

Abstract
Background
Cachexia‐anorexia syndrome is a complex metabolic condition characterized by skeletal muscle wasting, reduced food intake and prominent involvement of systemic and central inflammation. Here, the gut barrier function was investigated in pancreatic cancer‐induced cachexia mouse models by relating intestinal permeability to the degree of cachexia. We further investigated the involvement of the gut–brain axis and the crosstalk between tumour, gut and hypothalamus in vitro.
Methods
Two distinct mouse models of pancreatic cancer cachexia (KPC and 4662) were used. Intestinal inflammation and permeability were assessed through fluorescein isothiocyanate dextran (FITC‐dextran) and lipopolysaccharide (LPS), and hypothalamic and systemic inflammation through mRNA expression and plasma cytokines, respectively. To simulate the tumour–gut–brain crosstalk, hypothalamic (HypoE‐N46) cells were incubated with cachexia‐inducing tumour secretomes and LPS. A synthetic mimic of C26 secretome was produced based on its secreted inflammatory mediators. Each component of the mimic was systematically omitted to narrow down the key mediator(s) with an amplifying inflammation. To substantiate its contribution, cyclooxygenase‐2 (COX‐2) inhibitor was used.
Results
In vivo experiments showed FITC‐dextran was enhanced in the KPC group (362.3 vs. sham 111.4 ng/mL, P < 0.001). LPS was increased to 140.9 ng/mL in the KPC group, compared with sham and 4662 groups (115.8 and 115.8 ng/mL, P < 0.05). Hypothalamic inflammatory gene expression of Ccl2 was up‐regulated in the KPC group (6.3 vs. sham 1, P < 0.0001, 4662 1.3, P < 0.001), which significantly correlated with LPS concentration (r = 0.4948, P = 0.0226). These data suggest that intestinal permeability is positively related to the cachexic degree. Prostaglandin E2 (PGE2) was confirmed to be present in the plasma and PGE2 concentration (log10) in the KPC group was much higher than in 4662 group (1.85 and 0.56 ng/mL, P < 0.001), indicating a role for PGE2 in pancreatic cancer‐induced cachexia.
Parallel to in vivo findings, in vitro experiments revealed that the cachexia‐inducing tumour secretomes (C26, LLC, KPC and 4662) amplified LPS‐induced hypothalamic IL‐6 secretion (419%, 321%, 294%, 160%). COX‐2 inhibitor to the tumour cells reduced PGE2 content (from 105 to 102 pg/mL) in the secretomes and eliminated the amplified hypothalamic IL‐6 production. Moreover, results could be reproduced by addition of PGE2 alone, indicating that the increased hypothalamic inflammation is directly related to the PGE2 from tumour.
Conclusions
PGE2 secreted by the tumour may play a role in amplifying the effects of bacteria‐derived LPS on the inflammatory hypothalamic response. The cachexia‐inducing potential of tumour mice models parallels the loss of intestinal barrier function. Tumour‐derived PGE2 might play a key role in cancer‐related cachexia‐anorexia syndrome via tumour–gut–brain crosstalk.
Keywords: Hypothalamic inflammation, Prostaglandin, Gut–brain axis, Cancer, Cachexia
Introduction
The cachexia‐anorexia syndrome presents as an unmet clinical complication of cancer, contributing to increased morbidity and mortality, and reduced quality of life. Cancer‐related cachexia is characterized by increased systemic inflammation. 1 One important feature is inflammation of the hypothalamus, which drives sickness behaviour during cachexia. We and others have previously shown that hypothalamic inflammation plays an important role in the development of cancer cachexia. 2
The hypothalamus plays a key role in regulating the autonomic nervous system that controls the body's homoeostasis and plays a critical role in the development of sickness behaviour. The cachexia‐anorexia syndrome shows resemblance to a prolonged sickness behaviour response. As a response to cancer‐induced systemic inflammation, the hypothalamus produces pro‐inflammatory cytokines that are thought to affect the activity of neuroendocrine circuits. 3 This inflammatory response of the hypothalamus is known to stimulate the hypothalamic–pituitary–adrenal axis. 4 Stimulation of the hypothalamic–pituitary–adrenal axis can in turn lead to an increased release of adrenocorticotropic hormone by the pituitary gland. Consequently, the secretion of glucocorticoids by the adrenal cortex is elevated and stimulates muscle catabolism via activation of the glucocorticoid receptor. 5 Additionally, hypothalamic serotonin increases during inflammation. It will inhibit the activity of orexinergic neurons and result in decreased appetite. Moreover, hypothalamic serotonin has been related to fatigue. 6 Together, hypothalamic inflammation is thus suggested to provoke loss of skeletal muscle mass via increasing muscle catabolism and indirectly via decreasing appetite and increasing fatigue. 4
This study focuses on the question how intestinal permeability affects hypothalamic inflammation during cancer. Our initial hypothesis was that tumour‐induced systemic inflammation can be sensed and amplified by the hypothalamus, whereas in parallel, the elevated systemic inflammatory status increases the intestinal permeability, leading to more pathogen‐associated molecular patterns (PAMPs) like lipopolysaccharide (LPS) entering the circulation. PAMPs can be sensed by the hypothalamus and contribute to the amplification of hypothalamic inflammation. 7 The studies described here aim to explore which inflammatory mediators play a role in the activity of the gut–brain axis during cancer cachexia. For this purpose, two well‐characterized pancreatic cancer mouse models inducing either high or low cachexia were used. KPC and 4662 are mouse pancreatic cancer cell lines developed from the same original cell line but differ in their cachexia‐inducing capacity. 8 , 9 Specifically, the 4662 cell line induces less severe cachexia in mice compared with the KPC cell line. 8 DSS was used as a positive control for colonic inflammation and increased intestinal permeability. To investigate whether the difference in the cachexia‐inducing effect of the two different pancreatic cell lines is related to the crosstalk between the gut and the hypothalamus, biomarkers for intestinal permeability and hypothalamic inflammation were measured.
Moreover, inflammatory mediators involved in the crosstalk between tumour, gut and hypothalamus were identified using an in vitro hypothalamic inflammatory challenge model based on the effect of combinations of PAMPs and different tumour secretomes.
Methods
Experimental animals and design
Animals
Male and female 18–27 g WT C57BL/6J (stock no. 000664) were purchased from Jackson Laboratories. Animals were aged between 9 and 13 weeks at the time of study and maintained at 28°C. The housing and data collection of mice were performed as previously described. 8 Experiments were conducted in accordance with the National Institutes of Health Guide for the Care and Use of Laboratory Animals and approved by the Animal Care and Use Committee of Oregon Health & Science University. This animal experiment was an exploratory trial. For the KPC and 4662 model, it was not described previously what intestinal parameters would be during cachexia. Therefore, interpretation of intestinal outcomes was restricted to the presence of cachexia (loss of body and muscle mass) in the KPC model. This meant that the cachectic parameters as shown in Figure 1 were the primary parameters and the other outcomes were secondary.
Figure 1.
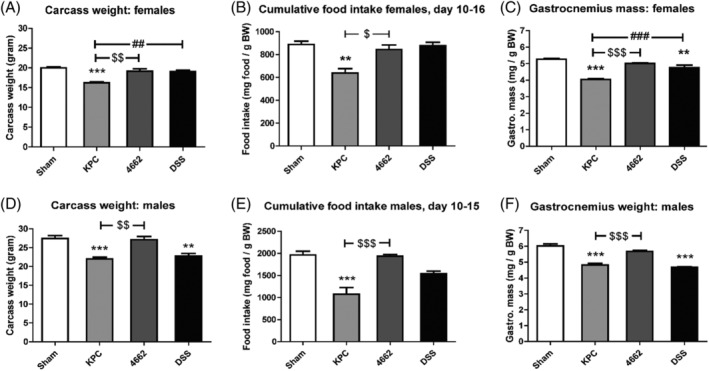
Carcass weight, food intake change and gastrocnemius weight in 4662‐induced and KPC‐induced cachexia. Carcass weight on day 16 in female mice (A) and male mice (D). Cumulative food intake during cachexia phase in female mice (B) and male mice (E). Gastrocnemius weight on day 16 in female mice (C) and male mice (F). Carcass weight was calculated by subtracting tumour weight from body weight on day of sacrifice. Gastrocnemius weight was related to initial body weight. For female mice, n = 3, 4, 4 in sham, KPC and 4662 groups, respectively. For male mice, n = 6, 4, 4 in sham, KPC and 4662 groups, respectively. *P < 0.05, ** P < 0.01, *** P < 0.001 compared with sham; $ P < 0.05, $$ P < 0.01, $$$ P < 0.001 compared with 4662; # P < 0.05, ## P < 0.01, ### P < 0.001 compared with DSS. One‐way ANOVA analysis of variance with post hoc Bonferroni's multiple comparison test.
Tumour cell culture implantation
C57BL/6 mice were inoculated orthotopically into the tail of the pancreas with either 1 million tumour cells in 40 μL PBS or an equal volume of cell‐free PBS. Tumour growth and implantation were performed as previously described. 8
The dextran sodium salt model as positive control
An animal model of dextran sodium salt (DSS‐)colitis was used as a positive control for colonic inflammation and increased intestinal permeability. Sham‐operated animals received 2.5% DSS in their drinking water for a total of 9 days, starting 1 week after their operation. On days 15 and 16, they were euthanized as described earlier. Animals receiving DSS exhibit weight loss, loose stool/diarrhoea and sometimes signs of rectal bleeding.
Intestinal permeability
Intestinal permeability was assessed by determining the transmucosal transport of 4000‐Da fluorescein isothiocyanate (FITC‐)dextran (Sigma‐Aldrich, St. Louis, MO), according to Johnson et al. 10 Mouse plasma LPS was measured by LPS ELISA (MyBioSource, San Diego, CA). We validated the kit in advance with plasma of additional sham‐operated control mice that were given an IP dose of LPS (delivered with 0.5% BSA as a carrier) or polyinosinic acid–polycytidylic acid (poly I:C) 2 h before being euthanized to test for the sensitivity and specificity of the kit. In the animal trial, the primary outcomes were the intestinal permeability‐related outcomes. Other parameters were secondary.
Blood and tissue collection
When the tumour‐bearing mice reached predesigned time points or predetermined criteria for euthanasia, they were given a gavage, and 4 h later, they got a dose of ketamine–xylazine–acepromazine cocktail to induce deep anaesthesia. Necropsy tissue including tumour, gastrocnemius, colon, cecum, hypothalamus, and plasma were collected as previously described. 11
Quantitative real‐time PCR
Intestinal and hypothalamic RNA was extracted using the RNeasy mini kit (Qiagen, Hilden, Germany) following the manufacturer's instructions. With use of TaqMan reverse transcription reagents and random hexamers, cDNA was transcribed. qPCR was run on an ABI 7300 (Applied Biosystems, Foster City, CA), with use of TaqMan universal PCR master mix and the following TaqMan mouse gene expressions assays (selection based on previous findings by our lab and others, which indicated importance in sickness behaviour, intestinal integrity and inflammation). Integrity and permeability genes in intestine: Tjp1, Cldn1, Muc2 and Ocln. Inflammatory response genes in the brain, such as Il1b, Lif, Cxcl2, Selp, Ccl2 (MCP‐1) and Il6, were compared between tumour‐bearing and sham‐operated mice by qRT‐PCR. The results were normalized to tissue‐appropriate control gene 18s (primers used are shown in Table S2).
The relative expression was calculated with the ΔΔCt method and normalized to the sham control. Normally distributed ΔCt values were used for the statistical analysis.
In vitro studies
Reagents
Lipopolysaccharide (O111:B4; LPS) was purchased from Sigma‐Aldrich (Schnelldorf, Germany). Recombinant murine IL‐6, CCL2 (MCP‐1) and LIF were from PeproTech (London, UK). Prostaglandin E2 and celecoxib were purchased from R&D Systems (Abingdon, UK).
Cells and culture
The mouse hypothalamic (mHypoE‐N46) cell line was purchased from CELLutions Biosystems (Burlington, Ontario, Canada). The mouse brain microglia (BV2) cell line was purchased from Banca Biologica e Cell Factory (Genova, Italy). The murine colon carcinoma (C26) cell line and Lewis lung carcinoma (LLC) cell line were purchased from ATCC (Teddington, UK). The mHypoE‐N46, C26 and LLC cells were grown in Dulbecco's Modified Eagle Medium (DMEM, Lonza, Verviers, Belgium); BV2 cells were grown in Roswell Park Memorial Institute 1640 medium (RPMI‐1640). The murine pancreatic cancer cell lines were generously provided by Dr Elizabeth Jaffee (KPC) and Robert Vonderheide (4662). 8 Both KPC and 4662 were cultured in low‐glucose DMEM (Gibco, Thermo Fisher, Waltham, MA). All media were supplemented with 10% foetal calf serum (FCS, Biowest, Ann Arbor, MI, USA) and 1% penicillin–streptomycin (Lonza, Verviers, Belgium) at 37°C in a humidified atmosphere with 5% CO2.
ELISA
Levels of IL‐6, LIF, CCL2 (MCP‐1) (all R&D Systems, Minneapolis, MN, USA) and PGE2 (Cayman Chemical, Ann Arbor, MI, USA) in cell culture media were detected using mouse ELISA kits according to manufacturer's instructions. All presented ELISA data were within the range of the respective standard curves. The samples were diluted to fall within the range of the standard curve when necessary.
Statistics analysis
All reported in vitro studies were averages of three or more independent experiments. Data were presented as mean ± SEMs. Statistical analysis was performed with Prism 5.0 (GraphPad). Differences between groups were assessed with one‐way or two‐way analysis of variance (ANOVA), followed by Bonferroni multiple comparison test. All data were checked for normality using the Shapiro–Wilk normality test. Significance was defined as a P value of less than 0.05. Spearman's correlation was conducted for in vivo experiments to compare correlations of both plasma cytokine levels and colon Muc2 gene expression with hypothalamic inflammatory genes expression.
Results
KPC allografts induced more intestinal permeability than 4662 allografts
First, we confirmed the KPC model's advanced cachexia status compared with the 4662 model. Figure 1A and 1D shows significant differences in carcass weights between KPC and sham (P < 0.001), and KPC and 4662 (P < 0.01). The loss in body mass might be explained by a decrease in food intake (Figure S1). Food intake during the final 6 days was reduced in KPC mice compared with sham mice (P < 0.05), whereas food intake did not differ between sham and 4662 mice (Figure 1B and 1E). Moreover, gastrocnemius weights in both female (Figure 1C) and male (Figure 1F) KPC mice were decreased compared with sham mice (P < 0.0001), whereas for 4662 mice, only a small significant decrease in female mice was present (P < 0.05). These findings suggest a profound cachexia phenotype in KPC mice and less clear signs of cachexia in 4662 mice.
We analysed macroscopic markers of intestinal health to determine if they differed between the different cachexia phenotypes KPC, 4662, when compared with sham and DSS‐injected mice as a positive control for intestinal inflammation. KPC, 4662 and DSS mice showed a reduced length of the colon compared with sham (Figure 2A and 2C) (P < 0.001, P < 0.01 and P < 0.001, respectively). Additionally, KPC, DSS and male 4662 showed significantly lower cecum mass weights (Figure 2D) (P < 0.0001), whereas in females, the cecum content weight between KPC and 4662 differed significantly (Figure 2B) (P < 0.05). Also in males, there was a significant difference in colon length between KPC and 4662 (58.4 ± 1.5 mm vs. 73.25 ± 2.93, P = 0.0025) and cecum content weight (5.87 ± 1.21 vs. 11.83 ± 0.65 mg/g BW, P = 0.0015) (Figure 2C and 2D). Upon macroscopic examination, the cecum of KPC appeared to be smaller and to have a thinner intestinal wall (cecum tissue weight), compared with control and 4662 (Figure 2E). These data indicate that the intestinal response to the KPC tumour is comparable with the acute DSS‐induced colitis colonic inflammation, whereas the response to 4662 is less severe.
Figure 2.
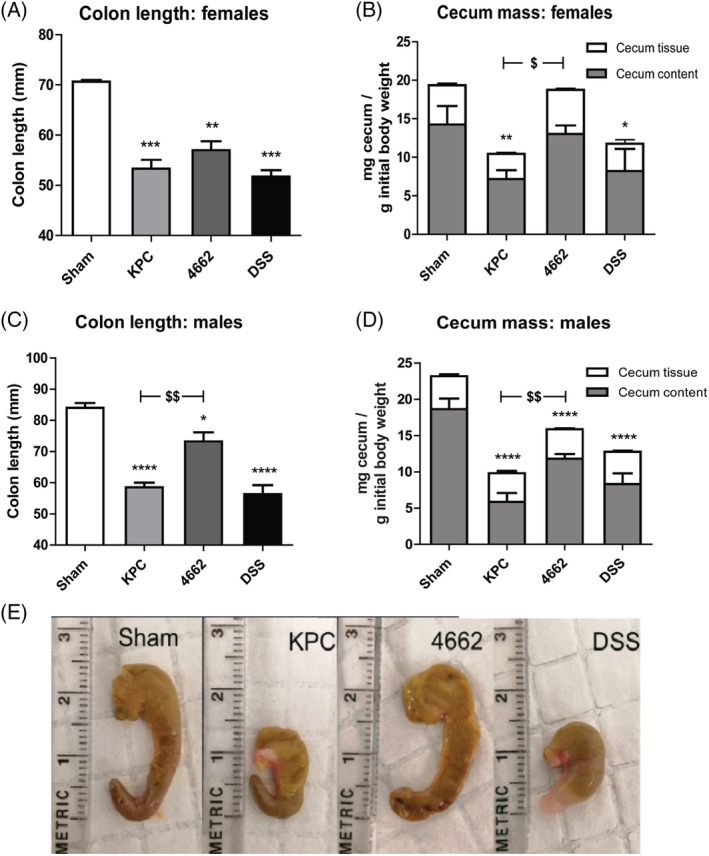
Colon length, cecum mass change and gross appearance of the cecum in sham, KPC, 4662 and DSS mice. For female mice, n = 3, 3, 4 in sham, KPC and 4,662 groups respectively. For male mice, n = 6, 4, 4 in sham, KPC and 4,662 groups respectively. *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001 compared with sham; $ P < 0.05, $$ P < 0.01 compared with 4662. One‐way ANOVA analysis of variance with post hoc Bonferroni's multiple comparison test.
Next, we evaluated the degree of systemic inflammation induced by KPC and 4662 allografts. Consistent with the macroscopic findings, we observed enhanced intestinal permeability in KPC mice, as measured by FITC‐dextran concentrations in the plasma (Figure 3A), with the intestinal permeability being more prominent in the KPC group (KPC 362.3 ± 48.7 ng/mL vs. 111.4 ± 38.56 in sham, P < 0.001). Trace levels of LPS were detectable in the plasma of control mice, and similar levels were detected in 4662 and DSS mice. LPS was significantly increased in KPC group (140.9 ± 8.6 ng/mL) compared with sham, 4662 and DSS (115.8 ± 2.9, 115.8 ± 1.8 and 107.5 ± 3.9; P < 0.05, P < 0.05, P < 0.01, respectively) (Figure 3B). Plasma IL‐6 was increased 4.3‐fold in KPC (P < 0.01) and 3.9‐fold in the 4662 group compared with the sham group (Figure 3C). Biomarkers of intestinal integrity and permeability were affected during PDAC in colon. As shown in Figure 3D, several transcripts of genes involved in intestinal integrity in the colon were significantly lower in the KPC group, compared with sham; Muc2 (P = 0.0411), Ocln (P = 0.0362) and Tjp1 (P = 0.0241). Cldn1 was not significantly affected in the KPC group. The DSS group did not show significant changes compared with sham. One possible reason is that the DSS group was given a relatively low concentration of DSS, which induced phenotype changes (Figure 2 ) but fewer changes in intestinal permeability and inflammation.
Figure 3.
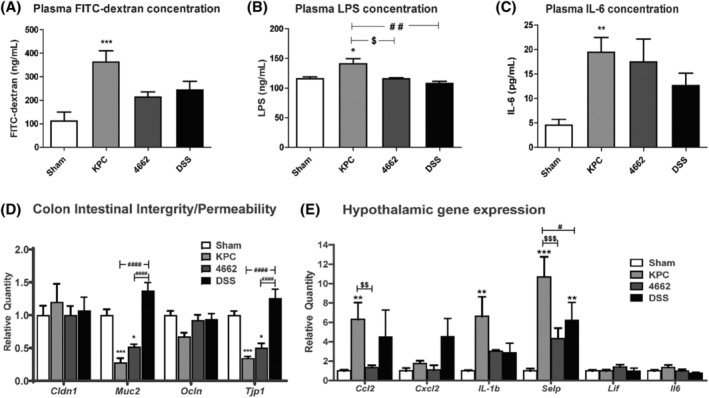
(A) Intestinal permeability (barrier dysfunction) as measured by FITC‐dextran, (B) endotoxin (LPS) concentration in the plasma and (C) plasma IL‐6 concentration (marker for systemic inflammation). Serum fluorescein dextran concentrations (λex = 485; λem = 535 nm) were measured 4 h after gavage. (D) qRT‐PCR analysis of biomarkers involved in intestinal permeability and integrity of tight junctions and mucous barrier (Cldn1, Muc2, Ocln and Tjp1) and (E) qRT‐PCR analysis of inflammatory cytokine and chemokine transcripts in the hypothalamus in KPC and 4662 PDAC‐bearing animals at 15–16 days after orthotopic implantation. mRNA levels are relative to sham group. N = 7–9, 7–8, 6–7, 4–6 in sham, KPC, 4662 and DSS groups, respectively. *P < 0.05, **P < 0.01, ***P < 0.001 compared with sham; $ P < 0.05, $$P < 0.01, $$$P < 0.001 compared with 4662. # P < 0.05, ## P < 0.01, ### P < 0.001, #### P < 0.0001 compared with DSS. One‐way ANOVA in (A, B and C) analysis of variance with post hoc Bonferroni's multiple comparison test. Two‐way ANOVA in (D and E) analysis of variance with post hoc Bonferroni's multiple comparison test.
Finally, hypothalamic inflammatory gene expression was tested. We previously showed an up‐regulation of inflammatory cytokine and chemokine transcripts in multiple brain regions important for behaviour and metabolism in the PDAC mouse model. 9 , 12 Here, we performed qRT‐PCR at the time when the animals reliably showed signs of anorexia and body weight loss (around 10 days). We looked at inflammatory markers that are up‐regulated in the hypothalamus during chronic systemic inflammation. In accordance with the previous studies, we found up‐regulations in the KPC group of Ccl2 (P < 0.01), Il‐1b (P < 0.01) and Selp (P < 0.001) and no significant changes in regulation for Cxcl2, Lif and Il6. The transcript of Ccl2, a chemokine associated with monocyte infiltration, was highly upregulated in the hypothalamus in the KPC condition (6.3 vs. sham 1, P < 0.01), but not in 4662. The Ccl2 expression in the DSS group also showed an upward trend (4.5 vs. sham 1, P = 0.1725) similar to that of the KPC group. Besides, gene expression of Ccl2 in the KPC group was significantly correlated with LPS concentration (r = 0.4948, P = 0.0226). Selp, a gene encoding P selectin, has been linked to the development of cancer cachexia. The transcript of Selp was significantly upregulated in both KPC group (10.7 vs. sham 1, P < 0.001) and DSS group (6.2 vs. sham 1, P < 0.01), but not in 4662. Taken together, these data suggest hypothalamic inflammatory gene expression was higher in the KPC condition than that in the 4662 condition. As a control, the level of inflammation triggered by DSS was intermediate between these two groups, suggesting that KPC allografts induce more intestinal permeability and systemic inflammation than 4662 allografts.
Secretome of cachexia‐inducing tumour cell lines amplified LPS‐induced hypothalamic inflammation
To imitate the influence of decreased integrity of the gut, a sub‐inflammatory level (31.6 ng/mL) of LPS was added to hypothalamic (HypoE‐N46) cells. This sub‐inflammatory level was set at the level at which microglial cells like BV2 did not respond yet (Figure 4B), whereas the hypothalamic cells did (Figure 4A). IL‐6 was secreted by the hypothalamic cells at these levels, whereas the glial BV2 cells did not respond. To investigate the synergistic interaction between tumour factors and LPS, hypothalamic cells were incubated with the secretome of several different cachexia‐inducing tumour cell lines. Figure 4C shows that a low concentration of LPS (31.6 ng/mL) or the addition of different tumour secretomes (C26 colon carcinoma (C26), Lewis lung cancer cells (LLC), KPC and 4662) induced secretion of IL‐6 in the hypothalamic cells. The combination of LPS with either one of the secretomes resulted in an amplified inflammatory response in the hypothalamic cells compared with LPS incubation (P < 0.0001, P < 0.001, P < 0.01, respectively). For C26, for example, the reaction was amplified up to 400%.
Figure 4.
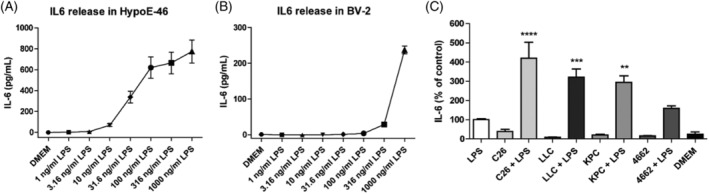
(A) IL‐6 secretion of mHypoE‐N46 cells incubated with different concentrations LPS (0–1000 ng/mL). (B) IL‐6 secretion of BV2 cells incubated with different concentrations LPS (0–1000 ng/mL). (C) IL‐6 secretion of mHypoE‐N46 cells incubated with LPS‐supplemented culture medium (LPS), C26, LLC, KPC and 4,662 tumour secretome (C26, LLC, KPC, 4662) and combined with LPS‐supplemented culture medium (+LPS) and regular culture medium (DMEM). The IL‐6 values are given as mean concentration in pg/mL (A,B) and corrected for total protein concentration and presented as percentage of LPS (%). *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001. One‐way ANOVA analysis of variance with post hoc Bonferroni's multiple comparison test.
Although both the KPC and 4662 cell lines were shown to induce an IL‐6 release, which was increased by addition of a low concentration of LPS; compared with 4662, the secretome of cachexia‐prone KPC cells induced a more significant amplification of IL‐6 secretion when combined with LPS, resulting in an amplification of up to 300%.
C26 tumour secretome mimic significantly amplified IL‐6 release of hypothalamic cells incubated with a low level of LPS
The C26 secretome resulting in the highest effect was analysed for the presence of inflammatory cytokines described to be involved in cachexia. Based on the 72‐h secretion profile, a C26 tumour medium mimic (C26 Mic) was designed with 110 pg/mL IL‐6, 100 pg/mL LIF, 30000 pg/mL CCL2 and 6500 pg/mL PGE2. To further explore which tumour factors were involved in the amplified hypothalamic inflammation, different mimics were tested in which one individual factor (IL‐6, LIF, CCL2 and PGE2, respectively) was omitted. The complete mimic and the mimics in which IL‐6, LIF or CCL2 were omitted showed similar results as the C26 tumour secretome. Surprisingly, the mimic without PGE2 combined with LPS showed attenuated secretion (P < 0.0001). This response was almost equal to the IL‐6 secretion induced by only LPS (Figure 5A). The possible role of PGE2 was confirmed by the concentrations as measured later in the different tumour secretomes, relatively C26 30.6 ng/mL, LLC 5.4 ng/mL, KPC 8.5 ng/mL and 4662 1.7 ng/mL after 96‐h incubation.
Figure 5.
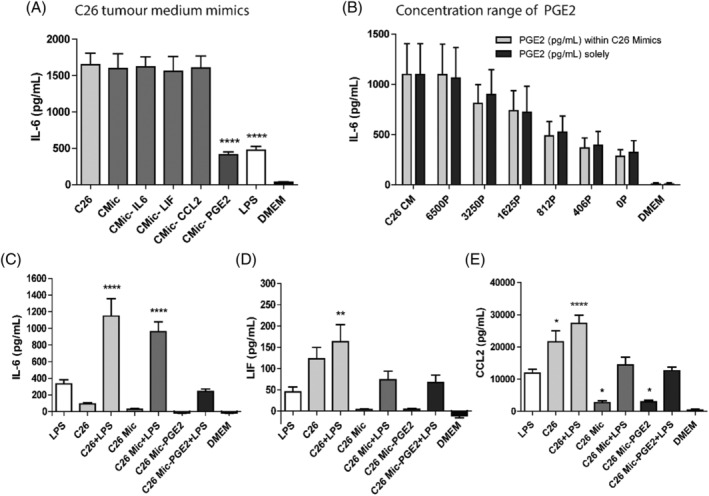
C26 tumour secretome mimic results in inflammatory responses in hypothalamic cells. (A) IL‐6 secretion of mHypoE‐N46 cells incubated with C26 tumour secretome (C26) and variations on the C26 tumour secretome mimic, combined with LPS‐supplemented culture medium (+LPS), compared with the positive control (C26). (B) IL‐6 secretion of mHypoE‐N46 cells incubated with different concentrations PGE2 (6500–0 pg/mL), combined with LPS. The different concentrations were either provided to the cells in regular culture medium (PGE2 solely) or culture medium containing the other C26 mimic components (PGE2 within C26 mimics). IL‐6 (C), LIF (D) and CCL2 (E) secretion of mHypoE‐N46 cells incubated with C26 tumour secretome or a C26 mimic, in the presence or absence of LPS, compared with the positive control (LPS). *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001. One‐way ANOVA analysis of variance with post hoc Bonferroni's multiple comparison test.
Subsequently, it was investigated if IL‐6 secretion was dependent on the PGE2 concentration. A range of 0–6500 pg/mL was tested. The maximum concentration resembled the PGE2 concentration in the C26 tumour secretome, which was the highest of the different tumour secretomes. The different PGE2 concentrations were either provided to the cells in regular culture medium (DMEM) or culture medium containing the other three components of the C26 mimic (IL‐6, LIF or CCL2) to confirm that the other compounds of the C26 mimic had no effect on IL‐6 secretion from hypothalamic cells. In both conditions, a clear dose dependency was observed and these dose response curves for IL‐6 secretion were similar when PGE2 was provided solely or as part of the mimic. Moreover, the IL‐6 secretion induced by the highest PGE2 concentration was similar to that of the C26 tumour secretome (Figure 5B). Collectively, these data indicate that PGE2 is an essential factor for amplifying hypothalamic inflammation in vitro.
To investigate if other inflammatory compounds were influenced in a similar way, the effect of the secretome and the mimic on secretion of other pro‐inflammatory mediators was investigated. The LIF (Figure 5D) and CCL2 (Figure 5E) release of hypothalamic cells showed no or less additive effects of the C26 tumour secretome and LPS. Moreover, LIF and CCL2 production were both not influenced by the mimic, indicating that for secretion of these inflammatory mediators, different pathways might be involved.
To further investigate the involvement of different inflammatory compounds, gene expression after different incubation times was investigated. In Figure 6 the relative expression of Il6, Ccl2, Cxcl2 and Selp were shown after 24 and 72 h of incubation. The amplifying effect of Il6 (Figure 6A) with combination of PGE2 and LPS (P < 0.01) was confirmed on mRNA expression level, both at 24 and 72 h. For Selp, the amplifying effect was present at 24 h in both KPC medium (P < 0.05) and PGE2 (P < 0.01) as a mimic when combined with LPS, and decreased after 72 h. For Ccl2 and Cxcl2, the amplifying effect was not significant after 72 h.
Figure 6.
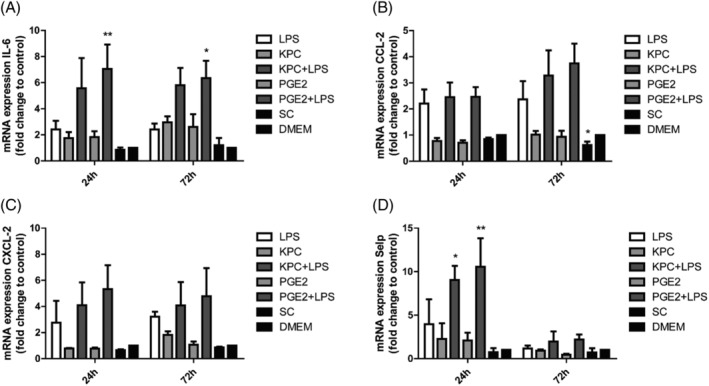
Relative expression of pro‐inflammatory genes by mHypoE‐N46 cells in reaction to KPC tumour medium or a matched concentration of PGE2 combined with LPS after 24‐h and 72‐h treatment. mRNA expression levels (A) Il6, (B) Ccl2, (C) Cxcl2 and (D) Selp were normalized to β‐actin and standardized to the negative control DMEM, compared with the positive control (LPS). *P < 0.05, **P < 0.01. Two‐way ANOVA analysis of variance with post hoc Bonferroni's multiple comparison test.
COX‐2 inhibition reduced inflammatory amplification effect of hypothalamic cells
Tumour cells could produce PGE2 via the enzyme COX‐2. Accordingly, we hypothesized that the tumour cells would secrete less PGE2 in the presence of the COX‐2 inhibitor celecoxib (CXB). The C26, LLC, KPC and 4662 tumour cells were cultured with or without CXB and optimal concentration was determined (Table S1 and Figure S2).
Hypothalamic cells were exposed to medium of tumour cells grown with CXB and to medium of the same tumour grown without CXB, but supplemented afterwards with CXB to discriminate between an effect caused by a reduced PGE2 production of the cancer cells, compared to a direct effect of CXB on the hypothalamic cells. All tumour secretomes amplified the IL‐6 secretion when combined with LPS (P < 0.001), whereas the corresponding tumour medium grown with CXB induced less IL‐6 secretion both in the absence and presence of LPS. If the secretome was taken from the regular tumour cells and then combined with celecoxib afterwards, no effect of celecoxib on hypothalamic IL‐6 production was measured. Additionally, when hypothalamic cells were incubated with CXB and LPS, the IL‐6 release was almost the same as isolated LPS treatment. These results indicate that CXB inherently has no effect on hypothalamic cells but decreases IL‐6‐induced inflammation via inhibition of PGE2 production by the tumour cells (Figure 7A).
Figure 7.
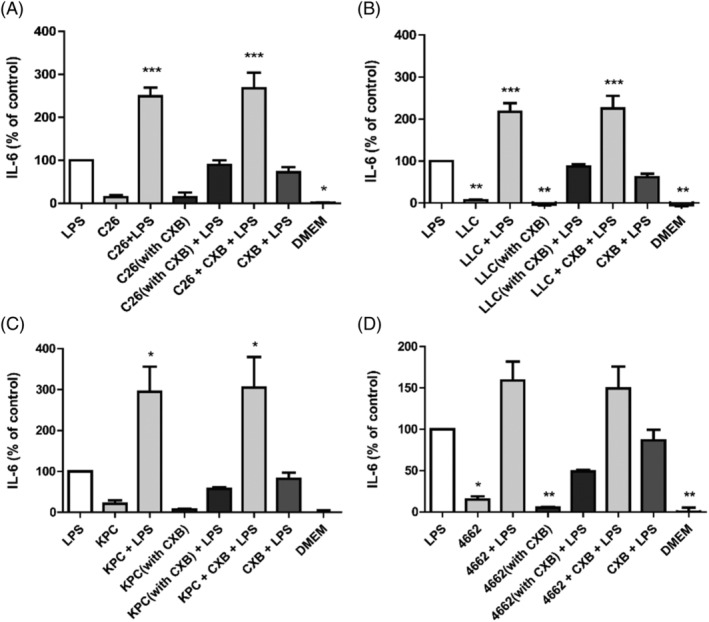
IL‐6 secretion of mHypoE‐N46 cells incubated with the tumour secretome of C26 cells (A), LLC cells (B), KPC cells(C) or 4662 cells (D), which were cultured with CXB. Hypothalamic cells were incubated with these medium combined with culture medium as well as LPS. As a control, cells were incubated with tumour secretome to which CXB was added after removal of the tumour cells combined with LPS (tumour med + CXB + LPS). Other control conditions included culture medium supplemented with LPS and CXB (LPS + CXB), culture medium supplemented with LPS (LPS) and regular culture medium (DMEM), compared with the positive control (LPS). *P < 0.05, **P < 0.01, ***P < 0.001. One‐way ANOVA analysis of variance with post hoc Bonferroni's multiple comparison test.
The magnitude of the effects resembled the levels of PGE2 produced by the tumour cells. Accordingly, the secretome of KPC cells resulted in an inflammatory response that was 300% larger than that of LPS (Figure 7C), whereas 4662 only amplified the response by around 160% (Figure 7D).
Finally, the PGE2 concentration was identified in animal models. As with prior studies, the KPC model induced more cachexia than the 4662 model with decreased food intake (P < 0.05), and lower gastrocnemius weight (P < 0.001). Tumour weight in KPC model was significantly increased compared with 4662 model (7.08 vs. 3.59% of baseline, P < 0.05). Moreover, it was confirmed that PGE2 was present in the plasma. PGE2 concentration in KPC group was 136.3 ng/mL, which was higher than in the control group (50.76 ng/mL), and up to 10‐fold compared with PGE2 in 4662 group (10.93 ng/mL) (Figure 8D). This provides further support for a role for PGE2 in pancreatic cancer‐induced cachexia.
Figure 8.
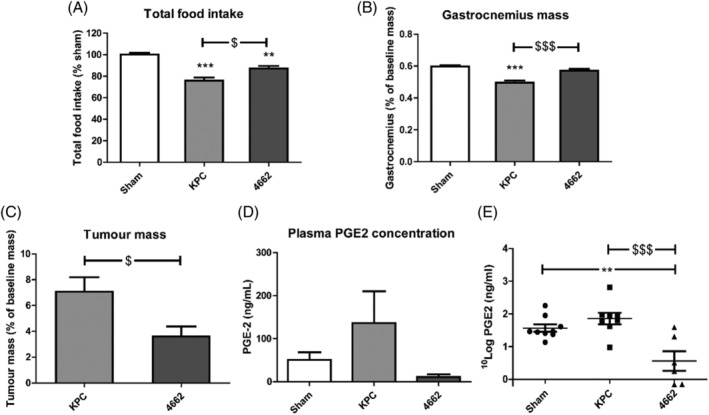
Total food intake, gastrocnemius and tumour weight and plasma PGE2 concentration in KPC and 4662 mice models. (A) Total food intake normalized to sham control group. Gastrocnemius mass (B) and tumour mass (C) normalized to initial body weight. The mice data for 4662 of total food intake and gastrocnemius mass in (A) and (B) has been published. 8 Terminal PGE2 concentration (D, E) in the plasma of mice models. N = 9, 8, 6 in sham, KPC and 4,662 groups respectively. *P < 0.05, **P < 0.01, ***P < 0.001 compared with sham; $ P < 0.05, $$P < 0.01, $$$ P < 0.001 compared with 4662. One‐way ANOVA analysis of variance with post hoc Bonferroni's multiple comparison test.
Discussion
Cachexia is a multi‐organ syndrome that is accompanied by systemic inflammation, as demonstrated in multiple clinical studies. 13 , 14 Several papers indicate that this inflammation is related to increased intestinal permeability 15 , 16 and acute sickness syndrome. 3 This suggests that as cachexia progresses, crosstalk between intestinal derived bacterial and tumour‐derived pro‐inflammatory factors might stimulate hypothalamic inflammation. Therefore, understanding the central role of the gut–brain axis in cancer cachexia 3 is of great interest. Herein, we observe that the degree of cancer cachexia is positively correlated with the sickness behaviour and systemic inflammation and remarkably also with increased gut permeability and LPS levels. According to the results from in vitro models, a combination of the tumour secretome and low levels of LPS significantly induced a hypothalamic inflammatory amplification. This amplification was mainly associated with PGE2 secreted by the tumour. In addition, the COX‐2 inhibitor celecoxib decreased this amplification. Although cancer cachexia is probably sum of different inflammatory pathways, here in our study, we present an important link between intestinal permeability and hypothalamic inflammation and suggest that the combination of PAMPs with prostaglandins plays a key role in the tumour–gut–brain crosstalk during cancer cachexia.
Two subtypes of a mouse model of pancreatic ductal adenocarcinoma (PDAC) were used to induce either a high degree (KPC) or a low degree of cachexia (4662). The KPC model is extensively characterized and has many features relevant to human disease. This is due in part to orthotopic implantation of these tumour cells into the pancreas, which has a few unique advantages: The tumours are organ specific, grow relatively large and are clinically pertinent, which is important to accurately model the tumour biology. 17 The KPC model also induces several important signs and symptoms including muscle catabolism and of central nervous system dysfunction as observed in humans, including muscle catabolism, in particular anorexia and lethargy. 3 , 11 , 12 Although the 4662 is obtained from the same tumour cell line as KPC, it induces a less severe cachexia phenotype. 8 The low‐level DSS‐induced moderate colitis model was used as a positive control to verify the degree of changes in intestinal phenotype and intestinal permeability in the KPC and 4662 models. KPC showed more profound intestinal changes than the model compound DSS, whereas 4662 showed milder phenotypic changes and less intestinal inflammatory changes compared with DSS. This suggests that (1) a difference in intestinal phenotypic changes and intestinal permeability might contribute to the difference between the cachexia found in KPC compared with 4662 and (2) that inflammation induced by gut‐derived bacterial compounds might play a role in this. These data support the reports of a diminished intestinal barrier function in two different mouse models of colon cancer cachexia and leukaemic mice with cachexia. 18 For colorectal and pancreatic cancer patients, there are some papers supporting changes in intestinal function and integrity, with emphasis on changes in microbiota composition and intestinal permeability. 19 , 20 Additionally, significantly higher hypothalamic inflammation in the KPC mice was observed, compared with 4662 and sham. This is the first time that intestinal health in low and high cachexia has been compared and linked to hypothalamic‐driven sickness behaviour. We suspect that there is an intricate and coordinated interaction between gut barrier function and cancer cells, probably mediated via tumour‐induced mediators, that causes an amplified hypothalamic inflammation and thus a more severe cachexia phenotype. It needs further investigation whether this crosstalk between gut and hypothalamus is linked to the appearance of anorexia. In this light, it is interesting if this interaction is related to one of the recently described anorexia‐related hypothalamic signalling pathways like Dilp8/INSL3‐Lgr3 21 and lipocalin2/MCR4 signalling. 8
To study the crosstalk between the hypothalamus and the gut in detail, an in vitro model was set up to mimic the gut–brain axis by testing on hypothalamic cells the effect of gut‐derived bacterial compounds in the presence of various types of cachexia‐inducing tumour secretomes. Starting from the theory that it is a signal initiating from the hypothalamus, it was tested which concentration of LPS still stimulated an inflammatory response in both hypothalamic neuronal cells and microglia cells. It turned out that the neuronal cells were most sensitive to LPS. This low concentration of LPS was used to imitate the effects of increased intestinal permeability (LPS leaking through tight junctions and into the bloodstream) on the hypothalamus. Four tumour cell lines that can induce different cachectic levels were used to examine the effect of different cancer types on the hypothalamus. Interestingly, we found a significant amplification of IL‐6 release by hypothalamic cells with all high‐cachexia‐associated tumour media combined with LPS. IL‐6 production in the brain upon peripheral infection has been reported to play a role in the development of sickness behaviour. 22 Additionally, IL‐6 was confirmed in a mouse tumour model to disrupt blood–brain barrier (BBB) permeability, which may further contribute to the transport of PGE2 across the BBB. 23 The degree of amplification was consistent with the observed severity of cancer‐induced cachexia in the different mouse models. For example, the KPC secretome induced a higher IL‐6 release than the 4662 secretome. As a highly selective COX‐2 inhibitor, celecoxib significantly inhibited PGE2 production of the cachexia‐inducing tumour cells and as a consequence, amplification of the hypothalamic IL‐6 secretion was absent. Expression of mRNA coding for other inflammatory mediators was changed in a similar way: amplified when KPC medium was combined with a low concentration of LPS. Moreover, the effect of KPC could be mimicked by PGE2, indicating a broad effect of PGE2 on hypothalamic inflammation.
Tumour‐secreted PGE2 has been reported for colon, breast and pancreatic cancers 24 and to have a significant impact on the elevated inflammatory state. 25 Some clinical trial data support a link between PGE2 and the development of anorexia and cachexia. 26 From this point of view, PGE2 is potentially an auxiliary indicator for diagnosis of cancer‐induced cachexia. However, the metabolic system of cancer cells is relatively complex. Within different cancer types, it can be hypothesized that in addition to PGE2, there are likely to be prostaglandins like PGE1 and PGE3 that affect the occurrence of magnified hypothalamic inflammation.
Moreover, PGE2 in the hypothalamus is a principal mediator of the LPS‐induced febrile response. 27 COX‐2 is expressed mostly in blood vessels and initially synthesized by macrophages of the LPS‐processing organs like the lung and liver. 28 In addition to PGE2 secreted in brain endothelial cells and microglia, PGE2 can be transported across the BBB. PGE2 membrane transporters are reported to be involved in regulating its concentration in the hypothalamus. LPS is reported to stimulate the production of prostaglandin transporter OATP2A1 (SLCO2A1), 29 thus activating PGE2 transport across the BBB. With this in mind, it would be interesting to further explore the role of prostaglandin transporters in hypothalamic inflammatory amplification.
The mediobasal hypothalamus is activated early in pancreatic cancer‐induced cachexia, including the appearance of microglial cells that have an activated morphology. 30 Microglial cells, as the resident macrophages of the central nervous system, are found abundantly in the hypothalamus. Because the mediobasal hypothalamus contains crucial neural circuits controlling food intake, energy expenditure and overall energy homoeostasis, it has a key role in the development of anorexia and cachexia. 31 We hypothesize that the combination of inflammatory mediators that are produced by the hypothalamic cells can attract and activate more microglial cells and in this way start a larger secondary inflammatory reaction. Upon this reaction, even a third reaction might follow, because microglial cells have been implicated as protective against cachexia with higher expression of anti‐inflammatory factors like arginase‐1, thus contributing to maintenance of body mass during chronic systemic inflammation. 30
A large body of evidence demonstrates a complex interplay between intestinal health, gut barrier function and host inflammatory responses during cachexia, with microbial pathogens and local inflammation compromising the intestinal barrier function, causing translocation of endotoxins that induce immune activation. 32 In view of the interaction of substances playing roles in the gut–brain axis communication, there would in theory be two possible strategies to reduce the inflammatory response during cachexia. The first option could consist of reducing hypothalamic secretion of inflammatory mediators like IL‐6, CCL2 and CXCL‐2 by selective COX‐2 inhibition, which seems a promising strategy to diminish sickness behaviour in cancer patients. This is, however, not applicable to clinical circumstances since COX‐2‐derived prostanoids such as prostacyclin contribute to the maintenance of vascular homoeostasis. Conventional selective COX‐2 inhibitors have been associated with adverse cardiovascular side effects. 33 Next to that, NSAIDs with mixed COX‐1 and COX‐2 selectivity have been reported to increase intestinal permeability. 34 PGE2 regulates proliferation of endothelial cells of the gastrointestinal tract, wherefore peripheral inhibition of its production may further aggravate gut health of the cancer patients. 35 It is therefore our hypothesis that interfering with a COX‐2 inhibitor will likely have both cachexia enhancing and reducing effects that will interfere with each other. This is probably why reported effects of NSAIDs on cancer cachexia are only marginal. Interfering in the process at hypothalamic receptor level might have more effect.
Targeting mPGES‐1, the terminal enzyme for PGE2 production, will lead to the specific reduction of PGE2 levels and might be considered safer. However, no mPGES‐1 inhibitors have yet been approved for clinical practice. 36 Therefore, studies targeting the PGE2 receptors might be an appropriate strategy to reduce sickness behaviour, where the PGE2 receptors involved in this amplified response remain to be identified. The EP receptors and specifically the EP3 receptor are abundant and widely distributed in the brain. In addition, inhibition of EP3 receptor activity has previously been reported to decrease PGE2‐induced reduction of lever‐pressing behaviour in rats. 37
On the other hand, hypothalamic IL‐6, CCL2 and CXCL‐2 secretion may also be reduced by lowering LPS levels. Because LPS initially enters the periphery from the gut, supporting intestinal functionality with, for example, nutritional fibre or probiotic supplementation might be beneficial. 38 To enhance treatment efficacy, PGE2‐lowering strategies could be combined with prebiotics and probiotics as they are considered to enhance intestinal barrier function and thus reduce LPS load. 39 A multitarget treatment aiming to reduce both PGE2 and LPS activity in the hypothalamus could also be considered a promising approach.
Several limitations should be considered when interpreting results of this study. Considering the configuration of the in vitro model, the C26 mimic was based on the categories and concentrations determined from previous experiments. Although PGE2 was determined to play a key role, the results may not reveal more detailed interactions between different mediators. Second, in our in vivo experiments we measured the entire hypothalamic tissue, but only one hypothalamic neuron cell line (hypoE‐46) was used for in vitro model. Follow‐up experiments with co‐cultures of hypothalamic neuronal and glial cells might be a promising attempt. Furthermore, for intestinal‐derived pro‐inflammatory factors, the research can be expanded to investigate the changes in gut microbiota composition, which can explore the impact of intestinal changes on the gut‐brain axis during cancer cachexia.
In conclusion, PGE2 was identified as a tumour‐secreted inflammatory mediator that contributes to amplified hypothalamic LPS‐induced inflammation during cancer cachexia. To our knowledge, this is the first time that an amplified response of hypothalamic cells to a combination of tumour‐derived and intestinal bacteria‐derived compounds has been described. These findings provide a novel insight into the interplay between gut and brain axis and therefore maybe a contribution to a better design for future multi‐ targeting therapeutic interventions for cachexia patients.
Conflict of interest
None declared.
Supporting information
Figure S1 Body weight over post‐implantation phase (A) and normalized food intake throughout post‐implantation phase (B) in sham, 4662, KPC and DSS mice. N = 3, 4, 4, 3 in Sham, KPC, 4662 and DSS groups respectively.
Figure S2 IL‐6 secretion of mHypoE‐N46 cells incubated with C26 (A), LLC (B), KPC (C) and 4662 (D) medium grown under different concentrations of CXB combined with LPS, compared to the regular incubation with tumour secretome and LPS. The CXB concentrations to which tumour cells were exposed, included: 20, 10, 1, 0.1, 0.01, 0.001, 0.0001 μM. * P < 0.05, ** P < 0.01, *** P < 0.001, **** P < 0.0001. One‐way ANOVA analysis of variance with post hoc Bonferroni's Multiple Comparison Test.
Table S1 PGE2 concentrations in the tumour secretome of C26, LLC, KPC and 4662 cells cultured with different concentrations of the selective COX‐2 inhibitor CXB. The CXB concentrations to which tumour cells were exposed, included: 20, 10, 1, 0.1, 0.01, 0.001, 0.0001 μM.
Table S2 Primers used for RT‐qPCR.
Acknowledgements
We thank Dr Elizabeth Jaffee, of Johns Hopkins University, for kindly providing C57BL/6 KPC epithelial PDAC cells derived in her laboratory. We additionally thank Josef Argilès for kindly providing Lewis lung carcinoma (LLC) cells. We acknowledge financial support by the China Scholarship Council to the first author. The authors certify that they comply with the ethical guidelines for publishing in the Journal of Cachexia, Sarcopenia and Muscle: update 2019. 40
Li X., Holtrop T., Jansen F. A. C., Olson B., Levasseur P., Zhu X., Poland M., Schalwijk W., Witkamp R. F., Marks D. L., and van Norren K. (2022) Lipopolysaccharide‐induced hypothalamic inflammation in cancer cachexia‐anorexia is amplified by tumour‐derived prostaglandin E2, Journal of Cachexia, Sarcopenia and Muscle, 13, 3014–3027, doi: 10.1002/jcsm.13093
Xiaolin Li, Tosca Holtrop, and Fleur A. C. Jansen contributed equally to this work
References
- 1. Argilés JM, Busquets S, Stemmler B, López‐Soriano FJ. Cancer cachexia: Understanding the molecular basis. Nat Rev Cancer 2014;14:754–762. [DOI] [PubMed] [Google Scholar]
- 2. Dwarkasing JT, Witkamp RF, Boekschoten MV, ter Laak MC, Heins MS, van Norren K. Increased hypothalamic serotonin turnover in inflammation‐induced anorexia. BMC Neurosci 2016;17:26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Burfeind KG, Michaelis KA, Marks DL. The central role of hypothalamic inflammation in the acute illness response and cachexia. Semin Cell Dev Biol 2016;54:42–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. van Norren K, Dwarkasing JT, Witkamp RF. The role of hypothalamic inflammation, the hypothalamic–pituitary–adrenal axis and serotonin in the cancer anorexia–cachexia syndrome. Curr Opin Clin Nutr Metab Care 2017;20:396–401. [DOI] [PubMed] [Google Scholar]
- 5. Braun TP, Szumowski M, Levasseur PR, Grossberg AJ, Zhu X. Muscle atrophy in response to cytotoxic chemotherapy is dependent on intact glucocorticoid signaling in skeletal muscle. PLoS ONE 2014;9:106489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Ryan JL, Carroll JK, Ryan EP, Mustian KM, Fiscella KMG. Mechanisms of cancer‐related fatigue. Oncologist 2007;12:22–34. [DOI] [PubMed] [Google Scholar]
- 7. Witkamp RF, van Norren K. Let thy food be thy medicine … when possible. Eur J Pharmacol 2018;836:102–114. [DOI] [PubMed] [Google Scholar]
- 8. Olson B, Zhu X, Norgard MA, Levasseur PR, Butler JT, Buenafe A, Burfeind KG, Michaelis KA, Pelz KR, Mendez H, Edwards J, Krasnow SM, Grossberg AJ, Marks DL. Lipocalin 2 mediates appetite suppression during pancreatic cancer cachexia. Nat Commun 2021;12:1–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Burfeind KG, Zhu X, Levasseur PR, Michaelis KA, Norgard MA, Marks DL. TRIF is a key inflammatory mediator of acute sickness behavior and cancer cachexia. Brain Behav Immun 2018;73:364–374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Johnson AMF, Costanzo A, Gareau MG, Armando AM, Quehenberger O, Jameson JM, Olefsky JM. High fat diet causes depletion of intestinal eosinophils associated with intestinal permeability. PLoS ONE 2015;10:e0122195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Michaelis KA, Zhu X, Burfeind KG, Krasnow SM, Levasseur PR, Morgan TK, Marks DL. Establishment and characterization of a novel murine model of pancreatic cancer cachexia. J Cachexia Sarcopenia Muscle 2017;8:824–838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Zhu X, Burfeind KG, Michaelis KA, Braun TP, Olson B, Pelz KR, Morgan TK, Marks DL. MyD88 signalling is critical in the development of pancreatic cancer cachexia. J Cachexia Sarcopenia Muscle 2019;10:378–390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Deans C, Wigmore SJ. Systemic inflammation, cachexia and prognosis in patients with cancer. Curr Opin Clin Nutr Metab Care 2005;8:265–269. [DOI] [PubMed] [Google Scholar]
- 14. Paul D. The systemic hallmarks of cancer. J Cancer Metastasis Treat 2020;6. [Google Scholar]
- 15. Bindels LB, Neyrinck AM, Loumaye A, Catry E, Walgrave H, Cherbuy C, Leclercq S, van Hul M, Plovier H, Pachikian B, Bermúdez‐Humarán LG, Langella P, Cani PD, Thissen JP, Delzenne NM. Increased gut permeability in cancer cachexia: Mechanisms and clinical relevance. Oncotarget 2018;9:18224–18238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Herremans KM, Riner AN, Cameron ME, Trevino JG. The microbiota and cancer cachexia. Int J Mol Sci 2019;20:6267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Herreros‐Villanueva M, Hijona E, Cosme A, Bujanda L. Mouse models of pancreatic cancer. World J Gastroenterol 2012;18:1286–1294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Bindels LB, Neyrinck AM, Claus SP, le Roy CI, Grangette C, Pot B, Martinez I, Walter J, Cani PD, Delzenne NM. Synbiotic approach restores intestinal homeostasis and prolongs survival in leukaemic mice with cachexia. ISME J 2016;10:1456–1470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Flemer B, Lynch DB, Brown JMR, Jeffery IB, Ryan FJ, Claesson MJ, O'Riordain M, Shanahan F, O'Toole PW. Tumour‐associated and non‐tumour‐associated microbiota in colorectal cancer. Gut 2017;66:633–643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Ge W, Hu H, Cai W, Xu J, Hu W, Weng X, Qin X, Huang Y, Han W, Hu Y, Yu J, Zhang W, Ye S, Qi L, Huang P, Chen L, Ding K, Wang LD, Zheng S. High‐risk stage III colon cancer patients identified by a novel five‐gene mutational signature are characterized by upregulation of IL‐23A and gut bacterial translocation of the tumor microenvironment. Int J Cancer 2020;146:2027–2035. [DOI] [PubMed] [Google Scholar]
- 21. Yeom E, Shin H, Yoo W, Jun E, Kim S, Hong SH, Kwon DW, Ryu TH, Suh JM, Kim SC, Lee KS, Yu K. Tumour‐derived Dilp8/INSL3 induces cancer anorexia by regulating feeding neuropeptides via Lgr3/8 in the brain. Nat Cell Biol 2021;23:172–183. [DOI] [PubMed] [Google Scholar]
- 22. Burton MD, Sparkman NL, Johnson RW. Inhibition of interleukin‐6 trans‐signaling in the brain facilitates recovery from lipopolysaccharide‐induced sickness behavior. J Neuroinflammation 2011;8:1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Kim J, Chuang HC, Wolf NK, Nicolai CJ, Raulet DH, Saijo K, Bilder D. Tumor‐induced disruption of the blood‐brain barrier promotes host death. Dev Cell 2021;56:2712–2721.e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Wang D, Dubois RN. Prostaglandins and cancer. Gut 2006;55:115–122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Ricciotti E, Fitzgerald GA. Prostaglandins and inflammation. Arterioscler Thromb Vasc Biol 2011;31:986–1000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Wang W, Andersson M, Lõnnroth C, Svanberg E, Lundholm K. Prostaglandin E and prostacyclin receptor expression in tumor and host tissues from MCG 101‐bearing mice: A model with prostanoid‐related cachexia. Int J Cancer 2005;115:582–590. [DOI] [PubMed] [Google Scholar]
- 27. Engström L, Ruud J, Eskilsson A, Larsson A, Mackerlova L, Kugelberg U, Qian H, Vasilache AM, Larsson P, Engblom D, Sigvardsson M, Jönsson JI, Blomqvist A. Lipopolysaccharide‐induced fever depends on prostaglandin E2 production specifically in brain endothelial cells. Endocrinology 2012;153:4849–4861. [DOI] [PubMed] [Google Scholar]
- 28. Steiner AA, Ivanov AI, Serrats J, Hosokawa H, Phayre AN, Robbins JR, Roberts JL, Kobayashi S, Matsumura K, Sawchenko PE, Romanovsky AA. Cellular and molecular bases of the initiation of fever. PLoS Biol 2006;4:e284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Nakamura Y, Nakanishi T, Shimada H, Shimizu J, Aotani R, Maruyama S, Higuchi K, Okura T, Deguchi Y, Tamai I. Prostaglandin transporter OATP2A1/SLCO2A1 is essential for body temperature regulation during fever. J Neurosci 2018;38:5584–5595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Burfeind KG, Zhu X, Norgard MA, Levasseur PR, Huisman C, Michaelis KA, Olson B, Marks DL. Microglia in the hypothalamus respond to tumor‐derived factors and are protective against cachexia during pancreatic cancer. Glia 2020;68:1479–1494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Valdearcos M, Myers MG, Koliwad SK. Hypothalamic microglia as potential regulators of metabolic physiology. Nat Metab 2019;1:314–320. [DOI] [PubMed] [Google Scholar]
- 32. Puppa MJ, White JP, Sato S, Cairns M, Baynes JW, Carson JA. Gut barrier dysfunction in the Apc Min/+ mouse model of colon cancer cachexia. Biochim Biophys Acta ‐ Mol Basis Dis 2011;1812:1601–1606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Schjerning AM, McGettigan P, Gislason G. Cardiovascular effects and safety of (non‐aspirin) NSAIDs. Nat Rev Cardiol 2020;17:574–584. [DOI] [PubMed] [Google Scholar]
- 34. Maseda D, Ricciotti E. NSAID–Gut Microbiota Interactions. Front Pharmacol 2020;11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Wang D, Wang H, Brown J, Daikoku T, Ning W, Shi Q, Richmond A, Strieter R, Dey SK, DuBois RN. CXCL1 induced by prostaglandin E2 promotes angiogenesis in colorectal cancer. J Exp Med 2006;203:941–951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Bergqvist F, Morgenstern R, Jakobsson PJ. A review on mPGES‐1 inhibitors: From preclinical studies to clinical applications. Prostaglandins Other Lipid Mediat 2020;147:106383. [DOI] [PubMed] [Google Scholar]
- 37. Yamaguchi T, Kubota T, Watanabe S, Yamamoto T. Activation of brain prostanoid EP3 receptors via arachidonic acid cascade during behavioral suppression induced by Δ8‐tetrahydrocannabinol. J Neurochem 2003;88:148–154. [DOI] [PubMed] [Google Scholar]
- 38. van Krimpen SJ, Jansen FAC, Ottenheim VL, Belzer C, van der Ende M, van Norren K. The effects of pro‐, pre‐, and synbiotics on muscle wasting, a systematic review—Gut permeability as potential treatment target. Nutrients 2021;13:1115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Pham VT, Seifert N, Richard N, Raederstorff D, Steinert R, Prudence K, Mohajeri MH. The effects of fermentation products of prebiotic fibres on gut barrier and immune functions in vitro. PeerJ 2018;2018:e5288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. von Haehling S, Morley JE, Coats AJS, Anker SD. Ethical guidelines for publishing in the Journal of Cachexia, Sarcopenia and Muscle: update 2021. J Cachexia Sarcopenia Muscle 2021;12:2259–2261. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1 Body weight over post‐implantation phase (A) and normalized food intake throughout post‐implantation phase (B) in sham, 4662, KPC and DSS mice. N = 3, 4, 4, 3 in Sham, KPC, 4662 and DSS groups respectively.
Figure S2 IL‐6 secretion of mHypoE‐N46 cells incubated with C26 (A), LLC (B), KPC (C) and 4662 (D) medium grown under different concentrations of CXB combined with LPS, compared to the regular incubation with tumour secretome and LPS. The CXB concentrations to which tumour cells were exposed, included: 20, 10, 1, 0.1, 0.01, 0.001, 0.0001 μM. * P < 0.05, ** P < 0.01, *** P < 0.001, **** P < 0.0001. One‐way ANOVA analysis of variance with post hoc Bonferroni's Multiple Comparison Test.
Table S1 PGE2 concentrations in the tumour secretome of C26, LLC, KPC and 4662 cells cultured with different concentrations of the selective COX‐2 inhibitor CXB. The CXB concentrations to which tumour cells were exposed, included: 20, 10, 1, 0.1, 0.01, 0.001, 0.0001 μM.
Table S2 Primers used for RT‐qPCR.