

Abstract
Background
Muscle atrophy, leading to muscular dysfunction and weakness, is an adverse outcome of sustained period of glucocorticoids usage. However, the molecular mechanism underlying this detrimental condition is currently unclear. Pyruvate dehydrogenase kinase 4 (PDK4), a central regulator of cellular energy metabolism, is highly expressed in skeletal muscle and has been implicated in the pathogenesis of several diseases. The current study was designed to investigated and delineate the role of PDK4 in the context of muscle atrophy, which could be identified as a potential therapeutic avenue to protect against dexamethasone‐induced muscle wasting.
Methods
The dexamethasone‐induced muscle atrophy in C2C12 myotubes was evaluated at the molecular level by expression of key genes and proteins involved in myogenesis, using immunoblotting and qPCR analyses. Muscle dysfunction was studied in vivo in wild‐type and PDK4 knockout mice treated with dexamethasone (25 mg/kg body weight, i.p., 10 days). Body weight, grip strength, muscle weight and muscle histology were assessed. The expression of myogenesis markers were analysed using qPCR, immunoblotting and immunoprecipitation. The study was extended to in vitro human skeletal muscle atrophy analysis.
Results
Knockdown of PDK4 was found to prevent glucocorticoid‐induced muscle atrophy and dysfunction in C2C12 myotubes, which was indicated by induction of myogenin (0.3271 ± 0.102 vs 2.163 ± 0.192, ****P < 0.0001) and myosin heavy chain (0.3901 ± 0.047 vs. 0.7222 ± 0.082, **P < 0.01) protein levels and reduction of muscle atrophy F‐box (10.77 ± 2.674 vs. 1.518 ± 0.172, **P < 0.01) expression. In dexamethasone‐induced muscle atrophy model, mice with genetic ablation of PDK4 revealed increased muscle strength (162.1 ± 22.75 vs. 200.1 ± 37.09 g, ***P < 0.001) and muscle fibres (54.20 ± 11.85% vs. 84.07 ± 28.41%, ****P < 0.0001). To explore the mechanism, we performed coimmunoprecipitation and liquid chromatography‐mass spectrometry analysis and found that myogenin is novel substrate of PDK4. PDK4 phosphorylates myogenin at S43/T57 amino acid residues, which facilitates the recruitment of muscle atrophy F‐box to myogenin and leads to its subsequent ubiquitination and degradation. Finally, overexpression of non‐phosphorylatable myogenin mutant using intramuscular injection prevented dexamethasone‐induced muscle atrophy and preserved muscle fibres.
Conclusions
We have demonstrated that PDK4 mediates dexamethasone‐induced skeletal muscle atrophy. Mechanistically, PDK4 phosphorylates and degrades myogenin via recruitment of E3 ubiquitin ligase, muscle atrophy F‐box. Rescue of muscle regeneration by genetic ablation of PDK4 or overexpression of non‐phosphorylatable myogenin mutant indicates PDK4 as an amenable therapeutic target in muscle atrophy.
Keywords: PDK4, myogenin, ubiquitin–proteasomal system, phosphorylation, glucocorticoids, muscle atrophy
Introduction
Muscle atrophy, the wasting or loss of muscle tissue, is a debilitating consequence of aging and malnutrition, as well as several diseases including diabetes, obesity, cancer cachexia and long‐term corticosteroid therapy. 1 , 2 , 3 Glucocorticoids (GCs) are the most prescribed drugs used as an anti‐inflammatory and immunosuppressive agent. 4 However, it exhibits a wide range of dose‐limiting side effects such as muscle atrophy, osteoporosis, hyperglycaemia, insulin resistance, abnormal fat deposition and hypertension. 4 GC‐induced loss of muscle mass causes atrophy of type II muscle fibres and elicit muscle wasting by increasing the rate of protein catabolism and decreasing the rate of protein anabolism. 5
The catabolic action of GC mainly leads to myofibrillar protein degradation, which depends on ubiquitin–proteasome system (UPS) and autophagy. 3 GCs have been demonstrated to promote the induction of gene expression of several genes involved in the UPS. 3 Muscle‐specific E3‐ubiquitin‐ligases muscle ring finger1 (MuRF1)/Trim63 and muscle atrophy‐F‐box MAFbx/Atrogin‐1 are two such genes regulated by GC and regarded as the key markers of muscle atrophy. 3 , 6 To further underline the importance of the UPS system in the context of GC therapy‐induced muscle atrophy, several previous studies have demonstrated that inhibition of either of these two muscle‐specific E3‐ligases, Atrogin‐1/MAFbx or MuRF1/Trim63, protects against muscle atrophy following starvation, GC therapy and cardiac‐cachexia. 7 , 8 On the other hand, myogenin (MYOG), a muscle‐specific basic‐helix–loop–helix (bHLH) transcription factor, has been implicated in the coordination of skeletal muscle development and repair. 9 Mice lacking Myog lacks mature secondary skeletal muscle fibres throughout the body, leading to severe defects in skeletal muscle development and suffering from perinatal lethality. 9
Among the four pyruvate dehydrogenase kinase isoforms (PDK1‐4) that regulate the mitochondrial pyruvate dehydrogenase (PDH) activity via phosphorylation, PDK4 is expressed at a higher level in the heart and muscle. 10 PDK4 has the highest basal activity among the PDKs and has been demonstrated to be dramatically elevated during starvation and in the skeletal muscle of insulin resistance patients as well as in several pathological conditions related to muscle atrophy such as cancer, sepsis and amyotrophic lateral sclerosis. 11 , 12 Genetic ablation or pharmacological inhibition of Pdk4 has shown beneficial effects in statin‐induced myopathies in mice and in in vitro cancer cachexia model. 2 , 13 However, the exact involvement of PDK4 in the context of muscle atrophy remains unclear.
Here, we tested the hypothesis that upregulation of PDK4 during pathological conditions is a principal driving factor of muscle atrophy. Based on this concept, we investigated the mechanism underlying PDK4‐driven muscle atrophy and explored the possibility of targeting PDK4 to reverse muscle loss and induce muscle regeneration in a mouse model of GC‐induced muscle atrophy, paving way for a potential therapeutic option for this debilitating condition.
Material and methods
Animal models
All mouse experiments were approved by the Animal Care and Use Committee of Daegu‐Gyeongbuk Innovation Medical Foundation (DGMIF‐20111601‐00). Gastrocnemius anterior (GA) and tibialis anterior (TA) muscle tissues were harvested from male 12‐weeks‐old ob/ob, 18‐weeks‐old CRH‐Tg C57BL/6J mice and 6‐weeks‐old BALB/c mice (cancer cachexia model) under fed‐condition. Dexamethasone (DEX)‐induced muscle atrophy models were developed as described previously. 3 Briefly, 8‐week‐old male wild‐type (WT) and Pdk4 −/− C57BL/6J mice were randomly allocated to four groups (n = 8 in each group). Control mice were administrated with PEG‐solution (10 mL/kg body weight, 30% in 0.9% saline); DEX‐group was administered with DEX (25 mg/kg dissolved in PEG 400 solution), intraperitoneally, every other day for 10 days. Mouse body weight was recorded daily, and the grip‐strength was recorded three times using the grip‐strength test‐meter (Jeung Do Bio and Plant Co. Ltd, Seoul, South Korea). Twenty‐four hours after the last intraperitoneal injection, mice were sacrificed. GA, TA, extensor digitorum longus (EDL) and soleus (SOL) muscles were isolated for further analyses. For adenoviral infection, mice were anaesthetized using isoflurane and GFP or MYOGAA adenoviruses (3 × 1011 p.f.u) were injected intramuscularly in GA muscle for 2 days. In the cancer cachexia model, CT‐26 cells (3 × 106 cells per mouse) were subcutaneously injected into the left flanks of male BALB/c mice as described in a previous study. 14 Detailed mice information is listed in Table S1 .
Human tissue
Human skeletal (gluteus‐maximus) muscle was collected from the remnant muscle tissues during receiving hip replacement surgery after approval from the Kyungpook National University Hospital (IRB No. KNUH 2020‐11‐028‐001). All patients who underwent hip replacement surgery were classified according to steroid administration. Control muscle biopsy samples were from patients who did not use steroids but needed surgery for other reasons. Detailed patients' information is listed in Table S2 .
Cell culture and differentiation
Primary human normal and type‐2‐diabetic skeletal myoblast (SKM‐D‐F/SKB‐F‐SL; ZenBio), were maintained in skeletal muscle cell growth medium (#SKM‐M; ZenBio) and differentiated into myotubes with differentiation medium (#SKM‐D; ZenBio). C2C12 myoblasts, AD293Tcell line (ATCC). C2C12 myoblast stably expressing PDK4‐FLAG and VXY control were maintained in DMEM high glucose media (#LM001‐05; Welgene). Colon cancer (CT‐26) cells were maintained in RPMI‐1640 media (#SH30027.01; HyClone) supplemented with 10% foetal bovine serum (#SH30084.03; HyClone) and antibiotic‐antimycotic (#15240‐062; Gibco). C2C12 myoblast were differentiated into myotubes with DMEM containing 2% horse serum (Gibco) for 6 days. For in vitro cancer cachexia‐induced muscle atrophy model, the C2C12 myotubes were exposed to conditioned medium (CM). CM is defined as 1:1 ratio of media collected from confluent CT‐26 culture and C2C12 maintenance media.
Plasmid and adenoviral constructs
C‐terminus HA‐tagged MYOGS43A and MYOGT57A mutants were constructed from human MYOG cDNA construct (Addgene #78341) by replacing serine/threonine with alanine using the Site‐Directed Mutagenesis Kit (210518‐5; Agilent Technologies). PDK4‐FLAG plasmid was provided by Dr Robert A. Harris (Indiana University, USA). The AdEasy system was used to generate adenoviruses. 15 pcDNA3 and mock adenovirus served as a control for transient transfections and adenovirus transductions, respectively.
Cell culture transfection, adenovirus transduction, treatment condition and siRNA
On the third day of differentiation, cells were transfected with siRNA using RNAi Max reagent (#13778150; ThermoFisher Scientific) or transduced with adenovirus for 48 h. Then the cells were exposed to DEX for 24 h. For overexpression, cells were transfected with plasmid DNA using Lipofectamine 2000 (11668019; ThermoFisher Scientific). Predesigned siRNAs targeting mouse Mafbx (#67731), siControl (#SN‐1002) were from Bioneer (South Korea).
Coimmunoprecipitation and immunoblotting
Co‐immunoprecipitation and immunoblotting were performed as described previously. 11 Mouse monoclonal pierce anti‐HA agarose (#26181, Cell Signaling Technology), protein A/G‐plus‐agarose (#SC2003; Santa Cruz Biotechnology) and mouse anti‐FLAG M2‐agarose (#A2220; Sigma) beads used for co‐immunoprecipitation. Proteins were analysed with their corresponding specific antibodies. Antibody information is listed in the Table S1 .
Immunofluorescence imaging
After differentiation for 6 days, cells were fixed in 4% paraformaldehyde (PFA) for 30 min, permeabilized (0.2% Triton X, 0.1M glycine in phosphate‐buffered saline) at RT for 15 min and incubated with the indicated primary antibodies overnight at 4°C. Then, the cells were washed, incubated with secondary antibodies conjugated with fluorophores (Alexa‐488/568) and mounted using VECTASHIELD mounting medium containing DAPI (H1200). Myogenin immunofluorescent intensity was from at least five images per well of 96‐well‐plates randomly selected then fluorescence was quantified using ImageXpress Micro Confocal and MetaXpress software (molecular devices). The fusion‐index and nuclei number were calculated as described previously. 16 Nuclei were counted from five images per well of 96‐well‐plates using ImageJ. Nuclear number assays were determined by calculating the percentage of nuclei incorporated into the MyHC positive myotubes with the indicated number of nuclei.
Liquid chromatography–mass spectrometry
For liquid chromatography– tandem mass spectrometry (LC–MS/MS), Coomassie‐stained sodium dodecyl‐sulfate polyacrylamide gel electrophoresis gel of affinity proteins were de‐stained and processed as follows. Briefly, protein bands were trypsin‐digested in‐gel. Tryptic digests were extracted and separated by online reversed‐phase chromatography using Eazy nano LC1200 ultra‐high performance LC followed by electrospray ionization at a flow rate of 300 nL min−1. Samples were eluted in split gradient followed column wash. The chromatography system was coupled with an Orbitrap Fusion Lumos mass spectrometer. The top 50 precursors were fragmented, and spectra were acquired in CID mode. Obtained spectra were validated by SEQUEST (Thermo Scientific, San Jose, CA, USA) interfaced with Mascot algorithm (Mascot 2.4, Matrix Science).
In vitro kinase assay
MYOG phosphorylation by PDK4 was analysed as described previously. 17 Briefly, PDK4 recombinant protein (provided by Dr Nam‐Ho Jeoung) and human recombinant MYOG protein (#H00004656‐P01; Novus Biological) were mixed in 1X‐kinase‐buffer (#9802S; Cell Signaling) and incubate 30 min at 30°C in the presence of 500‐μM ATP (A2383; Sigma). Samples were analysed by immunoblotting.
In situ proximity ligation assay
Proximity ligation assays (PLA) in AD293Tcells were performed as described previously. 11 Images were captured using an ImageXpress Micro Confocal (Molecular Devices).
Quantitative real‐time PCR
Total RNA was isolated from muscle tissue/cells by using QIAzol lysis reagent (#79306; Qiagen). A 2 μg of total RNA was used for cDNA synthesis (#K1622; Thermo Scientific), and qPCR was performed. Primers are listed in Table S1 .
Measurement of muscle fibre cross‐sectional area
Tissues were fixed in 4% PFA overnight and then embedded into paraffin; 4‐μm muscle sections were cut in cross‐section and haematoxylin and eosin staining was performed by standard staining procedures. Using a VS200 image analysis system (Olympus, Tokyo, Japan), a total of 25 microscopic fields were randomly selected from each slide. The cross‐sectional diameter of muscle fibres was measured using ImageJ (NIH).
Statistical analysis
GraphPad Prism 8 (La Jolla, CA, USA) was used for analysis. Student's t‐test and one‐way or two‐way analysis of variance (ANOVA) was performed with recommended comparison test from at least three independent experiments. P < 0.05 was considered statistically significant. Data are presented as mean ± SEM.
Results
PDK4 expression inversely correlates with MYOG expression in human and mouse models of muscle atrophy
Enhanced PDK4 expression is associated with impaired skeletal muscle regenerative capacity or muscle loss leading to multiple diseases. 18 , 19 Therefore, to test whether PDK4 is directly involved in the pathogenesis, we first investigated endogenous PDK4 expression changes in multiple models of muscle atrophy. PDK4 protein level was higher in GA and TA muscles from obese mice (ob/ob), corticotrophin‐releasing hormone transgenic (CRH‐Tg) mice and in isolated type‐2‐diabetic human myotubes (hT2DM) (Figures 1 A and S1A ). This was accompanied by a reduction in MYOG protein level, a key transcriptional regulator of myogenesis (Figures 1 A and S1A ). Enhanced PDK4 level was further evidenced in gluteus‐maximus (GM) muscles isolated from steroid‐treated patients. Similarly, we observed a reduction in MYOG level in these tissues when compared with healthy subjects (Figure 1 B ). Impaired muscle regeneration was confirmed by histology, as evidenced by a reduction in fibre diameter in steroid‐treated patients' samples (Figure 1 C ). Next, analysis of publicly available transcriptomics dataset (GEO: GSE169571) further demonstrated that among all the Pdk isoforms only Pdk4 expression was upregulated in quadriceps from GCs‐treated (dexamethasone) mice (Figure 1 D ). Interestingly, although the downstream transcriptional target genes of MYOG, myomarker (Mymk) and myomixer (Mymx), were reduced in dexamethasone (DEX)‐treated tissue samples, however, Myog gene expression remained unchanged, suggesting that the regulation of MYOG is at the posttranscriptional level.
Figure 1.
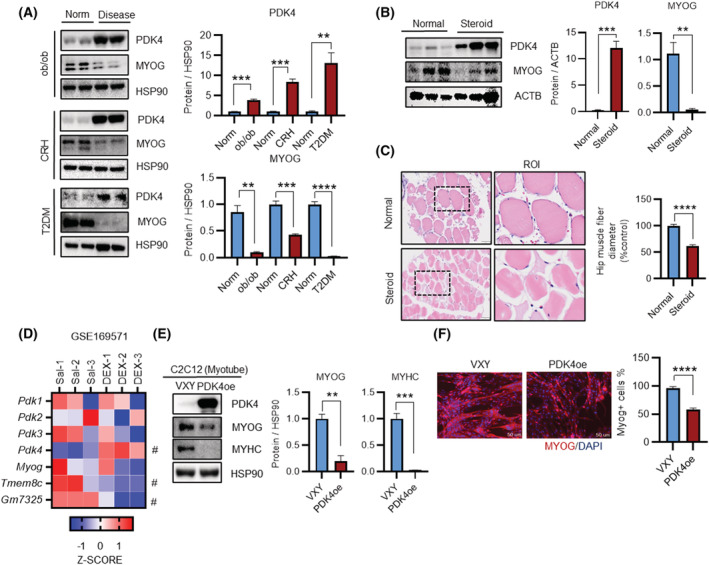
PDK4 is upregulated in human and mouse models of muscle atrophy. (A) Immunoblot analysis of indicated proteins in gastrocnemius anterior (GA) muscles from obese, corticotrophin releasing hormone (CRH)‐Tg mice, and Type 2 diabetes (T2D) human myotube. (Right) Quantification represents the levels of the indicated protein normalized to HSP90. (B) Immunoblot analysis of indicated proteins in human gluteus maximus muscles (GM) from a normal subject and steroid patients. (Right) Quantification represents the levels of the indicated protein normalized to ACTB. (C) Representative image of haematoxylin and eosin staining of myofibre cross section isolated from human gluteus maximus (hGM) muscles. A microscope with a ×20 objective was used to capture the images. (Right) the cross‐sectional diameter of human gluteus maximus (GM) muscles. (D) (GSE169571) RNA sequencing analysis of quadriceps muscles from saline and dexamethasone (DEX) treated mouse. (E) Immunoblot analysis of indicated proteins in VXY and FLAG‐tagged PDK4 overexpressing C2C12 myotubes stable cells. (Right) Quantification represents the levels of the indicated protein normalized to HSP90. (F) Representative image of MYOG immunofluorescence in VXY and PDK4‐flag expressing C2C12 myotubes stable cells. (Right) Quantification represents the levels of the MYOG + ve cell (Mean ± SEM). (C, F) Scale bar 50 μM. (A, D and E) n = 3. (F) n = 4; number of image capture sites 20 from each group. (B, C) n = 6 human per group. **P < 0.01; ***P < 0.001; ****P < 0.0001. Unpaired t‐test was used for all analyses.
To further explore the involvement of PDK4 in the muscle regeneration process, we generated a mouse C2C12 stable‐cell line overexpressing FLAG‐tagged PDK4 (Pdk4oe). Protein levels of MYOG and MYHC were significantly decreased in PDK4oe cells as compared to control (VXY) (Figure 1 E ). Furthermore, immunofluorescence analysis showed a reduction in MYOG as well as MYHC intensity with reduced cell fusion and lessened multinuclear myotubes formation in the Pdk4oe cells than those in the control (Figures 1 F and S1B,C ). Consistent with the publicly available RNAseq result, there was no significant difference in Myog mRNA level between Pdk4oe cells and control (Figure S1A ). Together, these data indicate that PDK4 is a potential driver for reducing MYOG expression levels in the muscle's regeneration process of different disease models.
PDK4 depletion prevents glucocorticoids‐induced muscle atrophy
Upon establishing a positive correlation between PDK4 expression and muscle atrophy in vivo, we next sought to utilize an in vitro GC‐induced model 20 by challenging cells with synthetic GC (DEX), to investigate the contribution of PDK4 towards the development of muscle atrophy. As reported previously, 19 , 21 PDK4 protein level were induced upon DEX‐stimulation in a dose‐dependent manner (Figure 2A ). In line with our earlier observation (Figure 1 ), protein levels of myogenesis markers, MYOG and MYHC, were downregulated whereas the muscle specific E3‐ubiquitin ligases MAFbx/ATROGIN‐1 and TRIM63/MURF‐1 levels were upregulated upon DEX‐stimulation (Figure 2 A ).
Figure 2.
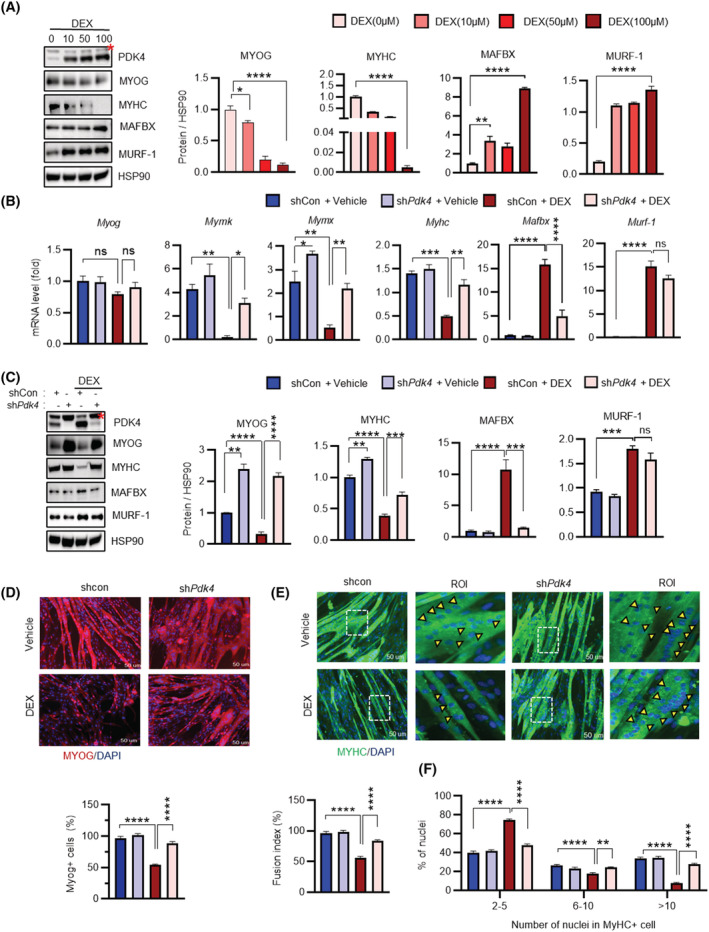
Knockdown of PDK4 prevents glucocorticoids‐induced muscle atrophy in vitro. (A) Immunoblot analysis of indicated protein in C2C12 myotubes after being treated with dexamethasone (DEX) for 24 h with different concentrations of 0, 10, 50 and 100 μM. (Right) Quantification represents the levels of the indicated protein normalized to HSP90. (B) qPCR analysis of indicated genes in C2C12 myotubes transfected with shPdk4 or shControl and treated with or without dexamethasone (DEX) (100 μM) for 24 h. (C) Immunoblot analysis of indicated proteins in C2C12 myotubes transfected with shPdk4 or shControl and treated with or without dexamethasone (DEX) (100 μM) for 24 h. (Right) Quantification represents the levels of the indicated protein normalized to HSP90. (D, E) Representative image of MYOG and MYHC immunofluorescence of C2C12 myotubes transfected with shPdk4 or shControl and treated with or without dexamethasone (DEX) (100 μM) for 24 h. (Below) Quantification represents the levels of the MYOG + ve cell and (Below) fusion index. (F) The distribution of nuclei per myotubes were calculated at Day 6 differentiation. Yellow arrowheads number of nuclei in one myotube. Mean ± SEM. (D and E) Scale bar 50 μM. (A–C) n = 3. (D, E) n = 4; number of image capture sites 20 from each group. *P < 0.05; **P < 0.01; ***P < 0.001; ****P < 0.0001. Dunnett's multiple comparisons test was used for the analysed panel (A). *P < 0.05; **P < 0.01; ***P < 0.001; ****P < 0.0001. Tukey's multiple comparisons test was used for the analysed panels (B–F). ROI, region of interest. Red * = nonspecific band.
Next, to determine whether PDK4 inhibition could reverse DEX‐induced muscle atrophy phenotype, we used adenovirus‐mediated targeted knockdown PDK4 (shPdk4) and chose optimal DEX‐stimulatory dose (100 μM) to mimic muscle atrophy in C2C12 myotubes. PDK4 knockdown was confirmed at protein level (Figure 2 C ). However, neither DEX‐treatment nor PDK4 knockdown led to any change in the protein level of the other PDK‐isoforms (Figure S2A ), suggesting that the effect of DEX on muscle atrophy might be solely a PDK4‐dependent phenomenon. DEX‐stimulation resulted in an increase in PDK4 level in mock virus‐infected (shControl) cells but not in shPdk4‐infected cells. Conversely, DEX‐triggered reduction in MYOG and MYHC protein levels were reversed in PDK4 depleted cells (Figure 2 C ) whereas no difference in MYOD expression was observed upon PDK4 depletion (Figure S2A ). Consequently, reduction in mRNA levels of key MYOG downstream transcriptional targets Mymk, Mymx and MyHC was reversed in PDK4 depleted cells (Figure 2B ). However, there was no significant change at the mRNA level of MYOG upon DEX‐stimulation, suggesting that PDK4‐induced downregulation of MYOG occurs at the posttranscriptional level. Additionally, immunofluorescence staining demonstrated that DEX‐triggered reduction in MYOG and MYHC staining, as well as reduction in cell fusion and multinucleated myotubes formation, was reversed upon knockdown of PDK4 (Figure 2 D–F ). We further observed that although both MAFbx and MURF‐1 protein levels were upregulated upon DEX‐stimulation, only MAFbx level was affected upon PDK4‐depletion. Additionally, PDK4 knockdown had no effect on the expression of E3‐ubiquitin ligases Fbxo‐30 and TRIM32 22 (Figure S2B ). Thus, we hypothesized that PDK4 depletion in the DEX‐induced muscle atrophy model might stabilize MYOG protein. Stabilization of MYOG, in turn, induces its downstream transcriptional targets that contribute towards the muscle regeneration process.
PDK4 is a glucocorticoid‐sensing kinase targeting MYOG for ubiquitin‐dependent degradation
Next, we sought to determine whether PDK4‐dependent downregulation of MYOG involves the UPS‐mediated degradation. To that end, we transiently overexpressed FLAG‐tagged PDK4 (PDK4‐FLAG) and HA‐tagged MYOG (HA‐MYOG) in the absence or presence of a 26S proteasomal inhibitor (MG132) in AD293T cells. Immunoblot analysis revealed a reduction in MYOG level upon co‐expression with PDK4, which was reversed upon MG132 co‐treatment (Figure 3 A ). Earlier, we have observed that PDK4 depletion led to enhanced MYOG expression even under basal conditions (Figure 2 C ). These observations indicated that PDK4‐induced MYOG degradation might involve the UPS. To examine MYOG ubiquitination in the absence or presence of PDK4, we used ubiquitin motif detecting antibody and performed immunoprecipitation with MYOG in the presence of MG132. A ladder of high molecular weight ubiquitinated MYOG products were detected in cells co‐transfected with PDK4, confirming that MYOG is targeted for degradation in the presence of PDK4 (Figure 3 B ).
Figure 3.
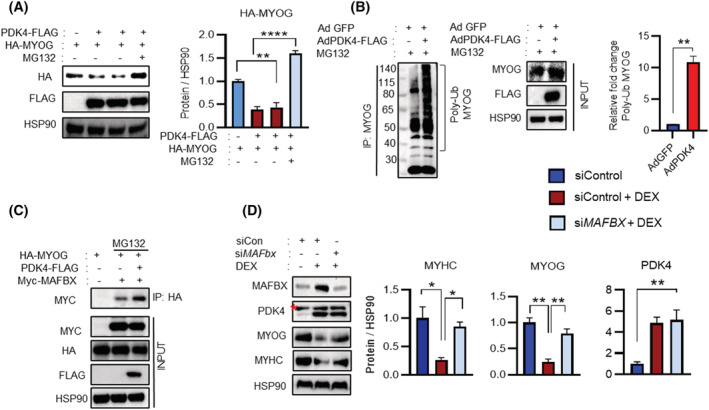
PDK4 mediates MYOG proteasomal degradation by ubiquitination. (A) Immunoblot analysis of indicated proteins in AD293T cells transfected with indicated constructs. After 48 h, cells were treated with or without MG132 10 μM for 6 h. (Right) Quantification represents the levels of the indicated protein normalized to HSP90. (B) C2C12 myotubes were transfected with the indicated Adenovirus (Ad‐Pdk4‐flag) and (Ad‐GFP) in the presence of MG132 10 μM for 6 h. Co‐immunoprecipitation (co‐IP) using MYOG antibody and immunoblot analysis of indicated proteins. (Right) Quantification represents the levels of the poly‐ub MYOG protein. (C) AD293T cells were transfected with indicated constructs; after 48 h, cells were treated with or without MG132 10 μM for 6 h. Co‐immunoprecipitation (co‐IP) using HA antibody and immunoblot analysis of indicated proteins. (D) Immunoblot analysis of indicated proteins in C2C12 myotubes transfected with control or Mafbx siRNA in the presence or absence of dexamethasone (DEX) 100 μM for 24 h. (Right) Quantification represents the levels of the indicated protein normalized to HSP90. Mean ± SEM. (A, D) n = 3. *P < 0.05; **P < 0.01; ***P < 0.001; ****P < 0.0001. Tukey's multiple comparisons test was used for the analysed panel (A) and unpaired t‐test was used for the analysed panel (B). Red * = nonspecific band.
A previous report indicated that MYOG is ubiquitinated by MAFbx, which belongs to the SCF‐complex. 23 Therefore, we investigated whether PDK4‐induced MYOG ubiquitination by recruitment of MAFbx. AD293Tcells were transiently transfected with PDK4, MYOG and MAFbx, an E3‐ubiquitin‐ligase and a key component of the SCF‐complex. Co‐immunoprecipitation (co‐IP) assays demonstrated that MYOG interacts with MAFbx, and this interaction was enhanced upon PDK4 expression and PDK4 overexpression resulted in MG132‐sensitive reduction of MYOG. (Figures 3 C and S3A ). However, PDK4 overexpression did not promote interaction between MYOG and another well‐known muscle specific E3‐ubiquitin ligase, MuRF‐1 (Figure S3B ), suggesting that PDK4‐induced degradation of MYOG is MAFbx dependent. To ascertain this observation in the context of muscle atrophy, we stimulated C2C12 myotubes with DEX following the siRNA‐mediated knockdown of MAFbx. Immunoblot analysis revealed that DEX‐induced downregulation of MYOG was reversed upon knockdown of MAFbx, without affecting the PDK4 protein level (Figure 3 D ). Together, these results indicate that PDK4 promotes MAFbx recruitment to catalyse muscle atrophy induced MYOG degradation.
MYOG is a novel substrate of PDK4
To validate MYOG as a potential substrate of PDK4, we performed a co‐IP assay in C2C12 myotubes stably overexpressing PDK4 and in AD293Tcells by overexpressing PDK4 and MYOG in the absence or presence of MG132. We observed that MYOG physically interacted with PDK4 (Figures 4 A and S4A ), and this interaction was further validated by PLA (Figure 4 B ). In vitro kinase assay revealed that MYOG was directly phosphorylated by recombinant PDK4 (Figure 4 C ), suggesting that MYOG is a novel phosphorylation substrate for PDK4. Using LC–MS/MS analysis, we identified two predicted candidate PDK4 phosphorylation sites, Ser43 and Thr57 in human MYOG (Figure S4B,C ), which lie in the transcriptional activation domain (TAD) of MYOG. 24 We used phosphomotif antibody (p‐S/T) directed against the phosphorylated serine and threonine residues to determine whether they recognize MYOG phosphorylated at Ser43 or Thr57 in AD293Tcells. HA‐tagged variants of MYOG cDNAs were transiently transfected, and co‐expression with PDK4 increased MYOG phosphorylation. Non‐phosphorylatable MYOGS43A, MYOGT57A, or double mutation (MYOGAA) abolished the interaction with the phosphomotif antibody (Figure 4 D ). Additionally, all these non‐phosphorylatable mutants stabilized MYOG protein even in the presence of PDK4 (Figure 4 D ). Cycloheximide (CHX) chase analysis of MYOG degradation in AD293Tcells co‐transfected with PDK4 further demonstrated the enhanced protein stability of the MYOGAA mutant (Figures 4 E and S4D ). Next, to determine whether non‐phosphorylatable MYOG mutant prevented UPS‐mediated MYOG degradation, C2C12 myotubes were transduced with adenovirus overexpressing MYOGAA mutant (Ad‐MYOGAA) or mock virus (Ad‐Mock). Treatment of the cells with DEX increased MYOG phosphorylation and ubiquitination as well as enhanced interaction between MYOG and MAFbx and all these effects were attenuated in the MYOGAA‐overexpressing cells (Figures 4 F and S4E,F ). Taken together, we have shown that MYOG protein stability is regulated by PDK4‐induced phosphorylation and recruitment of E3‐ligase MAFbx and that conditions that exacerbate muscle atrophy exaggerate PDK4‐induced MYOG phosphorylation and degradation.
Figure 4.
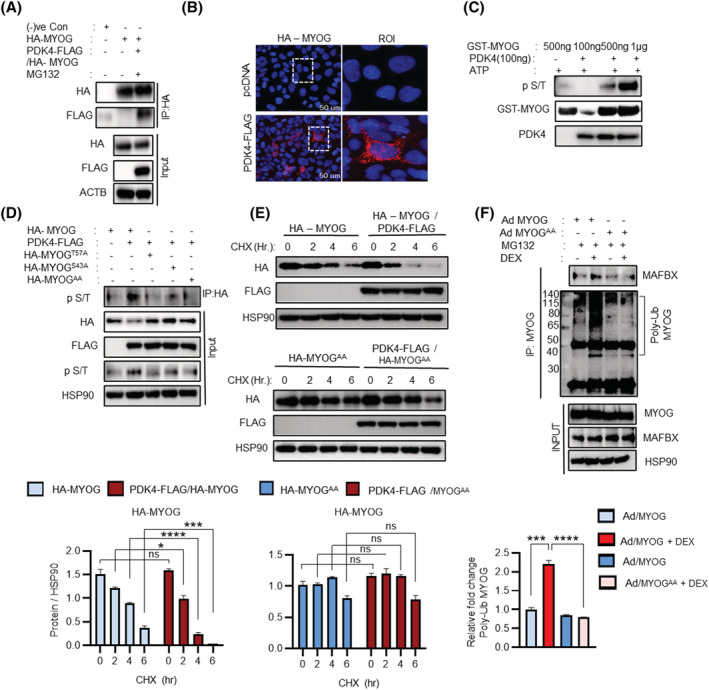
Identification of MYOG as a novel substrate of PDK4. (A) AD293T cells were transfected with indicated constructs. After 48 h, cells were treated with or without MG132 10 μM for 6 h. Co‐immunoprecipitation (co‐IP) using HA antibody and immunoblot analysis of indicated proteins. (B) AD293T cells were transfected with indicated constructs and performed in situ proximity ligation assay (PLA). (C) In vitro kinase assay. Using recombinant GST‐MYOG and PDK4 protein in the presence of ATP and the phosphorylation was detected by indicated antibody. (D) AD293T cells were transfected with indicated constructs for 48 h. Co‐immunoprecipitation (co‐IP) using HA antibody and immunoblot analysis of indicated proteins. (E) AD293T cells were transfected with indicated constructs. After 48 h, transfected cells were treated with cycloheximide (CHX) 100 μM and incubated cells for indicated time before harvest. Immunoblot analysis of indicated proteins. (Below) Quantification represents the levels of the indicated protein normalized to HSP90. (F) C2C12 myotubes were infected with adenovirus expressing MYOG (AdMYOG) and mutant MYOGᴬᴬ (AdMYOGᴬᴬ) in the presence or absence of dexamethasone (DEX) (100 μM) for 24 h, treated MG132 10 μM for 6 h. Co‐immunoprecipitation (co‐IP) using HA antibody and immunoblot analysis of indicated proteins. (Below) Quantification represents the levels of the poly‐ub MYOG protein. Mean ± SEM. (B) Scale bar 50 μM. (E) n = 3. *P < 0.05; **P < 0.01; ***P < 0.001; ****P < 0.0001. Unpaired t‐test was used for the analysed panels (E, F). ROI, region of interest.
Ectopic overexpression of non‐phosphorylatable MYOG mutant protects against glucocorticoid‐induced muscle atrophy
To understand the mechanistic significance of our findings, we initially investigated the effect of phospho‐mutant MYOG in cultured human myotubes by overexpressing Ad‐GFP or Ad‐MYOGAA. DEX‐stimulation enhanced PDK4 expression; however, only myotubes exposed to Ad‐GFP showed a marked reduction in MYHC. This reduction was reversed in Ad‐MYOGAA transduced myotubes, even upon DEX‐stimulation (Figure 5 A ). To our surprise, we observed that MAFbx expression was reduced in the MYOG mutant overexpressing myotubes, indicative of a hitherto unknown negative‐feedback‐loop where stabilization of MYOG protein potentially leads to a redundancy in the requirement of its E3‐ubiquitin‐ligase and thus a decrease in its expression. Immunofluorescence analysis further revealed enhanced MYHC staining in Ad‐MYOGAA transduction myotubes, increased cell fusion and multinucleated myotubes formation compared with Ad‐GFP myotubes upon DEX‐stimulation (Figure 5 B,C ), suggesting that phospho‐mutant MYOG can protect against the development of GCs‐induced muscle atrophy in isolated human myotubes.
Figure 5.
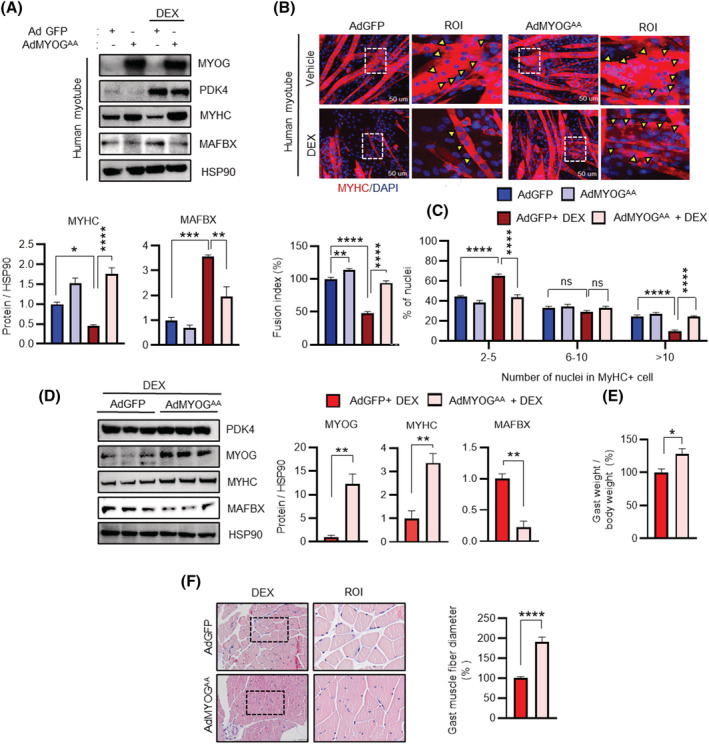
Ectopic overexpression of MYOG mutant protects against glucocorticoids‐induced muscle atrophy in human myotube and mice. (A) Immunoblots analysis of indicated proteins in human myotubes infected with adenovirus expressing mutant MYOGᴬᴬ (Ad MYOGᴬᴬ) or GFP (AdGFP). Myotubes were culture for 24 h in the presence or absence of dexamethasone (DEX) 100 μM. (Below) Quantification represents the levels of the indicated protein normalized to HSP90. (B) Representative image of MYHC immunofluorescence in human myotubes infected with adenovirus expressing mutant MYOGᴬᴬ (Ad MYOGᴬᴬ) or GFP (AdGFP) in the presence or absence of dexamethasone (DEX) 100 μM for 24 h. (Below) Quantification represents the levels of fusion index and the distribution of nuclei per myotube were calculated at Day 6 differentiation. (C) The distribution of nuclei per myotubes were calculated at Day 6 differentiation. Yellow arrowheads indicate the number of nuclei in one myotube. (D) Immunoblots analysis of indicated proteins in gastrocnemius anterior (GA) muscle. (Right) Quantification represents the levels of the indicated protein normalized to HSP90. (E) The ratio of the weight of gastrocnemius anterior (GA) to total body weight. (F) Representative image of haematoxylin and eosin staining of myofibre cross section from AdGFP or AdMYOGᴬᴬ injected in gastrocnemius anterior muscles from dexamethasone (DEX)‐treated mice. A microscope with a ×20 objective was used to capture the images. (Right) the cross‐sectional diameter of gastrocnemius anterior (GA) muscle. Mean ± SEM. (B) Scale bar 50 μM. (F) n = 5 for each group. (A, E) n = 3. (B, C) n = 4; number of image capture sites 20 from each group. *P < 0.05; **P < 0.01; ***P < 0.001; ****P < 0.0001. Unpaired t‐test was used for analyzed for panel (E–F). *P < 0.05; **P < 0.01; ***P < 0.001; ****P < 0.0001. Tukey's multiple comparisons test was used for analysed panels (A–C). ROI = region of interest.
Next, to examine the function of PDK4 dependent MYOG degradation in vivo, we ectopically overexpressed Ad‐GFP or Ad‐MYOGAA in the mouse model of DEX‐induced muscle atrophy. 3 Mice were administered DEX (25 mg/kg; i.p.) every other day for 10 days, adenoviruses were injected twice (on Day 4 and Day 5 from the first DEX injection) to the GA muscle. The knock‐in efficiency was confirmed at the level of both mRNA and protein of MYOG (Figures 5 D and S4A ). Mice transduced with Ad‐MYOGAA showed enhanced MYOG and MYHC protein levels even in the presence of DEX and reduced level of MAFbx (Figure 5D ). Consequently, analysis of the downstream transcriptional program that is known to be initiated by MYOG demonstrated that the mRNA levels of MyHC, Mymk and Mymx were induced in the GA muscle of Ad‐MYOGAA mice, compared with the Ad‐GFP cohorts (Figure S4A ). Additionally, in comparison with the Ad‐GFP‐transduced mice, Ad‐MYOGAA transduced mice were less prone to loss of GA muscle upon DEX challenge, as observed from the higher muscle weight in these mice (Figure 5 E ). Histochemical analysis of the GA muscle further revealed the protective effect of Ad‐MYOGAA against DEX‐induced muscle atrophy as observed from the markedly larger cross‐sectional diameter of muscle fibre when compared with Ad‐GFP cohorts (Figure 5 F ). Collectively, these results confirm that overexpression of non‐phosphorylatable MYOG mutant can rescue DEX‐induced muscle atrophy.
Genetic ablation of PDK4 enhances MYOG stability to attenuate glucocorticoid‐induced muscle atrophy
GC‐induced muscle atrophy is characterized by the apparent loss of fast‐twitch‐type‐II glycolytic muscle fibre with reduced or no impact on type‐I fibres. 25 GA, TA and EDL are predominantly fast‐twitch‐type‐II glycolytic muscle fibre whereas SOL is predominantly type‐I fibres muscle. 26 Therefore, we finally investigated the effect of systemic PDK4 deletion (Pdk4 −/− ) in GC‐induced muscle atrophy in various mouse muscle types (Figure 6 A ). DEX‐stimulation induced PDK4 and MAFbx protein levels in GA and TA but less so in EDL. No change was observed in SOL. Consequently, MYOG and MYHC protein levels were reduced in GA and TA upon DEX‐stimulation, whereas a slight reduction was observed in the EDL and no change was visible in SOL. In congruence with our earlier observations, this negative effect of DEX‐stimulation on myogenesis was reversed in Pdk4 −/− mice in GA, TA and EDL, but not in SOL (Figures 6 B and S6B ). Gene expression analysis further revealed that DEX‐stimulation failed to attenuate the myogenic programme upon ablation of PDK4 in GA, TA and EDL but not in SOL (Figure S6A ).
Figure 6.
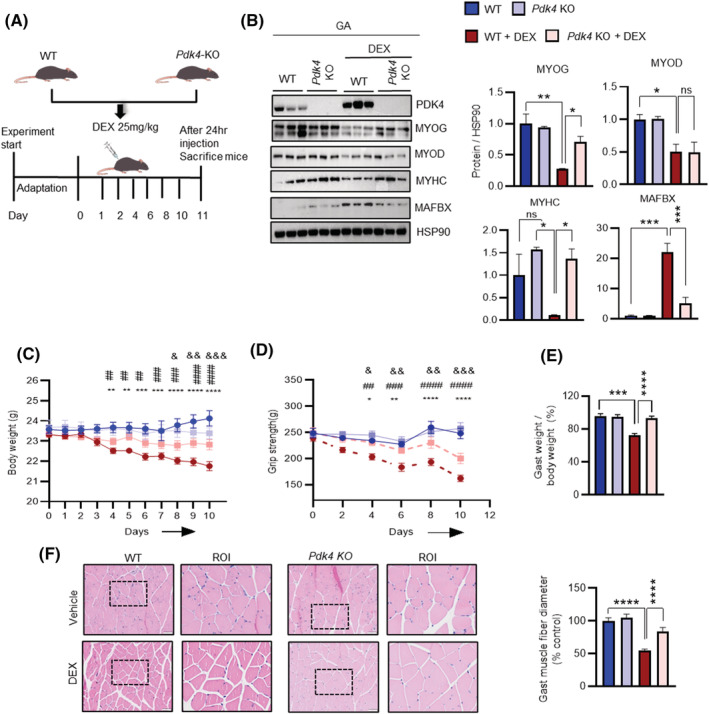
PDK4 ablation prevents muscle loss in glucocorticoids ‐induced muscle atrophy in mice. (A) Experimental scheme for dexamethasone‐induced muscle atrophy model in WT and Pdk4KO mice. (B) Immunoblots analysis of indicated proteins using gastrocnemius anterior (GA) muscle. (Right) Quantification represents the levels of the indicated protein normalized to HSP90. (C) Total body weight. (D) Grip strength test. (E) The ratio of gastrocnemius anterior (GA) muscle weight to body weight. (F) Representative haematoxylin and eosin staining of myofibre cross‐section of Gast. A microscope with a ×20 objective was used to capture the images. (Right) the cross‐sectional diameter of gastrocnemius anterior (GA) muscle. Mean ± SEM. (F) Scale bar 50 μM. (B) n = 3 for each group. (C–F) n = 6 for each group. & P < 0.05; && P < 0.01; &&& P < 0.001; &&&& P < 0.0001, Pdk4KO + DEX vs WT + DEX. # P < 0.05; ## P < 0.01; ### P < 0.001; #### P < 0.0001, Pdk4KO versus WT + DEX. *P < 0.05; **P < 0.01; ***P < 0.001; ****P < 0.0001, WT versus WT + DEX. (Body weight and grip strength). *P < 0.05; **P < 0.01; ***P < 0.001; ****P < 0.0001. Tukey's multiple comparisons test was used for the analysed panels (B, E, and F). ROI, region of interest.
To further evidence the phenotypic changes associated with muscle atrophy in the absence or presence of PDK4, we tracked the bodyweight of these animals throughout the course of the experimental regime. DEX‐stimulation induced significant weight loss in wild‐type cohorts (Pdk4 +/+ ), starting from Day 4 after the initial stimulation. This effect was markedly attenuated in Pdk4 −/− mice stimulated with DEX (Figure 6 C ). Additionally, we performed a grip strength test, an indicator of muscle strength, in these mice. DEX‐stimulation showed an early reduction of muscle strength, starting from Day 4, in Pdk4 +/+ cohorts but failed to affect muscle strength in Pdk4 −/− mice (Figure 6 D ). Similarly, PDK4 ablation protected against DEX‐mediated reduction in muscle index, as observed from the GA, TA and EDL mice (Figures 6 E and S6D ). Finally, we compared the size of myofibres in various mouse muscle types by measuring the cross‐sectional area after histochemical staining (Figures 6 F and S6D ). The cross‐sectional diameter of GA, TA and EDL muscle fibre in DEX‐stimulated mice were smaller than that from the control group whereas no change was observed in SOL (Figure 6 C,F ). However, Pdk4 deletion prevented the DEX‐mediated reduction of the cross‐sectional diameter of GA, TA and EDL muscle‐fibre (Figures 6 F and S6D ). Furthermore, we investigated the effect of PDK4 inhibition in cancer cachexia‐induced muscle atrophy. Tumour‐bearing mice and CM‐stimulation enhanced PDK4 expression and MAFbx protein levels in GA and TA whereas MYOG and MYHC were reduced (Figure S7A–C ). Congruently, this negative effect of CM‐stimulation on myogenesis was reversed upon PDK4 knockdown (Figure S7C ). Immunofluorescence staining demonstrated that CM‐triggered reduction in MYOG and MYHC staining, reduction in cell fusion and multinucleated myotubes formation were reversed upon PDK4 knockdown (Figure S7D–F ). These results indicate that PDK4 depletion is sufficient to attenuate atrophic muscle loss in‐vivo and this effect is mediated by stabilization of the master regulator of muscle myogenesis, MYOG.
Overall, here we have provided multiple lines of evidence showing that in GC and cancer cachexia‐induced muscle atrophy, upregulation of PDK4 triggers MYOG phosphorylation, leading to its ubiquitination and degradation by MAFbx. Conversely, PDK4 deficiency protects against muscle atrophy in vivo, thereby evoking the possibility of therapeutically targeting PDK4 as a viable option for muscle atrophy.
Discussion
The anabolic and catabolic processes are finely tuned to maintain muscle fibre size and skeletal muscle mass in normal physiology. An excess GC level mainly observed in pathological conditions such as obesity, 27 type‐2‐diabetes (T2D), 28 Cushing syndrome 29 and long term corticosteroid therapy 30 distort this balance, leading to an increase in muscle protein degradation and a reduction in muscle fibre size which subsequently triggers muscle‐wasting. 31 The catabolic action of GC that promotes myofibrillar protein degradation was shown to be induced via activation of the UPS and autophagy. 3 However, the underlying mechanism involved in GC‐induced muscle atrophy remained misty. Previously, GCs treatment was shown to decrease the protein levels of MYOG, a muscle‐specific bHLH transcription factor, which plays a crucial role in skeletal muscle differentiation and development by regulating the expression of Mymk, Mymx and MyHC, the key factors involved in muscle differentiation and fusion process. 32 Genetic deletion MYOG causes perinatal lethality in mice due to compromised skeletal muscle maturation. 9 , 33 Furthermore, a decrease in MYOG expression in high‐fat diet (HFD)‐feeding and STZ treatment in mice are associated with muscle atrophy, 16 whereas overexpression of MYOG rescues muscle dystrophy caused by motor neuron degeneration in nuclear receptor interaction protein (NRIP) deficient mice, 34 suggesting that downregulation of MYOG promotes muscle atrophy.
PDK4, a kinase that regulates mitochondrial pyruvate dehydrogenase (PDH) activity is predominantly expressed in skeletal muscle. 35 Its expression is further increased in cancer cachexia, obesity, T2D and by GC. In this study, we found that expression PDK4 is induced in the skeletal muscle of obese, corticotropin‐releasing hormone transgenic (CRH‐Tg) mice and the skeletal muscle cells isolated from T2DM‐patients as well as tumour‐bearing mice compared with the controls. We observed that increase in the PDK4 expression was inversely correlated with the expression levels of MYOG. In addition, we found that either overexpression or induction of PDK4 expression by DEX or CM, decreased MYOG protein level, whereas shRNA‐mediated knockdown of PDK4 prevented DEX and CM‐induced degradation of MYOG, suggesting that PDK4 plays a crucial role in regulating MYOG protein stability via a post‐translational modification‐dependent mechanism. Previously, calmodulin‐dependent kinase‐II (CaMKII) and protein kinase‐C (PKC) has been shown to regulate MYOG transcriptional activity by phosphorylating at T87 without affecting its stability. 36 Here, an in‐depth analysis revealed that PDK4 directly phosphorylates MYOG at two sites, serine 43 (S43) and threonine 57 (T57). We found that PDK4‐mediated phosphorylation of MYOG at these sites led to an induction of MYOG ubiquitination and degradation. In contrast, non‐phosphorylatable mutant of MYOG (A43 and A57) was resistant to ubiquitination and degradation mediated by DEX‐treatment or PDK4 overexpression (Figure 7 ). These results strongly indicate that PDK4‐mediated phosphorylation of MYOG mediates GC and CM‐induced MYOG degradation.
Figure 7.
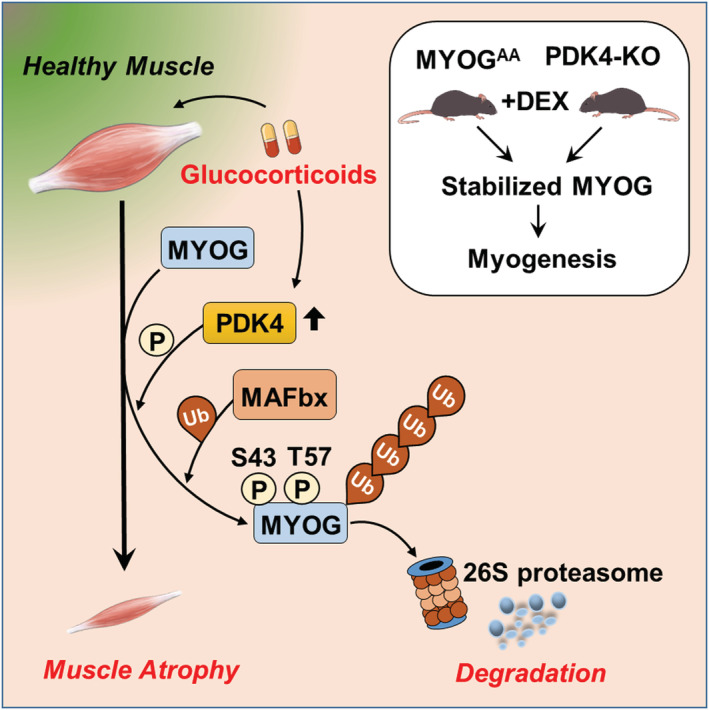
Proposed model for PDK4‐induced muscle atrophy.
MAFbx/Atrogin‐1, E3‐ubiquitin ligase responsible for promoting protein degradation during muscle atrophy 8 , 37 was shown to interact with MYOG to promote its proteasomal degradation. 23 Furthermore, fork‐head‐box protein O1 (FOXO1), a known transcription factor of PDK4, 38 , 39 was found to play a role in promoting muscle atrophy via transcriptional regulation of MAFbx to promote protein degradation during muscle atrophy. 8 , 37 We found that overexpression or DEX‐mediated induction of PDK4 enhanced the interaction between MYOG and MAFbx but not with the non‐phosphorylatable mutant of MYOG (MYOGAA). Conversely, knockdown of MAFbx rescued MYOG degradation in the presence of DEX, indicating that PDK4‐mediated phosphorylation of MYOG promotes its ubiquitination process and proteasomal degradation via recruitment of MAFbx. In contrast to an earlier study, 40 we observed interaction between MAFbx and MYOG. This apparent contrast can be a result of different experimental conditions.
Previous studies have highlighted the involvement of PDK4 in promoting muscle atrophy in amyotrophic‐lateral‐sclerosis (ALS) 12 and in cancer cachexia 2 whereas knockdown of PDK4 or pharmacological inhibition of PDK activity with dichloroacetate (DCA) was shown to prevent muscle atrophy in those conditions. However, the detailed molecular mechanism remains unknown. Interestingly, we found that knockdown of PDK4 preserves myotube size and prevent protein catabolism involved in DEX‐induced muscle atrophy in mice and CM‐induced muscle atrophy in vitro. Whereas genetic ablation of PDK4 or ectopic overexpression of MYOGAA attenuated DEX‐induced muscle atrophy, thereby implicating that PDK4 plays an important role in the pathogenesis of GCs‐induced muscle atrophy. Altogether, we identified a novel mechanism where PDK4 play a critical role in promoting GCs and CM‐induced muscle atrophy via modulating MYOG protein stability. Therefore, the PDK4‐MYOG axis represents a novel therapeutic target to alleviate GCs and CM‐induced muscle atrophy.
There are several limitations in this study. Although PDK4 is inducible under several pathological conditions, 11 , 12 however, it is intrinsically a short‐lived protein. This suggests that triggers of muscle atrophy might play a role in altering PDK4 protein stability, as observed by us and previous reports. 2 Genome‐wide association studies on muscle atrophy can identify potential mutations and/or single nucleotide variants within the PDK4 gene that can be attributed to play a causal role in the pathology. Furthermore, reduced muscle building owing to disruption in ribosome production plays a critical role in the process of muscle wasting. How PDK4 might regulate this process remains elusive and warrants future research. Finally, based on our current findings, therapeutic targeting of PDK4 can be a prospective route to counter muscle wasting and requires immediate focus.
Conflict of interest
Authors declare that they have no competing interests.
Supporting information
Figure S1. Upregulation of PDK4 determines myotubes atrophy.
Figure S2. Knockdown of PDK4 prevents DEX‐induced muscle atrophy in vitro
Figure S3. PDK4 recruit SCF‐complex E3‐Ubiqutin ligase MAFbx
Figure S4. Interaction of MYOG and PDK4
Figure S5. Overexpression of non‐phosphorylatable MYOG mutant rescue dexamethasone (DEX)‐induced muscle atrophy.
Figure S6. Genetic ablation of PDK4 rescue dexamethasone (DEX)‐induced muscle atrophy.
Figure S7. Knockdown of PDK4 prevents Cancer cachexia‐induced muscle atrophy in vitro
Table S1. Antibodies, Chemicals, Oligonucleotide, And Experimental Models Information.
Table S2. Details of patient information
Acknowledgements
This work was supported by the National Research Foundation of Korea (NRF) grant funded by the Korea government (MSIT) (Grant Number: 2022R1A2B5B03001929) (I‐KL); NRF grant funded by the Ministry of Science and ICT (NRF‐2021R1A5A2021614 and NRF‐2020R1C1C1012729) (J. ‐H. J.); NRF‐2021R1F1A1061393 (D. C.); NRF‐2022R1A2C1007857 (T. T.).
Sinam I. S., Chanda D., Thoudam T., Kim M.‐J., Kim B.‐G., Kang H.‐J., Lee J. Y., Baek S.‐H., Kim S.‐Y., Shim B. J., Ryu D., Jeon J.‐H., and Lee I.‐K. (2022) Pyruvate dehydrogenase kinase 4 promotes ubiquitin–proteasome system‐dependent muscle atrophy, Journal of Cachexia, Sarcopenia and Muscle, 13, 3122–3136, doi: 10.1002/jcsm.13100
Contributor Information
Jae‐Han Jeon, Email: jeonjh@knu.ac.kr.
In‐Kyu Lee, Email: leei@knu.ac.kr.
References
- 1. Fu X, Zhu M, Zhang S, Foretz M, Viollet B, Du M. Obesity impairs skeletal muscle regeneration through inhibition of AMPK. Diabetes 2016;65:188–200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Pin F, Novinger LJ, Huot JR, Harris RA, Couch ME, O'Connell TM, et al. PDK4 drives metabolic alterations and muscle atrophy in cancer cachexia. FASEB J 2019;33:7778–7790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Shen S, Liao Q, Liu J, Pan R, Lee SMY, Lin L. Myricanol rescues dexamethasone‐induced muscle dysfunction via a sirtuin 1‐dependent mechanism. J Cachexia Sarcopenia Muscle 2019;10:429–444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Vandewalle J, Luypaert A, de Bosscher K, Libert C. Therapeutic mechanisms of glucocorticoids. Trends Endocrinol Metab 2018;29:42–54. [DOI] [PubMed] [Google Scholar]
- 5. Schakman O, Kalista S, Barbé C, Loumaye A, Thissen JP. Glucocorticoid‐induced skeletal muscle atrophy. Int J Biochem Cell Biol 2013;45:2163–2172. [DOI] [PubMed] [Google Scholar]
- 6. Gumucio JP, Mendias CL. Atrogin‐1, MuRF‐1, and sarcopenia. Endocrine 2013;43:12–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Bowen TS, Adams V, Werner S, Fischer T, Vinke P, Brogger MN, et al. Small‐molecule inhibition of MuRF1 attenuates skeletal muscle atrophy and dysfunction in cardiac cachexia. J Cachexia Sarcopenia Muscle 2017;8:939–953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Sandri M, Sandri C, Gilbert A, Skurk C, Calabria E, Picard A, et al. Foxo transcription factors induce the atrophy‐related ubiquitin ligase atrogin‐1 and cause skeletal muscle atrophy. Cell 2004;117:399–412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Hasty P, Bradley A, Morris JH, Edmondson DG, Venuti JM, Olson EN, et al. Muscle deficiency and neonatal death in mice with a targeted mutation in the myogenin gene. Nature 1993;364:501–506. [DOI] [PubMed] [Google Scholar]
- 10. Zhang S, Hulver MW, McMillan RP, Cline MA, Gilbert ER. The pivotal role of pyruvate dehydrogenase kinases in metabolic flexibility. Nutr Metab (Lond) 2014;11:10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Thoudam T, Ha CM, Leem J, Chanda D, Park JS, Kim HJ, et al. PDK4 augments ER‐mitochondria contact to dampen skeletal muscle insulin signaling during obesity. Diabetes 2019;68:571–586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Palamiuc L, Schlagowski A, Ngo ST, Vernay A, Dirrig‐Grosch S, Henriques A, et al. A metabolic switch toward lipid use in glycolytic muscle is an early pathologic event in a mouse model of amyotrophic lateral sclerosis. EMBO Mol Med 2015;7:526–546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Mallinson JE, Constantin‐Teodosiu D, Glaves PD, Martin EA, Davies WJ, Westwood FR, et al. Pharmacological activation of the pyruvate dehydrogenase complex reduces statin‐mediated upregulation of FOXO gene targets and protects against statin myopathy in rodents. J Physiol 2012;590:6389–6402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Kim A, Im M, Ma JY. A novel herbal formula, SGE, induces endoplasmic reticulum stress‐mediated cancer cell death and alleviates cachexia symptoms induced by colon‐26 adenocarcinoma. Oncotarget 2018;9:16284–16296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Luo J, Deng ZL, Luo X, Tang N, Song WX, Chen J, et al. A protocol for rapid generation of recombinant adenoviruses using the AdEasy system. Nat Protoc 2007;2:1236–1247. [DOI] [PubMed] [Google Scholar]
- 16. Liu X, Qu H, Zheng Y, Liao Q, Zhang L, Liao X, et al. Mitochondrial glycerol 3‐phosphate dehydrogenase promotes skeletal muscle regeneration. EMBO Mol Med 2018;10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Hong AW, Guan KL. Non‐radioactive LATS in vitro kinase assay. Bio Protoc 2017;7:e2391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. McAinch AJ, Cornall LM, Watts R, Hryciw DH, O'Brien PE, Cameron‐Smith D. Increased pyruvate dehydrogenase kinase expression in cultured myotubes from obese and diabetic individuals. Eur J Nutr 2015;54:1033–1043. [DOI] [PubMed] [Google Scholar]
- 19. Connaughton S, Chowdhury F, Attia RR, Song S, Zhang Y, Elam MB, et al. Regulation of pyruvate dehydrogenase kinase isoform 4 (PDK4) gene expression by glucocorticoids and insulin. Mol Cell Endocrinol 2010;315:159–167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Stitt TN, Drujan D, Clarke BA, Panaro F, Timofeyva Y, Kline WO, et al. The IGF‐1/PI3K/Akt pathway prevents expression of muscle atrophy‐induced ubiquitin ligases by inhibiting FOXO transcription factors. Mol Cell 2004;14:395–403. [DOI] [PubMed] [Google Scholar]
- 21. Gopal K, Saleme B, al Batran R, Aburasayn H, Eshreif A, Ho KL, et al. FoxO1 regulates myocardial glucose oxidation rates via transcriptional control of pyruvate dehydrogenase kinase 4 expression. Am J Physiol Heart Circ Physiol 2017;313:H479–H490. [DOI] [PubMed] [Google Scholar]
- 22. Aniort J, Stella A, Philipponnet C, Poyet A, Polge C, Claustre A, et al. Muscle wasting in patients with end‐stage renal disease or early‐stage lung cancer: common mechanisms at work. J Cachexia Sarcopenia Muscle 2019;10:323–337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Jogo M, Shiraishi S, Tamura TA. Identification of MAFbx as a myogenin‐engaged F‐box protein in SCF ubiquitin ligase. FEBS Lett 2009;583:2715–2719. [DOI] [PubMed] [Google Scholar]
- 24. Schwarz JJ, Chakraborty T, Martin J, Zhou JM, Olson EN. The basic region of myogenin cooperates with two transcription activation domains to induce muscle‐specific transcription. Mol Cell Biol 1992;12:266–275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Wang Y, Pessin JE. Mechanisms for fiber‐type specificity of skeletal muscle atrophy. Curr Opin Clin Nutr Metab Care 2013;16:243–250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Eng CM, Smallwood LH, Rainiero MP, Lahey M, Ward SR, Lieber RL. Scaling of muscle architecture and fiber types in the rat hindlimb. J Exp Biol 2008;211:2336–2345. [DOI] [PubMed] [Google Scholar]
- 27. Akalestou E, Genser L, Rutter GA. Glucocorticoid metabolism in obesity and following weight loss. Front Endocrinol (Lausanne) 2020;11:59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Chiodini I, Adda G, Scillitani A, Coletti F, Morelli V, di Lembo S, et al. Cortisol secretion in patients with type 2 diabetes: relationship with chronic complications. Diabetes Care 2007;30:83–88. [DOI] [PubMed] [Google Scholar]
- 29. Reincke M. Cushing syndrome associated myopathy: it is time for a change. Endocrinol Metab (Seoul) 2021;36:564–571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Kanda F, Okuda S, Matsushita T, Takatani K, Kimura KI, Chihara K. Steroid myopathy: pathogenesis and effects of growth hormone and insulin‐like growth factor‐I administration. Horm Res 2001;56:24–28. [DOI] [PubMed] [Google Scholar]
- 31. Braun TP, Marks DL. The regulation of muscle mass by endogenous glucocorticoids. Front Physiol 2015;6:12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Sassoon D, Lyons G, Wright WE, Lin V, Lassar A, Weintraub H, et al. Expression of two myogenic regulatory factors myogenin and MyoD1 during mouse embryogenesis. Nature 1989;341:303–307. [DOI] [PubMed] [Google Scholar]
- 33. Nabeshima Y, Hanaoka K, Hayasaka M, Esuml E, Li S, Nonaka I, et al. Myogenin gene disruption results in perinatal lethality because of severe muscle defect. Nature 1993;364:532–535. [DOI] [PubMed] [Google Scholar]
- 34. Chen HH, Tsai LK, Liao KY, Wu TC, Huang YH, Huang YC, et al. Muscle‐restricted nuclear receptor interaction protein knockout causes motor neuron degeneration through down‐regulation of myogenin at the neuromuscular junction. J Cachexia Sarcopenia Muscle 2018;9:771–785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Holness MJ, Bulmer K, Gibbons GF, Sugden MC. Up‐regulation of pyruvate dehydrogenase kinase isoform 4 (PDK4) protein expression in oxidative skeletal muscle does not require the obligatory participation of peroxisome‐proliferator‐activated receptor alpha (PPARalpha). Biochem J 2002;366:839–846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Blagden CS, Fromm L, Burden SJ. Accelerated response of the myogenin gene to denervation in mutant mice lacking phosphorylation of myogenin at threonine 87. Mol Cell Biol 2004;24:1983–1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Cid‐Díaz T, Leal‐López S, Fernández‐Barreiro F, González‐Sánchez J, Santos‐Zas I, Andrade‐Bulos LJ, et al. Obestatin signalling counteracts glucocorticoid‐induced skeletal muscle atrophy via NEDD4/KLF15 axis. J Cachexia Sarcopenia Muscle 2021;12:493–505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Furuyama T, Kitayama K, Yamashita H, Mori N. Forkhead transcription factor FOXO1 (FKHR)‐dependent induction of PDK4 gene expression in skeletal muscle during energy deprivation. Biochem J 2003;375:365–371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Puthanveetil P, Wang Y, Wang F, Kim MS, Abrahani A, Rodrigues B. The increase in cardiac pyruvate dehydrogenase kinase‐4 after short‐term dexamethasone is controlled by an Akt‐p38‐forkhead box other factor‐1 signaling axis. Endocrinology 2010;151:2306–2318. [DOI] [PubMed] [Google Scholar]
- 40. Lagirand‐Cantaloube J, Cornille K, Csibi A, Batonnet‐Pichon S, Leibovitch MP, Leibovitch SA. Inhibition of atrogin‐1/MAFbx mediated MyoD proteolysis prevents skeletal muscle atrophy in vivo. PLoS ONE 2009;4:e4973. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Upregulation of PDK4 determines myotubes atrophy.
Figure S2. Knockdown of PDK4 prevents DEX‐induced muscle atrophy in vitro
Figure S3. PDK4 recruit SCF‐complex E3‐Ubiqutin ligase MAFbx
Figure S4. Interaction of MYOG and PDK4
Figure S5. Overexpression of non‐phosphorylatable MYOG mutant rescue dexamethasone (DEX)‐induced muscle atrophy.
Figure S6. Genetic ablation of PDK4 rescue dexamethasone (DEX)‐induced muscle atrophy.
Figure S7. Knockdown of PDK4 prevents Cancer cachexia‐induced muscle atrophy in vitro
Table S1. Antibodies, Chemicals, Oligonucleotide, And Experimental Models Information.
Table S2. Details of patient information