

Abstract
Epstein-Barr virus (EBV) expresses two highly abundant noncoding RNAs called EBV-encoded RNA 1 (EBER1) and EBER2, which are preserved in all clinical isolates of EBV, thus underscoring their essential function in the viral life cycle. Recent epitranscriptomics studies have uncovered a vast array of distinct RNA modifications within cellular as well as viral noncoding RNAs that are instrumental in executing their function. Here we show that EBER2 is marked by pseudouridylation, and by using HydraPsiSeq the modification site was mapped to a single nucleotide within the 3′ region of EBER2. The writer enzyme was identified to be the snoRNA-dependent pseudouridine synthase Dyskerin, which is the catalytic subunit of H/ACA small nucleolar ribonucleoprotein complexes, and is guided to EBER2 by SNORA22. Similar to other noncoding RNAs for which pseudouridylation has a positive effect on RNA stability, loss of EBER2 pseudouridylation results in a decrease in RNA levels. Furthermore, pseudouridylation of EBER2 is required for the prolific accumulation of progeny viral genomes, suggesting that this single modification in EBER2 is important for efficient viral lytic replication. Taken together, our findings add to the list of RNA modifications that are essential for noncoding RNAs to implement their physiological roles.
Keywords: EBER2, Epstein-Barr virus, HydraPsiSeq, noncoding RNA, pseudouridylation
INTRODUCTION
Epstein-Barr virus (EBV) is a highly successful human lymphotropic gamma-1 herpesvirus that infects ∼95% of the adult world population. Upon infection, EBV mainly persists in a latent (dormant) stage inside host cells and expresses only a minimal set of viral genes to remain under the radar of the host immune system. Among the genes expressed during latency are two noncoding (nc)RNAs called EBV-encoded RNA 1 (EBER1) and EBER2, which are both nuclear transcripts of similar length (∼170 nt) (Lerner et al. 1981; Howe and Steitz 1986). One of the most striking features of the EBERs is their conspicuously high copy number, which is on par with some of the most abundant host noncoding RNAs that play key roles in vital cellular processes (Lee 2021). The fact that EBV requires such high copy numbers of ∼106 and ∼2.5 × 105 for EBER1 and EBER2, respectively, strongly suggests that both noncoding transcripts are instrumental players during the viral life cycle. Furthermore, whole-genome sequencing studies of clinical isolates of EBV showed that both EBERs are absolutely preserved in every examined EBV strain with only minor polymorphisms (Moss and Steitz 2013; Xu et al. 2019), arguing that EBERs fulfill a vital function during viral propagation and transmission in nature. While the molecular function of EBER1 remains to be elucidated, the mode of action of EBER2 has been studied in more detail. EBER2 acts as a guide RNA to recruit the host transcription factor PAX5 to the so-called terminal repeat regions on the EBV genome, and this recruitment results in transcriptional repression of the nearby latent membrane protein (LMP) genes (Lee et al. 2015; Lee and Steitz 2015). Moreover, depletion of EBER2 negatively impacts the replication of viral genomes to be packaged into progeny virions, demonstrating that EBER2 is also pivotal for viral lytic replication. EBER2 has also been shown to regulate cellular targets, such as UCHL1, to impart a growth advantage for EBV-infected cells (Li et al. 2021).
Host noncoding RNAs that are found in high abundance, such as rRNAs, tRNAs, and snRNAs, have in common that they carry various RNA modifications, some of which assist in enhancing their stability and others are critical for exerting their molecular function (Roundtree et al. 2017; Bohnsack and Sloan 2018). Of the over 170 distinct RNA modifications that have been identified to date, pseudouridine (Ψ) was the first to be discovered and is also the most abundant one. Pseudouridine is the C5-glycoside isomer of uridine in which the C5 and N1 positions of uridine are interconverted, and this modification is catalyzed by either standalone pseudouridine synthase (PUS) enzymes or an RNA-dependent machinery. The latter utilizes box H/ACA small nucleolar (sno)RNAs to locate their substrates through complementary base-pairing and four core proteins (Dyskerin [DKC1], GAR1, NHP2, and NOP10), of which DKC1 is the catalytic subunit (Rintala-Dempsey and Kothe 2017; Zhao et al. 2018). Pseudouridylation sites in several ncRNAs overlap with functionally critical regions, such as the peptidyl transferase center in rRNAs, the eponymous TΨC loop in tRNAs, or the branch-site recognition sequence in U2 snRNA (Ge and Yu 2013; Machnicka et al. 2013). This particular modification is thought to stabilize RNA–RNA interactions by increasing the rigidity of the phosphodiester backbone and base stacking ability, and depletion of pseudouridylation at these critical sites severely compromises noncoding RNA function (Spenkuch et al. 2014).
Pseudouridine retains the base-pairing properties of uridine and is thus indistinguishable from uridine in reverse transcription reactions. However, pseudouridine can be derivatized using carbodiimides, such as CMCT (1-cyclohexyl-(2-morpholinoethyl)carbodiimide metho-p-toluene sulfonate), to produce a stable Ψ-CMC adduct that unlike uridine can no longer undergo Watson–Crick base-pairing and thus be detected in subsequent primer extension assays as premature stops. This chemical principle has been leveraged for uncovering pseudouridylation sites at a transcriptome-wide level by coupling CMCT-treatment with next-generation sequencing, which have shown that pseudouridylation events are not restricted to noncoding RNAs but occur frequently in mRNAs as well (Carlile et al. 2014; Schwartz et al. 2014; Li et al. 2015).
We have previously shown that EBER1 harbors only one type of RNA modification, that is, 5-methylcytosine, and that this modification occurs at a single specific nucleotide in almost all EBER1 molecules (Henry et al. 2020). Here we show that EBER2 is also modified and contains a single Ψ residue, which is deposited by the snoRNA-dependent machinery. Of physiological relevance, loss of pseudouridylation in EBER2 results in decreased viral lytic replication of progeny EBV genomes. Our study thus provides another example for how RNA modifications contribute significantly to RNA function.
RESULTS
EBER2 is modified by pseudouridylation
EBER2 was purified from EBV-positive BJAB-B1 cells to near homogeneity and examined by LC–MS/MS for any RNA modification as previously described (Henry et al. 2020). The only noticeable peak was detected for pseudouridine, and quantification of this peak estimated the abundance of pseudouridine modifications at ∼0.17 for each EBER2 molecule. To corroborate our mass spectrometry results, we sought to confirm the presence of pseudouridine in EBER2 by antibody detection. To this end, native EBER2 purified from EBV-positive cells as well as in vitro-transcribed EBER2, which is devoid of any modification, was subjected to western blot analysis using an anti-pseudouridine antibody (Itoh et al. 1989). Native EBER2 indeed showed the presence of pseudouridine whereas synthetic EBER2 did not (Fig. 1A), thus confirming our mass spectrometry results.
FIGURE 1.

EBER2 is pseudouridylated at U160. (A) Antibody-mediated detection of pseudouridylation in EBER2. Native EBER2, purified from EBV-infected cells, as well as in vitro-transcribed EBER2, was probed with an anti-Ψ antibody (monoclonal antibody APU-6) to detect the presence of pseudouridine and verify our mass spectrometry results. Northern blot analysis for EBER2 was carried out with the same blot to verify comparable loading. (B) HydraPsiSeq analysis maps the pseudouridylation site to U160 of EBER2. HydraPsiSeq was carried out in parallel with native and in vitro-transcribed EBER2. Protected U160 is shown by an arrow. Gray bars indicate cleavage at non-U residues. (C) Secondary structure of EBER2; the pseudouridylation site Ψ160 is highlighted.
The HydraPsiSeq technique was recently introduced, which is a method for mapping pseudouridine modifications with nucleotide resolution by taking advantage of next-generation sequencing (Marchand et al. 2020). HydraPsiSeq relies on the protection from hydrazine/aniline-mediated cleavage at pseudouridine sites, while unmodified uridines are subject to cleavage. The ratio of cleavage to non-cleavage events visualized by deep sequencing allows for quantification of pseudouridylation within a transcript. We performed HydraPsiSeq with both native and synthetic (unmodified) EBER2 in order to map U residues protected from hydrazine/aniline cleavage which may represent pseudouridine residues. Overall, the U-cleavage profiles were very similar for native and unmodified EBER2 transcripts. However, one position, U160, was found to be protected in native EBER2 (Fig. 1B, top panel) and cleaved almost at normal level in the unmodified transcript (Fig. 1B, bottom panel). Pseudouridylation of U160 is attested by reduced but not totally abolished hydrazine cleavage at this site. Calculation of relative pseudouridylation level at U160 was based on the observed U-protection profile in the ±2 nt neighboring region (U153–U159–U150–U162–U167). Since U167 is too close to the 3′ end of EBER2 and not covered by sequencing, calculation of the HydraPsiSeq quantitative score has limited precision. The estimated protection level is consistent with a pseudouridylation frequency of ∼40%–80% at U160 based on the results obtained by two technical replicates. The higher estimate of modification determined by HydraPsiSeq compared to the retrieved frequency by LC–MS/MS analysis (∼0.17 Ψ per molecule) may be due to the fact that pseudouridine peaks are generally small in LC–MS/MS analyses and may result in an underestimation. On the basis of these data, U160 is the only site mapped by HydraPsiSeq to be tentatively, and at least partially, pseudouridylated (Fig. 1C).
To further validate HydraPsiSeq mapping, we applied primer extension assays following CMCT-treatment of RNA. As pseudouridine, unlike some other RNA modifications, maintains its ability for Watson–Crick base-pairing and is thus a silent modification in reverse transcription reactions, its detection requires prior derivatization with CMCT, which adds a bulky adduct to the N3 position of pseudouridine and induces a premature stop during reverse transcription. Given the close proximity of the HydraPsiSeq-mapped pseudouridylation site to the 3′ end of EBER2, primer extension assays required the addition of a 3′ linker that could act as a primer for reverse transcription (Fig. 2A). Moreover, as full-length EBER2 is not amenable to reverse transcription if a primer annealing to its very 3′ end is used (only internal primers hybridizing to the two accessible regions within EBER2 [nts 47–70 and 101–124] allow for efficient reverse transcription [Lee et al. 2015]), EBER2 needed to be digested internally with RNase H at its major loop region prior to CMCT-treatment and primer extension. Using this alternative chemical approach to HydraPsiSeq, we were able to confirm that U160 of EBER2 is indeed pseudouridylated based on the premature RT stop when using CMCT-treated EBER2 (Fig. 2B). To ascertain that EBER2 pseudouridylation is present in a wide array of EBV-positive cell lines, especially in physiologically relevant settings, such as patient-derived cells, we performed primer extension assays with EBER2 from BJAB-B1, HH514-16, Raji, and Jijoye cells. We were able to confirm the presence of EBER2 Ψ160 in all of these cell lines (Fig. 2C). Taken together, our observations demonstrate that a substantial portion of the cellular pool of EBER2 is pseudouridylated at position U160.
FIGURE 2.

Detection of EBER2 pseudouridylation by primer extension. (A) Experimental outline for verifying pseudouridylation by primer extension assay following CMCT-treatment. A linker is ligated to the 3′ end of EBER2, which is then digested with RNase H at the major loop region (red arrowhead). EBER2 is treated with CMCT to form a covalent adduct at pseudouridylation sites that will elicit a premature stop in reverse-transcription reactions. (B) A representative primer extension assay with digested EBER2 is shown, which corroborates the presence of pseudouridylation (arrow). Sequencing ladders utilizing dideoxy nucleotides are included for orientation. Please note that not each nucleotide of EBER2 is displayed at equal band intensity, which is a common observation with direct RNA sequencing using reverse transcription and dideoxy nucleotides. (C) EBER2 pseudouridylation is found in a wide array of EBV-positive cell lines. EBER2 isolated from BJAB-B1, HH514-16, Raji, and Jijoye cells was subjected to CMCT treatment followed by primer extension assay. In vitro-transcribed EBER2 was also examined as a negative control.
Dyskerin is the writer enzyme of EBER2 Ψ160
We next sought to identify the enzyme responsible for the pseudouridylation of EBER2. To this end, we generated knockdown cell lines by CRISPR interference for all nuclear PUS enzymes, such as PUS1, PUS3, PUS4/TRUB1, PUS7, PUS7L, RPUSD4, and DKC1. Our approach entailed transduction with a lentiviral construct expressing deactivated dCas9 together with a PUS-specific single-guide (sg)RNA. We were able to generate stable knockdown cell lines for all PUS enzymes as verified by qRT-PCR (Fig. 3A), except for the essential gene DKC1. Unlike the standalone PUS enzymes, which display normal cell proliferation upon knockdown, the snoRNA-dependent DKC1 is essential for cell viability (He et al. 2002), and a Tet-inducible knockdown system, in which dCas9 is expressed from a Doxycycline (Dox)-responsive promoter to conditionally knock down DKC1, was generated. Western blot as well as qRT-PCR analyses confirmed that DKC1 was efficiently depleted starting at day 4 of Dox-addition to the culture medium (Fig. 3A,B), and all experiments described herein were performed after 5 d of Dox-addition. We then repeated primer extension assays for EBER2 with RNA isolated from each knockdown cell line, which showed that detection of Ψ160 is reduced consistently only after DKC1 depletion and not for any other PUS enzyme (Fig. 3C), indicating that the box H/ACA snoRNA-dependent machinery is responsible for modifying EBER2.
FIGURE 3.

The pseudouridine synthase Dyskerin is the writer of EBER2 Ψ160. (A) All nuclear PUS enzymes were depleted in BJAB-B1 cells using CRISPR interference; their specific depletion was verified by qRT-PCR (asterisks). These cell lines were transduced with a lentiviral vector that expresses the dCas9–KRAB fusion protein and a PUS-specific sgRNA. The results are the average from three independently established knockdown cell lines for each PUS enzyme, and error bars are standard deviation. (B) As a stable knockdown cell line for the essential DKC1 protein could not be established, Tet-inducible knockdown cell lines were generated. Western blot analysis showed that efficient DKC1 depletion was observed after 4 d of Dox-addition to the culture medium. Anti-Nucleolin antibody served as a loading control. All experiments were conducted after 5 d of Dox-addition to allow for sufficient depletion of DKC1. (C) Primer extension assays of EBER2 purified from various PUS-depleted cells after CMCT treatment. A decrease in pseudouridylation at U160 of EBER2 is observed upon DKC1 depletion (arrow). Quantification of three independent experiments is shown in the graph (asterisk indicates P < 0.001 in a Student's t-test).
To substantiate the notion that DKC1 is the enzyme that catalyzes pseudouridylation of EBER2, we performed CLIP-seq for DKC1 in EBV-infected cells. Given the fact that CLIP identifies direct RNA–protein interactions only (Moore et al. 2014), our observation that the 3′ region (nts 128–168) of EBER2, which overlaps with the pseudouridylation site at U160, binds to DKC1 supports their enzyme-substrate relationship (Fig. 4A). To assess the specificity of the DKC1 peaks in our CLIP assay, we examined what other transcripts displayed binding sites for DKC1. As expected, the most prominent peaks were found within box H/ACA snoRNAs, of which 63 out of the 108 listed in the LBME database (Lestrade and Weber 2006) exhibited peaks, while no peak was found over any box C/D snoRNA. Moreover, when analyzing for the presence of crosslink-induced mutation sites (CIMS), which are indicators of bona fide direct RNA–protein interactions, as they occur when reverse transcriptase encounters UV-crosslinked amino acid-RNA adducts and introduces mutations during the readthrough process while synthesizing cDNA, a notable CIMS was detected over the DKC1 footprint of EBER2 (Fig. 4A, bottom). While a CLIP peak was also observed over EBER1, the CIMS frequency is far lower than for EBER2, further supporting the notion that EBER2 is indeed a substrate of DKC1.
FIGURE 4.
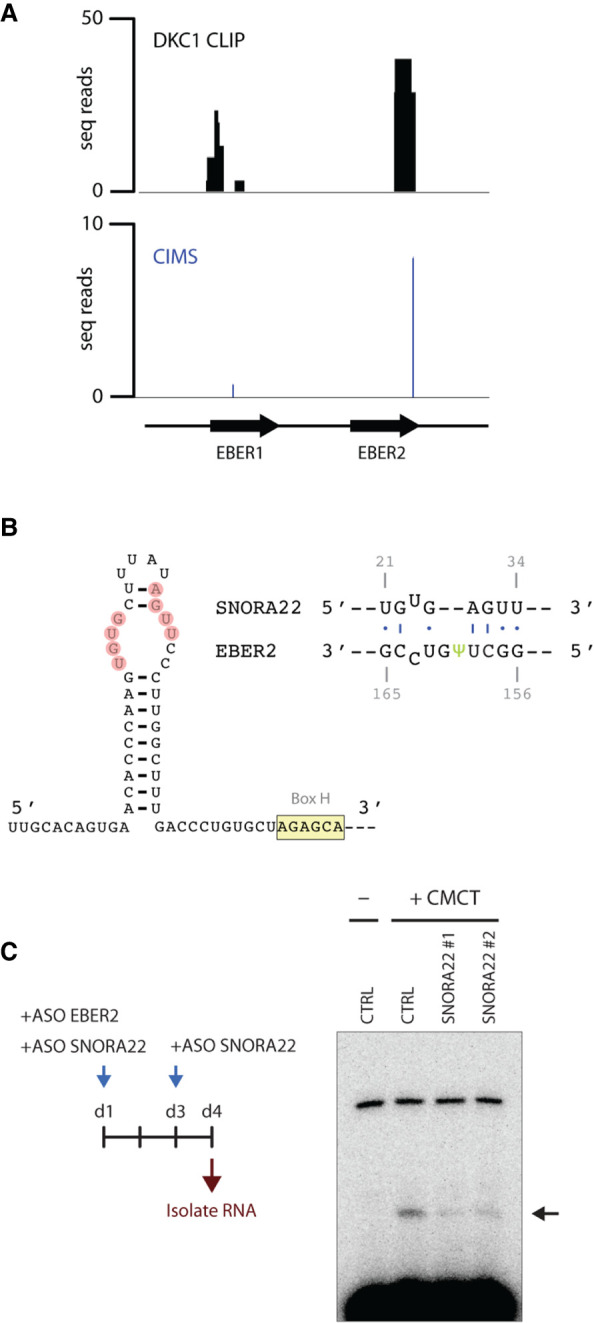
SNORA22 is the EBER2-specific H/ACA snoRNA that guides DKC1. (A) DKC1 binds directly to EBER2 in vivo. CLIP-seq for DKC1 was carried out in EBV-positive BJAB-B1 cells to examine direct binding of EBER2 as a substrate. A footprint region at the 3′ end of EBER2 (nucleotides 128–168), which overlaps with the pseudouridylation site at position U160, was detected. A crosslink-induced mutation site (CIMS), which is indicative of a bona fide direct RNA–protein interaction, was also observed within the footprint region. (B) A predicted RNA duplex between EBER2 and SNORA22 is shown surrounding the pseudouridylation site at U160. SNORA22 nucleotides engaging in RNA–RNA interaction are highlighted, and the conserved Box H is indicated. Please see Supplemental Figure 1A–D for further information. (C) Knockdown of SNORA22 decreases the abundance of EBER2 Ψ160. Left panel shows the experimental outline for depleting EBER2 and SNORA22 prior to CMCT treatment and primer extension assay. Knockdown of SNORA22 with two distinct ASOs results in lowered levels of premature RT stops.
To identify the H/ACA snoRNA that guides DKC1 to EBER2, we performed the 2CIMPL technique (Le Sage et al. 2020) for DKC1, which is an application designed to identify RNA hybrids associated with a protein of interest (Supplemental Fig. 1A). An experimental approach was necessary because a computational search among the snoRNAs listed in the LBME database did not retrieve a candidate hit for EBER2 that exhibited at least four complementary bases on each side of the pseudouridylation site, suggesting that the base-pairing interactions between EBER2 and the snoRNA guide may be less extensive and/or imperfect. Our 2CIMPL assay generated 15.6 million total mappable reads after PCR collapsing, of which 1190 reads mapped to EBER2 and 670 reads were RNA–RNA hybrids. Two hundred forty-one of these hybrids contained sequences of H/ACA snoRNA and the vast majority (240/241) were hybrids with SNORA22, suggesting that SNORA22 may be the EBER2-specific guide. We searched for putative base-pairing interactions within SNORA22 that could form a duplex with the region surrounding the pseudouridylation site in EBER2 and identified a region within a bulge in the 5′ region (Fig. 4B; Supplemental Fig. 1B,C). We next attempted to identify ASOs that could be used to knock down SNORA22 in cell culture using RNase H-mediated digestion and identified two efficient knockdown ASOs (Supplemental Fig. 1E). We then nucleofected these ASOs into BJAB-B1 cells to deplete SNORA22 and performed primer extension assays after CMCT treatment to examine whether depletion of SNORA22 affects EBER2 pseudouridylation levels. We concomitantly depleted EBER2 as well using ASOs to discard already modified, long-lived EBER2 molecules, which would skew our analysis, to only consider newly transcribed EBER2 RNA in the absence of SNORA22 (Fig. 4C, left). Knockdown with either SNORA22 ASO resulted in a decrease in premature stops in our primer extension assays, indicating that SNORA22 is indeed involved in the pseudouridylation process of EBER2 (Fig. 4C, right). Taken together, our results indicate that DKC1 is the writer for EBER2 and is guided by SNORA22 to locate its substrate.
Pseudouridylation of EBER2 increases its stability
Pseudouridylation has previously been shown to affect the stability of several cellular noncoding RNAs. We asked whether pseudouridylation of EBER2 would have a similar effect on its stability. To this end, we isolated RNA from each PUS-depleted cell line and measured the overall EBER2 levels by northern blot analysis. Only in DKC1-knockdown cells were EBER2 levels approximately twofold reduced, whereas no change was apparent in the other PUS-knockdown cells (Fig. 5A), further supporting the notion that DKC1 is indeed the writer enzyme for EBER2. The reduced EBER2 levels were verified with a second DKC1-knockdown cell line (Fig. 5B). We tested the possibility of whether a single nucleotide change from U to C at position 160 (U160C), which can no longer be pseudouridylated, would result in lower RNA stability as well. However, outside the context of EBV infection, this single nucleotide mutation did not display any changes in stability compared to its wild-type counterpart in transfection assays (Supplemental Fig. 1F). We also examined whether the reduced EBER2 levels may possibly be caused by a lowered transcription rate upon DKC1 knockdown. For this, we performed pulse labeling using tritiated uridine to metabolically label nascent RNAs. Conditional DKC1-knockdown cells were exposed to Dox for 5 d, whereupon tritiated uridine was added to the culture medium for a brief period of 1 h. Following total RNA isolation, EBER1 and EBER2 were specifically selected using antisense oligonucleotides and subjected to autoradiography after polyacrylamide gel electrophoresis. No change was observed in the level of nascent transcripts of EBER1 or EBER2 in wild-type and DKC1-knockdown cells (Fig. 5C), suggesting that the decrease in overall EBER2 level upon DKC1 knockdown is not due to a lowered transcription rate but due to lowered RNA stability.
FIGURE 5.

Loss of pseudouridylation decreases EBER2 stability. (A) Total RNA isolated from various PUS-knockdown cells was subjected to northern blot analysis. EBER2 levels were reduced only in cells depleted for DKC1. Northern blot analysis for EBER1 served as a loading control. Quantification of three independent experiments is shown in the graph (asterisk indicates P < 0.001). (B) Two independent DKC1-knockdown cell lines were examined for decreased EBER2 levels. Abundance was determined by qRT-PCR and using EBER1 levels for normalization. Results are the average of three independent experiments; error bars represent standard deviation. (C) Lower EBER2 levels are not caused by a decreased transcription rate. DKC1-knockdown cells grown 5 d in the presence of Dox were cultured for 1 h with tritiated uridine followed by ASO-mediated selection of EBER1 and EBER2. The level of nascent labeled EBER2, as shown in the representative autoradiograph, remained unchanged after DKC1 knockdown, indicating the same rate of transcription in both wild-type and knockdown cells.
Pseudouridylation of EBER2 is essential for efficient viral lytic replication of EBV genomes
It was previously shown that the physiological functions of EBER2 lie in the transcriptional regulation of EBV latent genes LMP1, LMP2A, LMP2B, and the cellular target UCHL1 as well as in mediating high levels of viral lytic replication of EBV progeny genomes (Lee et al. 2015, 2016; Li et al. 2021). We examined whether DKC1 knockdown, and thus ablation of EBER2 pseudouridylation, affects LMP and UCHL1 gene expression by real-time PCR analysis. Other than a decrease in EBER2, which confirmed our northern blot result, no effect on gene expression of the other latent EBV genes or UCHL1 were observed after DKC1 depletion (Fig. 6A). To examine whether viral lytic replication is affected, we generated Tet-inducible DKC1-knockdown cell lines with the EBV-positive and viral replication-competent cell line HH514-16 (Rabson et al. 1983). The lytic cycle can be induced in this cell line by adding the histone deacetylase inhibitor sodium butyrate to the culture medium, which will activate the expression of the immediate-early gene BZLF1 that encodes the replication activator and latent-to-lytic switch protein Zebra. We verified that DKC1 was efficiently knocked down after Dox addition and that the viral lytic cycle was successfully activated by sodium butyrate treatment in our conditional knockdown cells (Fig. 6B). As growth curve analysis with HH514-16 cells following DKC1 knockdown showed that cell proliferation was adversely affected not until 6 d after Dox-addition (Fig. 6C), we ascertained that viral lytic replication measurements were taken at day 5 of knockdown induction before any effect on cell growth was apparent. Moreover, metabolic labeling of nascent protein production did not show any apparent differences between control and DKC1 knockdown cells on day 5 of Dox-treatment (Fig. 6B, bottom panel). Sodium butyrate treatment elicited a robust activation of the lytic cycle and accumulation of virions in the culture supernatant under wild-type conditions but showed a marked reduction in viral lytic replication under conditions of DKC1 depletion (Fig. 6D). Similarly, intracellular EBV genome levels were also reduced upon DKC1 depletion in both inducible knockdown cell lines. Taken together, our results suggest that pseudouridylation of EBER2 facilitates its function in promoting viral lytic replication of EBV genomes.
FIGURE 6.

Pseudouridylation of EBER2 is essential for efficient lytic replication of the EBV genome. (A) Pseudouridylation of EBER2 is not required for the regulation of EBV latent membrane protein (LMP) genes LMP1, LMP2A, and LMP2B. EBV latent genes were examined by qRT-PCR following DKC1 knockdown. Except for EBER2, none of the other latent genes tested here was affected. The mRNA level of UCHL1, a cellular EBER2-regulated gene, was also not affected. RNA levels were normalized to GAPDH mRNA abundance and are the average of three independent experiments; error bars represent standard deviation. (B) Two Tet-inducible DKC1-knockdown cell lines were established with the EBV-positive replication-competent cell line HH514-16. DKC1 depletion after 5 d of Dox-addition and induction of EBV lytic replication using sodium butyrate (NaB) was verified. Western blot analysis was performed for DKC1 to monitor knockdown as well as for Zebra to confirm robust induction of the lytic cycle by NaB. Anti-Nucleolin antibody served as a loading control. After Dox-addition, cells were also treated with a brief pulse of 35S-methionine to detect nascent protein production. No apparent difference was observed in control and DKC1-knockdown cells. (C) A representative growth curve for the inducible HH514-16 knockdown cell line is shown. A proliferation defect was observed only after 6 d of Dox-addition to the culture medium. Values are the mean of three measurements; error bars indicate standard deviation. (D) Viral lytic replication is decreased upon DKC1 knockdown. Left panel shows experimental outline. (Middle panel) Viral genome abundance in virions harvested from the culture supernatant was determined after DKC1 depletion (+Dox) and induction of lytic replication (+NaB). The EBV genome level was normalized to a spike-in control to account for sample loss during viral genome isolation. (Right panel) Measurement of intracellular EBV genome abundance normalized to the GAPDH locus. Results are the mean of three independent experiments; error bars represent standard deviation.
DISCUSSION
As multiple cellular noncoding RNAs that reside at high copy numbers have been shown to be equipped with various RNA modifications, we examined whether the equally abundant viral noncoding RNAs EBER1 and EBER2 are also modified. We recently showed that EBER1 carries a single 5-methylcytosine modification, which negatively affects its stability (Henry et al. 2020), and here we show that EBER2 similarly is also modified at a single nucleotide by pseudouridylation (Fig. 1). We mapped the pseudouridylation site by applying the CMCT-RT protocol and the recently developed method HydraPsiSeq, and conclude that DKC1, which is the catalytic subunit of the box H/ACA snoRNA-dependent machinery, in concert with SNORA22 is the writer for EBER2. Similar to other cellular noncoding RNAs, pseudouridylation of EBER2 increases its stability and, of physiological relevance, promotes efficient viral lytic replication of progeny EBV genomes.
Unlike standalone pseudouridine synthase enzymes, DKC1 is part of the snoRNA-guided complex that targets substrate RNAs through complementary base-pairing with a specific snoRNA; the duplex-forming regions are typically located within bulges at defined positions within the secondary structure of snoRNAs (Supplemental Fig. 1B). Using the 2CIMPL approach, we attempted to identify the EBER2-specific guide RNA and uncovered SNORA22 as a candidate box H/ACA snoRNA. We were able to locate nucleotide stretches upstream of the box H that showed a putative RNA duplex-forming region with EBER2's pseudouridylation site. The fact that the RNA hybrids retrieved by 2CIMPL are composed of the region surrounding the pseudouridylation site and the predicted base-pairing region of SNORA22 support the formation of this particular duplex (Supplemental Fig. 1D). Moreover, depletion of SNORA22 resulted in a decrease in detectable EBER2 Ψ160 by primer extension assays, indicating that DKC1 in concert with SNORA22 is responsible for this modification.
As DKC1, and thus the box H/ACA snoRNP machinery, modifies a plethora of RNAs, including mRNAs, snoRNAs, tRNAs, and rRNAs, we cannot rule out the possibility that the defect in viral lytic replication observed after DKC1 depletion is not due to abrogating EBER2 pseudouridylation but caused by a side effect of another RNA or a secondary effect of cell cycle arrest. However, our growth curve analysis following the time course of DKC1 knockdown as well as the examination of nascent protein production showed that the observed phenotype in lytic replication was examined well within the time frame of normal cell proliferation and before cell growth was affected by DKC1 depletion (Fig. 6B,C), thus supporting the notion that this phenotype is likely due to the loss of EBER2 pseudouridylation. We would like to point out that the effect on lytic replication was observed in a viral reactivation system that depends on non-specific chemical stimulation and in a cell culture system, and will have to be validated in a more physiological setting involving naturally infected primary B or epithelial cells.
EBER2 has previously been shown to be involved in transcription regulation and to be required for efficient viral lytic replication (Lee et al. 2015; Li et al. 2021). The transcriptional regulation of LMP genes is mediated by EBER2 via its binding to the TR regions of the EBV genome, which are located in close vicinity (either adjacent or in the intronic regions) of the LMP genes, and the regulation of viral lytic replication was also thought to be contingent upon EBER2 association with viral chromatin (Lee and Steitz 2015). However, our results indicate that these two phenotypes are likely mechanistically uncoupled, as preventing DKC1-mediated pseudouridylation has no effect on LMP gene expression but manifest itself in defects in lytic replication (Fig. 6A,D). How a single modification in EBER2 can modulate one pathway and not the other will have to be addressed in future studies that further dissect the molecular mechanism of EBER2 involvement in activating viral lytic replication. The regulation of and by EBER2 appears to be more intricate than anticipated and include several layers, as expression of a non-pseudouridylatable version of EBER2 (U160C mutant) by itself in a heterologous system had no effect on RNA stability (Supplemental Fig. 1F), suggesting that other factors present in the context of EBV infection are required to enhance EBER2 stability.
MATERIALS AND METHODS
Isolation of EBER2
EBER2 was in vitro-transcribed using T7 polymerase followed by PAGE-purification or isolated from total RNA from EBV-positive BJAB-B1 cells using biotinylated antisense oligonucleotides (ASOs) as described previously (Henry et al. 2020). In brief, 250 µg total RNA resuspended in 100 µL TE buffer was added to 20 µL ASO-beads (for all oligonucleotide sequences please see Supplemental Table 1), 100 µL H2O, 100 µL Denaturant buffer (100 mM HEPES pH 7.5, 8 M urea, 200 mM NaCl, 2% SDS), and 300 µL 2× Hybridization buffer (1.12 M urea, 1.5 M NaCl, 10× Denhardt's solution, 10 mM EDTA) following incubation for 4 h at RT on a rotator. Beads were washed three times with Wash buffer (10 mM HEPES pH 7.5, 250 mM NaCl, 2 mM EDTA, 1 mM EGTA, 0.1% N-lauroylsarcosine, 0.2% SDS), and RNA was eluted from the beads by adding 200 µL tetraethylammonium chloride (TEACl) buffer (10 mM Tris pH 7.4, 2.4 M TEACl, 0.05% Tween-20) and incubating the beads for 5 min at 40°C. RNA in the supernatant was extracted with phenol–chloroform and resolved on a 10% denaturing polyacrylamide gel prior to PAGE purification. An amount of 50 ng of purified synthetic or native EBER2 was used for each HydraPsiSeq experiment as previously described (Marchand et al. 2020). In brief, RNA was subjected to random hydrazine cleavage, followed by aniline-driven phosphodiester bond scission at all unmodified U residues (Helm et al. 2021). Pseudouridine (as well as some 5-substitute U residues, such as m5U) is resistant to hydrazine cleavage (Marchand et al. 2022). Resulting RNA fragments were converted into a sequencing library and sequenced on HiSeq1000 Illumina sequencer in SR50 (single-read 50 nt) mode. After trimming, reads were mapped to the EBER2 reference sequence and quantitative HydraPsiSeq scores were calculated from the normalized U-cleavage profile. Relative protection for all uridine residues was calculated using quantitative PsiScore, conceptually similar to ScoreC used in the RiboMethSeq protocol (Pichot et al. 2020).
For detection of pseudouridylation modification in native EBER2 by western blot analysis, a monoclonal antibody against pseudouridine (clone APU-6; MBL Life Science, cat. no D347-3) was used at a 1:100 dilution.
Primer extension assay following CMCT treatment of RNA
EBER2 was purified with biotinylated ASO-beads as described above from BJAB-B1, HH514-16, Raji, and Jijoye cells. EBER2 (250 ng) was ligated to a 3′ linker (RL3) by mixing 2 µL of 10× Reaction Buffer, 2 µL of 1 mg/mL BSA, 2 µL of 20 µM RL3, and 1 µL of T4 RNA ligase (Thermo EL0021) in a total reaction volume of 20 µL, and incubated overnight at 16°C. After phenol–chloroform extraction, the ligation product (EBER2-RL3) was digested with RNase H by adding 4 µL of 10× RNase H Buffer (NEB), 2 µL of 100 µM KD-ASO, 1 µL RNase inhibitor (NEB M0314), 2 µL RNase H (NEB M0297) in a total volume of 40 µL for 30 min at 37°C, followed by PAGE purification of the 3′ portion of EBER2-RL3.
EBER2-RL3 was treated with CMCT (Sigma, cat. no. C106402) by adding 20 µL of 1 M CMCT and 80 µL of BEU Buffer (50 mM Bicine, 7 M urea, 4 mM EDTA, pH 8.0–8.5) for 15 min at 37°C. After ethanol precipitation, RNA was incubated in Na2CO3 solution (50 mM Na2CO3, 2 mM EDTA, pH 10.8; adjusted by NaHCO3) for 1 h at 37°C followed by phenol–chloroform extraction. RNA was subjected to reverse transcription using gamma-ATP-labeled DP3 oligonucleotide in primer extension assays and subsequently resolved in a denaturing 12% urea polyacrylamide gel, which was dried prior to exposure to a phosphor imaging screen.
For primer extension assays after SNORA22 depletion, 2.5 × 106 BJAB-B1 cells were nucleofected with 5 µL each of 100 µM EBER2 and SNORA22 ASOs in Lonza's SF Buffer and program DS-120. After 48 h, nucleofection with SNORA22 ASOs was repeated. RNA was isolated after 72 h from the first nucleofection.
CLIP-seq and 2CIMPL assay
CLIP-seq (aka HITS-CLIP) was performed as previously described (Lee et al. 2017) using BJAB-B1 cells and anti-DKC1 antibody (Santa Cruz, cat. no. sc-373956). In brief, BJAB-B1 cells were irradiated with UV light twice at 400 mJ/cm2 on ice. Cells were lysed and nuclear extract was subjected to partial RNase digest prior to immunoprecipitation of DKC1–RNA adducts, which were ligated to a radioactively labeled 3′ linker, followed by SDS–PAGE and size selection of appropriately sized DKC1–RNA adducts. RNA footprints were separated from DKC1 protein by Proteinase K treatment and isolated by phenol–chloroform extraction. After ligation of a 5′ linker, DKC1-associated RNAs were reverse-transcribed, amplified by PCR, and subsequently converted into an Illumina-compatible library using the NEBNext Ultra DNA Library Kit (NEB). The library was deep sequenced with an iSeq100 system.
For 2CIMPL analysis, 2 × 107 BJAB-B1 cells were first irradiated with 254 nm UV light as for CLIP-seq followed by irradiation with 365 nm UV light in the presence of 50 µg/mL AMT for 30 min on ice. Cells were washed with PBS, and nuclear extract was prepared by lysing the cells in 500 µL Sucrose Lysis Buffer (10 mM Tris pH 8.0, 0.32 M Sucrose, 2 mM Mg-Acetate, 3 mM CaCl2, 0.1% NP-40). Cells were spun down in a tabletop centrifuge for 5 min at 3000 rpm, and the pelleted nuclei were lysed in 200 µL PXL buffer (1× PBS, 1% NP40, 0.5% deoxycholate, 0.1% SDS), incubated on ice for 5 min, and then spun at full speed for 10 min at 4°C. The supernatant was treated with 5 µL RQ1 DNase (Promega) for 5 min at 37°C, followed by partial RNase A digest for 5 min at 37°C by adding 0.25 or 0.025 µg RNase A. The reaction was stopped by adding 5 µL RNase inhibitor (RNasin Plus, Promega) and spun at full speed for 10 min. The reaction volume was adjusted to 400 µL with PXL buffer and precleared with 50 µL Protein G Dynabeads for 1 h at 4°C. An amount of 20 µL of DKC1 antibody-Dynabeads was added and incubated for 4 h, followed by three washes with PXL buffer and twice with PNK buffer (20 mM Tris pH 7.4, 10 mM MgCl2, 0.5% NP40). Beads was CIP-treated by adding 8 µL of 10× CutSmart Buffer, 2 µL RNasin, 3 µL CIP (NEB), and 67 µL H2O (total reaction volume of 80 µL) at 37°C for 20 min with constant shaking. Beads were then washed once with PXL buffer, once with PNK-EGTA buffer (20 mM Tris pH 7.4, 20 mM EGTA, 0.5% NP40), and twice with PNK buffer prior to polynucleotide kinase treatment by adding 8 µL of 10× PNK buffer (NEB), 5 µL of gamma-ATP, 2 µL RNasin, 4 µL T4 PNK (NEB), and 60 µL H2O (total reaction volume of 80 µL) at 37°C for 5 min, whereupon 1 µL of 10 mM cold ATP was added and incubated for an additional 20 min with constant shaking. Beads were washed once with PXL buffer and twice with PNK buffer, followed by overnight on-bead ligase reaction (10 µL 10× T4 RNA ligase 1 buffer (NEB B0216S), 10 µL 10 mM ATP, 2 µL RNasin, 5 µL T4 RNA ligase 1 (NEB M0204), 73 µL H2O) at 16°C with constant shaking. The beads were prepared for SDS–PAGE, the gel was transferred to a nitrocellulose membrane, and RNA was eluted from the membrane exactly as for CLIP-seq. Size-selected RNAs were irradiated for 8 min with 254 nm UV light on ice to reverse AMT-crosslinking and then ethanol-precipitated. The recovered RNAs were converted into an Illumina-compatible library using the NEBNext RNA Library Kit (NEB). The concentration of the library was determined using the NEBNext Library Quant Kit (NEB) and sequenced on the Illumina NextSeq platform. Data analysis was performed as described on our Github page at github.com/NaraLee-Lab/DKC1_2CIMPL. The next-generation sequencing files generated in this manuscript have been deposited in the Sequence Read Archive as BioProject ID PRJNA809673 (DKC1 CLIP-seq) and PRJNA809674 (2CIMPL).
Metabolic labeling of RNA and isolation of EBERs
Tet-inducible DKC1-knockdown BJAB-B1 cells were cultured either without or in the presence of 0.5 ng/mL Dox for 5 d. Cell count was adjusted to 106 cells per ml, and 20 µL (20 µCi) of 3H-uridine (PerkinElmer, cat. no. NET367250UC) was added to 2 mL of culture medium. After a 1-h pulse, RNA was isolated using TRIzol. EBER1 and EBER2 were isolated from total RNA using biotinylated ASOs as described above, resolved in a denaturing 10% polyacrylamide gel, and transferred to a nitrocellulose membrane, which was subjected to northern blot analysis using IR-dye coupled probes (Miller et al. 2018). Probe sequences are listed in Supplemental Table 1.
To metabolically label HH514-16 cells with 35S-methionine (PerkinElmer, cat. no. NEG772), cells were grown for 1 h in RPMI 1640 medium containing dialyzed FBS and no methionine or cysteine. After starvation, labeled amino acid mix was added to the culture medium (25 µL to 1.5 mL medium at 106 cells/mL). Cells were grown for 2 h before lysate was prepared using RIPA buffer.
Measuring viral genome abundance after induction of lytic replication
A Tet-inducible DKC1-knockdown cell line was established with the EBV-positive replication-competent HH514-16 cells (Rabson et al. 1983). To this end, dCas9–KRAB was cloned to be expressed under a Tet-regulated promoter in a lentiviral vector along with the sgRNA expression cassette targeting the DKC1 promoter (for a list of sgRNA sequences, please see Supplemental Table 1), followed by lentiviral transduction to generate a stable cell line. Doxycycline (Dox) was added to the culture medium at 0.5 µg/mL for the indicated time window, and 3 mM sodium butyrate (NaB) was added to activate the EBV lytic cycle. Twenty-four hours after NaB addition, viral lytic replication was assessed. Virions in the culture supernatant were isolated by passing the supernatant through a 0.45 µm filter, incubating it for 1 h at 37°C after adding 10 µg RNase A and 50 U DNase I to remove contaminating nucleic acids not packaged into virions, followed by ultracentrifugation at 28,000 rpm in a SW50.1 rotor for 2 h at 4°C. The pellet was resuspended in 600 µL lysis buffer (20 mM Tris pH 8.0, 10 mM EDTA, 100 mM NaCl, 0.5% SDS) for 10 min and treated with 100 µg Proteinase K for 2 h at 37°C. An amount of 10 ng of pcDNA3 vector was spiked in (to account for sample loss during recovery) before viral DNA was phenol–chloroform extracted. The EBV genome copy number was determined by qPCR using primers that target the EBV DS region and normalizing it to the Neomycin resistance cassette in the pcDNA3 vector.
To measure intracellular EBV genome abundance by qPCR, 2 × 106 cells were lysed in 300 µL of 50 mM Tris pH 8.0, 1% SDS, 10 mM EDTA, sonicated briefly, and spun at full speed for 10 min at 4°C. An amount of 50 µL of lysate was diluted with 450 µL of 16.7 mM Tris pH 8.0, 0.01% SDS, 1.1% Triton X-100, 165 mM NaCl, 1.2 mM EDTA, and 10 µg Proteinase K. After 6 h at 50°C, genomic DNA was extracted with phenol–chloroform and subjected to real-time PCR measurements.
SUPPLEMENTAL MATERIAL
Supplemental material is available for this article.
Supplementary Material
ACKNOWLEDGMENTS
We thank Annika Kotter for technical assistance. This work was supported by the National Institutes of Health (NIH grant number R21 AI151073) to N.L., and by ViroMOD FRCR funding from the Grand Est Region (France) to Y.M. M.H. was funded by the Deutsche Forschungsgemeinschaft (DFG, German Research Foundation)–TRR-319–TP A05 and SPP1784, HE 3397/13-2.
Footnotes
Article is online at http://www.rnajournal.org/cgi/doi/10.1261/rna.079219.122.
REFERENCES
- Bohnsack MT, Sloan KE. 2018. Modifications in small nuclear RNAs and their roles in spliceosome assembly and function. Biol Chem 399: 1265–1276. 10.1515/hsz-2018-0205 [DOI] [PubMed] [Google Scholar]
- Carlile TM, Rojas-Duran MF, Zinshteyn B, Shin H, Bartoli KM, Gilbert WV. 2014. Pseudouridine profiling reveals regulated mRNA pseudouridylation in yeast and human cells. Nature 515: 143–146. 10.1038/nature13802 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ge J, Yu YT. 2013. RNA pseudouridylation: new insights into an old modification. Trends Biochem Sci 38: 210–218. 10.1016/j.tibs.2013.01.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- He J, Navarrete S, Jasinski M, Vulliamy T, Dokal I, Bessler M, Mason PJ. 2002. Targeted disruption of Dkc1, the gene mutated in X-linked dyskeratosis congenita, causes embryonic lethality in mice. Oncogene 21: 7740–7744. 10.1038/sj.onc.1205969 [DOI] [PubMed] [Google Scholar]
- Helm M, Schmidt-Dengler MC, Weber M, Motorin Y. 2021. General principles for the detection of modified nucleotides in RNA by specific reagents. Adv Biol (Weinh) 5: e2100866. 10.1002/adbi.202100866 [DOI] [PubMed] [Google Scholar]
- Henry BA, Kanarek JP, Kotter A, Helm M, Lee N. 2020. 5-methylcytosine modification of an Epstein-Barr virus noncoding RNA decreases its stability. RNA 26: 1038–1048. 10.1261/rna.075275.120 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Howe JG, Steitz JA. 1986. Localization of Epstein-Barr virus-encoded small RNAs by in situ hybridization. Proc Natl Acad Sci 83: 9006–9010. 10.1073/pnas.83.23.9006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Itoh K, Mizugaki M, Ishida N. 1989. Detection of elevated amounts of urinary pseudouridine in cancer patients by use of a monoclonal antibody. Clin Chim Acta 181: 305–315. 10.1016/0009-8981(89)90236-2 [DOI] [PubMed] [Google Scholar]
- Lee N. 2021. The many ways Epstein-Barr virus takes advantage of the RNA tool kit. RNA Biol 18: 759–766. 10.1080/15476286.2021.1875184 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee N, Steitz JA. 2015. Noncoding RNA-guided recruitment of transcription factors: a prevalent but undocumented mechanism? Bioessays 37: 936–941. 10.1002/bies.201500060 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee N, Moss WN, Yario TA, Steitz JA. 2015. EBV noncoding RNA binds nascent RNA to drive host PAX5 to viral DNA. Cell 160: 607–618. 10.1016/j.cell.2015.01.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee N, Yario TA, Gao JS, Steitz JA. 2016. EBV noncoding RNA EBER2 interacts with host RNA-binding proteins to regulate viral gene expression. Proc Natl Acad Sci 113: 3221–3226. 10.1073/pnas.1601773113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee N, Le Sage V, Nanni AV, Snyder DJ, Cooper VS, Lakdawala SS. 2017. Genome-wide analysis of influenza viral RNA and nucleoprotein association. Nucleic Acids Res 45: 8968–8977. 10.1093/nar/gkx584 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lerner MR, Andrews NC, Miller G, Steitz JA. 1981. Two small RNAs encoded by Epstein-Barr virus and complexed with protein are precipitated by antibodies from patients with systemic lupus erythematosus. Proc Natl Acad Sci 78: 805–809. 10.1073/pnas.78.2.805 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le Sage V, Kanarek JP, Snyder DJ, Cooper VS, Lakdawala SS, Lee N. 2020. Mapping of influenza virus RNA–RNA interactions reveals a flexible network. Cell Rep 31: 107823. 10.1016/j.celrep.2020.107823 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lestrade L, Weber MJ. 2006. snoRNA-LBME-db, a comprehensive database of human H/ACA and C/D box snoRNAs. Nucleic Acids Res 34: D158–D162. 10.1093/nar/gkj002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li X, Zhu P, Ma S, Song J, Bai J, Sun F, Yi C. 2015. Chemical pulldown reveals dynamic pseudouridylation of the mammalian transcriptome. Nat Chem Biol 11: 592–597. 10.1038/nchembio.1836 [DOI] [PubMed] [Google Scholar]
- Li Z, Baccianti F, Delecluse S, Tsai MH, Shumilov A, Cheng X, Ma S, Hoffmann I, Poirey R, Delecluse HJ. 2021. The Epstein-Barr virus noncoding RNA EBER2 transactivates the UCHL1 deubiquitinase to accelerate cell growth. Proc Natl Acad Sci 118: e2115508118. 10.1073/pnas.2115508118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Machnicka MA, Milanowska K, Osman Oglou O, Purta E, Kurkowska M, Olchowik A, Januszewski W, Kalinowski S, Dunin-Horkawicz S, Rother KM, et al. 2013. MODOMICS: a database of RNA modification pathways–2013 update. Nucleic Acids Res 41: D262–D267. 10.1093/nar/gks1007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marchand V, Pichot F, Neybecker P, Ayadi L, Bourguignon-Igel V, Wacheul L, Lafontaine DLJ, Pinzano A, Helm M, Motorin Y. 2020. HydraPsiSeq: a method for systematic and quantitative mapping of pseudouridines in RNA. Nucleic Acids Res 48: e110. 10.1093/nar/gkaa769 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marchand V, Bourguignon-Igel V, Helm M, Motorin Y. 2022. Analysis of pseudouridines and other RNA modifications using HydraPsiSeq protocol. Methods 203: 383–391. 10.1016/j.ymeth.2021.08.008 [DOI] [PubMed] [Google Scholar]
- Miller BR, Wei T, Fields CJ, Sheng P, Xie M. 2018. Near-infrared fluorescent northern blot. RNA 24: 1871–1877. 10.1261/rna.068213.118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moore MJ, Zhang C, Gantman EC, Mele A, Darnell JC, Darnell RB. 2014. Mapping Argonaute and conventional RNA-binding protein interactions with RNA at single-nucleotide resolution using HITS-CLIP and CIMS analysis. Nat Protoc 9: 263–293. 10.1038/nprot.2014.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moss WN, Steitz JA. 2013. Genome-wide analyses of Epstein-Barr virus reveal conserved RNA structures and a novel stable intronic sequence RNA. BMC Genomics 14: 543. 10.1186/1471-2164-14-543 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pichot F, Marchand V, Ayadi L, Bourguignon-Igel V, Helm M, Motorin Y. 2020. Holistic optimization of bioinformatic analysis pipeline for detection and quantification of 2′-O-methylations in RNA by RiboMethSeq. Front Genet 11: 38. 10.3389/fgene.2020.00038 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rabson M, Heston L, Miller G. 1983. Identification of a rare Epstein-Barr virus variant that enhances early antigen expression in Raji cells. Proc Natl Acad Sci 80: 2762–2766. 10.1073/pnas.80.9.2762 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rintala-Dempsey AC, Kothe U. 2017. Eukaryotic stand-alone pseudouridine synthases—RNA modifying enzymes and emerging regulators of gene expression? RNA Biol 14: 1185–1196. 10.1080/15476286.2016.1276150 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roundtree IA, Evans ME, Pan T, He C. 2017. Dynamic RNA modifications in gene expression regulation. Cell 169: 1187–1200. 10.1016/j.cell.2017.05.045 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwartz S, Bernstein DA, Mumbach MR, Jovanovic M, Herbst RH, Leon-Ricardo BX, Engreitz JM, Guttman M, Satija R, Lander ES, et al. 2014. Transcriptome-wide mapping reveals widespread dynamic-regulated pseudouridylation of ncRNA and mRNA. Cell 159: 148–162. 10.1016/j.cell.2014.08.028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spenkuch F, Motorin Y, Helm M. 2014. Pseudouridine: still mysterious, but never a fake (uridine)!. RNA Biol 11: 1540–1554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu M, Yao Y, Chen H, Zhang S, Cao SM, Zhang Z, Luo B, Liu Z, Li Z, Xiang T, et al. 2019. Genome sequencing analysis identifies Epstein-Barr virus subtypes associated with high risk of nasopharyngeal carcinoma. Nat Genet 51: 1131–1136. 10.1038/s41588-019-0436-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao Y, Dunker W, Yu YT, Karijolich J. 2018. The role of noncoding RNA pseudouridylation in nuclear gene expression events. Front Bioeng Biotechnol 6: 8. 10.3389/fbioe.2018.00008 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.