol Physiol 311: L517–L524, 2016. doi: 10.1152/ajplung.00069.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Pirruccello JP, Chaffin MD, Chou EL, Fleming SJ, Lin H, Nekoui M, Khurshid S, Friedman SF, Bick AG, Arduini A, Weng LC, Choi SH, Akkad AD, Batra P, Tucker NR, Hall AW, Roselli C, Benjamin EJ, Vellarikkal SK, Gupta RM, Stegmann CM, Juric D, Stone JR, Vasan RS, Ho JE, Hoffmann U, Lubitz SA, Philippakis AA, Lindsay ME, Ellinor PT. Deep learning enables genetic analysis of the human thoracic aorta. Nat Genet 54: 40–51, 2022. doi: 10.1038/s41588-021-00962-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental Fig. S1: https://doi.org/10.6084/m9.figshare.19787476.
Supplemental Fig. S2: https://doi.org/10.6084/m9.figshare.19787437.
Supplemental Fig. S3: https://doi.org/10.6084/m9.figshare.19787494.
Supplemental Fig. S4: https://doi.org/10.6084/m9.figshare.19787497.
Supplemental Fig. S5: https://doi.org/10.6084/m9.figshare.21187738.
Supplemental Fig. S6: https://doi.org/10.6084/m9.figshare.21187804.
Supplemental Fig. S7: https://doi.org/10.6084/m9.figshare.21187822.
Data Availability Statement
Raw data for RNA sequencing are available as a supplemental file (http://doi.org/10.6084/m9.figshare.19787626).