

Abstract
Orthosteric activation of CB1 is known to cause a plethora of adverse side effects in vivo. Allosteric modulation is an exciting therapeutic approach and is hoped to offer improved therapeutic potential and a reduced on-target side effect profile compared to orthosteric agonists. This study aimed to systematically characterize the in vitro activity of the positive allosteric modulator ZCZ011, explicitly considering its effects on receptor regulation. HEK293 cells expressing hCB1 receptors were used to characterize ZCZ011 alone and in combination with orthosteric agonists. Real-time BRET approaches were employed for G protein dissociation, cAMP signaling, and β-arrestin translocation. Characterization also included ERK1/2 phosphorylation (PerkinElmer AlphaLISA) and receptor internalization. ZCZ011 is an allosteric agonist of CB1 in all pathways tested, with a similar signaling profile to that of the partial orthosteric agonist Δ9-tetrahydrocannabinol. ZCZ011 also showed limited positive allosteric modulation in increasing the potency and efficacy of THC-induced ERK1/2 phosphorylation, β-arrestin translocation, and receptor internalization. However, no positive allosteric modulation was observed for ZCZ011 in combination with either CP55940 or AMB-FUBINACA, in G protein dissociation, nor cAMP inhibition. Our study suggests that ZCZ011 is an allosteric agonist, with effects that are often difficult to differentiate from those of orthosteric agonists. Together with its pronounced agonist activity, the limited extent of ZCZ011 positive allosteric modulation suggests that further investigation into the differences between allosteric and orthosteric agonism is required, especially in receptor regulation end points.
Keywords: cannabinoid receptor 1, allosteric modulator, allosteric agonist
Introduction
Phytocannabinoids from Cannabis sativa, endogenous cannabinoids, and synthetic cannabinoids act primarily through two cannabinoid receptor subtypes: cannabinoid receptor 1 (CB1) and 2 (CB2).1−3 Ligands that activate CB1 are currently being investigated as potential therapeutics in a range of central nervous system (CNS) disorders including neuropathic and inflammatory pain, Huntington’s disease, multiple sclerosis, epilepsy, and Parkinson’s disease.4 The traditional approach to developing therapeutics at CB1 has centered around compounds that bind to the orthosteric binding site which is common to the endocannabinoids 2-arachidoloylglycerol (2-AG), anandamide (AEA), and the phytocannabinoid Δ9-trans-tetrahydrocannabinol (THC). However, the therapeutic utility of CB1 is limited by psychoactivity, and more recently, significant toxicological effects such as seizures, hypothermia, and tachycardia have been observed following recreational use of high efficacy synthetic cannabinoids.5,6
One potential approach to reducing on-target adverse effects is the use of allosteric modulators. Allosteric ligands are compounds that bind to a receptor at a site that is topographically distinct from the orthosteric binding site.7 They can increase (positive allosteric modulation) or decrease (negative allosteric modulation) the binding and/or signaling of orthosterically bound ligands. “Pure” allosteric modulators have no activity in the absence of orthosteric ligands and, therefore, have the potential to exogenously regulate endocannabinoid action in specific regions/tissues implicated in disease states. Allosteric modulators are thought to be therapeutically advantageous; they display a “ceiling effect” (that is, the degree of cooperativity is saturable), have higher receptor subtype specificity, and are also hoped to cause reduced tolerance and receptor downregulation,8 though to date there is limited evidence to support this latter attribute. In contrast, allosteric agonists are ligands that induce receptor activation and downstream signaling cascades independently; that is, in the absence of an orthosteric ligand.9
The first cannabinoid positive allosteric modulators reported in literature are from the 2-phenylindole structural class and include GAT211 (the racemic mixture of GAT229 and GAT228) and ZCZ011 (Figure 1A, a racemic mixture for which enantiomers have not been separately characterized).10,11 GAT229 and GAT228 are reported to have different functional outcomes in vitro, where GAT229 is a pure positive allosteric modulator and GAT228 is an allosteric agonist.10 This may also be the case for ZCZ011, although separation of the enantiomers is required to investigate this further.
Figure 1.

Concentration–response curves showing allosteric agonism of ZCZ011 and orthosteric agonism of THC, AMB-FUBINACA, and CP55940 in HEK293 cells. (A) Chemical structure of ZCZ011, (B) inhibition of cAMP stimulated by 5 μM forskolin and cannabinoid ligands, (C) dissociation of Gαi3 and Gβγ subunits as determined by TRUPATH BRET assay stimulated by cannabinoid ligands, (D) phosphorylation of ERK1/2 by cannabinoid ligands, (E) internalization of hCB1, and (F) translocation of β-arrestin-2 by cannabinoid ligands. Representative data is expressed as mean ± SD of technical duplicates or triplicates in the same assay. Each experiment was repeated five times (n = 5), and mean parameters are reported in the text or Table 1.
ZCZ011 was first reported by Ignatowska-Jankowska et al.11 as an ago-PAM (positive allosteric modulator that displays intrinsic efficacy in the absence of an orthosteric ligand). In vitro, ZCZ011 increased binding of [3H]CP55940 and potentiated signaling of AEA in [35S]GTPγS binding, β-arrestin recruitment, and ERK phosphorylation (in PathHunter β-arrestin cells and CHO-hCB1 cells, respectively). When tested alone, ZCZ011 was found to be an agonist in the DiscoverX cAMP assay, a weak agonist in β-arrestin recruitment and ERK phosphorylation, but paradoxically caused no [35S]GTPγS binding.11In vivo, ZCZ011 alone (40 mg/kg) reversed mechanical and cold allodynia in neuropathic pain (chronic constriction injury model), without causing any cannabimimetic effects (catalepsy, hypothermia, locomotor depression, or conditioned place preference). The effects were found to be of long duration with no development of tolerance after repeated administration. In combination with CP55940, ZCZ011 potentiated antinociception while also augmenting the adverse CB1-mediated effects such as catalepsy and hypothermia.11 Following the initial characterization of ZCZ011, it has been reported as a positive allosteric modulator of CP55940 in cAMP accumulation and β-arrestin recruitment, with intrinsic efficacy in both pathways.12,13
There are currently two proposed binding sites for the 2-phenylindole structural class of positive allosteric modulators, the first based on the crystal structure of ZCZ011 in combination with the orthosteric agonist CP5594014 and the second being the GAT229 binding site (proposed on the basis of in silico docking data15). Both structures suggest that ZCZ011 would stabilize an active conformation of CB1 with subtle differences to an orthosteric agonist. It therefore remains unclear whether allosteric agonists will trigger the same signaling and desensitization pathways as orthosteric agonists. Such data is of particular interest given the suggestion that allosteric ligands may show reduced desensitization and tolerance.8 We therefore aimed to perform an in vitro characterization of ZCZ011 at CB1 in the presence and absence of orthosteric agonists, with a focus on receptor regulation pathways (β-arrestin recruitment and receptor internalization). Classical signaling pathways were also included for comparison.
Results and Discussion
Ago-PAMs such as GAT211, GAT229, ZCZ011, and a series of structural analogues, have been found to activate CB1in vivo and induce antinociception, decrease intraocular pressure (IOP), and slow the progression and severity of an animal model of Huntington’s Disease. Interestingly these in vivo effects have been observed in the absence of cannabimimetic effects associated with orthosteric activation of CB1, such as tolerance, catalepsy, hypothermia, and dependence.11,16−20 To date, no in vitro mechanism has been defined that explains the differences between orthosteric and allosteric agonism. This study provides a comprehensive in vitro characterization of the allosteric agonist ZCZ011, with a focus on receptor regulation end points, and compares the allosteric signaling profile to those of classical orthosteric cannabinoids. Orthosteric agonists were chosen to represent a range of efficacy profiles, THC is generally considered to be a partial agonist, and AMB FUBINACA is a high efficacy synthetic cannabinoid. For some assays where THC and AMB-FUBINACA produce vastly different profiles (e.g., G protein dissociation and β-arrestin translocation), CP55940 was also included as a more moderate efficacy ligand.
Signaling Profile of the Allosteric Agonist ZCZ011 in the Absence of Orthosteric Agonist
cAMP Inhibition
Canonical signaling of CB1 is through Gαi proteins, which decrease cAMP levels by inhibition of adenylate cyclase. ZCZ011 alone caused concentration-dependent inhibition of forskolin stimulated cAMP with an Emax of 63.7 ± 1.7% inhibition of cAMP (curve span; n = 5, Figure 1B), equi-efficacious with the previously described full agonist AMB-FUBINACA (Emax = 66.6 ± 2.8%; n = 5) and of slightly but significantly higher efficacy than the partial agonist THC (Emax = 56.1 ± 1.9%; n = 5, p < 0.05) (Figure 1B). Though a high efficacy ligand in this pathway, ZCZ011 was significantly less potent (pEC50 6.53 ± 0.10; n = 5) than both THC and AMB-FUBINACA (pEC50 = 8.17 ± 0.11, and 9.57 ± 0.09 respectively; n = 5, Figure 1B). This finding aligns with the initial characterization11 and two more recent studies,12,13 which all report ZCZ011 to be an efficacious agonist in cAMP inhibition. Real-time kinetic traces of cAMP inhibition can be found in Figures S1 and S2.
G Protein Dissociation
As the cAMP pathway has high receptor reserve in the HEK cell line used in the current study, low receptor occupancy is sufficient to cause full cAMP inhibition.21 Therefore, this pathway cannot easily distinguish between partial and full agonists,21 as evidenced by the high efficacy of THC in Figure 1B. As the reserve “bottleneck” in this pathway is thought to be the adenylate cyclase, an assay measuring G protein dissociation may more accurately report ligand efficacy.22 G protein dissociation was therefore investigated in HEK-hCB1 cells using a real-time TRUPATH BRET assay23 in the hope that this pathway would better reflect differing compound efficacies. The Gαi3 biosensor was chosen, as this pathway shows a robust response with a broad range of ligand efficacies for CB1.
ZCZ011 caused G protein dissociation in a concentration-dependent manner (pEC50 = 6.11 ± 0.07), Figure 1C), with lower potency than both AMB-FUBINACA and CP55940 (Table 1). Interestingly, THC did not induce robust G protein dissociation in this assay, indicating comparatively lower efficacy (Figure 1C). ZCZ011 was therefore found to be a higher efficacy ligand (Emax = 132.60 ± 11.12) than THC in this pathway, equi-efficacious with CP55940, and less efficacious than AMB-FUBINACA (Figure 1C, Table 1). Real-time kinetic data can be found in Figures S1 and S3. Our study is the first to report that ZCZ011 is an efficacious agonist of G protein dissociation using novel TRUPATH BRET biosensors, which contrasts with a related G protein activity assay paradigm; [35S]GTPγS binding experiments suggested that ZCZ011 did not induce G protein activation.11 In contrast to the G protein dissociation assay, the GTPγS experiment is carried out in cell membranes, and the assay is highly sensitive to NaCl and Mg2+ concentrations such that the assay can be optimized to be more sensitive to agonism or inverse agonism through buffer components.24 It is therefore possible that the previous GTPγS assays11 lacked sensitivity to the agonist activity of ZCZ011.
Table 1. Potencies (pEC50) and Efficacies (Emax) of G Protein Dissociation by Cannabinoid Ligands at CB1a.
| ligand | pEC50 | Emax (ΔBRET.sec) |
|---|---|---|
| THC | ND | ND |
| THC + 1 μM ZCZ011 | ND | ND |
| AMB-FUBINACA | 8.67 ± 0.05† | 176.00 ± 9.98† |
| AMB-FUBINACA + 1 μM ZCZ011 | 8.42 ± 0.02* | 189.70 ± 7.31 |
| CP55940 | 7.74 ± 0.08† | 150.80 ± 8.15 |
| CP55940 + 1 μM ZCZ011 | 7.57 ± 0.11 | 176.20 ± 7.30* |
Data is presented as mean ± SEM of five independent biological replicates, with Emax defined as the top of the curve (maximal response). Statistical tests to compare orthosteric ligand responses in the absence and presence of ZCZ011 were performed in GraphPad Prism using paired t tests (* < 0.05). Statistical tests to compare ZCZ011 (alone) to orthosteric agonists (alone) were performed in GraphPad Prism using a repeated measures one-way ANOVA with Tukey’s posthoc test († < 0.05). ND indicates values that were not determined due to the low signal size.
ERK1/2 Phosphorylation
Canonical signaling of CB1 includes phosphorylation of the mitogen-activated protein kinases ERK1/2, attributed to both Gα and Gβγ signaling.25−27 This signaling paradigm was investigated using the AlphaLISA Surefire pERK kit, where all responses were normalized to pERK response in the presence of vehicle (0%) or 1 μM CP55940 (100%) at 3 min 30 s, the time point for peak pERK activity.
ZCZ011 induced phosphorylation of ERK1/2 at concentrations greater than 1 μM, with 10 μM ZCZ011 inducing the same level of phosphorylation as 10 μM THC (37.1 ± 7.1%, and 30.3 ± 3.4% respectively, n = 5, Figure 1D), although the curve may not have saturated due to its low potency in this assay. ZCZ011 was also found to be an agonist with similar potency and efficacy in this pathway when initially characterized.11
Receptor Internalization
Concentration–response curves for CB1 internalization were generated over a 60 min time course of agonist exposure in the absence and presence of ZCZ011. ZCZ011 alone produced concentration-dependent receptor internalization with significantly lower potency (pEC50 = 5.87 ± 0.06) but higher efficacy (Emax = 0.0156 ± 0.0024 min–1) compared to THC (Figure 1E, Table 4). ZCZ011 alone (1 μM) induced CB1 internalization with a similar rate to that of 1 μM THC (with half-lives of 44.86 ± 8.86 min and 44.77 ± 3.68 min, respectively).
Table 4. Potencies (pEC50) and Efficacies (Emax) of CB1 Internalization by Cannabinoid Ligands over 60 mina.
| ligand | pEC50 | Emax (min–1, span) |
|---|---|---|
| THC | 7.66 ± 0.08 | 0.0075 ± 0.0010 |
| THC + 100 nM ZCZ011 | 7.76 ± 0.13 | 0.0148 ± 0.0004* |
| THC + 1 μM ZCZ011 | 7.89 ± 0.03 | 0.0260 ± 0.0018* |
Data is presented as mean ± SEM of five independent biological replicates, with Emax expressed as the span of the curve. Statistical tests (comparing THC alone to all other conditions) were performed in GraphPad Prism using a repeated measures one-way ANOVA with Dunnett’s posthoc tests; p values indicated as * < 0.05.
A novel finding of our study is that ZCZ011 induces efficacious internalization of CB1. Together with the β-arrestin translocation data, this provides insight into the receptor regulation occurring through allosteric activation of CB1. A theoretical advantage of allosteric modulators is that tolerance may be reduced compared to exogenous orthosteric agonists.8In vivo data has shown that chronic administration of ZCZ011 alone (40 mg/kg) causes antiallodynia with no loss of effect over time (no development of tolerance), although tolerance was not explored for ZCZ011 in combination with an exogenous orthosteric agonist.11 Data from the current study shows that ZCZ011 induces translocation of β-arrestin 2 to the membrane and efficacious receptor internalization in the absence of orthosteric ligand (Figure 3, 1D). Furthermore, ZCZ011 induces CB1 internalization with greater efficacy than THC, an orthosteric ligand that is known to cause receptor downregulation and produce tolerance in vivo.28 In this light, the lack of tolerance produced by ZCZ011 in vivo is surprising.11 It is important to note, however, that in vivo experiments cannot be performed in the absence of the endogenous cannabinoids (AEA and 2-AG), meaning that it is not possible to distinguish between the agonism of ZCZ011 alone and its positive allosteric modulation of endocannabinoids.
Figure 3.

Concentration–response curves showing β-arrestin 1 (A, C, E) or 2 (B, D, F) translocation by cannabinoid ligands in HEK293-WT cells transiently transfected with hCB1, compared to simulations of additive agonism. Data has been normalized to appropriate controls and pooled across five independent biological replicates (n = 5). Data is expressed as mean ± SEM.
ZCZ011 was found to be less potent and less efficacious in all pathways tested when compared to the full agonist AMB-FUBINACA. In contrast, it was less potent but equi-efficacious to the partial agonist CP55940 in β-arrestin 2 translocation and G protein dissociation pathways. In comparison to the phytocannabinoid THC, ZCZ011 was found to be of higher efficacy in cAMP inhibition, G protein dissociation, and receptor internalization (Tables 1 and 5) and equi-efficacious in ERK1/2 phosphorylation and β-arrestin translocation (Tables 1 and 4). The apparent inconsistency of ZCZ011 appearing more efficacious compared to THC in all G protein dependent pathways except for ERK1/2 phosphorylation could be due to poor definition of the top of the curve due to the low potency of ZCZ011 in this pathway. However, in all signaling pathways, ZCZ011 was less potent than THC.
Table 5. Conclusions from Comparison of Empirical Data to Simulations of Additive Agonism for Orthosteric Ligands in Combination with ZCZ011a.
| signaling pathway | THC | CP55940 | AMB-FUBINACA |
|---|---|---|---|
| cAMP | inconsistent (negative) | – | – |
| G protein dissociation | – | inconsistent (negative) | inconsistent (negative) |
| ERK1/2 phosphorylation | inconsistent (positive) | – | not inconsistent |
| β-arrestin translocation | inconsistent (positive) | not inconsistent | not inconsistent |
| receptor trafficking | inconsistent (positive) | – | – |
Comparisons between empirical data and in silico simulations of additive agonism have been qualitatively classified as “not inconsistent” (could be explained by additive agonism) and “inconsistent” (negative = less-than-additive effect, positive = greater-than-additive effect).
The ability of ZCZ011 to induce canonical signaling associated with orthosteric activation of CB1 with equal/greater efficacy than that of THC is notable for the future development of exogenous therapeutics targeting CB1. This finding is surprising given ZCZ011’s apparent lack of classical tetrad effects when administered alone, in contrast to the on-target tetrad effects produced by orthosteric agonists.11,12 Following in vivo dosing of 40 mg/kg (i.p.), ZCZ011 has been found in whole brain homogenates at concentrations ranging from 19 nM to 9.82 μM,11,29 suggesting the concentrations utilized in the current study are physiologically attainable and relevant. As ZCZ011 behaves as a classical CB1 agonist in HEK cells, but has a distinct in vivo profile, it is clear we need a greater understanding of how CB1 activation leads to the production of on-target adverse effects. All in vivo experimentation is performed in rodents; it is therefore possible that subtle species differences in CB1 (between mice and humans) is contributing to the differences in in vitro and in vivo findings. Investigation into the mechanism by which ZCZ011 effects rodent CB1 compared to human CB1 should be done to validate these findings.
Translocation of β-Arrestin 1 and 2 to the Cell Membrane
Due to literature suggesting that allosteric modulators may result in reduced drug-induced tolerance,8,30,31 this study aimed to characterize regulation and desensitization pathways of ZCZ011. Translocation of β-arrestins (1 and 2) to the membrane is thought to be involved in regulation of receptor number. G protein coupled receptor kinases (GRKs) phosphorylate CB1, and this causes an increase in recruitment of β-arrestin 1 and 2 to the receptor. This study measures translocation of β-arrestin 1 and 2 to the membrane using a real-time BRET assay in HEK cells transiently transfected with pplss-3HA-hCB1 and BRET assay components as previously described.32,33
Similar to THC, ZCZ011 alone did not cause β-arrestin 1 translocation. ZCZ011 induced β-arrestin 2 translocation with equivalent potency and efficacy to THC (pEC50 = 5.09 ± 0.09, Emax = 64.17 ± 8.09, Table 3) was found to be equi-efficacious (but less potent) than the partial agonist CP55940 and of lower efficacy than AMB-FUBINACA (Figure 1F, Table 3). Real-time kinetic data showing translocation of β-arrestin 2 by ZCZ011 can be found in Figure S1. This data aligns well with previous literature, in which ZCZ011 was found to be an agonist in β-arrestin 2 recruitment (though β-arrestin 1 recruitment was not previously investigated).11−13
Table 3. Potencies (pEC50) and Efficacies (Emax) of β-Arrestin 1 and 2 Translocation by Ligands at CB1a.
| β-arrestin 1 |
β-arrestin 2 |
|||
|---|---|---|---|---|
| ligand | pEC50 | Emax (span) | pEC50 | Emax (span) |
| THC | 5.98 ± 0.58 | 6.31 ± 2.13b | 5.59 ± 0.23 | 39.84 ± 4.02 |
| THC + 1 μM ZCZ011 | 6.72 ± 0.54 | 18.63 ± 3.75*b | 6.62 ± 0.34* | 43.56 ± 3.57 |
| THC + 10 μM ZCZ011 | 6.83 ± 0.56 | 34.42 ± 5.36*b | 6.89 ± 0.09* | 54.53 ± 3.07 |
| CP55940 | 7.22 ± 0.22 | 20.84 ± 3.20 | 7.69 ± 0.04† | 99.27 ± 14.93 |
| CP55940 + 1 μM ZCZ011 | 7.61 ± 0.13 | 28.64 ± 2.31 | 7.76 ± 0.06 | 108.00 ± 16.83 |
| CP55940 + 10 μM ZCZ011 | 7.51 ± 0.12 | 33.06 ± 2.94* | 7.94 ± 0.14 | 111.30 ± 13.75 |
| AMB-FUBINACA | 7.52 ± 0.06 | 60.59 ± 4.72 | 7.96 ± 0.05† | 195.30 ± 28.44† |
| AMB-FUBINACA + 1 μM ZCZ011 | 7.80 ± 0.08* | 58.92 ± 3.65 | 8.27 ± 0.10* | 161.20 ± 19.63* |
| AMB-FUBINACA + 10 μM ZCZ011 | 8.04 ± 0.07* | 53.05 ± 3.29 | 8.44 ± 0.05* | 163.50 ± 20.65* |
Data is presented as mean ± SEM of five independent biological replicates, with Emax expressed as the span of the curve. Statistically significant differences when comparing the orthosteric ligand in the absence and presence of ZCZ011 were performed in GraphPad Prism using repeated measures one-way ANOVA with Dunnett’s multiple comparisons test (* < 0.05). Statistically significant differences when comparing ZCZ011 to orthosteric ligands alone were performed in GraphPad Prism using a mixed effects analysis with multiple comparisons († < 0.05). ND indicates values that were not determined.
A concentration response curve was not accurately fit, therefore response at a 10 μM orthosteric agonist was used.
Analysis of Allosteric Modulation by ZCZ011
Current literature surrounding allosteric agonists in combination with orthosteric agonists is prone to misinterpretation due to experimental design and analysis failing to consider the magnitude of allosteric agonism in the absence of an orthosteric ligand.34 One example of this misinterpretation is when PAM concentration–response curves are performed in the presence of the EC20 orthosteric agonist, without considering the activity of the allosteric compound alone. This can lead to the inability to distinguish allosteric agonism from allosteric effects that are specific to the presence of an orthosteric agonist,17,35−37 resulting in the incorrect designation of allosteric agonists as allosteric modulators. A similar example is when statistical analysis is performed on single points within a concentration–response, to compare the response of an orthosteric ligand alone to its response in the presence of a fixed concentration of allosteric ligand (e.g., Garai et al.37). This is problematic because allosteric compounds with intrinsic activity will cause a baseline shift in the orthosteric agonist concentration–response curve, which will not be accounted for in this type of analysis. When potency and efficacy values are interpolated and compared without presentation of the curves themselves, the ability to draw conclusions is impinged.35,37
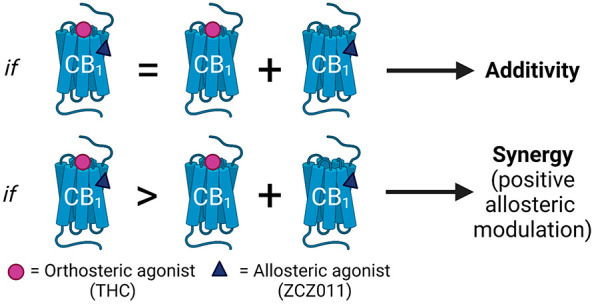
Allosteric agonists in combination with an orthosteric agonist produce signaling profiles that are difficult to interpret, and because more than one type of interaction may occur, allosteric agonism (additive effects) must be differentiated from positive allosteric modulation (synergistic effects). If the orthosteric ligand is a full agonist and no increase in maximal response is possible, the addition of an allosteric ligand with agonist properties will cause an increase in the baseline but no change to Emax (as in Figure 3F). This results in a decreased span of response and may therefore manifest as an apparent increase in the potency of the orthosteric agonist, although this is actually an artifact of the increased baseline (Figure 3F, Table 3). This has been seen throughout allosteric modulator literature, where increases in potency are reported as evidence for positive allosteric modulation despite shifted baselines.10 Similarly, for partial orthosteric agonists (for which the response produced is not pathway-maximal; as for THC and CP55940 in several pathways in this study), the addition of an allosteric agonist will increase both the bottom of the curve and Emax. This increase in Emax appears to signal an increase in the efficacy of the orthosteric agonist, but the span of the orthosteric ligand response is not actually increased, rather, the curve no longer starts at zero. To capture the effect of allosteric agonism and positive allosteric modulation in combination with both high and low efficacy ligands, we selected orthosteric ligands for their established variable efficacies (rather than prioritizing endogenous ligands, which are likely more relevant for considering the effects of allosteric ligands in vivo).
Due to the complex signaling profiles of allosteric agonists in combination with orthosteric agonists, a nonclassical analysis method is needed to interpret these findings. To distinguish allosteric agonism from allosteric modulation, we simulated “additive agonism” profiles for each pathway investigated. These profiles entailed two agonists binding to noninteracting sites within the same system. By creating overlay plots of additive agonism simulations with empirical results, we were able to qualitatively assess whether the increases in potency and/or efficacy we observed could be explained simply by additive agonism, or whether a different mechanistic explanation (such as positive allosteric modulation) was required to explain the data.
cAMP Inhibition
To investigate the effect ZCZ011 has on cAMP inhibition, THC in the absence and presence of several concentrations of ZCZ011 was explored. Due to the large receptor reserve in the cAMP pathway, partial agonists induce close to maximal cAMP inhibition. For this reason, only the partial agonist THC was investigated in combination with ZCZ011 as full agonist responses would not be further potentiated. Real-time kinetic data of cAMP inhibition by THC in combination with ZCZ011 is reported in Figure S2. The span of the THC concentration–response curve was progressively reduced with increasing ZCZ011-concentrations due to the agonism of ZCZ011 alone at these concentrations. However, the maximum extent (Emax) of the response was unchanged from THC alone (Emax = 47.0 ± 2.6%; n = 5) with the addition of 10 nM, 100 nM, and 1 μM ZCZ011 (Emax = 49.4 ± 3.9%, 44.3 ± 1.2%, and 41.7 ± 1.2%, respectively; n = 5) (Figure 2A). At concentrations of 10 nM and 100 nM, ZCZ011 decreased the potency of THC (n = 5) from pEC50 = 8.42 ± 0.11 to 7.93 ± 0.03 and 8.05 ± 0.01, respectively (Figure 2A). In combination with 1 μM ZCZ011, the potency of THC was unchanged (pEC50 = 8.63 ± 0.06; n = 5). While the experimental data showed that ZCZ011 decreased THC potency compared to THC alone, simulation of additive agonism for cAMP inhibition suggested that costimulation would increase THC potency (Figure 2A). The incongruence of these data sets can be interpreted to mean that the experimental data is inconsistent with the effects of ZCZ011 and THC acting either additively or synergistically in this pathway (Table 5).
Figure 2.

Concentration–response curves showing the G protein dependent signaling of ZCZ011 in combination with orthosteric agonists in HEK293-hCB1 cells compared to simulations of additive agonism. (A) Inhibition of cAMP stimulated by 5 μM forskolin and THC in the absence and presence of ZCZ011, (B–D) dissociation of Gαi3 and Gβγ subunits as determined by TRUPATH BRET assay stimulated by THC (B), AMB-FUBINACA (C), or CP55940 (D) alone and in the presence of 1 μM ZCZ011. (E–F) Phosphorylation of ERK1/2 by AMB-FUBINACA (E), or THC (F). Data has been normalized to appropriate controls and pooled across five independent biological replicates (n = 5). Data is expressed as mean ± SEM.
G Protein Dissociation
The TRUPATH BRET assay allowed us to more closely investigate the allosteric modulation of G protein dissociation by ZCZ011 in the presence of an orthosteric agonist. Real-time kinetic data showing G protein dissociation of orthosteric agonists in combination with ZCZ011 is reported in Figure S3. In combination with THC, positive allosteric modulation was not observed; there was no change in response potency or span with the addition of ZCZ011 (Figure 2B), which may reflect the very weak agonism of THC in this pathway. In combination with CP55940, 1 μM ZCZ011 caused no change in potency, but it did significantly increase the Emax of the response (Figure 2D, Table 1). The full agonist AMB-FUBINACA in combination with 1 μM ZCZ011 showed decreased potency and equivalent Emax compared to AMB-FUBINACA alone (Figure 2C, Table 1). Simulations of G protein dissociation for both CP55940 and AMB-FUBINANCA in combination with ZCZ011 predicted an increase in potency of the orthosteric ligand, but the experimental results (Figure 2C/D, Table 1) revealed no change (for CP55940) or a decrease in potency (for AMB-FUBINACA; Table 5). As the additive agonism simulations for cAMP inhibition and G protein dissociation predicted changes in potency and/or efficacy that were greater than the effects shown in the experimental data, no evidence was adduced for a positive interaction between these ligands. Instead, a possible negative interaction was suggested between ZCZ011 and the orthosteric ligand in these pathways.
ERK1/2 Phosphorylation
In combination with the partial agonist THC, an inactive concentration of ZCZ011 (1 μM) caused an increase in potency and efficacy of the response (Figure 2F, Table 2). However, in combination with the full agonist AMB-FUBINACA, 1 μM ZCZ011 did not cause an increase in potency or efficacy (Figure 2E, Table 2). On comparison with additive agonism simulations, empirical data showed that ZCZ011 induced a greater-than-additive increase in THC potency and efficacy in ERK1/2 phosphorylation (Figure 2F, Table 2). Likewise, THC efficacy in the presence of ZCZ011 (compared to THC alone) was greater than that simulated by additive agonism (Figure 2F, Table 2), indicating a synergistic effect consistent with positive allosteric modulation. In contrast, for AMB-FUBINACA-induced ERK1/2 phosphorylation in combination with ZCZ011, additive agonism simulations were not inconsistent with the experimental data, suggesting that additive agonism is sufficient to explain this data (Figure 2E, Table 2).
Table 2. Potencies (pEC50) and Efficacies (Emax) of ERK1/2 Phosphorylation by Cannabinoid Ligands at CB1a.
| ligand | pEC50 | Emax |
|---|---|---|
| THC | 6.69 ± 0.22 | 31.0 ± 3.3 |
| THC + 1 μM ZCZ011 | 7.6 ± 0.17* | 77.7 ± 17.7* |
| AMB-FUBINACA | 8.22 ± 0.08 | 129.2 ± 8.8 |
| AMB-FUBINACA + 1 μM ZCZ011 | 8.53 ± 0.17 | 148.6 ± 14.2 |
Data is presented as mean ± SEM of five independent biological replicates, with Emax expressed as the span of the curve (percentage of 1 μM CP55940 stimulation). Statistically significant differences when comparing the orthosteric ligand in the absence and presence of ZCZ011 were performed in GraphPad Prism using a paired t test, with p values indicated as * < 0.05.
Translocation of β-Arrestin 1 and 2 to the Cell Membrane
As allosteric ligands are predicted to have decreased tolerance in vivo, translocation of β-arrestin 1 and 2 to the membrane was investigated in the presence of orthosteric agonists with a range of efficacies. Real-time kinetic BRET traces showing translocation of β-arrestin 1 and 2 can be found in Figures S4 and S5, respectively. To determine whether ZCZ011 increases the efficacy of THC, the response of 10 μM THC in the absence and presence of ZCZ011 (1 μM and 10 μM) was compared. Both 1 and 10 μM ZCZ011 in combination with THC (10 μM) caused an increase in β-arrestin 1 translocation compared with THC alone (Table 3, Figure 3A). ZCZ011 (10 μM) in combination with the partial agonist CP55940 increased the efficacy of β-arrestin 1 translocation, although potency of the response was unchanged (Table 3, Figure 3C). In combination with the full agonist AMB-FUBINACA, efficacy of β-arrestin 1 translocation was unchanged in combination with 1 μM or 10 μM ZCZ011, likely an indication that the AMB-FUBINACA Emax could not be further increased. However, due to the slight baseline shift caused by ZCZ011 alone, both 1 μM and 10 μM ZCZ011 increased the apparent potency of AMB-FUBINACA (Table 3, Figure 3E).
ZCZ011 at 10 μM increased the potency (pEC50) and efficacy of THC in β-arrestin 2 translocation when used in combination, but at 1 μM ZCZ011, only potency was significantly increased (Table 3, Figure 3B). ZCZ011 (1 μM and 10 μM) in combination with CP55940 caused no change in potency or efficacy when compared to CP55940 alone (Table 3, Figure 3D). However, unlike with CP55940, in combination with 1 μM and 10 μM ZCZ011, the potency of AMB-FUBINACA in β-arrestin 2 translocation was significantly increased. ZCZ011 significantly decreased the span of the AMB-FUBINACA-induced β-arrestin 2 translocation concentration response curve; however, this is due to the agonism of ZCZ011 alone as ZCZ011 did not alter the maximum response (top of the curve) produced (Table 3, Figure 3F). Simulations of additive agonism for THC-induced β-arrestin 1 and 2 translocation were inconsistent with experimental data in that ZCZ011 was observed to increase the efficacy of THC to a greater extent than predicted by additive agonism simulation, suggesting a synergistic relationship consistent with positive allosteric modulation. ZCZ011 also increased the efficacy of CP55940-induced β-arrestin 1 translocation and the potency of AMB-FUBINACA-induced β-arrestin 1 and 2 translocation (Figure 3, Table 3). However, these increases were not inconsistent with simulations of additive agonism, and therefore additivity may be sufficient to explain these ZCZ011 effects.
Receptor Internalization
To further investigate the effect of allosteric modulation on receptor regulation, CB1 internalization assays were performed. Only THC was utilized in this assay, as the speed of internalization of CP55940 and AMB-FUBINACA were too rapid to reliably detect an increase through positive allosteric modulation.33 At 1 h, the potency of receptor internalization by THC in combination with ZCZ011 (100 nM and 1 μM) was unchanged when compared to THC alone (Figure 4, Table 4). However, both 100 nM and 1 μM ZCZ011 in combination with THC significantly increased the efficacy of THC, indicating positive allosteric modulation occurring in this pathway (Figure 4, Table 4). Next the time course of internalization was examined for both ligands at 1 μM. Both 100 nM and 1 μM ZCZ011 in combination with 1 μM THC significantly increased the rate of internalization by THC (from 44.77 ± 3.68 to 22.39 ± 1.06 and 12.51 ± 0.84 min, respectively) (Figure 4).
Figure 4.

Internalization of CB1 induced by cannabinoid ligands in HEK293-hCB1 cells. (A) Kinetic concentration–response data were analyzed by MRT (see Methods) and are presented as 1/MRT, where data has been normalized to the response of THC alone and pooled across five independent biological replicates (n = 5). (B) Kinetic hCB1 internalization responses to high drug concentrations, presented over 60 min, where representative data are expressed as mean ± SD of technical duplicates (n = 5 data are shown in Table 4). (C) Internalization half-lives are expressed as mean ± SEM of five independent biological replicates (n = 5). * indicates a significant difference when compared to 1 μM THC.
Overall, the “simulation versus experimental data” comparisons provided little evidence for ZCZ011 acting as a positive allosteric modulator in combination with CP55940 and AMB-FUBINACA in any pathway examined, in contrast to previous literature reports to date.11−13 However, greater-than-additive responses, consistent with positive allosteric modulation, were observed with ZCZ011 in combination with THC for ERK1/2 phosphorylation, β-arrestin translocation, and receptor internalization (Table 5).
Interestingly, a negative interaction was observed upon comparing empirical cAMP inhibition and G protein dissociation with simulations of additive agonism for all orthosteric ligands. This suggests that ZCZ011 is not a positive allosteric modulator of G protein dissociation. Negative allosteric modulation or competitive antagonism could explain the negative interaction observed, although more investigation is required. This negative interaction is interesting because ZCZ011 was found to be a positive allosteric modulator of ERK1/2 phosphorylation, a signaling pathway known to be mediated by G proteins.25−27 These contrasting findings could be attributed to a mutual inconsistency in pERK and G protein dissociation assay sensitivities, such that low levels of pERK may not be detectable (and if so, apparent PAM effects observed could be overestimated), or that PAM effects were underestimated in G protein dissociation assays due to limits in detecting high efficacy responses. As Gαi3 was the only G protein investigated in the G protein dissociation assay, it is possible that ZCZ011 potentiates the dissociation of a different Gα subtype thus causing an increase in ERK1/2 phosphorylation. However, as the Gαi3 dissociation results align with the cAMP inhibition data, it is unlikely that any noncanonical signaling is affecting this data.
A novel finding of our study is that ZCZ011 is a positive allosteric modulator of the efficacy and rate of THC-induced receptor internalization, as comparing experimental data with simulated profiles showed that ZCZ011 had a greater-than-additive effect on internalization (Figure 4, Table 4). This is an interesting finding considering allosteric modulation is proposed to result in decreased tolerance;8 potentiation of receptor internalization suggests that this may not be the case. However, in vivo, the key orthosteric ligand will be an endogenous cannabinoid. As the endocannabinoid AEA has similar efficacy to THC,38 and 2-AG has similar efficacy to AMB-FUBINACA, we hypothesize that, in vivo, in the presence of endogenous cannabinoid, ZCZ011 would exhibit similar effects to those observed in combination with THC or AMB-FUBINACA, although clearly this merits direct examination.
The terminology in the literature surrounding allosteric agonists is sometimes ambiguous, where allosteric compounds are automatically referred to as allosteric modulators irrespective of their modulator activity and are therefore being referred to as “ago-PAMs”. However, this term seems to poorly describe ligands like ZCZ011: moderately efficacious allosteric agonists that display agonism at lower concentrations, with limited modulatory activity at higher concentrations in the presence of some orthosteric ligands. Literature describing allosteric operational model analysis states that the principal difference between an allosteric agonist and an allosteric modulator is that allosteric agonists possess intrinsic efficacy, therefore resulting in increased basal response.39 ZCZ011 meets the allosteric agonist criteria due to its intrinsic efficacy at concentrations lower than those where its activity is PAM-like. However, it is important to emphasize that PAM effects can still be detected in some assays, though they appear to be ligand and/or context-dependent.
In conclusion, ZCZ011 is an efficacious allosteric agonist with a similar in vitro signaling profile to that of THC. ZCZ011 was found to be a PAM of THC-induced ERK1/2 phosphorylation, β-arrestin translocation, and receptor internalization, although no data in this study supports the designation of ZCZ011 as a positive allosteric modulator of CP55940 or AMB-FUBINACA, nor for G protein signaling for any ligand. The difference in PAM effects observed for different orthosteric agonists may simply be due to the low efficacy of THC compared to the synthetic cannabinoids, such that ZCZ011 is able to potentiate signaling to a greater extent. Due to the reported absence of CB1 agonist effects in vivo with administration of ZCZ011,11 our study suggests further investigation is needed to understand the mechanism by which the classical tetrad of cannabimimetic effects are produced following CB1 activation. Our study is the first to report that ZCZ011 efficaciously drives receptor internalization in the absence of an orthosteric ligand. The relationship between in vivo tolerance to cannabinoids and the molecular mechanisms underpinning receptor regulation (classically entailing desensitization and internalization) also requires further investigation, considering the inconsistency between the internalization data we report in this study and the lack of tolerance seen in vivo.11
Methods
Drugs
Racemic ZCZ011 was provided as a generous gift from Professor Ruth Ross (University of Toronto) and was constituted and stored at 10 mM in DMSO. THC was purchased from THC Pharm GmbH (Germany) and stored at 31.6 mM in absolute ethanol. AMB-FUBINACA was provided as a gift from Sam Banister (University of Sydney) and stored at 10 mM in DMSO. CP55940 was purchased from Cayman Chemical Company (Ann Arbor, MI, USA) and stored at 10 mM in absolute ethanol. All drugs were aliquoted into single use aliquots and stored at −80 °C. Forskolin was also purchased from Cayman Chemical Company and was stored at 31.6 mM in DMSO in large, multiuse aliquots.
Cell Lines and Maintenance
All assays were performed in human embryonic kidney 293 (HEK293) cells. Wild type HEK293 cells transiently transfected with pplss-3HA tagged hCB1 (construct first reported in Finlay et al.21) were used for the β-arrestin membrane translocation assay, and HEK293 cells stably expressing triple-hemagglutinin-tagged hCB1 (Cawston et al.40) were used for all other assays. HEK293 cells were cultured in high glucose Dulbecco’s Modified Eagle Medium (DMEM) containing 10% fetal bovine serum (FBS) and maintained in 75 cm2 vented-cap flasks in a humidified 37 °C incubator with 5% CO2. Stable hCB1-expressing HEK293 cells were cultured in 250 μg/mL zeocin as previously described.40
BRET-CAMYEL cAMP Assay
Cellular cAMP was measured using a kinetic bioluminescence resonance energy transfer (BRET) assay, CAMYEL.40,41
Briefly, HEK/3HA-hCB1 cells were seeded into 10 cm culture dishes (Corning, Corning, NY) at a density of 6 × 106 cells/dish and incubated at 37 °C for 24 h to achieve 50–60% confluence. Medium was then replaced and cells were transfected with 5 μg pcDNA3.1-His-CAMYEL using linear polyethylenimine (30 μg PEI, Polysciences, Warrington, PA) in a 1:6 DNA:PEI ratio. A transfection mixture was prepared in 150 mM NaCl and dispensed into a culture dish dropwise. Cells were then incubated for 24 h.40,41 Transfected cells were lifted and replated at 50,000 cells/well in a poly d-lysine (high molecular weight PDL, Sigma-Aldrich) coated, white 96-well CulturPlates (PerkinElmer), and incubated overnight. To assay, the medium was aspirated and cells were washed with PBS and preincubated for 30 min in a BRET assay medium (phenol red-free DMEM containing 25 mM HEPES, supplemented with 1 mg/mL fatty-acid free bovine serum albumin; BSA, MP Biomedicals, Auckland, NZ). Coelenterazine-h (final concentration of 5 μM, prepared 10× in assay medium; Nanolight Technology, Prolume Ltd.) was then dispensed, and plates were transferred to a LUMIstar Omega plate reader (BMG Labtech GmbH, Ortenberg, BW, Germany) and incubated in the dark at 37 °C for 5 min before addition of drugs. Drug components (agonist or vehicle, allosteric ligand or vehicle, and forskolin or vehicle) were prepared at 10× concentration and combined in a polypropylene V-well plate for dispensing. Drugs were then added together to the assay plate with a multichannel pipet (final stimulation volume of 100 μL). Luminescence was detected simultaneously at 475 and 535 nm in real-time using the LUMIstar for approximately 20 min. Inverse BRET ratios (460/535 nm) were calculated in Omega MARS software (V3.1 R5, BMG Labtech GmbH), and data was analyzed using GraphPad Prism v8 (GraphPad Software Inc., La Jolla, CA, USA). Kinetic BRET traces were transformed into concentration response curves using the in-built area under the curve (AUC) analysis in GraphPad Prism v8 and normalized to forskolin (100%) and vehicle (0%).
β-Arrestin Membrane Translocation Assay
The β-arrestin membrane translocation assay used was first described by Donthamsetti et al.42 and adapted by Ibsen et al.32 and Finlay et al.33 Wildtype HEK293 cells were seeded into 10 cm dishes and incubated for 24 h at 37 °C prior to transfection to gain 50–60% confluency. Complete medium was then replaced, and a transfection mixture was prepared containing (per dish): 2 μg mem-Linker-Citrine-SH3 pcDNA3.1+, 50 ng Rluc8-β-arrestin pcDNA3.1+ (either human β-arrestin 1 or 2, depending on the assay), 1.6 μg pplss-3HA-hCB 1pEF4a, and 350 ng empty pcDNA3.1+. Plasmid solutions were initially diluted in sterile Milli-Q water, before being diluted to the desired concentration in OptiMEM (Thermo Fisher Scientific) and combined with 36 μg of PEI-Max (a 1:9 DNA:PEImax ratio, polyethylenimine 25K, Polysciences). The transfection mixture was incubated at room temperature for 20 min before dropwise addition to 10 cm dishes and overnight incubation. Transfected cells were then plated at 60,000 cells/well in PDL (Sigma-Aldrich) coated white 96-well CulturPlates (PerkinElmer) and incubated for 24 h.
To assay, cells were washed with PBS and preincubated for 30 min in BRET assay medium. Coelenterazine-h (5 μM) was then added to the cells and luminescence at 475 and 535 nm was simultaneously detected in the LUMIstar for approximately 5 min to establish a baseline BRET ratio. A 10× concentrated drug was then added to the cells, and plates were returned to the LUMIstar and luminescence was detected for a further 25 min. BRET ratios (535/475 nm) were calculated in Omega MARS software, and data was analyzed using GraphPad Prism v8. Predrug addition traces (coelenterazine-h incubation) were averaged and subtracted from the postdrug addition BRET ratio data at matched time points for each drug condition resulting in ΔBRET ratios. “Vehicle” BRET ratios were then subtracted from matched drug responses within each read cycle (baseline correction), and the area under the curve analysis was performed using the in-built AUC analysis in GraphPad Prism v8.
BRET-TRUPATH G Protein Dissociation Experiment
G protein dissociation was measured using the TRUPATH kit (a gift from Bryan Roth, Addgene kit #1000000163) first described by Olsen et al.23
Briefly, 6 × 106 HEK/3HA-hCB1 cells were seeded into 10 cm culture dishes and incubated at 37 °C for 24 h to achieve 50–60% confluency. Complete culture medium was replaced, and cells were transfected with a DNA mixture comprising 1 μg Gαi3-Rluc8 pcDNA5/FRT/TO, 1 μg Gβ3 pcDNA3.1, and 1 μg Gγ9-GFP2 pcDNA3.1. Plasmids were diluted in OptiMEM and incubated for 20 min at room temperature with 27 μg PEI-max (1:9 DNA:PEImax ratio), before being added dropwise to cells and incubated for 24 h. Transfected cells were then lifted and plated at 60,000 cells/well into white PDL-coated 96-well plates and incubated for 24 h.
To assay, cells were washed with PBS and preincubated for 30 min in a BRET assay medium. Coelenterazine-400a (Nanolight, final concentration 5 μM) was then added to the cells and luminescence was simultaneously detected with BRET2 filters (410 and 515 nm) in the LUMIstar in the dark at 37 °C for approximately 5 min. Drugs were prepared at 10× concentration in BRET assay medium and added to cells followed by a further 25 min real-time detection in the LUMIstar. BRET ratios (515/410 nm) were calculated in Omega MARS software, and data was analyzed using GraphPad Prism v8. “Vehicle” BRET ratios were then subtracted from matched conditions (baseline-corrected) and in-built AUC analysis was performed in GraphPad Prism v8 to obtain concentration–response curves.
AlphaLISA SureFire pERK Experiment
Phosphorylated ERK1/2 was detected using an AlphaLISA SureFire pERK kit (PerkinElmer). Briefly, HEK/3HA-hCB1 cells were plated at 20,000–25,000 cells/well into the inner 60 wells of clear PDL-coated 96-well plates (Corning, NY) and cultured for 24 h. The medium was then aspirated and replaced with 50 μL/well serum-free DMEM, supplemented with 1 mg/mL BSA. Cells were then incubated for a further 16 h prior to stimulation. Drugs were prepared at 2× the concentration in serum-free DMEM supplemented with 1 mg/mL BSA and warmed to 37 °C before addition to the assay plate while partially submerged in a 37 °C water bath. The stimulation time point was 3 min 30 s, after which the plate was rapidly moved to ice, well contents aspirated, and AlphaLISA lysis buffer dispensed. Detection of pERK was performed in accordance with the manufacturer’s instructions, whereby the acceptor beads were dispensed prior to donor beads. Plates were read in a CLARIOstar (BMG Labtech GmbH) and analyzed in GraphPad Prism where concentration response curves were normalized to 1 μM CP55940 (100%) and vehicle (0%).
Receptor Internalization Assays
Receptor internalization was investigated using the assay first described by Grimsey et al.43 and further developed by Finlay et al.44 and Zhu et al.45 HEK/3HA-hCB1 cells were plated at 40,000 cells/well into the inner 60 wells of a clear PDL-coated 96-well plate (Corning, NY) and cultured for 24 h. To assay, complete medium was replaced with serum-free DMEM supplemented with 1 mg/mL BSA (assay medium) and incubated for 30 min at 37 °C. Primary mouse anti-HA.11 clone 16B12 monoclonal antibody (BioLegend, San Diego, CA, USA; cat. no. 901503; RRID: AB_2565005) was diluted in assay medium (1:500, BioLegend, San Diego, CA), warmed to 37 °C, and then added to cells 30 min prior to drug stimulation. Antibody-containing medium was then aspirated, cells were washed with assay medium, and drug dilutions were added to appropriate wells at specific time points in a staggered fashion so that all time points would complete at the same time. To stop internalization, plates were placed on ice for 5 min before the contents of the wells were aspirated and replaced with secondary antibody (AlexaFluor goat antimouse highly cross-adsorbed 488/594 diluted 1:300, Thermo Fisher Scientific, Waltham, MA). Plates were then placed on a plate rocker for 30 min at room temperature, before the secondary antibody was aspirated, and plates were washed twice with assay medium. Cells were fixed using 4% paraformaldehyde (PFA) in 0.1 M phosphate buffer for 10 min, before wells were aspirated and washed twice with PBS. Cells were treated with 4 mg/mL Hoescht 33258 (diluted 1:500 in PBS + 0.2% Triton X-100, PBS-T) for 15 min, and then washed twice with PBS-T before being left for storage and imaging in PBS-T supplemented with 0.4 mg/mL merthiolate.
Plates were imaged using an ImageXpress Micro XLS High-Content System automatic microscope and analyzed using MetaXpress v6.2.3.733 (Molecular Devices, San Jose, CA). One phase decay nonlinear regression curves were fit to the data, and half-life values were extrapolated to compare rates of internalization. To create concentration–response curves from the kinetic data, inverse mean residence time (MRT) was calculated for time-course fluorescence data as previously described by Zhu et al.45 MRT is defined as the ratio of the area under the first moment curve (AUMC) and the area under the curve (AUC) of concentration versus time. Inverse MRT was used to account for the slow constitutive internalization of CB1 relative to agonist-induced internalization, therefore producing more accurate estimates.45
Comparison of Additive Agonism Simulations with Empirical Data
As ZCZ011 is an agonist in all pathways tested, to investigate whether observed changes in potency and/or efficacy of the orthosteric agonist response was due to allosteric modulation we compared empirical data with simulations of additive agonism (Table 5). Potency and efficacy parameters were obtained from data for each ligand alone, and simulations were performed for two agonists acting at two distinct sites within the same system. This approach simply consisted of combining two Emax models, where the binding of one drug at one site does not affect the binding of the second drug, and therefore, simulations represented additivity rather than allosteric modulation. Additive agonism simulations used the following equation:
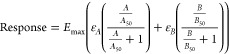 |
where A refers to the first ligand (in this case orthosteric), and B refers to the second ligand (in this case ZCZ011), Emax refers to the maximum system response, estimated by full agonist response, ε refers to the intrinsic efficacy of the respective ligand; empirical data was pooled (n = 5) and normalized to the response of a full agonist (ε = 1), relative ε values were determined for each ligand, A/B50 refers to the potency of each ligand, and total responses were constrained to the system Emax. Nonlinear regression was performed on simulated data to generate curves to compare curve shape to empirical data.
Overlay plots were then created to qualitatively compare empirical data to simulations of additive agonism. Overlay plots showing additivity simulations that fall within the experimental error of empirical data suggest that the empirical data is not inconsistent with additive agonism. However, this does not imply any particular mechanism. If data are inconsistent with additive agonism, depending on the nature of the inconsistency this could indicate positive allosteric modulation, or a negative interaction: if changes in efficacy and/or potency are greater than the additive agonism simulation, this would indicate positive allosteric modulation, and conversely, if the changes are less than that simulated by additive agonism, this would indicate a negative interaction.
Data Analysis
All data processing was performed using GraphPad Prism v8. Sigmoidal concentration response curves were fit using three parameter nonlinear regression curves, and data are presented as mean ± SEM, unless otherwise stated. Statistical analyses were performed on extrapolated data from nonlinear regression analyses, using parameters derived from five independent experiments. Repeated measures one-way ANOVA or unpaired t tests were performed on potencies and efficacies as reported in-text, and appropriate posthoc tests were used when significant results were obtained (p < 0.05). Figures presented in this manuscript are representative, in line with statistical recommendations, in order to avoid misestimation of response parameters from combination of data from independent experiments.46
Experimental plate maps were randomized throughout the study; however, blinding was not feasible.
Acknowledgments
The authors acknowledge funding from the University of Otago School of Biomedical Sciences. H.G. was supported by a University of Otago Doctoral Scholarship. Graphical abstract was created with BioRender.com.
The data that support the findings of this study are available from the corresponding author upon reasonable request.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsptsci.2c00160.
Real-time kinetic traces from BRET assays showing ZCZ011 alone in cAMP inhibition, G protein dissociation, and β-arrestin 2 translocation (Figure S1), positive allosteric modulation of cAMP inhibition (Figure S2), G protein dissociation (Figure S3), β-arrestin 1 (Figure S4), and β-arrestin 2 (Figure S5) translocation (PDF)
Author Contributions
H.G. designed and performed experiments, analyzed data, and wrote the paper. R.A.R. and I.R.G. developed and generously provided ZCZ011 to be used for all experiments. S.B.D. contributed to data analysis and assisted with simulations. D.B.F. designed and performed experiments, contributed to data analysis, and reviewed drafts of the paper. M.G. designed experiments, obtained funding, and reviewed drafts of the paper. All authors reviewed and approved the final version of the paper.
The authors declare the following competing financial interest(s): Michelle Glass is a consultant for Aelis Farma and has in the past completed work for medicinal cannabis companies Rua Bioscience and Soma Therapeutics. Ruth Ross and Ian Grieg are inventors on patent applications filed by the Universities of Toronto and Aberdeen on CB1 allosteric modulators.
Special Issue
Published as part of the ACS Pharmacology & Translational Science virtual special issue "GPCR Signaling".
Supplementary Material
References
- Gaoni Y.; Mechoulam R. Isolation, Structure, and Partial Synthesis of an Active Constituent of Hashish. J. Am. Chem. Soc. 1964, 86 (8), 1646–1647. 10.1021/ja01062a046. [DOI] [Google Scholar]
- Matsuda L. A.; Lolait S. J.; Brownstein M. J.; Young A. C.; Bonner T. I. Structure of a Cannabinoid Receptor and Functional Expression of the Cloned CDNA. Nature 1990, 346 (6284), 561–564. 10.1038/346561a0. [DOI] [PubMed] [Google Scholar]
- Munro S.; Thomas K. L.; Abu-Shaar M. Molecular Characterization of a Peripheral Receptor for Cannabinoids. Nature 1993, 365 (6441), 61–65. 10.1038/365061a0. [DOI] [PubMed] [Google Scholar]
- Haspula D.; Clark M. A. Cannabinoid Receptors: An Update on Cell Signaling, Pathophysiological Roles and Therapeutic Opportunities in Neurological, Cardiovascular, and Inflammatory Diseases. International Journal of Molecular Sciences 2020, 21, 7693. 10.3390/ijms21207693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pertwee R. G. The Diverse CB 1 and CB 2 Receptor Pharmacology of Three Plant Cannabinoids: Δ 9 -Tetrahydrocannabinol, Cannabidiol and Δ 9 -Tetrahydrocannabivarin. Br. J. Pharmacol. 2008, 153 (2), 199–215. 10.1038/sj.bjp.0707442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buckner J. D.; Heimberg R. G.; Matthews R. A.; Silgado J. Marijuana-Related Problems and Social Anxiety: The Role of Marijuana Behaviors in Social Situations. Psychol. Addict. Behav. 2012, 26 (1), 151–156. 10.1037/a0025822. [DOI] [PubMed] [Google Scholar]
- Kenakin T. Allosteric Theory: Taking Therapeutic Advantage of the Malleable Nature of GPCRs. Curr. Neuropharmacol. 2007, 5 (3), 149–156. 10.2174/157015907781695973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wootten D.; Christopoulos A.; Sexton P. M. Emerging Paradigms in GPCR Allostery: Implications for Drug Discovery. Nat. Rev. Drug Discovery 2013, 12, 630–644. 10.1038/nrd4052. [DOI] [PubMed] [Google Scholar]
- Gentry P. R.; Sexton P. M.; Christopoulos A. Novel Allosteric Modulators of G Protein-Coupled Receptors. J. Biol. Chem. 2015, 290, 19478–19488. 10.1074/jbc.R115.662759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laprairie R. B.; Kulkarni P. M.; Deschamps J. R.; Kelly M. E. M.; Janero D. R.; Cascio M. G.; Stevenson L. A.; Pertwee R. G.; Kenakin T. P.; Denovan-Wright E. M.; Thakur G. A. Enantiospecific Allosteric Modulation of Cannabinoid 1 Receptor. ACS Chem. Neurosci. 2017, 8 (6), 1188–1203. 10.1021/acschemneuro.6b00310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ignatowska-Jankowska B. M.; Baillie G. L.; Kinsey S.; Crowe M.; Ghosh S.; Owens R. A.; Damaj I. M.; Poklis J.; Wiley J. L.; Zanda M.; Zanato C.; Greig I. R.; Lichtman A. H.; Ross R. A. A Cannabinoid CB 1 Receptor-Positive Allosteric Modulator Reduces Neuropathic Pain in the Mouse with No Psychoactive Effects. Neuropsychopharmacology 2015, 40 (13), 2948–2959. 10.1038/npp.2015.148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tseng C.-C.; Baillie G.; Donvito G.; Mustafa M. A.; Juola S. E.; Zanato C.; Massarenti C.; Dall’angelo S.; Harrison W. T. A.; Lichtman A. H.; Ross R. A.; Zanda M.; Greig I. R. The Trifluoromethyl Group as a Bioisosteric Replacement of the Aliphatic Nitro Group in CB 1 Receptor Positive Allosteric Modulators. J. Med. Chem. 2019, 62, 5049–5062. 10.1021/acs.jmedchem.9b00252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saleh N.; Hucke O.; Kramer G.; Schmidt E.; Montel F.; Lipinski R.; Ferger B.; Clark T.; Hildebrand P. W.; Tautermann C. S. Multiple Binding Sites Contribute to the Mechanism of Mixed Agonistic and Positive Allosteric Modulators of the Cannabinoid CB1 Receptor. Angew. Chem., Int. Ed. Engl. 2018, 57 (10), 2580–2585. 10.1002/anie.201708764. [DOI] [PubMed] [Google Scholar]
- Yang X.; Wang X.; Xu Z.; Wu C.; Zhou Y.; Wang Y.; Lin G.; Li K.; Wu M.; Xia A.; Liu J.; Cheng L.; Zou J.; Yan W.; Shao Z.; Yang S. Molecular Mechanism of Allosteric Modulation for the Cannabinoid Receptor CB1. Nat. Chem. Biol. 2022, 18, 831–840. 10.1038/s41589-022-01038-y. [DOI] [PubMed] [Google Scholar]
- Hurst D. P.; Garai S.; Kulkarni P. M.; Schaffer P. C.; Reggio P. H.; Thakur G. A. Identification of CB1 Receptor Allosteric Sites Using Force-Biased MMC Simulated Annealing and Validation by Structure-Activity Relationship Studies 2019, 10, 1216. 10.1021/acsmedchemlett.9b00256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slivicki R. A.; Iyer V.; Mali S. S.; Garai S.; Thakur G. A.; Crystal J. D.; Hohmann A. G. Positive Allosteric Modulation of CB 1 Cannabinoid Receptor Signaling Enhances Morphine Antinociception and Attenuates Morphine Tolerance Without Enhancing Morphine-Induced Dependence or Reward. Front. Mol. Neurosci. 2020, 13, 54. 10.3389/fnmol.2020.00054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garai S.; Kulkarni P. M.; Schaffer P. C.; Leo L. M.; Brandt A. L.; Zagzoog A.; Black T.; Lin X.; Hurst D. P.; Janero D. R.; Abood M. E.; Zimmowitch A.; Straiker A.; Pertwee R. G.; Kelly M.; Szczesniak A.-M.; Denovan-Wright E. M.; Mackie K.; Hohmann A. G.; Reggio P. H.; Laprairie R. B.; Thakur G. A. Application of Fluorine-and Nitrogen-Walk Approaches: Defining the Structural and Functional Diversity of 2-Phenylindole Class of CB1 Receptor Positive Allosteric Modulators HHS Public Access. J. Med. Chem. 2020, 63 (2), 542–568. 10.1021/acs.jmedchem.9b01142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cairns E. A.; Szczesniak A.-M.; Straiker A. J.; Kulkarni P. M.; Pertwee R. G.; Thakur G. A.; Baldridge W. H.; Kelly M. E. M. The In Vivo Effects of the CB 1-Positive Allosteric Modulator GAT229 on Intraocular Pressure in Ocular Normotensive and Hypertensive Mice. J. Ocul. Pharmacol. Ther. 2017, 33 (8), 582–590. 10.1089/jop.2017.0037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Datta U.; Kelley L. K.; Middleton J. W.; Gilpin N. W. Positive Allosteric Modulation of the Cannabinoid Type-1 Receptor (CB1R) in Periaqueductal Gray (PAG) Antagonizes Anti-Nociceptive and Cellular Effects of a Mu-Opioid Receptor Agonist in Morphine-Withdrawn Rats. Psychopharmacology (Berl). 2020, 237 (12), 3729–3739. 10.1007/s00213-020-05650-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trexler K. R.; Eckard M. L.; Kinsey S. G. CB1 Positive Allosteric Modulation Attenuates Δ9-THC Withdrawal and NSAID-Induced Gastric Inflammation. Pharmacol., Biochem. Behav. 2019, 177, 27–33. 10.1016/j.pbb.2018.12.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finlay D. B.; Cawston E. E.; Grimsey N. L.; Hunter M. R.; Korde A.; Vemuri V. K.; Makriyannis A.; Glass M. Gαs Signalling of the CB1 Receptor and the Influence of Receptor Number. Br. J. Pharmacol. 2017, 174 (15), 2545–2562. 10.1111/bph.13866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sadana R.; Dessauer C. W. Physiological Roles for G Protein-Regulated Adenylyl Cyclase Isoforms: Insights from Knockout and Overexpression Studies. Neurosignals. 2009, 17 (1), 5–22. 10.1159/000166277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olsen R. H. J.; DiBerto J. F.; English J. G.; Glaudin A. M.; Krumm B. E.; Slocum S. T.; Che T.; Gavin A. C.; McCorvy J. D.; Roth B. L.; Strachan R. T. TRUPATH, an Open-Source Biosensor Platform for Interrogating the GPCR Transducerome. Nat. Chem. Biol. 2020, 16 (8), 841–849. 10.1038/s41589-020-0535-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glass M.; Northup J. K. Agonist Selective Regulation of G Proteins by Cannabinoid CB 1 and CB 2 Receptors. Mol. Pharmacol. 1999, 56 (6), 1362–1369. 10.1124/mol.56.6.1362. [DOI] [PubMed] [Google Scholar]
- Dalton G. D.; Howlett A. C. Cannabinoid CB1 Receptors Transactivate Multiple Receptor Tyrosine Kinases and Regulate Serine/Threonine Kinases to Activate ERK in Neuronal Cells. Br. J. Pharmacol. 2012, 165 (8), 2497–2511. 10.1111/j.1476-5381.2011.01455.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dalton G. D.; Carney S. T.; Marshburn J. D.; Norford D. C.; Howlett A. C. CB1 Cannabinoid Receptors Stimulate Gβγ-GRK2-Mediated FAK Phosphorylation at Tyrosine 925 to Regulate ERK Activation Involving Neuronal Focal Adhesions. Front. Cell. Neurosci. 2020, 14, 176. 10.3389/fncel.2020.00176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galve-Roperh I.; Rueda D.; Gomez del Pulgar T.; Velasco G.; Guzman M. Mechanism of Extracellular Signal-Regulated Kinase Activation by the CB(1) Cannabinoid Receptor. Mol. Pharmacol. 2002, 62 (6), 1385–1392. 10.1124/mol.62.6.1385. [DOI] [PubMed] [Google Scholar]
- Lichtman A.; Martin B. Cannabinoid Tolerance and Dependence. Handb. Exp. Pharmacol. 2005, 168 (168), 691–717. 10.1007/3-540-26573-2_24. [DOI] [PubMed] [Google Scholar]
- Poklis J. L.; Clay D. J.; Ignatowska-Jankowska B. M.; Zanato C.; Ross R. A.; Greig I. R.; Abdullah R. A.; Mustafa M. A.; Lichtman A. H.; Poklis A. HPLC-MS-MS Determination of ZCZ-011, A Novel Pharmacological Tool for Investigation of the Cannabinoid Receptor in Mouse Brain Using Clean Screen FAStä Column Extraction. J. Anal Toxicol 2015, 39 (5), 353–358. 10.1093/jat/bkv015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu X.; Tian X.; Guo X.; He Y.; Chen H.; Zhou J.; Wang Z. J. AMPA Receptor Positive Allosteric Modulators Attenuate Morphine Tolerance and Dependence. Neuropharmacology 2018, 137, 50–58. 10.1016/j.neuropharm.2018.04.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DiCello J. J.; Carbone S. E.; Saito A.; Pham V.; Szymaszkiewicz A.; Gondin A. B.; Alvi S.; Marique K.; Shenoy P.; Veldhuis N. A.; Fichna J.; Canals M.; Christopoulos A.; Valant C.; Poole D. P. Positive Allosteric Modulation of Endogenous Delta Opioid Receptor Signaling in the Enteric Nervous System Is a Potential Treatment for Gastrointestinal Motility Disorders. Am. J. Physiol. - Gastrointest. Liver Physiol. 2022, 322 (1), G66–G78. 10.1152/ajpgi.00297.2021. [DOI] [PubMed] [Google Scholar]
- Ibsen M. S.; Finlay D. B.; Patel M.; Javitch J. A.; Glass M.; Grimsey N. L. Cannabinoid CB1 and CB2 Receptor-Mediated Arrestin Translocation: Species, Subtype, and Agonist-Dependence. Front. Pharmacol. 2019, 10 (APR), 1–18. 10.3389/fphar.2019.00350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finlay D. B.; Manning J. J.; Søes Ibsen M.; Macdonald C. E.; Patel M.; Javitch J. A.; Banister S. D.; Glass M. Do Toxic Synthetic Cannabinoid Receptor Agonists Have Signature in Vitro Activity Profiles? A Case Study of AMB-FUBINACA. ACS Chem. Neurosci 2019, 10 (10), 4350–4360. 10.1021/acschemneuro.9b00429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manning J. J.; Green H. M.; Glass M.; Finlay D. B. Pharmacological Selection of Cannabinoid Receptor Effectors: Signalling, Allosteric Modulation and Bias. Neuropharmacology 2021, 193, 108611. 10.1016/j.neuropharm.2021.108611. [DOI] [PubMed] [Google Scholar]
- Schaffer P. C.; Kulkarni P. M.; Janero D. R.; Thakur G. A. Focused Structure-Activity Relationship Profiling around the 2-Phenylindole Scaffold of a Cannabinoid Type-1 Receptor Agonist-Positive Allosteric Modulator: Site-III Aromatic-Ring Congeners with Enhanced Activity and Solubility. Bioorg. Med. Chem. 2020, 28 (21), 115727. 10.1016/j.bmc.2020.115727. [DOI] [PubMed] [Google Scholar]
- Garai S.; Leo L. M.; Szczesniak A. M.; Hurst D. P.; Schaffer P. C.; Zagzoog A.; Black T.; Deschamps J. R.; Miess E.; Schulz S.; Janero D. R.; Straiker A.; Pertwee R. G.; Abood M. E.; Kelly M. E. M.; Reggio P. H.; Laprairie R. B.; Thakur G. A. Discovery of a Biased Allosteric Modulator for Cannabinoid 1 Receptor: Preclinical Anti-Glaucoma Efficacy. J. Med. Chem. 2021, 64 (12), 8104–8126. 10.1021/acs.jmedchem.1c00040. [DOI] [PubMed] [Google Scholar]
- Garai S.; Schaffer P. C.; Laprairie R. B.; Janero D. R.; Pertwee R. G.; Straiker A.; Thakur G. A. Design, Synthesis, and Pharmacological Profiling of Cannabinoid 1 Receptor Allosteric Modulators: Preclinical Efficacy of C2-Group GAT211 Congeners for Reducing Intraocular Pressure. Bioorg. Med. Chem. 2021, 50, 116421. 10.1016/j.bmc.2021.116421. [DOI] [PubMed] [Google Scholar]
- Pertwee R. G.; Ross R. A. Cannabinoid Receptors and Their Ligands. Prostaglandins. Leukot. Essent. Fatty Acids 2002, 66 (2–3), 101–121. 10.1054/plef.2001.0341. [DOI] [PubMed] [Google Scholar]
- Jakubík J.; Randáková A.; Chetverikov N.; El-Fakahany E. E.; Doležal V. The Operational Model of Allosteric Modulation of Pharmacological Agonism. Sci. Rep. 2020, 10 (1), 14421. 10.1038/s41598-020-71228-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cawston E. E.; Redmond W. J.; Breen C. M.; Grimsey N. L.; Connor M.; Glass M. Real-Time Characterization of Cannabinoid Receptor 1 (CB1) Allosteric Modulators Reveals Novel Mechanism of Action. Br. J. Pharmacol. 2013, 170 (4), 893–907. 10.1111/bph.12329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang L. I.; Collins J.; Davis R.; Lin K. M.; DeCamp D.; Roach T.; Hsueh R.; Rebres R. A.; Ross E. M.; Taussig R.; Fraser I.; Sternweis P. C. Use of a CAMP BRET Sensor to Characterize a Novel Regulation of CAMP by the Sphingosine 1-Phosphate/G13 Pathway. J. Biol. Chem. 2007, 282 (14), 10576–10584. 10.1074/jbc.M609695200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donthamsetti P.; Quejada J. R.; Javitch J. A.; Gurevich V. V.; Lambert N. A. Using Bioluminescence Resonance Energy Transfer (BRET) to Characterize Agonist-Induced Arrestin Recruitment to Modified and Unmodified G Protein-Coupled Receptors. Curr. Protoc. Pharmacol. 2015, 70 (1), 1. 10.1002/0471141755.ph0214s70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grimsey N. L.; Narayan P. J.; Dragunow M.; Glass M. A Novel High-Throughput Assay for the Quantitative Assessment of Receptor Trafficking. Clin. Exp. Pharmacol. Physiol. 2008, 35, 1377–1382. 10.1111/j.1440-1681.2008.04991.x. [DOI] [PubMed] [Google Scholar]
- Finlay D. B.; Joseph W. R.; Grimsey N. L.; Glass M. GPR18 Undergoes a High Degree of Constitutive Trafficking but Is Unresponsive to N-Arachidonoyl Glycine. PeerJ. 2016, 4 (3), e1835 10.7717/peerj.1835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu X.; Finlay D. B.; Glass M.; Duffull S. B. Model-Free and Kinetic Modelling Approaches for Characterising Non-Equilibrium Pharmacological Pathway Activity: Internalisation of Cannabinoid CB1 Receptors. Br. J. Pharmacol. 2019, 176 (14), 2593–2607. 10.1111/bph.14684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hall D. A.; Langmead C. J. Matching Models to Data: A Receptor Pharmacologist’s Guide. Br. J. Pharmacol. 2010, 161 (6), 1276–1290. 10.1111/j.1476-5381.2010.00879.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.