

Abstract
Background
Critical illness myopathy (CIM) is a consequence of modern critical care resulting in general muscle wasting and paralyses of all limb and trunk muscles, resulting in prolonged weaning from the ventilator, intensive care unit (ICU) treatment and rehabilitation. CIM is associated with severe morbidity/mortality and significant negative socioeconomic consequences, which has become increasingly evident during the current COVID‐19 pandemic, but underlying mechanisms remain elusive.
Methods
Ten neuro‐ICU patients exposed to long‐term controlled mechanical ventilation were followed with repeated muscle biopsies, electrophysiology and plasma collection three times per week for up to 12 days. Single muscle fibre contractile recordings were conducted on the first and final biopsy, and a multiomics approach was taken to analyse gene and protein expression in muscle and plasma at all collection time points.
Results
(i) A progressive preferential myosin loss, the hallmark of CIM, was observed in all neuro‐ICU patients during the observation period (myosin:actin ratio decreased from 2.0 in the first to 0.9 in the final biopsy, P < 0.001). The myosin loss was coupled to a general transcriptional downregulation of myofibrillar proteins (P < 0.05; absolute fold change >2) and activation of protein degradation pathways (false discovery rate [FDR] <0.1), resulting in significant muscle fibre atrophy and loss in force generation capacity, which declined >65% during the 12 day observation period (muscle fibre cross‐sectional area [CSA] and maximum single muscle fibre force normalized to CSA [specific force] declined 30% [P < 0.007] and 50% [P < 0.0001], respectively). (ii) Membrane excitability was not affected as indicated by the maintained compound muscle action potential amplitude upon supramaximal stimulation of upper and lower extremity motor nerves. (iii) Analyses of plasma revealed early activation of inflammatory and proinflammatory pathways (FDR < 0.1), as well as a redistribution of zinc ions from plasma.
Conclusions
The mechanical ventilation‐induced lung injury with release of cytokines/chemokines and the complete mechanical silencing uniquely observed in immobilized ICU patients affecting skeletal muscle gene/protein expression are forwarded as the dominant factors triggering CIM.
Keywords: critical illness myopathy, mechanical ventilation, membrane exitability, muscle paresis, myosin loss
Introduction
Critical illness myopathy (CIM) is associated with severe morbidity/mortality and significant negative socioeconomic consequences. 1 , 2 This has become increasingly evident during the current COVID‐19 pandemic and coupled to the increased demand for intensive care unit (ICU) treatment and mechanical ventilation. 3 CIM is characterized by general muscle paresis of all limb and trunk muscles, prolonged weaning from the ventilator, but craniofacial muscles, sensory and cognitive functions are typically less affected. However, CIM has for many years been lumped together with muscle paresis of neurogenic origin, such as critical illness polyneuropathy, myasthenia gravis and Guillain–Barré syndrome, due to misinterpretation of electrophysiological signals. 1 Nevertheless, CIM has a primary myogenic origin and was initially regarded as a rare event of limited clinical relevance. We now know that it is the most frequent type of acquired muscle paresis in ICU patients, reported in 25–58% of the general ICU population and in 100% of some ICU subpopulations (see Friedrich et al. and Rodriguez et al. 1 , 4 ). Critical care is one of the costliest disciplines in modern health care, and the costs associated with prolonged weaning due to impaired respiratory muscle function account for ~30% of overall ICU costs or a staggering $64 billion in the United States annually 5 prior to the COVID‐19 pandemic. The older the mechanically ventilated patient, the less favourable are the economic and clinical outcomes. 6 Old age is associated with additional weaning problems, expensive ICU treatment, prolonged post‐ICU rehabilitation, duration of hospital stay and impaired quality of life for years after hospital discharge. 7 Further, old age and muscle wasting are the two factors that most strongly predict morbidity and mortality in the ICU.
It is highly likely that common components of the ICU treatment per se are directly involved in the pathogenesis of CIM as there is heterogeneity of underlying disease and pharmacological treatment among patients exhibiting similar CIM outcomes. Post‐synaptic neuromuscular blockade, sepsis and high‐dose corticosteroid hormone treatment have been suggested to trigger CIM. 1 However, CIM is observed in the absence of all these three factors both in ICU patients and in experimental studies, and the factors triggering CIM and underlying mechanisms remain elusive. In a previous longitudinal clinical intervention study in seven neuro‐ICU patients, all patients developed CIM in response to controlled mechanical ventilation (CMV) and immobilization for 7–11 days in the absence of sepsis, neuromuscular blockers and high‐dose corticosteroid hormone treatment. 8 That is, all patients had a pathological low myosin:actin ratio at the end of the observation period. 8 In experimental studies using a rat ICU model where rats were exposed to long‐term CMV and immobilization, all rats showed a preferential myosin loss and severely impaired force generation capacity at CMV durations longer than 5 days. 9 CMV is typically avoided due to the ventilator‐induced lung injury (VILI) caused by repetitive alveolar strain physically deforming structural elements and the activation inflammatory signalling. 10 , 11 Assisted ventilation is the preferred mode of ventilation due to less severe pulmonary damage. The lung injury associated with mechanical ventilation and the immobilization observed in deeply sedated or pharmacologically paralyzed ICU patients are the only factors ICU patients with CIM have in common, and it is hypothesized that they represent the most important factors triggering CIM.
Neuro‐ICU patients represent a more homogenous clinical ICU population than the general ICU population with less complex clinical histories. Furthermore, neuro‐ICU patients with an insufficient central ventilator drive and high sedation levels are more frequently exposed to CMV. The current longitudinal study was therefore undertaken to analyse muscle and plasma in neuro‐ICU patients exposed to immobilization and CMV for 12 days with repeated muscle biopsies, electrophysiological measurements and blood collection three times per week beginning on the first day of mechanical ventilation. A time‐course multiomics approach was taken to analyse muscle and plasma samples together with measurements of regulation of muscle contraction at the muscle fibre level together with electrophysiological methods frequently used in the diagnosis of CIM. We hypothesized a time‐dependent development of CIM with a progressive loss of the molecular motor protein myosin during the observation period.
Materials and methods
Patients
A detailed presentation of anthropometric data, gender, age, clinical outcome and comorbidities is presented in Table 1 and Materials and Methods section in the supporting information. During the ICU hospitalization, there was no evidence of sepsis or severe sepsis (sepsis with organ dysfunction) according to the 2001 SCCM/ESICM/ACCP/ATS/SIS International Sepsis Definitions Conference. 12 Muscle samples, electrophysiological measurements and blood were collected on the first day of mechanical ventilation in the neuro‐ICU and followed by consecutive biopsies three times per week (Monday, Wednesday and Friday). Written and verbal informed consent was obtained from a close relative, and the study was approved by the ethics committee at the Karolinska Hospital (Dnr 2016/242‐31/2). All patients had a serious central nervous system lesion; many patients had been exposed to emergency surgery and mechanically ventilated within 24 h after hospital admission (Table 1 ). We have therefore also compared gene expression in the patients on the first day on the ventilator with six healthy control (68–82 years) subjects enrolled in one of our previous studies. 13 All patients were given physical therapy 5 days/week to prevent joint contractures.
Table 1.
Clinical background in the 12 neuro‐ICU patients included in this study
| Patient ID, gender, age | Admission diagnosis | Days in ICU/in hospital | Severity score | Days on MV | Sepsis in ICU | Corticosteroid treatment (home/ICU) | Relevant comorbidities | ICU prognosis | Intra‐hospital death | Discharged from hospital (AH/home) | 30 day survival | Still in ICU after 30 days | 365 day survival | Number of tissue collections |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1. (623), M, 51 | TBI | 17/17 |
SAPS 3: 68 SOFA max: 9 |
10 | No | Yes/No | No | Discharged | No | Yes/AH | Alive | No | Alive | 6 |
| 2. (626), M, 72 | Acute SDH | 33/33 |
SAPS 3: 64 SOFA max: 10 |
19 | No | No/Yes | Diabetes | Discharged | No | Yes/AH | Alive | No | Deceased | 6 |
| 3. (628), F, 44 | SAH | 8/8 |
SAPS 3: 75 SOFA max: 10 |
8 | No | No/Yes | No | Deceased | Yes | No | Deceased | No | Deceased | 2 |
| 4. (629), M, 59 | Unspecified intracranial injury | 20/32 |
SAPS 3: 77 SOFA max: 8 |
12 | No | No/No | No | Discharged | No | Yes/AH | Alive | No | Alive | 6 |
| 5. (632), F, 26 | Multitrauma | 20/30 |
SAPS 3: 68 SOFA max: 11 |
10 | No | No/No | No | Discharged | No | Yes/AH | Alive | No | Alive | 6 |
| 6. (639), F, 83 | SAH | 58/58 |
SAPS 3: 72 SOFA max: 9 |
37 | No | No/No | No | Discharged | Yes | Yes/AH | Alive | Yes | Deceased | 6 |
| 7. (640), F, 40 | SAH | 18/26 |
SAPS 3: 62 SOFA max: 12.8 |
4 | No | No/No | Endometriosis | Discharged | No | Yes/AH | Alive | No | Alive | 2 |
| 8. (662), M, 62 | AVM | 15/15 |
SAPS 3: 58 SOFA max: 8 |
14 | No | No/Yes | Diabetes, epilepsy | Deceased | Yes | No | Deceased | No | Deceased | 6 |
| 9. (646), M, 76 | TBI | 12/37 |
SAPS 3: 69 SOFA max: 10 |
12 | No | No/No | Hypertension | Discharged | No | Yes/AH | Alive | No | Alive | 6 |
| 10. (661), F, 64 | SAH | 16/16 |
SAPS 3: 59 SOFA max: 10 |
10 | No | No/No | Rheumatoid arthritis | Discharged | No | Yes/AH | Alive | No | Alive | 4 |
| 11. (X), F, 47 | C2 fracture, aneurysm | 6/6 |
SAPS 3: 73 SOFA max: 11 |
6 | No | No/No | Previous stroke | Deceased | Yes | No | Deceased | No | Deceased | 1 |
| 12. (657), M, 58 | SAH | 24/24 |
SAPS 3: 75 SOFA max: 15 |
12 | No | No/No | Prostate cancer | Discharged | No | Yes/AH | Alive | No | Alive | 5 |
Abbreviations: AH, another hospital; AVM, arteriovenous malformation; ICU, intensive care unit; MV, mechanical ventilation; SAH, subarachnoid haemorrhage; SAPS 3, Simplified Acute Physiology Score 3; SDH, subdural haematoma; SOFA, Sequential Organ Failure Assessment; TBI, traumatic brain injury.
Muscle samples
Muscle samples were obtained from the tibialis anterior (TA). The first and the following four were taken from the same TA muscle and the final from the contralateral TA muscle on the contralateral side. Muscle samples from seven neuro‐ICU patients (ID: 623, 626, 629, 632, 639, 646 and 657; Table 1 ) were subject to RNAseq and Olink proteomics.
Library preparation and transcriptome sequencing
Total TA muscle RNA was extracted using RNeasy® Fibrous Tissue Mini Kit. For details, see Materials and Methods section in the supporting information.
Olink proteomics assay
Protein lysates were subject to Olink® Target 96 Inflammation panel (Olink Proteomics AB, Uppsala, Sweden) using Proximity Extension Assay (PEA) technology (see Materials and Methods section in the supporting information).
Differential gene expression and enrichment analyses
For time‐course RNAseq and plasma proteomics, differentially expressed genes (DEGs) and differentially expressed proteins (DEPs) from each cluster were subject to over‐represented (enrichment) analysis by ClusterProfiler, 23 respectively. The terms from Gene Ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG) enrichment analyses were considered significantly enriched when false discovery rate (FDR) was <0.1. For first biopsy versus control RNAseq analyses, gene set enrichment analysis (GSEA) was performed on the corresponding DEGs by ClusterProfiler (see Materials and Methods section in the supporting information).
Myofibrillar protein expression
Actin and myosin quantification was determined by 12% sodium dodecyl sulphate–polyacrylamide gel electrophoresis (SDS‐PAGE) on two 10 μm cryo‐cross sections from percutaneous muscle biopsies or from approximately half of the microbiopsy. The myosin heavy chain (MyHC) isoform expression was determined in the single muscle fibres analysed for single‐cell contractile measurements on sensitive 6% SDS‐PAGE (see Materials and Methods section in the supporting information).
Established methods to measure plasma Zn levels, single muscle fibre contractile properties, in vivo muscle torque and electrophysiology are presented in detail in the supporting information.
Statistics
A one‐way analysis of variance (ANOVA) was used to compare statistical differences between multiple groups and the paired t test when comparing pre‐ and post‐values in the same patient. Comparisons were made between the observations when patients were immobilized and exposed to CMV at the neuro‐ICU. Additional analyses were also performed including a healthy control group to identify changes taking place prior to ICU admission during initial surgical treatment. Pearson correlation analysis was performed to identify the correlation between DEPs and myosin:actin ratio. Values are given as means ± SD.
Results
Patients
A detailed presentation of the patients is given in Table 1 . Two of the 12 patients died early during mechanical ventilation and were not included in the analyses. Of the remaining 10 patients, electrophysiological measurements, blood collection and muscle biopsies were obtained on multiple occasions until the day when a weaning trial from CMV was performed and in seven patients at all six time points.
Myosin expression and electrophysiology
A progressive preferential myosin loss was observed in all neuro‐ICU patients during the observation period (Figure 1A and Table 2 ). One neuro‐ICU patient had a borderline pathological 14 myosin:actin ratio (1.6, Figure 1 A ) on the first day of mechanical ventilation, whereas the others had the expected ratio with twice as much myosin as actin. 14 In spite of a low initial myosin:actin ratio, this patient showed the same progressive myosin loss during the observation period as the other patients.
Figure 1.
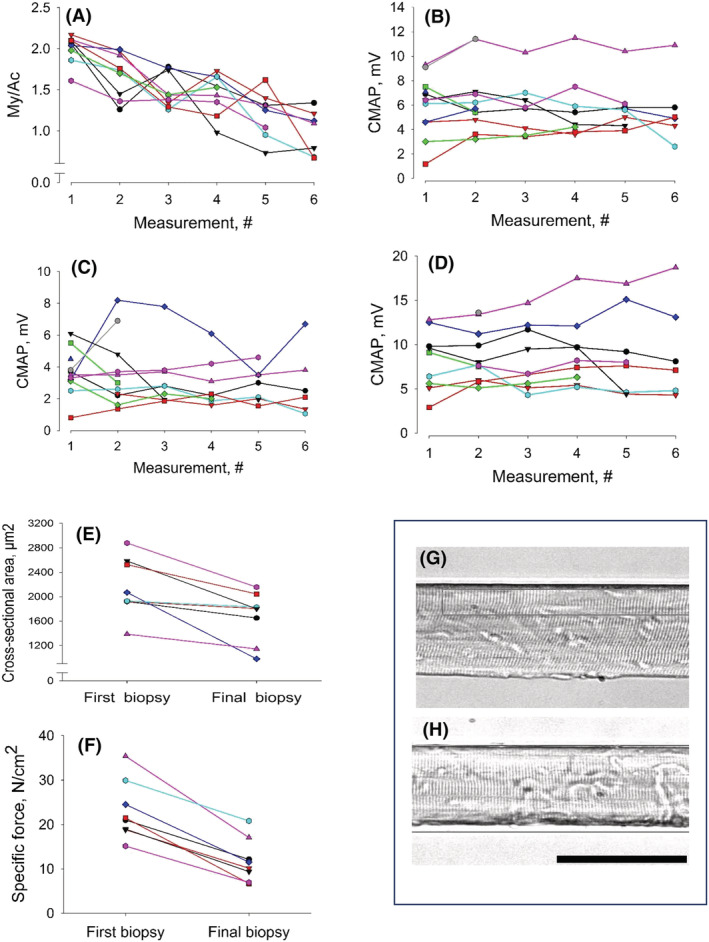
Myosin:actin ratios, compound muscle action potential (CMAP) amplitudes and single muscle fibre contractile measurements: (A) myosin:actin ratio and CMAP amplitude in response to supramaximal stimulation of (B) the median, (C) fibular and (D) tibial nerves at the six different measurements during the 12 day observation period. (E) Single muscle fibre cross‐sectional area, (F) specific force and representative single muscle fibres from (G) the first and (H) final muscle biopsy. Each patient is given the same colour in (A)–(F). The horizontal bar denotes 100 μm.
Table 2.
Myosin:actin ratio and the CMAP amplitude (mV), mCV (m/s) and distal Lat. (ms) in motor nerves (fibular, tibial and median nerves) together with sCV (m/s) and sAmp in median and sural nerves measured at six different time points during the 12 day observation period
| Variable | 1st | 2nd | 3rd | 4th | 5th | 6th | P value |
|---|---|---|---|---|---|---|---|
| Myosin:actin ratio | 2.0 ± 0.253,4,5,6 | 1.7 ± 0.35,6 | 1.5 ± 0.21,6 | 1.4 ± 0.21 | 1.2 ± 0.31,2 | 0.9 ± 0.21,2,3 | <0.001 |
| CMAP (median nerve) (mV) | 6.0 ± 2.4 | 6.5 ± 2.7 | 5.8 ± 2.3 | 5.8 ± 2.6 | 5.9 ± 2.0 | 5.6 ± 2.8 | n.s. |
| mCV (median nerve) (m/s) | 55.7 ± 3.1 | 56.7 ± 3.2 | 56.6 ± 6.0 | 52.3 ± 4.2 | 56.9 ± 5.1 | 55.8 ± 3.1 | n.s. |
| Lat. (median nerve) (ms) | 3.8 ± 0.9 | 3.5 ± 0.7 | 3.4 ± 0.7 | 3.5 ± 0.6 | 3.5 ± 0.7 | 3.3 ± 0.7 | n.s, |
| sCV (median nerve) (m/s) | 53.7 ± 9.6 | 56.8 ± 8.9 | 58.1 ± 8.5 | 55.9 ± 6.9 | 57.2 ± 8.1 | 57.3 ± 7.2 | n.s. |
| sAmp (median nerve) (μV) | 16.7 ± 12.9 | 20.4 ± 15.9 | 17.9 ± 10.2 | 15.5 ± 9.5 | 15.2 ± 10.5 | 15.8 ± 6.5 | n.s. |
| CMAP (fibular nerve) (mV) | 3.4 ± 1.7 | 3.2 ± 2.1 | 3.0 ± 2.1 | 2.9 ± 1.7 | 2.5 ± 1.4 | 2.7 ± 2.3 | n.s. |
| mCV (fibular nerve) (m/s) | 46.1 ± 5.6 | 44.1 ± 8.1 | 43.0 ± 9.0 | 40.9 ± 8.1 | 43.3 ± 6.9 | 46.0 ± 5.0 | n.s. |
| Lat. (fibular nerve) (ms) | 4.8 ± 1.4 | 4.8 ± 1.9 | 4.5 ± 1.5 | 4.5 ± 1.4 | 5.2 ± 2.5 | 4.8 ± 1.7 | n.s. |
| CMAP (tibial nerve) (mV) | 8.2 ± 3.4 | 8.7 ± 2.9 | 8.4 ± 3.7 | 9.1 ± 3.9 | 8.8 ± 4.8 | 9.4 ± 5.6 | n.s. |
| mCV (tibial nerve) (m/s) | 46.9 ± 5.0 | 48.1 ± 4.8 | 49.6 ± 4.7 | 46.3 ± 4.7 | 48.1 ± 3.1 | 48.4 ± 5.6 | n.s. |
| Lat. (tibial nerve) (ms) | 4.7 ± 1.4 | 3.9 ± 0.5 | 4.6 ± 1.9 | 4.2 ± 0.8 | 4.5 ± 1.2 | 4.2 ± 0.5 | n.s. |
| sCV (sural nerve) (m/s) | 57.6 ± 10.0 | 59.0 ± 5.1 | 57.3 ± 8.3 | 59.6 ± 4.8 | 55.8 ± 7.3 | 61.1 ± 5.1 | n.s. |
| sAmp (sural nerve) (μV) | 12.9 ± 5.7 | 15.2 ± 12.0 | 9.8 ± 4.3 | 11.1 ± 5.8 | 12.1 ± 6.0 | 12.7 ± 6.6 | n.s. |
Note: Values are means ± standard deviation. Statistically significant differences according to one‐way ANOVA are indicated as indices (not statistically significant [n.s.]). Abbreviations: CMAP, compound muscle action potential; Lat., latency; mCV, motor nerve conduction velocity; sAmp, sensory nerve amplitude; sCV, sensory nerve conduction velocity. Statistically significant differences between days are indicated by superscript numbers.
The amplitude of the compound muscle action potential (CMAP), motor conduction velocity and distal latencies upon supramaximal stimulation of the fibular, median or tibial nerves did not change during the observation period (Figure 1B–D and Table 2 ). Sensory nerve conduction velocities and amplitudes in the mixed median nerve and the sensory sural nerve were unaffected during the 12 days (Table 2 ). Spontaneous EMG activity (fibrillation potentials and positive sharp waves) was recorded in upper and lower extremity muscles in 9 of the 12 patients and was first observed at second to fourth examination biopsy and persisted throughout the observation period.
Single muscle fibre size and function
Average muscle fibre cross‐sectional area (CSA) and maximum single muscle fibre force normalized to CSA (CSA declined 30% [P < 0.007] and specific force 50% [P < 0.0001], respectively, during the 12 day observation period) (Figure 1 E,F ). Overall force generation capacity was even lower than these values because 9–21% of tested muscle fibres did not generate any force upon maximum calcium activation in 5 of the 10 patients and not included in the analyses. The loss in specific force was strongly linked to the preferential loss of the molecular motor protein myosin in response to the ICU condition. In the patient with underlying malignancy and low myosin:actin ratio on Day 1, specific force was low from the beginning (15 ± 7 N/cm2) but showed a similar ~50% decline (P < 0.001) to 7 ± 2 N/cm2 in parallel with the progressive myosin loss.
Skeletal muscle gene expression patterns and enriched biofunctions during immobilization and mechanical ventilation
A total of 2597 genes were identified as DEGs and annotated across the six time points. According to clustering analysis of gene expression pattern, DEGs were divided into four distinctly different clusters: Cluster 1 (773 DEGs; 29.8%), Cluster 2 (373 DEGs; 14.4%), Cluster 3 (496 DEGs; 19.1%) and Cluster 4 (955 DEGs; 36.8%) (Figure 2 A ).
Figure 2.
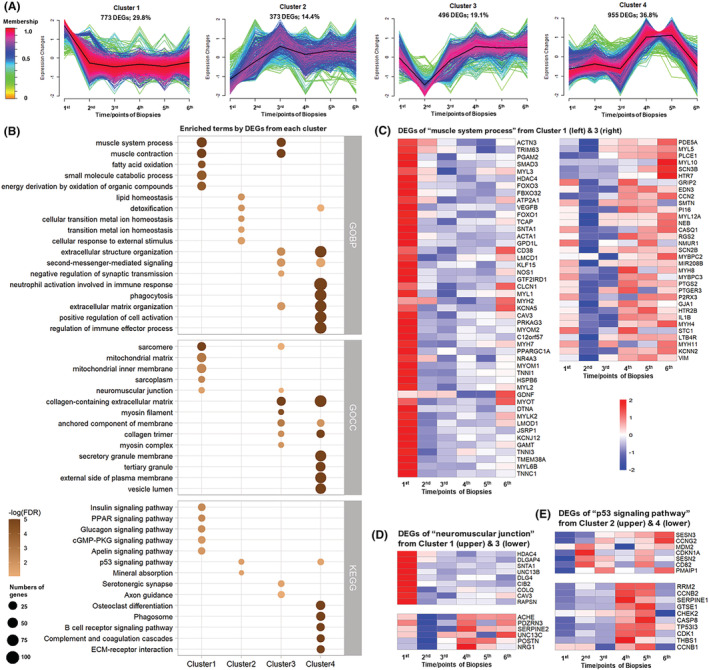
Clustered expression patterns of differentially expressed genes (DEGs) from the neuro‐intensive care unit (ICU) patients during 12 day observation and the functional enrichment analysis: (A) time‐course series in the seven neuro‐ICU patients from whom muscle samples were collected at six time points during the 12 day period. Membership varying from green to red colour indicates the degree of genes suitable to the cluster. (B) Comparison of top 5 enriched GObp, GOcc and Kyoto Encyclopedia of Genes and Genomes (KEGG) terms among four clusters. Heatmap of DEGs in (C) the GObp term ‘muscle system process’ from Cluster 1 (left half) and Cluster 3 (right half), (D) the GOcc term ‘neuromuscular junction’ from Cluster 1 (upper half) and Cluster 3 (lower half) and (E) the KEGG term ‘p53 signaling pathway’ from Cluster 2 (upper half) and Cluster 4 (lower half). Terms are ranked by −log (false discovery rate [FDR]). Dot size represents the number of DEGs involved in each term.
In Cluster 1, gene expressions decreased from the first to the second biopsy and remained depressed during the whole observation period. According to functional enrichment analysis, many DEGs were highly related to muscle system and function, as reflected in GObp terms like ‘muscle system process’ and ‘muscle contraction’ and GOcc terms including ‘sarcomere’, ‘sarcoplasm’, ‘sarcoplasmic reticulum’ and ‘neuromuscular junction’, as well as the KEGG terms ‘Calcium signaling pathway’ and so forth. Multiple DEGs were highly related to metabolism and energy homeostasis, as reflected in GObp terms including ‘fatty acid oxidation’, ‘ATP metabolic process’ and ‘cellular carbohydrate metabolic process’ and GOcc terms like ‘mitochondrial matrix’ and ‘mitochondrial inner membrane’. This was further confirmed by KEGG terms including ‘insulin signaling pathway’, ‘Arginine biosynthesis’ and ‘AMPK signaling pathway’ (Figure 2 B ). Combined with the gene expression pattern of Cluster 1, these enriched terms suggest a dramatic down‐regulation of muscle function and energy metabolism throughout the 12 day observation period.
In Cluster 2, gene expressions showed an opposite pattern compared with Cluster 1, with multiple terms related to protein degradation like ‘regulation of autophagy’ and ‘signal transduction by p53 class mediator’, and KEGG term ‘p53 signaling pathway’(Figure 2 B ), suggesting the upregulation of autophagy‐mediated protein degradation.
In Cluster 3, an early decline in gene expression was observed from the first to the second biopsy, followed by a gradual increase throughout the observation period. Notably, transcriptional upregulation of both adult and embryonic myosin heavy and light chain isoforms was observed in the final muscle biopsy (Figure 2 C ). Many DEGs were highly related to mechano‐electro‐transduction, as reflected in GObp terms like ‘extracellular structure organization’, ‘second‐messenger‐mediated signaling’ and ‘neuron projection guidance’ and GOcc terms like ‘neuromuscular junction’ (Figure 2 D ) and ‘collagen‐containing extracellular matrix’, as well as the KEGG term ‘serotonergic synapse’ (Figure 2 B ). Together with the gene expression pattern in Cluster 3, these enriched terms suggest a recovery or compensatory mechanism from third to final biopsy.
In Cluster 4, a more complex expression pattern was observed, gene expressions were relatively stable during the initial three biopsies, followed by increased levels until the fifth biopsy and return to the original values in the final muscle biopsy taken from the contralateral TA muscle. The majority of the DEGs were highly related to immune responses (Figure 2 B ). A KEGG term ‘p53 signaling pathway’ overlapped with Cluster 2. The detailed information of involved DEGs from both clusters is shown in Figure 2 E . Despite the upregulation of these pathways until the fifth muscle biopsy, these changes were back to control levels on the final biopsy after 12 days.
Skeletal muscle gene expression and enriched biofunctions compared with healthy controls
A total of 2611 DEGs were identified and annotated when comparing healthy controls on the first day of mechanical ventilation. A healthy control group was added because it cannot be excluded that some early events during surgical procedures prior to ICU admission may have affected gene expression. The DEGs were highly related to RNA processing and homeostasis, as reflected in the top‐ranked GObp terms ‘ribonucleoprotein complex biogenesis’ (normalized enrichment score [NES]: 2.66) and ‘ncRNA processing’ (NES: 2.46), GOcc term ‘ribonucleoprotein complex’ (NES: 2.40) and KEGG terms ‘Ribosome’ (NES: 2.05) and so forth (Figure 3 A,B ). There were also many DEGs related to inflammatory responses and the subsequent biological processes, as reflected in the enrichment terms like ‘inflammatory response’ (GObp; NES: 2.73; Figure 3 C ), ‘IL‐17 signaling pathway’ (KEGG; NES: 2.18), ‘NF‐kappa B signaling pathway’ (KEGG; NES: 1.93), ‘p53 signaling pathway’ (KEGG; NES: 2.03; Figure 3 D ) and ‘Apoptosis’ (KEGG; NES: 1.75) in the patients.
Figure 3.
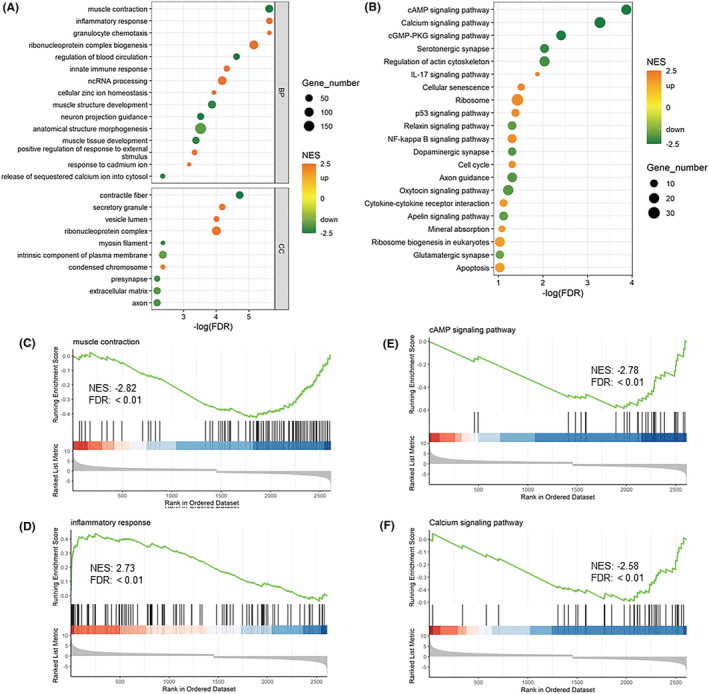
Gene set enrichment analysis (GSEA) of differentially expressed genes (DEGs) from first biopsy versus control: (A) the top GObp and GOcc terms and (B) Kyoto Encyclopedia of Genes and Genomes (KEGG) terms were selected for demonstration; GSEA plot of the gene set enrichment analysis for (C) ‘muscle contraction (GObp)’, (D) ‘inflammatory response (GO bp)’, (E) ‘cAMP signaling pathway (KEGG)’ and (F) ‘calcium signaling pathway (KEGG)’. NES, normalized enrichment score
Many DEGs were highly related to muscular adaptations, neuromuscular junction, excitation–contraction coupling and extracellular matrix such as ‘muscle contraction’ (GObp; NES: −2.82; Figure 3 E ), ‘muscle tissue development’ (GObp; NES: −2.32), ‘contractile fiber’ (GOcc; NES: −2.73), ‘myosin filament’ (GOcc; NES: −2.34), neuromuscular junction, ‘neuron projection guidance’ (GObp; NES: −2.49), ‘presynapse’ (GOcc; NES: −2.14), ‘serotonergic synapse’ (KEGG; NES: −2.26), ‘axon guidance’ (KEGG; NES: −1.94), ‘release of sequestered calcium ion into cytosol’ (GObp; NES: −2.34), ‘calcium signaling pathway’ (KEGG; NES: −2.58) and ‘extracellular matrix’ (GOcc; NES: −1.91). Thus, the negative NES of these terms suggests an inhibited or downregulated status of these biological processes in ICU patients. Comparisons of neuro‐ICU patient biopsies versus healthy controls were also made by adding health controls to time‐course RNAseq study. Similar molecular alterations were observed to that in the time‐course analyses from the first to sixth biopsy (Figures S1–S7 ). Specifically, functional enrichment analyses indicated that ubiquitin–proteasome system was slightly upregulated from first biopsy to second biopsy, followed by a slow downregulation (Figure S1 , Cluster 2.1). On the other hand, the autophagy–lysosome system was upregulated from first biopsy to third biopsy and followed by maintained levels (Figure S1 , Cluster 2.2). The general level of these two protein degradation systems was higher than in healthy controls (Figures S1 , S2 and S4 ). Involved DEGs with fold change along with duration are shown in Figures S3 and S5 . KEGG pathways ‘lysosome’ and ‘proteasome’ are shown in Figures S6 and S7 , respectively.
Biomarkers in plasma—Zinc ion homeostasis and inflammatory cytokines
Zinc ion homeostasis and its redistribution between plasma and muscle tissue have been forwarded as an important biomarker for preferential myosin loss in cancer cachexia related to intracellular zinc accumulation induced by ZIP14. 15 Plasma zinc ion concentration was lower than the normal range in all subjects at the time of the first biopsy, followed by a gradual increase with increasing duration, and zinc levels returned to normal levels in some but not all patients at the end of the observation period (Figure 4 A ). A series of DEGs related to ‘cellular zinc ion homeostasis’ showed a higher expression level in the TA muscle at the time of the first biopsy than in healthy controls and followed by decreased levels in parallel with increasing zinc plasma levels, such as in the SLC39A14 (ZIP14) gene responsible for zinc ion uptake (Figure 4 B ). These results imply a redistribution of zinc ions between plasma and muscle tissue in neuro‐ICU patients. Furthermore, 80 of 92 proteins in the Olink Inflammation panel were detected across all plasma samples. Of these, 28 proteins were identified as DEPs at the six time points. According to the clustering analysis, 26 DEPs were grouped within a Cluster 1 (26 DEPs; 92.9%) and the remaining two DEPs (IL10 and NTF3) in Cluster 2 (Figure 5 A,B ). Specifically, the protein levels of the 26 DEPs increased with increasing duration from the first to the fifth time point and followed by modest reduction at the final plasma sample. The majority of DEPs in this cluster were primarily involved in ‘Cytokine‐cytokine receptor interaction’ (KEGG), ‘IL‐17 signaling pathway’ (KEGG), ‘NF‐kappa B signaling pathway’ (KEGG) and ‘IL6 JAK STAT3 signaling’ (hallmark) pathways (Figure 5 C ). Six DEPs exhibited an over 10‐fold increase with ventilation duration, including EIF4EBP1, AXIN1, MMP1, SIRT2, STAMBP and CXCL5 (Table S1 ). According to correlation analyses, 18 DEPs correlated negatively with the myosin/actin ratio (Figure 4 A ). The topmost correlated DEPs were CCL25, TWEAK, CX3CL1 and TGFB1 (Figure S8 ).
Figure 4.
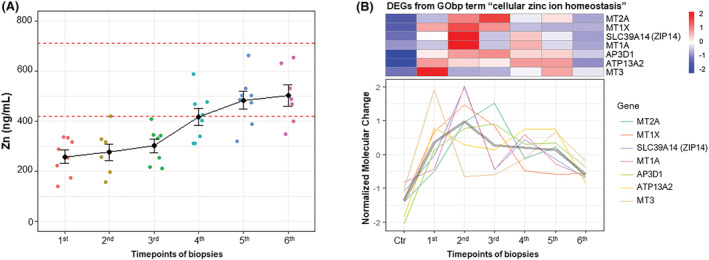
Zinc ion homeostasis in neuro‐intensive care unit (ICU) patients: the alteration tendency of (A) plasma zinc concentration and (B) differentially expressed genes (DEGs) related to ‘cellular zinc ion homeostasis’ in tibialis anterior (TA) muscle tissue at six time points of biopsies from neuro‐ICU patients compared with healthy control
Figure 5.
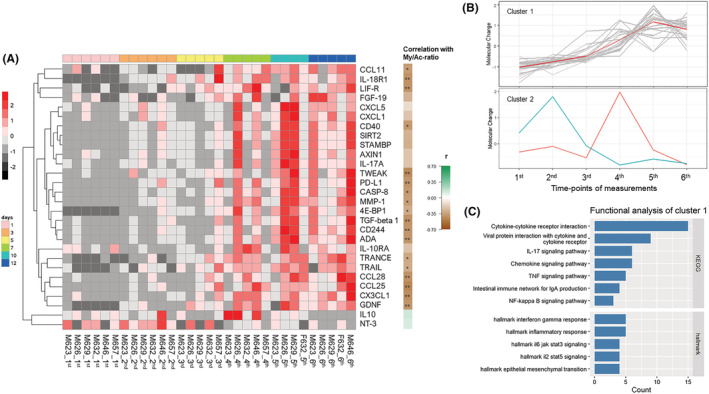
Clustered expression patterns of plasma differentially expressed proteins (DEPs) from the neuro‐intensive care unit (ICU) patients: (A) heatmap in time‐course series of 28 DEPs, (B) DEP alterations of Cluster 1 and Cluster 2 at the six time points of biopsies and (C) enriched KEGG terms of DEPs from Cluster 1 at the six time points of biopsies in seven neuro‐ICU patients
Recovery after hospital discharge
Follow‐up of in vivo muscle function was done in two patients (F64 and M58 in Table 1 three times during the year following hospital discharge. Both patients had a low voluntary knee‐extensor muscle strength compared with age‐matched health controls irrespective speed of movement at the initial measurement, but progressive strength improvements were observed in the following measurements (Figure S10 ). In the female patient, voluntary strength improved ~60% irrespective speed of movement, but was still lower than in age‐matched healthy controls. In the male subject, 60% and 100% improvements were observed at speed of movements corresponding to 30 and 180 deg/s, respectively, and muscle function was within the normal range 12 months after hospital discharge. The strength improvements were paralleled by an increased 6 min walk test from 295 to 425 m in the male patient and from 280 to 315 m in the female subject.
Discussion
In this prospective longitudinal study, repeated muscle biopsies, electrophysiological measurements and blood collection were obtained six times in neuro‐ICU exposed to 12 day long‐term CMV and immobilization. All patients showed a progressive preferential myosin loss, the hallmark of CIM. Single muscle fibre contractile measurements on the first and final days of the observation period showed a dramatic loss in muscle fibre size and maximum force generation normalized to fibre size resulting in global muscle function decline exceeding 65% after 12 day critical care. Genomic and proteomic analyses of muscle and plasma showed activation of signalling pathways involved in downregulation of contractile protein synthesis, and activation of autophagy, ubiquitin proteasomal and Gadd45a protein degradation pathways. In addition, pathological low zinc ion plasma concentrations were observed in all neuro‐ICU patients together with upregulation of ZIP14 and other genes related to ‘cellular zinc ion homeostasis’. This is of specific interest because the myosin loss in patients with metastatic cancer has also been shown to be related to zinc ion homeostasis regulated by TNF‐α and TGF‐β induced ZIP14, resulting in zinc ion uptake from plasma and accumulation in muscle cells. Although a progressive loss of myosin was observed in all neuro‐ICU patients during the observation period, CMAP amplitudes and motor or sensory nerve conduction velocities were not affected, that is, electrophysiological parameters frequently used in CIM diagnosis.
Cluster 4 in the RNAseq analyses showed a complex expression pattern, being stable during the initial three biopsies, followed by increased levels of genes related to immune responses until the fifth biopsy and return to the original values in the final muscle biopsy taken from the contralateral TA muscle. This complex expression pattern suggests an inflammatory response related to repeated muscle sampling from the same muscle in accordance with previous studies on the effects of repeated muscle sampling. 16 Low amplitude or absent CMAP amplitudes together with normal or subnormal motor nerve conduction velocities have been reported in ICU patients with CIM secondary to a decreased muscle membrane excitability related to defective sodium channel regulation. 4 , 17 , 18 , 19 , 20 , 21 In the current study, motor and sensory nerve conduction velocities and amplitudes upon supramaximal stimulation were not affected during the 12 day observation period. This was an unexpected finding because a decline in CMAP amplitude was anticipated to precede the myosin loss. In pigs exposed to CMV and immobilization for 5 days, CMAP amplitude declined dramatically at a duration too short to induce a significant preferential myosin loss. 22 , 23 In the porcine model, the most pronounced decline in CMAP amplitude was observed in pigs exposed to endotoxin induced sepsis in combination with high‐dose systemic corticosteroid hormone treatment. 22 , 23 The decreased CMAP amplitude was associated with a significant down‐regulation of voltage‐gated sodium channels in response to 5 day exposure to the ICU condition and endotoxin‐induced sepsis. 23 , 24 In non‐septic rats exposed to the ICU condition, we have observed a progressive preferential myosin loss in response to the ICU condition for 5 days, whereas voltage‐gated sodium channels remained unaffected (Lee et al. in manuscript). None of the neuro‐ICU patients followed in this study were septic, and there was no consistent downregulation of voltage‐gated sodium channels during the 12 day observation period. Thus, the preferential myosin loss and the decreased muscle membrane excitability appear to be regulated by independent mechanisms. A sepsis‐induced myopathy may affect membrane excitability independently of mechanisms triggering the myosin loss, an explanation why some, but not all, ICU patients with myosin loss show decreased CMAP amplitudes. This is supported by experimental studies of a sepsis‐induced myopathy in a porcine ICU model where muscle membrane abnormalities suggesting membrane depolarization and/or sodium channel inactivation occurred within 6 h of peritonitis. 25
Spontaneous EMG activity was observed in all neuro‐ICU patients in this study, that is, at a duration significantly shorter than the spontaneous EMG activity recorded after axonal motor neuron loss. This is in accordance with the early spontaneous activity recorded in a porcine ICU model where pigs were sedated with either propofol or thiopental infusion for 5 days. Motor nerve conduction velocities did not change over time, but there was a significant increase in the amount of limb muscle spontaneous EMG activity in all propofol‐treated animals, but no spontaneous EMG activity was recorded in response to 5 day thiopental sedation. 26 The spontaneous EMG activity in the propofol‐treated animals was paralleled by histological observations of muscle membrane lesions as well as increased creatine kinase plasma levels. 26 All neuro‐ICU patients in this study were sedated with propofol, and the spontaneous EMG activity recorded in the neuro‐ICU patients is suggested to be due to a muscle membrane lesion induced by the sedation. Spontaneous EMG activity (primarily fibrillation potentials) was observed in both proximal and distal upper and lower extremity muscles with the extensor digitorum communis muscle showing most abundant amount of spontaneous EMG activity (compared with deltoid, first dorsal interosseous and vastus lateralis muscles). In spite of the spontaneous EMG activity recorded in the neuro‐ICU patients, the whole muscle CMAP amplitudes were not affected. Furthermore, CIM is observed in ICU patients not sedated with propofol, and CIM with myosin loss is observed in experimental studies without propofol sedation. Propofol is therefore most probably not an obligatory factor triggering CIM but may have additive negative effects on the neuromuscular function.
RNAseq analyses revealed a series of molecular alterations highly related to muscle atrophy, weakness and disorders, such as (i) downregulation of DEGs involved in muscle system, including sarcomeric unit, myofibre structure, neuromuscular junctions, energy production, mitochondrial functions and calcium signalling pathways. Time‐course RNAseq analyses revealed subsequent upregulation of a portion of these DEGs, suggesting a recovery and/or compensatory process during 12 day mechanical ventilation and immobilization. Notably, some of the recovered DEGs were embryonic and neonatal myosin isoforms (e.g., MYH8 and MYL5; Figure 2 C ) not found in adult skeletal muscle, indicating the initiation of a reassembly process in accordance with previous observations. 27 The upregulation of the histone deacetylase 4 (HDAC4) involved in muscle development further support the initiation of thick filament reassembly. A similar HDAC4 upregulation has been reported in rats exposed to long‐term (9–14 days) immobilization and CMV, 28 and (ii) upregulation of DEGs and pathways involved in protein degradation, such as two major systems—autophagy–lysosome and ubiquitin–proteosome system. In addition, a significant 18‐fold upregulation of the growth arrest and DNA damage‐inducible 45a protein (Gadd45a) was observed, a critical mediator of stress‐induced muscle atrophy, inducing muscle atrophy via repressing anabolic genes. 29
The myosin loss in patients with metastatic cancer has been shown to be related to the intracellular zinc ion homeostasis regulated by the metal‐ion transporter ZRT/IRT‐like protein 14 (ZIP14) induced by TNF‐α and TGF‐β, resulting in zinc ions uptake from plasma and accumulation in differentiated muscle cells causing MyHC loss. 15 In the present study, pathological low zinc ion concentrations were observed in all neuro‐ICU patients in parallel with higher expression levels of the metal iron transporter ZIP14 and other genes related to ‘cellular zinc ion homeostasis’ early during the observation period (Figure 4 ). In spite of an elevated expression of ZIP14 and related genes, zinc plasma levels increased gradually in all ICU patients and reached normal levels in some, albeit not all, patients at the end of the observation period. However, from the second day of CMV, all neuro‐ICU patients were given intravenous treatment with different metal ions including zinc, offering an explanation to the discrepancy between the expression of zinc transporters and zinc plasma levels. Redistribution of zinc ions from plasma to skeletal muscle may provide a mechanism underlying the myosin loss in CIM pathophysiology. In this context, it is interesting to note that the patient with the initial low myosin:actin ratio had an underlying malignancy and in accordance with the preferential myosin loss reported in clinical and experimental studies on the muscle wasting associated with cancer cachexia. 15 , 30 , 31
Sepsis and high doses of corticosteroids have negative effects on skeletal muscle, but they do not represent prerequisites in the CIM pathophysiology. The main identifiable factors ICU patients with CIM have in common are immobilization and mechanical ventilation. The complete mechanical silencing unique for deeply sedated or pharmacologically paralyzed mechanically ventilated ICU patients, that is, the lack of both external load related to passive mechanical loading by weight bearing and the internal load induced by activation of contractile proteins, is forwarded as one significant contributing factor. 9 Experimental and clinical studies show that passive or electrical stimulation of skeletal muscle only partially reduce the muscle atrophy and myosin loss associated with CIM, and another factor also plays a significant role in the CIM pathophysiology (in review). The preferential myosin loss was observed in all neuro‐ICU patients followed with repeated muscle biopsies in response to prolonged CMV and immobilization. In a previous study from our group, we observed a similar preferential myosin loss in all seven neuro‐ICU patients exposed to long‐term CMV. 8 In the general ICU population, on the other hand, approximately one third of the patients develop CIM, 5 , 32 that is, in a population more frequently exposed to assisted mechanical ventilation than neuro‐ICU patients frequently lacking a strong central ventilatory drive. Assisted mechanical ventilation is the preferred mode of ventilation, when there is a central ventilatory drive, because it induces less lung injury, known as the VILI. In experimental studies in our lab, all rats exposed to CMV for 5 days and longer developed the CIM phenotype with a preferential myosin loss in limb muscles of the fast, slow and mixed type. 9 , 28 , 33 , 34 , 35 , 36 , 37
It is therefore hypothesized that the lung injury induced by CMV and the systemic release of factors with a negative effect on peripheral organs, including respiratory and limb muscles, play an important role in triggering CIM pathophysiology. This is supported by the similar increase of inflammatory and proinflammatory factors in blood in this study as in our previous experimental study. 38 The positive effects of anti‐inflammatory interventions on function of respiratory and limb muscles in rats exposed to long‐term CMV 39 , 40 , 41 as well as the systemic administration of bone marrow‐derived mesenchymal stromal cells targeting lung tissue to reduce VILI and loss of muscle mass and function give further support for the ventilator lung injury being one important factor in a complex biological system resulting in CIM.
In conclusion, a dramatic loss in muscle fibre size and function in parallel with a preferential myosin loss was observed in all neuro‐ICU patients exposed to long‐term CMV, suggesting a link between immobilization, VILI and concomitant release of inflammatory factors impacting on skeletal muscle protein degradation pathways. The zinc homeostasis regulated by ZIP14 and other factors regulating zinc levels are forwarded as a potential factor underlying the preferential myosin loss associated with CIM and cancer cachexia and triggered by circulating cytokines (e.g., TGF‐β) and chemokines, which may provide important early biomarker of CIM as well as a potential target for future intervention strategies. Finally, the altered membrane excitability frequently observed in patients with CIM was not linked to the hallmark of CIM, that is, the preferential myosin loss, and membrane excitability is suggested to relate to mechanisms frequently coinciding with CIM, such as a sepsis‐induced myopathy.
Conflicts of interest
The authors declare no financial or nonfinancial competing interests. Written and verbal informed consent was obtained from a close relative, and the study was approved by the ethics committee at the Karolinska Hospital (Dnr 2016/242‐31/2). The study conforms to the ethical guidelines of the Journal of Cachexia, Sarcopenia and Muscle. 42
Funding
This study was supported by the Swedish Research Council (Vetenskapsrådet), Erling‐Persson Foundation (Familjen Erling‐Perssons Stiftelse), Stockholm City Council (Stockholms läns landsting, Alf), CIF Sweden and Karolinska Institutet.
Supporting information
Figure S1. Clustered expression patterns of DEGs from the neuro‐ICU patients versus healthy control: Time‐course series in the sevel neuro‐ICU patients from whom muscle samples were collected at six time‐points during the 12‐day period. Membership varying from green to red color indicates the degree of genes suitable to the cluster.
Figure S2. Comparision of enriched GObp terms among 5 clusters: The top 10 GObp terms of each clusters were selected for comparision and demonstration. Terms are ranked by ‐log (FDR). Dot size represents the number of DEGs invovled in each term.
Figure S3. Heatmap of DEGs invovled in enriched GObp terms related to proteolysis. DEGs of “Macroautophagy” from Cluster 2.1 (a) and Cluster 2.2 (b); (c) DEGs of “Protein polyubiquitination” from Cluster 2.2; (d) DEGs of “Regulation of protein ubiquitination” from Cluster 2.2.
Figure S4. Comparison of enriched KEGG terms among 5 clusters: The top GObp terms with FDR < 0.1 of each clusters were selected for comparision and demonstration. Terms are ranked by ‐log (FDR). Dot size represents the number of DEGs invovled in each term.
Figure S5. Heatmap of DEGs invovled in enriched KEGG terms related to proteolysis. DEGs of “Lysosome” from Cluster 2.1 (a) and Cluster 4 (b); (c) DEGs of “Proteasome” from Cluster 2.2; (d) DEGs of “Apoptosis” from Cluster 2.2.
Figure S6. KEGG graph of “Lysosome” with DEGs from both Cluster 2.1 (left half) and Cluster 4 (right half) (D5 vs healthy control).
Figure S7. KEGG graph of “Proteasome” with DEGs from Cluster 2.2 (D3 vs healthy control).
Figure S8. Topmost correlated DEPs with myosin/actin ratio.
Figure S9. Knee‐extensor muscle maximum torque. Maximum torque measured three times within 12 months after hospital discharge at a slow (30 deg./s, solid line) and a faster (180 deg./s, broken line) speed of movement in a male (open circles) and female (filled circles) patient.
Table S1. Fold change of 28 DEPs at the six different timepoints.
Cacciani N., Skärlén Å., Wen Y., Zhang X., Addinsall A. B., Llano‐Diez M., Li M., Gransberg L., Hedström Y., Bellander B.‐M., Nelson D., Bergquist J., and Larsson L. (2022) A prospective clinical study on the mechanisms underlying critical illness myopathy—A time‐course approach, Journal of Cachexia, Sarcopenia and Muscle, 13, 2669–2682, doi: 10.1002/jcsm.13104
References
- 1. Friedrich O, Reid MB, Van den Berghe G, Vanhorebeek I, Hermans G, Rich MM, et al. The sick and the weak: neuropathies/myopathies in the critically ill. Physiol Rev 2015;95:1025–1109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Zilberberg MD, Luippold RS, Sulsky S, Shorr AF. Prolonged acute mechanical ventilation, hospital resource utilization, and mortality in the United States. Crit Care Med 2008;36:724–730. [DOI] [PubMed] [Google Scholar]
- 3. Wunsch H. Mechanical ventilation in COVID‐19: interpreting the current epidemiology. Am J Respir Crit Care Med 2020;202:1–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Rodriguez B, Larsson L, Z'Graggen WJ. Critical illness myopathy: diagnostic approach and resulting therapeutic implications. Curr Treat Options Neurol 2022;1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Zilberberg MD, Shorr AF. Prolonged acute mechanical ventilation and hospital bed utilization in 2020 in the United States: implications for budgets, plant and personnel planning. BMC Health Serv Res 2008;8:242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Kurek CJ, Cohen IL, Lambrinos J, Minatoya K, Booth FV, Chalfin DB. Clinical and economic outcome of patients undergoing tracheostomy for prolonged mechanical ventilation in New York state during 1993: analysis of 6,353 cases under diagnosis‐related group 483. Crit Care Med 1997;25:983–988. [DOI] [PubMed] [Google Scholar]
- 7. Esteban A, Anzueto A, Frutos‐Vivar F, Alía I, Ely EW, Brochard L, et al. Outcome of older patients receiving mechanical ventilation. Intensive Care Med 2004;30:639–646. [DOI] [PubMed] [Google Scholar]
- 8. Llano‐Diez M, Renaud G, Andersson M, Marrero HG, Cacciani N, Engquist H, et al. Mechanisms underlying ICU muscle wasting and effects of passive mechanical loading. Crit Care 2012;16:R209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Ochala J, Gustafson AM, Diez ML, Renaud G, Li M, Aare S, et al. Preferential skeletal muscle myosin loss in response to mechanical silencing in a novel rat intensive care unit model: underlying mechanisms. J Physiol 2011;589:2007–2026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Marini JJ, Crooke PS, Tawfik P, Chatburn RL, Dries DJ, Gattinoni L. Intracycle power and ventilation mode as potential contributors to ventilator‐induced lung injury. Intensive Care Med Exp 2021;9:55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Marini JJ, Rocco PRM, Gattinoni L. Static and dynamic contributors to ventilator‐induced lung injury in clinical practice. Pressure, energy, and power. Am J Respir Crit Care Med 2020;201:767–774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Levy MM, Fink MP, Marshall JC, Abraham E, Angus D, Cook D, et al. 2001 SCCM/ESICM/ACCP/ATS/SIS International Sepsis Definitions Conference. Crit Care Med 2003;31:1250–1256. [DOI] [PubMed] [Google Scholar]
- 13. Llano‐Diez M, Fury W, Okamoto H, Bai Y, Gromada J, Larsson L. RNA‐sequencing reveals altered skeletal muscle contraction, E3 ligases, autophagy, apoptosis, and chaperone expression in patients with critical illness myopathy. Skeletal muscle 2019;9:9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Marrero H, Stalberg EV, Cooray G, Corpeno Kalamgi R, Hedstrom Y, Bellander BM, et al. Neurogenic vs. myogenic origin of acquired muscle paralysis in intensive care unit (ICU) patients: evaluation of different diagnostic methods. Diagnostics (Basel) 2020;10:966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Wang G, Biswas AK, Ma W, Kandpal M, Coker C, Grandgenett PM, et al. Metastatic cancers promote cachexia through ZIP14 upregulation in skeletal muscle. Nat Med 2018;24:770–781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Malm C, Nyberg P, Engstrom M, Sjodin B, Lenkei R, Ekblom B, et al. Immunological changes in human skeletal muscle and blood after eccentric exercise and multiple biopsies. J Physiol 2000;529:243–262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Rich MM, Pinter MJ. Crucial role of sodium channel fast inactivation in muscle fibre inexcitability in a rat model of critical illness myopathy. J Physiol 2003;547:555–566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Tankisi H, de Carvalho M, Z'Graggen WJ. Critical illness neuropathy. J Clin Neurophysiol 2020;37:205–207. [DOI] [PubMed] [Google Scholar]
- 19. Z'Graggen WJ, Tankisi H. Critical illness myopathy. J Clin Neurophysiol 2020;37:200–204. [DOI] [PubMed] [Google Scholar]
- 20. Rich MM, Pinter MJ. Sodium channel inactivation in an animal model of acute quadriplegic myopathy. Ann Neurol 2001;50:26–33. [DOI] [PubMed] [Google Scholar]
- 21. Rich MM, Pinter MJ, Kraner SD, Barchi RL. Loss of electrical excitability in an animal model of acute quadriplegic myopathy. Ann Neurol 1998;43:171–179. [DOI] [PubMed] [Google Scholar]
- 22. Norman H, Kandala K, Kolluri R, Zackrisson H, Nordquist J, Walther S, et al. A porcine model of acute quadriplegic myopathy: a feasibility study. Acta Anaesthesiol Scand 2006;50:1058–1067. [DOI] [PubMed] [Google Scholar]
- 23. Ochala J, Ahlbeck K, Radell PJ, Eriksson LI, Larsson L. Factors underlying the early limb muscle weakness in acute quadriplegic myopathy using an experimental ICU porcine model. PLoS One 2011;6:e20876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Aare S, Radell P, Eriksson LI, Chen YW, Hoffman EP, Larsson L. Role of sepsis in the development of limb muscle weakness in a porcine intensive care unit model. Physiol Genomics 2012;44:865–877. [DOI] [PubMed] [Google Scholar]
- 25. Ackermann KA, Bostock H, Brander L, Schroder R, Djafarzadeh S, Tuchscherer D, et al. Early changes of muscle membrane properties in porcine faecal peritonitis. Crit Care 2014;18:484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Lonnqvist PA, Bell M, Karlsson T, Wiklund L, Hoglund AS, Larsson L. Does prolonged propofol sedation of mechanically ventilated COVID‐19 patients contribute to critical illness myopathy? Br J Anaesth 2020;125:e334–e336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Norman H, Zackrisson H, Hedstrom Y, Andersson P, Nordquist J, Eriksson LI, et al. Myofibrillar protein and gene expression in acute quadriplegic myopathy. J Neurol Sci 2009;285:28–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Kalamgi RC, Salah H, Gastaldello S, Martinez‐Redondo V, Ruas J, Fury W, et al. Mechano‐signalling pathways in an experimental intensive critical illness myopathy model. J Physiol 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Ebert SM, Dyle MC, Kunkel SD, Bullard SA, Bongers KS, Fox DK, et al. Stress‐induced skeletal muscle Gadd45a expression reprograms myonuclei and causes muscle atrophy. J Biol Chem 2012;287:27290–27301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Acharyya S, Ladner KJ, Nelsen LL, Damrauer J, Reiser PJ, Swoap S, et al. Cancer cachexia is regulated by selective targeting of skeletal muscle gene products. J Clin Invest 2004;114:370–378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Banduseela V, Ochala J, Lamberg K, Kalimo H, Larsson L. Muscle paralysis and myosin loss in a patient with cancer cachexia. Acta Myol 2007;26:136–144. [PMC free article] [PubMed] [Google Scholar]
- 32. Zilberberg MD, de Wit M, Pirone JR, Shorr AF. Growth in adult prolonged acute mechanical ventilation: implications for healthcare delivery. Crit Care Med 2008;36:1451–1455. [DOI] [PubMed] [Google Scholar]
- 33. Norman H, Nordquist J, Andersson P, Ansved T, Tang X, Dworkin B, et al. Impact of post‐synaptic block of neuromuscular transmission, muscle unloading and mechanical ventilation on skeletal muscle protein and mRNA expression. Pflugers Arch 2006;453:53–66. [DOI] [PubMed] [Google Scholar]
- 34. Ochala J, Larsson L. Effects of a preferential myosin loss on Ca2+ activation of force generation in single human skeletal muscle fibres. Exp Physiol 2008;93:486–495. [DOI] [PubMed] [Google Scholar]
- 35. Renaud G, Llano‐Diez M, Ravara B, Gorza L, Feng HZ, Jin JP, et al. Sparing of muscle mass and function by passive loading in an experimental intensive care unit model. J Physiol 2013;591:1385–1402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Corpeno R, Dworkin B, Cacciani N, Salah H, Bergman HM, Ravara B, et al. Time course analysis of mechanical ventilation‐induced diaphragm contractile muscle dysfunction in the rat. J Physiol 2014;592:3859–3880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Kalamgi RC, Larsson L. Mechanical signaling in the pathophysiology of critical illness myopathy. Front Physiol 2016;7:23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Lyu Q, Wen Y, Zhang X, Addinsall AB, Cacciani N, Larsson L. Multi‐omics reveals age‐related differences in the diaphragm response to mechanical ventilation: a pilot study. Skeletal Muscle 2021;11:11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Akkad H, Cacciani N, Llano‐Diez M, Corpeno Kalamgi R, Tchkonia T, Kirkland JL, et al. Vamorolone treatment improves skeletal muscle outcome in a critical illness myopathy rat model. Acta Physiol (Oxf) 2018;e13172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Cacciani N, Salah H, Li M, Akkad H, Backeus A, Hedstrom Y, et al. Chaperone co‐inducer BGP‐15 mitigates early contractile dysfunction of the soleus muscle in a rat ICU model. Acta Physiol (Oxf) 2020;229:e13425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Salah H, Li M, Cacciani N, Gastaldello S, Ogilvie H, Akkad H, et al. The chaperone co‐inducer BGP‐15 alleviates ventilation‐induced diaphragm dysfunction. Sci Translat Med 2016;8:350ra103. [DOI] [PubMed] [Google Scholar]
- 42. von Haehling S, Morley JE, Coats AJS, Anker SD. Ethical guidelines for publishing in the Journal of Cachexia, Sarcopenia and Muscle: update 2021. J Cachexia Sarcopenia Muscle 2021;12:2259–2261. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Clustered expression patterns of DEGs from the neuro‐ICU patients versus healthy control: Time‐course series in the sevel neuro‐ICU patients from whom muscle samples were collected at six time‐points during the 12‐day period. Membership varying from green to red color indicates the degree of genes suitable to the cluster.
Figure S2. Comparision of enriched GObp terms among 5 clusters: The top 10 GObp terms of each clusters were selected for comparision and demonstration. Terms are ranked by ‐log (FDR). Dot size represents the number of DEGs invovled in each term.
Figure S3. Heatmap of DEGs invovled in enriched GObp terms related to proteolysis. DEGs of “Macroautophagy” from Cluster 2.1 (a) and Cluster 2.2 (b); (c) DEGs of “Protein polyubiquitination” from Cluster 2.2; (d) DEGs of “Regulation of protein ubiquitination” from Cluster 2.2.
Figure S4. Comparison of enriched KEGG terms among 5 clusters: The top GObp terms with FDR < 0.1 of each clusters were selected for comparision and demonstration. Terms are ranked by ‐log (FDR). Dot size represents the number of DEGs invovled in each term.
Figure S5. Heatmap of DEGs invovled in enriched KEGG terms related to proteolysis. DEGs of “Lysosome” from Cluster 2.1 (a) and Cluster 4 (b); (c) DEGs of “Proteasome” from Cluster 2.2; (d) DEGs of “Apoptosis” from Cluster 2.2.
Figure S6. KEGG graph of “Lysosome” with DEGs from both Cluster 2.1 (left half) and Cluster 4 (right half) (D5 vs healthy control).
Figure S7. KEGG graph of “Proteasome” with DEGs from Cluster 2.2 (D3 vs healthy control).
Figure S8. Topmost correlated DEPs with myosin/actin ratio.
Figure S9. Knee‐extensor muscle maximum torque. Maximum torque measured three times within 12 months after hospital discharge at a slow (30 deg./s, solid line) and a faster (180 deg./s, broken line) speed of movement in a male (open circles) and female (filled circles) patient.
Table S1. Fold change of 28 DEPs at the six different timepoints.