

ABSTRACT
Chimeric antigen receptor (CAR) T-cell therapy is a novel, customized immunotherapy that is considered a ‘living’ and self-replicating drug to treat cancer, sometimes resulting in a complete cure. CAR T-cells are manufactured through genetic engineering of T-cells by equipping them with CARs to detect and target antigen-expressing cancer cells. CAR is designed to have an ectodomain extracellularly, a transmembrane domain spanning the cell membrane, and an endodomain intracellularly. Since its first discovery, the CAR structure has evolved greatly, from the first generation to the fifth generation, to offer new therapeutic alternatives for cancer patients. This treatment has achieved long-term and curative therapeutic efficacy in multiple blood malignancies that nowadays profoundly change the treatment landscape of lymphoma, leukemia, and multiple myeloma. But CART-cell therapy is associated with several hurdles, such as limited therapeutic efficacy, little effect on solid tumors, adverse effects, expensive cost, and feasibility issues, hindering its broader implications.
KEYWORDS: CAR, CAR T-cell therapy, structure, generations, approval, clinical trials
Introduction
Adoptive cell therapy (ACT) is a type of immunotherapy manufactured to treat advanced stages of malignancies that are resistant to conventional therapies. ACT was first described 30 years ago and the principle of which was by taking advantage of the ability of the body’s own immune system, particularly T-cells, to recognize and kill cancer cells.1,2 Thus, ACTs are designed to improve immune cells’ ability to detect and destroy cancer cells, and thereby control cancer in the long term. Many types of ACTs have been discovered so far, including chimeric antigen receptor (CAR) T-cells, cytotoxic T lymphocytes (CTLs), engineered T-cell receptor (TCR) T-cells, tumor-infiltrating lymphocytes (TILs), virus-specific T-cells (VST), cytokine-induced killer cells (CIK), T reg, and natural killer (NK) cell therapies.1
CAR T-cell therapy is one of the most promising types of ACT that involves the adoptive transfer of T-cells expressing artificial receptors to cancer patients for therapeutic purposes.1,3 T-cells that orchestrate the immune response and directly kill the infected or cancerous cells are the backbone of CAR T-cell therapy.4 It utilizes the patient’s own (autologous) or donor-derived (allogenic) T-cells to genetically engineer and express recombinant proteins on the cell surface known as chimeric antigen receptors (CARs). CARs are specifically altered to recognize, target, and kill virtually any cancer cell expressing extracellular antigens, independently of major histocompatibility complex (MHC) or human leukocyte antigen (HLA).5 CAR-engineered T-cells are designed to comprise random or defined compositions of CD4+ and CD8+ naive and memory T-cells.6 When compared to products made from unselected or random T-cells with a variety of phenotypic compositions, CAR-T-cell products made from defined T-cell subsets can offer uniform efficacy. Moreover, combining the most potent CD4+ and CD8+ CAR-expressing subsets with optimal ratios produces synergistic antitumor activities in vivo.7
CAR T-cell therapy, formerly known as T-bodies, was first elucidated in 1987 in Japan by Yoshihisa Kuwana and coworkers by combining parts of an antibody with the TCR.8 In 1989, the concept of CAR T was independently illustrated by Gideon Gross and Zelig Eshhar in Israel.9,10 Since then, CAR T-cell therapy has evolved steadily over the last few decades, going through five generations since its inception to the most recent and advanced fifth generation, which offers improved therapeutic outcomes with lower toxicity. Multiple preclinical and clinical studies have been undertaken to explore the therapeutic roles of CAR T-cells in a variety of human diseases, including malignancies. Several types of CAR-T-cell therapy are currently either commercially approved for use or under clinical trials to evaluate their efficacy in treating various cancer types. An accumulating body of evidence shows that CAR T-cell therapy produces impressive clinical outcomes in advanced hematological malignancies such as leukemia and lymphoma, with a few of them currently receiving approval.11 This review primarily focused on CAR T-cell therapy, in the hope of gaining current updates on CAR generations, approved therapies, and ongoing clinical trials around CAR T-cell therapy. We also briefly highlighted the potential challenges associated with CAR T-cell therapy and the possible solutions, pinpointing future perspectives and key areas of knowledge gaps for future research.
Structural design of chimeric antigen receptor (CAR)
CAR is a hybrid receptor engineered to possess three structural domains, namely an ectodomain, a transmembrane domain, and an endodomain (Figure 1).12,13
Figure 1.
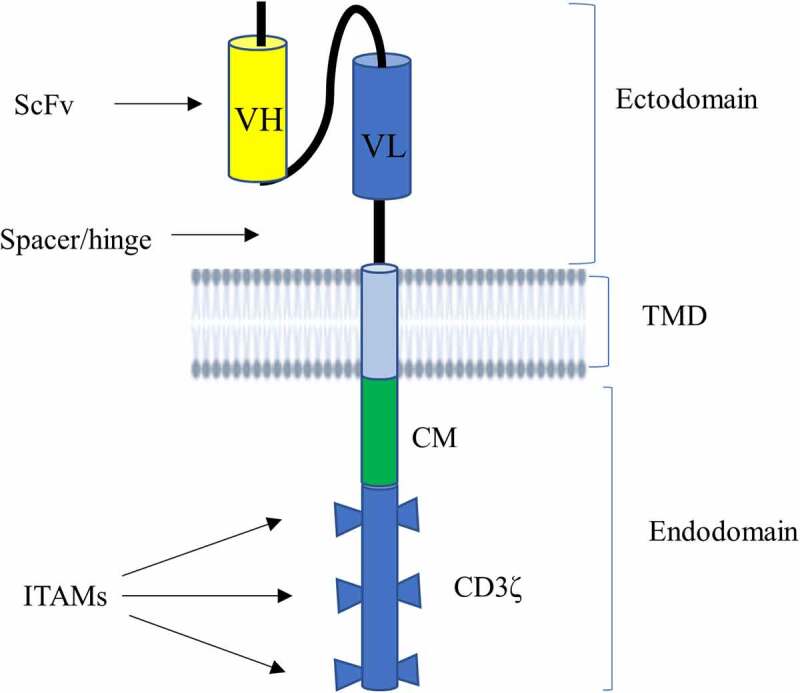
Schematic illustration of CAR structure. CAR contains an ectodomain possessing antigen recognition domain known as scFv and a short portion connecting it to TMD called hinge region (or spacer). scFv is made from VL and VH of an antibody that is connected to each other by a flexible linker. It also harbors a lipophilic alpha-helical domain spanning the plasma membrane known as TMD. CAR has also an endodomain comprising a CD3ζ that entails three ITAMs responsible for transmitting a primary signal, and CM mediating secondary or costimulatory signals.
Abbreviations: CAR, chimeric antigen receptor; CM, costimulatory molecule; ITAMs, immunoreceptor tyrosine-based activation motifs, scFv, single-chain variable fragment; TMD, transmembrane domain; VL, variable regions of the light; VH, variable regions of the heavy chain.
Ectodomain: It is the extracellular component of the CAR that contains an antigen recognition (binding) domain and a hinge region. The antigen-binding scaffold typically harbors a single-chain variable fragment (ScFv) formed from the variable regions of the light (VL) and heavy (VH) chains of an antibody. ScFv, which is mainly derived from murine, humanized, or human antibody sequences, aids CAR T-cell binding to potential target antigens and confers CAR specificity.12,14 This part is engineered to detect tumor antigens, such as CD19, BCMA, CD20, and CD30 independent of antigen processing and presentation by HLA.5 Besides the antibody-based binding domains (ScFv), the antigen recognition domain can also be non-antibody-based constructs, such as engineered binding scaffolds and natural ligands and receptors, to specifically recognize antigens. Engineered binding scaffolds are often created by randomly changing a portion of a stable protein domain. Then, using a variety of display technologies, including phage display, yeast display, and ribosome display, the best binders are chosen from the resulting library.15 The most often employed types of engineered binding scaffolds are DARPin, affibody, nanobody, Sso7d, monobody/adnectin, and anticalin. These binding domains can be derived from human and non-human organisms, such as bacteria, archaea, plants, or even artificially prepared proteins.15 In addition to engineered binding moieties, natural ligands or receptors can also serve as alternative binding domains to ScFv in CARs, involving natural killer group 2 member D (NKG2D), A20FMDV2 peptide, lymphocyte function-associated antigen 1 (LFA-1)-CARs, interleukin 13 (IL-13) mutein-CARs, and TCR variable region (TCRv).12,15–17 The hinge region (also called spacer), which is a short portion of the ectodomain that is primarily generated from immunoglobulin G (IgG), and occasionally from the hinges of CD28 and CD8, is present in the ectodomain in addition to the antigen recognition domain. It serves as a bridge between the ectodomain and the TMD and hence the ectodomain and the endo-domain. It is primarily designed to promote antigen attachment, flexibility, and synapse formation between the CAR T-cells and target T-cells.18 The longer the hinges, the more flexibility and access to membrane-proximal epitopes the CAR has, whereas the shorter the spacers, the less flexibility and the more it targets the antigen’s distal epitopes.19–21
Transmembrane domain (TMD): TMD is a single pass lipophilic alpha-helical domain of CAR that traverses the plasma membrane of the CAR T-cells. This domain connects the ectodomain to the endodomain; helps to express, anchor, and stabilize the CAR to the cell membrane; and enables proper CAR T-cell signaling.13,22 Overall, the TMD of CAR controls membrane integration and expression level. TMDs have, however, garnered less attention in systematic investigations of CAR design compared to other domains. In most instances, TMD is designed from which the adjacent hinge or endodomains are derived, including CD4, CD8α, CD28, or CD3ζ. But there is a high possibility of cross-talk of natural receptor TMDs with naive T-cell components, and this hampers the rational design and efficacy of CARs. Elazar et al. recently discovered de novo-designed receptor TMDs known as programmable membrane proteins (proMPs) that can tune engineered CAR receptor functions. proMPs are new design tools that have completely new sequences and form transmembrane homo-oligomers to generate novel programmed CAR (proCAR) constructs. The proCAR constructs endow T-cells with a predictable range of in vivo functional potencies while significantly attenuating inflammatory cytokine release compared to natural CD28 TMD containing CAR. It has also been shown that TMD modifications do not directly affect the antigen-binding or signaling domains of CAR, suggesting that this strategy could help develop CAR T-cell therapies with optimal safety and efficacy profiles.23
Endodomain: An endodomain, also known as an intracellular signaling domain or cytoplasmic tail, is the third domain of CAR that is found in the interior of CAR T-cells. CARs are engineered based on the natural architecture of TCR by incorporating several functional units. It has a TCR (CD3ζ) co-receptor with three immunoreceptor tyrosine-based activation motifs (ITAMs) as its principal functional unit to convey primary signals, making CD3ζ the key transmitter of signals from the TCR.22 This domain also incorporates costimulatory molecules (CMs) such as CD28, CD27, CD134 (OX40), and CD137 (4‐1BB).24 CMs are essential for delivering secondary signals when CAR comes into contact with a tumor antigen. This secondary or co-stimulatory signaling improves the effector functions of T-cells, such as anti-tumor activities, by reducing T-cell exhaustion and perpetuating T-cell signaling.12
Although CARs are designed to mimic TCR signaling, they differ greatly from the natural or conventional TCR in terms of architecture, antigen recognition domain, and intracellular signaling.25,26 The conventional TCR is made up of a heterodimer of two highly variable chains, either α and β chains (95%) or γ and δ chains (5%) that consist of a variable (V) and a constant (C) domain.26 Three highly variable complementary determining region loops are located at the most distal ends of each V domain of the TCR α and β chains to interact directly with the peptide-MHC complex (pMHC).27 Thus, TCR is an MHC-dependent receptor that recognizes antigens or peptides presented only by conventional or unconventional MHC for αβ and γδ heterodimers, respectively. On the contrary, the variable domains in the TCR-α and-β chains are replaced with those of immunoglobulin heavy or light chains from hapten-specific antibodies, typically with an ScFv in the CAR structure.28 ScFv recognizes cell surface antigens in an MHC-independent manner, allowing a wider range of antigens to be targeted. As discussed above, CAR incorporates TMD and endodomain in addition to the ScFv-containing ectodomain, making a single CAR protein act as a TCR, CD3, CD3, and CD3 all at once.
Antigen density threshold necessary for CARs to evoke T-cell effector functions is higher than the natural TCR. Unlike TCRs that bind agonist pMHC with micromolar affinity, CARs bind their MHC-independent ligands with nanomolar affinities.29,30 Salter et al. also found that TCRs are at least 100-fold more sensitive to antigen than CAR.25 CARs require thousands of surface antigen molecules for strong signaling, in contrast to native TCRs, which can activate T-cells after recognizing as few as 1–10 agonist pMHC complexes.31,32 This increased antigen density threshold of CAR results in tumor escape associated with low target antigen expression and limited efficacy in the treatment of cancer. HLA-independent T-cell (HIT) receptors have recently been demonstrated to effectively tackle against the obstacle of treating cancer by targeting tumors with low antigen density. Thus, HIT receptors provide a consistently high antigen sensitivity and mediate tumor recognition, even above the most sensitive construct of CAR. They are found to be excellent for targeting sparsely expressed cancer cell surface antigens.33
Moreover, CAR T- cells require receptor oligomerization and clustering during intracellular signaling unlike T-cells.34,35 While both assemble multi-component signaling complexes and form immunological synapses upon antigen engagement, the synapses of CAR are less reliant on intercellular adhesion molecule-1 (ICAM-1)/LFA-1 interactions and exhibit disorganized patterns of LCK localization when compared to TCR.36,37 The TCR adaptor protein linker of activated T-cells (LAT), as well as important T-cell signaling proteins such as CD3δ, CD3ε, and CD3γ, which make up a component of the T-cell co-receptor, is either not phosphorylated or are only weakly phosphorylated by CAR activation.25
Generations of CAR T-cell therapy
Since their discovery in the late 1980s, CAR T-cell therapies have progressed significantly in an attempt to increase activation, persistence, proliferation, safety, and efficacy.22 CAR T-cell therapies have gone through five generations in the last thirty years, with modifications to the endo-domain structure and the number of CMs used (Figure 2).38,39 Although their basic conformation and other domains have remained the same since their inception, the structure, composition, and function of the intracellular domain of the CAR receptor have changed significantly across these generations.40 The modifications of the endodomain greatly advance CAR T-cell technology and improve T-cell activation, proliferation, efficacy, and persistence as the generation grew. Besides, CMs have been added to the newer generations of CAR T-cells to enhance their ability to expand rapidly and survive longer after infusion.
Figure 2.
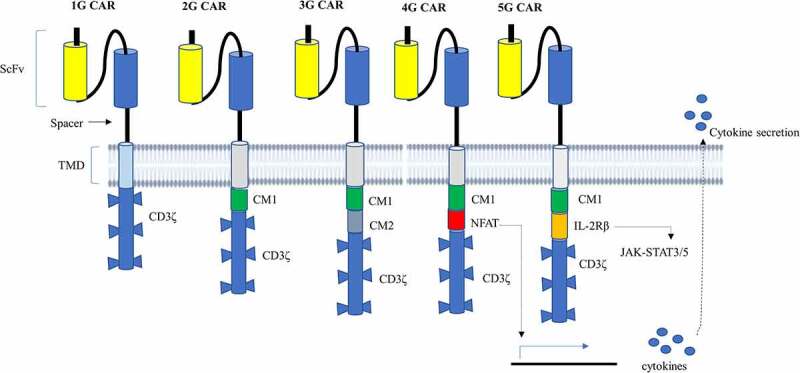
Diagrammatic representation of the structure of CAR from the first generation to the fifth generation. 1G CAR contains only a CD3ζ in its signaling domain with no CM. 2G CAR harbors CD3ζ and one CM, allowing dual signaling pathway.3g CAR, on the other hand, combines CD3ζ and several CMs that deliver multiple signaling. 4G CAR resembles 2G CAR with the incorporation of an additional NFAT-responsive cassette that expresses cytokines playing anti-cancer activities. Thus, 4G CAR has triple signals from primary CD3ζ, CM, and expressed transgenic proteins. 5G CAR is based on 2G CAR by adding membrane receptors IL-2Rβ that provides a binding site for STAT3 and activates the JAK-STAT signaling domain. 5G CAR has also a synergistic activation of triple signals from CD3ζ, CMs, and cytokine-inducing JAK-STAT3/5 pathway.
Abbreviations: IL-2R, interleukin receptor; Jak, Janus kinase; NFAT, nuclear factor of the activated T-cell; STAT, signal transducer and activator of transcription.
First-generation (1G) CAR T-cell
It was first developed in 1993 with an extracellular domain containing scFv and a cytoplasmic domain containing a CD3ζ (FcεRIγ) signaling domain but without extra CM. This induces the TCR signaling pathway, mediating cytokine (e.g., IL-2) secretion in an HLA-independent fashion.41 Owing to the absence of CM and cytokine-mediated signaling, 1G CAR T-cells are observed to have less T-cell proliferation, inadequate cytokine release, and poor in vivo persistence of T-cell responses.42 As a result, the antitumor activity of the 1G CARs is reduced, and hence it is currently considered obsolete.22,43
Second-generation (2G) CAR T-cell
In addition to intracellular CD3ζ domains, a 2G CAR T-cell carries CMs such as CD28, CD134 (OX-40), or CD137 (4‐1BB), resulting in dual signaling pathways mediated by CD3ζ and CMs.44–48 All of the FDA-approved products that are currently available on the market are 2G CAR T-cells, which contain a CM in addition to CD3ζ. The presence of CMs in 2G CARs enhances T-cell activation, proliferation, survival, cytokine production, cytotoxicity, and sustained response due to CAR cells’ resistance to apoptosis and increased in vivo life span.44,49–51 According to several studies, due to its delayed activation, a CAR T-cell construct possessing CD137 has a longer persistence and durable response but a weaker tonic signaling compared to those containing CD28 or CD134. CD28-based CAR cell therapy, on the other hand, is linked with greater T-cell expansion, survival, memory cell formation, and phosphorylation, resulting in robust signaling and quicker response.14,52–55 The 2G CAR T-cell therapies targeting CD19 are currently entering clinical practice and have been found to be highly effective against B cell malignancies.56 More recently, obecabtagene autoleucel (obe-cel), a new form of CD19 CAR genetically engineered with CAT-41BB-Z, is in clinical trials and showing outstanding results in some adult patients with recurrent B-Cell acute lymphoblastic lymphoma (ALL).57 Despite significant improvements, the persistence and relapse associated with CAR T-cells using a single CM have not been resolved, prompting the development of third-generation CARs.11
Third-generation (3G) CAR cells
It is made by combining CD3ζ and several CMs, such as CD28, CD137 (41BB), CD134 (OX-40), NKG2D, CD27, TLR2, or inducible T-cell co-stimulator (ICOS), to create integrated CAR T-cell constructs like CD3ζ-CD28-OX40, CD3ζ-CD28-41BB, CD3ζ-ICOS-4-1BB, and CD3ζ-TLR2-CD28.22,58–62 Among the various constructions of 3G CAR T-cell products, CD3ζ-CD28-41BB-based CAR-T-cells are currently the most commonly used construct. Multiple CMs employed in 3G CAR cells are essential to overcome the constraints of each CM used in 2G CARs. Thus, 3G CARs incorporate two CMs that may exhibit short-term efficacy with potent and quicker tumor elimination, such as CD28, as well as durable clinical responses, as in 4-1BB.63 Preclinical data demonstrated that 3G CARs showed better performance in treating some types of cancer than 2G CAR-T-cells, with excellent safety profiles, in vivo proliferation, perseverance, and anticancer activity.60 Consistently, a study by Ramos et al. showed that the CD19-directed 3G CAR T-cells had better expansion and longer persistence than CD19-targeted 2G CAR cells.56 However, there is a higher incidence of severe side effects and a faster CAR T-cell exhaustion associated with 3G CAR T-cells than with 2G CAR cells, owing to the over activation of multiple CM mediated signals.64,65
Fourth-generation (4G) CAR
It is also known as T-cell redirected for universal cytokine-mediated killing (TRUCK), universal CAR (UniCAR-T), or armored CAR-T-cells.22,66,67 4G CAR primarily resembles the constructs of 2G CAR T-cells, with huge modification of its intracellular signaling domain. This involves further incorporation of a nuclear factor of the activated T-cell (NFAT)-responsive cassette possessing transgenic immune modifiers (proteins) such as cytokines (IL-2, IL-5, IL-12, IFN-γ) and CMs such as CD28, OX-40, or 4‐1BB.68,69 NFATs are constructed transcription factors to regulate inducible or constitutive expression of transgenic proteins and their delivery to the targeted tumor site upon CAR cell activation, hence creating a more favorable tumor microenvironment for the immune responses. Following antigen-activated CAR-signaling, the activation of the NFAT promoter sequence results in the production of cytokines that kill cancer by activating innate immune cells.43,69,70 Consequently, 4G CAR T-cells have been proven to play a great role in modulating the tumor microenvironment by overcoming the challenge of antigen loss within tumor cells.71 Armored CAR-T-cells further enhance T-cell expansion, persistence, memory cells, and anti‐tumor activity, while they greatly reduce systemic toxicity. In addition, they are beneficial to reestablish post-infusion immune system of the patients. Despite these great improvements, 4G CARs still have markedly reduced efficacy against solid tumors and are linked to some adverse events due to on-target off-tumor activation of TRUCK T-cells and release of the transgenic cytokine in healthy tissues.69
Fifth-generation (5G) CAR
The structure of CAR-T-cells is continually under improvement, with the fifth-generation CAR-T-cell, also known as the next generation, currently in active development in the hope of tackling the bottlenecks of earlier generations of CAR T-cell therapy.
This generation involves significantly improved conventional CARs, which are monovalent CARs that exclusively target one particular antigen, as well as advanced CARs that are beyond conventional CARs by incorporating additional structures into the traditional CAR to recognize more than one antigen or low antigen density targets.
Conventional (monovalent) CAR
Next-generation conventional CAR is a novel CAR designed based on 2G CAR by integrating additional membrane receptors and T-cell engagers such as the JAK-STAT signaling domain into the endodomain. The binding site of 2G CAR is modified to contain a truncated beta chain of the IL-2 receptor (IL-2 Rβ) in the signaling domain and to bind with the STAT3 transcription factor.72 When antigen binds to this receptor, simultaneous activation of triple signaling by CD3ζ, CMs, and cytokine-inducing the JAK – STAT3/5 pathway occurs. These three activated signals work together to improve T-cell activation, proliferation, and persistence.73 5G CAR is the most advanced generation of CAR T-cells that are manufactured to have a better safety profile and a wider therapeutic window. Like 4G CAR, it is also an effective tool to create a conducive tumor micro-environment and restore a patient’s immunity after infusion.11 But next-generation conventional CARs exhibit limited efficacy in infiltrating and trafficking into solid tumors, with an unresolved problem of adverse effects. The structures, typical features, and limitations of the different generations of conventional CARs are summarized in Table 1.
Table 1.
A summary table on the generations of car T-cell and their structure, feature, and limitations.
| Generations | Endodomain | Typical features | Limitations | Refer. |
|---|---|---|---|---|
| 1G CAR T-cells | Contains only CD3ζ without any CM | It has only one signaling and is capable of T-cell activation only | Inadequate T-cell proliferation, cytokine release, T-cell persistence, and antitumor activity |
11,22,41–43 |
| 2G CAR T-cells | Composes CD3ζ and CMs (CD28, CD134/OX-40, or CD137/4‐1BB) | Provide two signals via CD3ζ and CM; offer better T-cell activation, proliferation, and persistence than 1G CAR T-cells | The problem of persistence and relapse | 11,14,44–55,58,74 |
| 3G CAR T-cells | Combines CD3ζ and multiple CMs (CD28, CD137/41BB, CD134/OX-40, NKG2D, CD27, TLR2 or ICOS, | Characterized by multiple signaling via CD3ζ and two CMs. Gives better safety profiles, proliferation, perseverance, and anti-tumor functions than 2G CAR T-cells | Higher incidence of severe side effects such as CRS and faster T-cell exhaustion |
56,58−60 |
| 4G CAR T-cells | Harbors CD3ζ, NFAT-responsive cassette containing cytokines (IL-2, IL-5, IL-12, IFNγ) and CMs (CD28, OX-40 or 4‐1BB) | It has primary CD3ζ, costimulatory signals, and the expression of transgenic proteins. It potentiates T-cell expansion, persistence, and anti-tumor capacity by overcoming tumor antigen loss by cytokine activation at the tumor site and modulating the tumor milieu. It is also essential to regain the patient’s immune systems and reduce toxicity to more than 3G | Poor cancer-killing potential in solid tumors; Side effects such as on-target off-tumor activation of TRUCK T-cells and release of the transgenic cytokine in healthy tissues; Double modification of T-cells |
43,68−71 |
| 5G CAR T-cells | Incorporates CD3ζ and additional membrane receptors: IL-2Rβ and STAT3 | Provide better T-cell activation, proliferation, solid tumor infiltration, and persistence by cytokine-inducing JAK/STAT signaling. It also has a better safety profile and a wider therapeutic window. It creates a more favorable tumor milieu and reestablishes the immune system after infusion | They are still as effective as blood cancer in infiltrating and trafficking into solid tumors and are associated with some side effects |
11,72,73 |
Abbreviations: CAR, Chimeric antigen receptor; CMs, costimulatory molecules; NFAT, nuclear factor of the activated T-cell; STAT, Signal transducer and activator of transcription; IL-2, Interleukin 2; JAK, Janus kinase.
Beyond conventional (advanced) CARs
Additional advancements beyond the conventional CARs, called Boolean logic gated CAR T-cells, have recently been developed to promote the CAR T-cells’ specificity, control their activities, as well as to overcome limitations associated with conventional CARs.75,76 They are cutting-edge CAR technologies engineered to improve the cancer-specificity of CAR T-cells, thereby increasing the efficacy and reducing adverse toxicities of the therapy. There are many forms of logic gating: the most common being AND-, OR- NOT, and IF-Better logic gates.76,77
AND logic gate CAR
Also known as split-recognition CAR, it is constructed by separating the various activation signals, such that one receptor includes the primary activation signal (CD3ζ), while the other construct contains the co-stimulatory domains, such as CD28 and 4-1BB. AND gate CAR systems are only active when two antigens are present on a cancer cell, whereby T-cells are transduced with a CD3ζ CAR directed toward one antigen and a co-stimulatory receptor (CCR) targeted toward a second antigen.78,79
OR logic gate
There may be multiple possible input signals in an OR logic gate, and any of them can trigger the desired outcome. It is an example of a multi-antigen approach requiring the recognition of either one or more of the targeted CAR T-cell antigens. Multi-antigen CAR T-cells are constructed using bicistronic CAR, tandem (tan) CAR, or loopCAR methods by introducing two different CARs into the same T-cell, unlike the monovalent conventional CAR T-cells that target only one antigen. This is very helpful to address the challenge of antigen escape associated with the tumor.76 Bicistronic or dual CARs are engineered to transduce two CAR constructs into the same T-cell, allowing them to target multiple tumor-associated antigens (TAAs). CAR T-cells can also be engineered to express CARs targeting three or more antigens, such as triCARs or quad-CARs. On the other hand, tanCAR is composed of two distinct scFv binding domains that are joined in tandem in a single CAR to target two different TAAs. It shows synergistic antitumor activity when both antigens are simultaneously recognized. LoopCAR is also a recent CAR design that has a looped structure of tanCAR constructs.80,81
NOT logic gate
Also referred to as inhibitory CARs (iCARs), they are tailored to incorporate inhibitory signaling rather than activation domains as the internal signaling component of the off-target CAR construct. Inhibitory signals will prevent the activation of the CAR T-cell when in the presence of off-target antigen.82 The NOT logic gate recognizes antigens that are expressed on normal tissue but absent on tumor tissue, and are coupled to the signaling domain of an inhibitory coreceptor such as PD-1 or CTLA-4. iCAR is expressed along with CARs that specifically target the antigen of interest to circumvent autoreactivity to bystander tissues. This makes it a very important strategy to prevent the occurrence of on-target, off-tumor toxicity.
IF-Better gate CAR
It is a novel CAR construct in which killing is initiated by high CAR target expression alone, but not by low CAR target expression unless a chimeric costimulatory receptor (CCR) target is present. The interaction of CCR with target antigen increases avidity and costimulation, which facilitates higher CAR sensitivity that is purposefully restricted to targeT-cells expressing both antigens. ADCLEC.syn1, which is a novel combinatorial CAR construct consisting of an ADGRE2-targeting 28z1XX-CAR and a CLEC12A-targeting CCR, operates based on IF-Better gating. A preclinical study done in acute myeloid leukemia (AML) indicated that the IF-Better gate performs better than a single-CAR T-cell and a dual-CAR T-cell, increasing anti-tumor activity and preventing tumor escapes in AML.83 Overall, a single CAR T-cell construct can use one or a combination of two or more varieties of Boolean logic gated CAR designs as discussed below.
SUPRA CAR system
It is a split, universal, and programmable CAR system that is composed of a soluble antigen-binding domain (zipFv) and a universal signal transduction receptor (zipCAR) expressed on T-cells to improve specificity and controllability.84 The zipFv has a leucine zipper and an scFv, whereas the zipCAR has intracellular signaling domains and an extracellular cognate zipper that specifically binds to the zipper on the zipFv. These zippers bridge the binding between the target antigen and zipCAR-expressing T-cells and elicit T-cell responses. This CAR design responds to combinatorial antigen in targeT-cells and enables ON/OFF switching for fine-tuning of T-cell activation and AND logic gate. SUPRA CAR was further developed to have a separate inhibitory domain by adding NOT logic to their abilities.85
Synthetic Notch (synNotch) receptor
It induces the expression of effector proteins upon the activation of this receptor by recognition of one antigen. This receptor applies an ‘if-then’ strategy, which means if the synNotch receptor recognizes its antigen, the expression of a CAR specific to another antigen is activated.86 Thus, when the synNotch receptor binds to its receptor, protease-mediated proteolysis releases an intracellular transcription regulation factor that initiates the transcription of a CAR against a second antigen sequentially. The synNotch CAR T-cells specifically kill dual-antigen-bearing cells once they are armed and activated while sparing single-antigen cells unaffected in vivo.87–89
Reversed (Rev) CAR
It applies AND and OR logic gating and contains only a small peptide epitope as an extracellular domain instead of the scFv that is found in conventional CARs.89 This novel platform reduces the CAR size, avoids nonspecific antigen binding, and avoids antigen-independent tonic signaling induced by scFv dimerization. It also allows the control of CAR T-cell activity, offers gated targeting strategies, and can be tailored to any tumor antigen and tumor type. RevCARs are inactive by themselves but become active and specifically redirected to tumor cells only when there is a bispecific antibody-based target module (RevTM). RevTM has two scFvs: one to detect short peptide epitope of RevCARs and the other to recognize TAAs. Due to the extremely short half-life of RevTM, the RevCAR T-cells can be switched on and off depending on the dosage of RevTM. Moreover, this CAR platform can adapt to any type of tumor antigen by simply changing RevTM. Several RevCARs incorporations into the same T-cell are possible due to the small size of RevCARs, allowing gated tumor targeting that reduces adverse toxicities.90
Avidity (Avid) CARs
It is another CAR variant that is constructed by incorporating a CAR dimerization domain and at least two low-affinity antigen-binding domains. AvidCARs can be ON-switch AvidCARs that only trigger CAR T-cell activation when both antigen-binding domains bind their target antigen and when a dimerization molecule is given, and AND logic AvidCARs in which both antigen-binding domains recognize two antigens to induce CAR T-cell activation with no need for a dimerization molecule. AvidCARs are generally designed to amplify the avidity of the antigen-binding domain with its target antigen and thereby further enhance the specificity and localization of CAR T-cell activation and anti-tumor functions.91
The synthetic T-cell receptor and antigen receptor (STAR)
STAR, which combines the specificity of a CAR and the internal signaling machinery of an endogenous TCR, has also recently been engineered to tackle the challenge of treating solid tumors using CAR T-cell therapy.92 This novel technology was found to be an interesting option to treat solid tumors, with superior or equipotent performance in controlling the tumor, and does not trigger tonic signaling that causes T-cell exhaustion compared to CAR T-cells. STAR mediates strong and sensitive TCR-like signaling upon stimulation by antigen binding. STAR T-cells demonstrate higher antigen sensitivity than CAR T-cells and may minimize the risk of antigen loss – induced tumor relapse in clinical use. These cells also exhibit less susceptibility to dysfunction, a lower risk of toxicity, and better proliferation than conventional CAR T-cells.93
Procedures of CAR T-cell production
In general, the overall process of CAR T-cell manufacturing in the laboratory takes 2 to 8 weeks and encompasses four stepwise phases: T-cell extraction, genetic engineering, expansion, and CAR T-cell adoptive transfer(Figure 3).40,94
Figure 3.
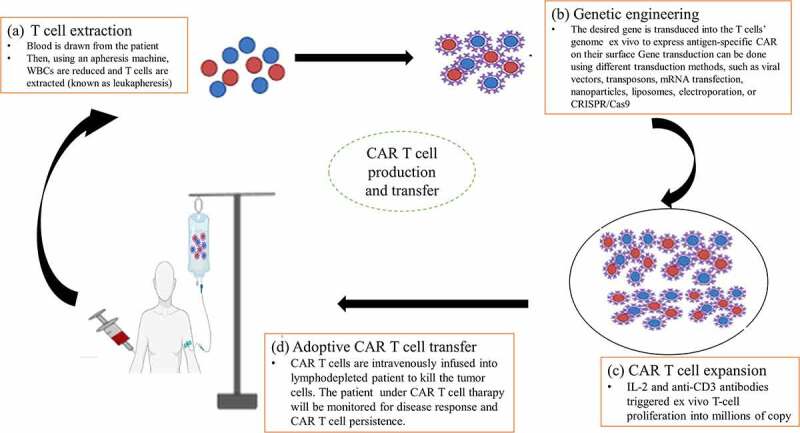
Procedures of CAR T-cell production. (a) T-cell extraction, (b) Genetic engineering (c) CAR T-cell expansion, and (d) Adoptive CAR T-cell transfer.
T-cell extraction
The first step in CAR T-cell development is the collection of T-cells through leukocyte apheresis (leukapheresis). Blood can be drawn from the patient or another healthy individual, for autologous and allogeneic extraction, respectively. Then, using an apheresis machine (blood cell separator), white blood cells are reduced to isolate T-cells while the remaining blood is reintroduced into circulation.95,96 After leukapheresis, the product is enriched for T-cells by washing the cells out of the apheresis buffer containing anticoagulants using a cell washer, such as the Haemonetics Cell Saver. Subsequently, elutriation is used to carry out the T-cell enrichment through counterflow centrifugation. Elutriation separates cells by size and density to reduce unwanted contaminating cells (such as granulocytes, red blood cells, and platelets) and preserve T-cell viability.97 Magnetic cell separation is the next step that is performed to select T-cell subsets at the level of CD4/CD8 composition using specific antibody bead conjugates or markers. The T-cell extraction and enrichment usually take 2 to 3 hours.98
Genetic engineering
After T-cell enrichment, they are genetically engineered in the cell processing center (or laboratory) in the way they express antigen-specific CAR receptors. The desired gene encoding CAR is genetically transduced into the T-cells’ genome using integrating viral vectors, transposons, or mRNA transfection.99,100 T-cell-enriched apheresis products are cultured in a sterile bioreactor in the presence of CAR encoding vectors to deliver the desired genes and cell-based artificial antigen-presenting cells (aAPCs). Besides, genes can be transferred using non-viral transduction methods such as nanoparticles, liposomes, electroporation, or CRISPR/Cas9 technology. Viral vectors, which can be retroviruses, lentiviruses, adenoviruses, or adenovirus-associated viruses, are very safe and the most widely used method of gene transduction. The desired gene carried by viral vectors reaches into the T-cell’s genome first by viral binding and fusion with the T-cell’s membrane and then the CAR gene gets integrated into the genome of T-cells. Eventually, genes are transcribed and translated to produce CARs, which are then inserted into the plasma membrane of T-cells to form artificial but living cells now known as CAR T-cells in the laboratory.99 More recently, a more advanced new gene-editing tool known as CRISPR/Cas9 has been introduced to integrate the CAR gene into specific sites in the genome of T-cells.101,102 Many patients with advanced disease complain about the long waiting time because they develop insurmountable progression or die before CAR-T infusion. Currently, a rapid manufacturing process of CAR T cells, known as the in vivo gene delivery system, has been introduced. Novel CD3-targeted lentiviruses (CD3-LVs) are an in vivo gene delivery system by genetically modifying human T-cells (in vivo) without prior activation. Agonistic CD3-specific ScFv on lentiviral vector particles delivers genes selectively into T-cells and activates T-cell expansion. Thus, CD3-LVs allow in vivo gene delivery without the need for blood processing and generate CAR T-cells directly in vivo.103 Preclinical analysis in a tumor mouse model by Agrawal et al. has also demonstrated the in vivo generation of CD19-specific CAR T-cells using the CD8-targeted lentiviral vector (CD8-LV).104 Moreover, Pfeiffer et al. revealed the feasibility of in vivo reprogramming of human CD8+ CAR T-cells against CD19+ cells.105 Compared to the current manufacturing procedures, the in vivo gene delivery system is less complex, time-consuming, and costly, as well as maintains efficacy since there is no manipulation before infusion.103
CAR T-cell expansion
After gene alteration, CAR-engineered T-cells are allowed to actively proliferate in the laboratory. Beads coated with anti-CD3/anti-CD28 monoclonal antibodies or aAPCs in combination with feeder cells and growth factors, such as IL-2 are key factors used for T-cell activation and ex vivo expansion. T-cells can proliferate logarithmically in a perfusion bioreactor in the presence of aAPCs and IL-2 within weeks. The beads or aAPCs can then be removed from the culture through magnetic separation.106 The WAVE Bioreactor, G-Rex, CliniMACS Prodigy, and the Cocoon are bioreactor culture systems that offer optimal gas exchange requirements and culture mixing required to expand a large number of cells for clinical use.107 G-Rex is a bioreactor that allows the expansion of cells from low seeding densities. Gas-permeable membranes are used in this bioreactor, enabling the flask to be placed right into a cell culture incubator. But it is an open culture system in which the flask of G-Rex must be opened during cell inoculation to manually pipette fluids and cells in and out under aseptic conditions. Recently, functionally closed versions of bioreactors have been developed to allow the free movement of fluids and cells in and out of the bioreactor. The Xuri cell expansion system (previously known as WAVE Bioreactor) is a functionally closed process for CAR T-cell manufacturing. It is based on the WAVE bioreactor platform that uses a rocking motion for optimal mixing and gas transfer to expand the CD19-directed CAR T-cell therapy.107 The CliniMACS Prodigy is a closed automated platform that carries out T-cell preparation, enrichment, activation, genetic manipulation, expansion, final formulation, and sampling in a single device.108 The Cocoon (Lonza) is another fully automated manufacturing machine in a closed system, which eliminates any handling of the product during manufacturing.109 These systems are very important for the reproducible and fast delivery of fresh cells for the treatment of patients while maintaining Good Manufacturing Practice (GMP) compliance. They simplify the manufacturing processes with minimal involvement of users. These automated platforms can also be used to manufacture CAR T-cells in large quantities for off-the-shelf use at lower cleanroom standards.108,110 Eventually, after the completion of the cell expansion process, the cell culture must be purified (washed) to isolate T-cells containing the desired gene and concentrated to a volume that can be infused into the patient. Cells are then formulated in an appropriate cryopreservation medium containing dimethyl sulfoxide (DMSO) and cryopreserved. After being cryopreserved in an infusible medium in the laboratory, the concentrated CAR T-cell products are then transferred to and thawed at the facility where the patient will get treatment.111 The procedure of ex vivo T-cell activation and expansion, along with viral transduction, can take 10 days to several weeks to complete.
CAR T-cell adoptive transfer
Ex vivo expanded CAR T-cells are intravenously infused into the patient’s venous blood via a needle, which typically takes an hour or more. However, before the infusion of the recombinant CAR T-cells, the circulating leukocytes in the patient must be depleted (referred to as lymphodepletion) using chemotherapy or radiotherapy.112 Lymphodepletion (also known as preconditioning regimen) is essential to get rid of other immune cells to make space for newly produced CAR-T-cells.113 This upregulates cytokine production while downregulating resource competition, thereby enhancing the proliferation and survival of the transferred CAR T-cells.114 Several regimens are commonly used for lymphodepletion, including cyclophosphamide (Cy), fludarabine (Flu), and bendamustine, alone or in combination. The lymphodepleting chemotherapy is given for 2–14 days prior to the infusion of CAR T-cells. Cyclophosphamide-based lymphodepletion regimens that last for 3–5 days are the most frequently employed before CAR T-cell infusion. The addition of fludarabine to lymphodepleting chemotherapy has been linked to increased CAR T-cell expansion and persistence and extension of disease-free survival in patients with ALL.115,116 Total body irradiation or radioimmunotherapy such as I-131 apamistamab, which is currently under phase III clinical trial, also holds a hope in lymphodepletion (Table 2). In addition, pentosan, clofarabine (30 mg/m2 daily for 5 days), Cy 440 mg/m2/day and etoposide 100 mg/m2/day for 2 days, cytarabine 300 mg/m2 (single dose) and etoposide 150 mg/m2 (single dose), methotrexate 1 g/m2 on day 1 and cytarabine 1 g/m2 every 12 hours on days 2 and 3, Cy 300 mg/m2 every 12 hours on days 1–3, vincristine 1.5 mg/m2 (maximum 2 mg) on day 3, and adriamycin 50 mg/m2 on day 3 were employed as lymphodepleting regimens before CAR T-cell therapy and showed effective outcomes.117,121
Table 2.
Major lymphodepleting regimens for CAR T-cell therapy and their dosages, advantages, and disadvantages.
| Regimen | Dosage | Advantage | Disadvantage | Refer. |
|---|---|---|---|---|
| Cy | 300 mg/m2 every 12 hours for 3 days | It effectively prolongs the persistence of infused cells and increases the effectiveness of treatment | It could cause adverse effects, ranging from mild to severe symptoms | 117 |
| Bendamustine | 90 mg/m2/day for 2 days | It is an effective lymphodepletion regimen before tisa-cel in R/R lymphoma. It also reduces hematological toxicities and infectious complications compared with Flu/Cy. Lower CRS and neurotoxicity than Flu/Cy. It shows higher neutrophil, hemoglobin, and platelet counts. | It may result in side effects such as fevers, chills, itching, skin rash, nausea, vomiting, fatigue, and more serious side effects infertility and liver injury. | 116 |
| Flu | 25 mg/m2/day for 3-5days | Flu improved CAR–T-cell persistence and PFS compared to Cy alone | No cell persistence in the second round of CAR T-cell treatment, myelosuppression, risk of infection, and neurotoxicity | 115 |
| Bendamustine/Flu | Bendamustine 70 mg/m2/day + Flu 30 mg/m2/day for 3 days | Provide longer persistence of CAR T-cells than Cy/Flu. It markedly increases the level of IL-15 and IL-17 than bendamustine alone. It enhances PFS compared with bendamustine alone or Cy/Flu. Good efficacy and safety profile | It may be associated with CRS | 118 |
| Cy/Flu | Cy 500 mg/m2/day + Flu 30 mg/m2/day for 2–5 days | Increase cell expansion and longer persistence both in CD4 and CD8 cells. It also improves clinical outcomes | This leads to more profound lymphopenia, higher rates of hematological toxicities | 115,116 |
| I-131 apamistamab | 75 mCi | It is a new CD45-targeting antibody radiation-conjugate given as a single-dose outpatient administration; It is a specific, safer, and more effective alternative; less toxic than chemotherapy-based lymphodepletion; prevents CRS | Radiation associated risks | 119,120 |
Abbreviations: CRS, cytokine release syndrome; Cy, Cyclophosphamide; Flu, Fludarabine; PFS, progression-free survival.
After the adoptive transfer of CAR T-cells, patient close monitoring to evaluate for side effects and disease response using CT scanning, bone marrow biopsies, and peripheral blood flow cytometry is required. Additionally, CAR T-cell persistence can be monitored by immunochemistry of bone marrow biopsy, RT-PCR, and flow cytometry of blood and bone marrow aspirate.122
Mechanism of action of CAR T-cell therapy
CAR T-cell therapy is a patient-specific, living, self-replicating immune-boosting drug.60 CARs are designed to program T-cells to seek out tumor cells expressing specific proteins on their surface as infectious agents. Accordingly, the engineered CAR T-cells express CAR receptors on their surface and allow the T-cells to detect and bind to specific antigens on the surface of tumor cells. This stimulates intracellular signaling, which causes the T-cells to become activated and destroy the cancer cells.95 CAR T-cells, in contrast to other ACTs such as TIL and TCR therapies, are attacker cells outfitted with synthetic CAR receptors that detect antigens and eradicate cancer cells expressing specific surface antigens without the use of HLA. In other words, regardless of HLA presentation, CARs have the ability to bind to and target a wide spectrum of antigens such as proteins, gangliosides, carbohydrates, or any other compounds presented on cancer cells. This renders more cancer cells vulnerable to CAR T-cell attacks by overcoming the immunosuppressive milieu, making it a more versatile therapeutic technology than other HLA-dependent ACTs.5,123
The cytotoxic mechanism of CAR T-cells resembles the signaling pathway of natural T-cells.6 Following the infusion of CAR T-cells into a patient, they come into contact with tumor antigens via the scFv of CAR receptors. CAR T-cells can target tumor surface antigens such as CD19, B cell maturation antigen (BCMA), CD20, CD30, and many others, with CD19 being the most studied antigen target, followed by BCMA.122–126 Right after tumor antigen engagement of CARs, the CAR T-cells undergo conformational changes and become activated. Specifically, the components of the intracellular domain form microclusters that involve a centripetal movement to form the core region of the immunological synapse, allowing the recruitment and phosphorylation of the downstream cascading proteins, including CD3ζ and CMs. The activated CAR T-cells then undergo extensive proliferation and differentiation, which is essential for the effector functions or cancer-killing activities of the CAR T-cells.123,127
CAR T-cells mediate tumor-killing activities by employing several synergistic mechanisms, involving the perforin-granzyme system, the death ligand–death receptors, and the recruitment of other components of the immune system (Figure 4). Primarily, CAR T-cells eradicate cancer cells by using the perforin-granzyme mediated cytolytic mechanism. Upon recognition of surface antigens on a target T-cell and activation of CAR T-cells, a fast calcium-mediated degranulation or release of the cytotoxic effector proteins (perforin and granzymes) from the lytic granules of the CAR T-cells occurs. After their release, perforin creates transmembrane pores on the plasma membranes of the target T-cells to allow cytotoxic granzymes access into the cytoplasm of target T-cells. Granzymes are serine proteases that are the main players in the cytolysis of cancer cells by CAR T-cells. These enzymes destroy antigen-positive cancer cells by stimulating both caspase-dependent and caspase-independent apoptotic pathways. Ultimately, the dead cancer cells will be rapidly removed by the nearby phagocytic cells.128–130
Figure 4.
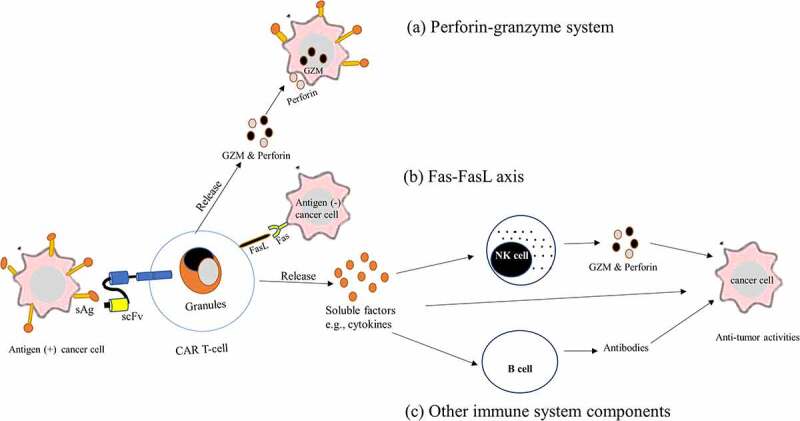
Mechanism of cancer-killing by CAR T-cell therapy. CAR T-cells detect tumor antigens via its scFv of CAR and become activated and kill cancer cells by using several mechanisms. (a) perforin-granzyme system. Activated CAR T-cells quickly release perforin and GZM from their lytic granules, and then perforin creates membrane pores on cancer cells, allowing GZMs entrance into the cytoplasm of the cancer cells and killing them. (b) the death ligand–death receptors such as the Fas-FasL axis: binding of Fas in the cancer cell membrane to the FasL present on activated CAR T-cells, induce apoptotic cancer cell death. (c) Recruitment of other components of the immune system. Activated CAR-T-cells release soluble factors such as cytokines upon CAR engagement with the target antigen. Secreted cytokines infiltrate tumor cells and cause inflammation eliminating cancer cells, and activate other immune cells such as B-cells and NK cells at the tumor site that aid in the anti-tumor activities.
Abbreviations: FasL, Fas ligand; GZM, granzymes; NK cells, natural killer cells; sAg, surface antigen; scFv, single chain variable fragment
Besides, the CAR T-cells make use of death ligand–death receptors such as the Fas–Fas ligand axis and TNF-related apoptosis-inducing ligand (TRAIL) systems to mediate their cytolytic effector functions. Fas-FasL is a perforin-independent mechanism of cytotoxicity that takes place upon the binding of Fas in the target T-cell membrane to the FasL present on activated CAR T-cells. These Fas–FasL interactions induce caspase 8, which in turn activates the downward stream of the apoptotic pathway to execute cancer cell killing.131 Unlike the perforin-granzyme axis, the Fas-FasL system is found to be a slow process that is required to target antigen-negative tumor cells within the antigen-positive tumor microenvironment. But available evidence indicates that human CAR T-cell therapy can kill cancer cells without the use of Fas–FasL killing mechanism.48,128 Strikingly, the TRAIL effector system appears to play a key role in tumor eradication by CAR T-cells. A collective body of data indicated that CAR T-cells exert anti-tumor activity through TRAIL-induced apoptosis of cancer cells.132
The other tumor cell killing mechanism of CAR T-cell therapy is the recruitment of other components of the immune system by the activated CAR T-cells to come into the tumor cells and destroy cancer cells.112 This entails increasing the production of cytokines and growth factors to infiltrate tumor cells and cause inflammation, eliminating cancer cells.133 Cytokines can also be released by the dead cancer cells and further increase CAR T-cell expansion that kills cancer cells.134 Moreover, CAR T-cells function by cytokine-mediated recruitment of other immune cells such as B-cells and NK cells at the tumor site, potentiating anti-tumor capacity. This results in an effective eradication of the tumor cells that are not directly detected by CAR T-cells in a non-HLA restricted manner.3,22,60 Overall, CAR T-cells have the potential to eradicate all cancer cells, either temporarily or permanently, a condition known as remission. CAR T-cells may remain in the body months after the infusion has been completed, preventing cancer recurrence and resulting in long-term remission for some kinds of blood cancer.117
FDA-approved CAR T-cell therapies
Several years have passed since the first characterization of CAR T-cell therapy to its first approval by different agencies. After many years of painstaking research in designing effective CAR T-cell therapies, some have recently received approval by the US Food and Drug Administration (FDA) and entered the mainstream of cancer therapy.135 The most successful results and approvals were obtained using CD19-and BCMA-directed CAR T-cell therapies in the treatment of blood cancers.122,124,125 In 2017, the first CAR T-cell treatment was approved for commercial use to treat patients with certain forms of B-cell malignancies.136 A total of six CAR T-cell therapies, which are all second-generation CAR T-cell products, have been licensed thus far by the FDA to enter the pharmaceutical markets to treat patients suffering from a wide range of aggressive hematological malignancies.138–142 Notably, favorable clinical responses have been observed with these therapies in refractory and relapsed (R/R) blood cancer such as B-cell lymphomas, leukemia, and multiple myeloma (MM).143–146 Although there are no documented contradictions on the currently approved CAR T-cell products in the manufacturers’ labeling, some reports suggested that patients with concurrent active infection or inflammatory illness should generally be deferred from receiving the therapy. Here below, all the FDA-approved CAR T-cell therapies are briefly discussed under two categories: CD19- and BCMA-targeted CAR T-cell therapies. Additionally, Table 3 provides a summary of all FDA-approved CAR T-cell products.
Table 3.
Summary of the FDA-approved adoptive CAR T-cell therapies in blood cancers.
| Generic name | Trade/brand name | Nickname | Structure* | Target antigen | Date of approval | Indications | Refer |
|---|---|---|---|---|---|---|---|
| Tisagenlecleucel | Kymriah™ | Tisa-cel | Ectodomain: Anti- CD19; Endodomain: CDζ-4-1BB | CD19 | 30 August 2017 | R/R B-ALL in patients aged between 3 and 25 years; adults and children with R/R large B-cell lymphoma such as DLBCL not otherwise specified or arising from FL, and HGBCL, and R/R FL after 2 or more lines of systemic therapy |
138,139,147–152 |
| Axicabtagene ciloleucel | Yescarta™ | Axi-cel | Ectodomain: Anti- CD19; Endodomain: CDζ-CD28 | CD19 | 18 October 2017 | Adult patients with R/R large B-cell lymphoma, such as DLBCL not otherwise specified or arising from indolent lymphoma, primary mediastinal large B-cell lymphoma, HGBCL, and R/R FL |
152,153 |
| Brexucabtagene autoleucel | Tecartus™ | Brexu-cel | Ectodomain: Anti- CD19; Endodomain: CDζ-CD28 | CD19 | 24 July 2020 | Adult patients with R/R MCL and B cell-ALL | 141,154−156 |
| Lisocabtagene maraleucel | Breyanzi™ | Liso-cel | Ectodomain: Anti- CD19; Endodomain: CDζ-4-1BB | CD19 | 5 February 2021 | Adult patients with R/R B cell lymphoma, such as DLBCL not otherwise specified or arise from indolent lymphoma, HGBCL, primary mediastinal large B-cell lymphoma, and grade 3b FL |
142,158–160 |
| Idecabtagene vicleucel | Abecma™ | Ide-cel | Ectodomain: Anti-BCMA; Endodomain: CDζ-4-1BB | BCMA | 26 March 2021 | Adult patients with R/R MM after four or more prior lines of therapy |
160,161 |
| Ciltacabtagene autoleucel | Carvykti™ | Cilta-cel | Ectodomain: Anti-BCMA; Endodomain: CDζ-4-1BB | BCMA | 28 February 2022 | Adult patients with R/R MM after four or more prior lines of therapy |
162,163 |
*All have second-generation CAR constructs.
Abbreviations: B-cell-ALL, B cell acute lymphoblastic leukemia, BCMA, B cell maturation antigen, DLBCL, diffuse large B-cell lymphoma; FL, follicular lymphoma; HGBCL, high-grade B cell lymphoma; MCL, mantle cell lymphoma; MM, multiple myeloma; R/R, relapsed/refractory.
CD19-targeted CAR T-cell therapy
CD19-targeted CAR T-cell therapies (CART-19) are genetically engineered by using CD19 as a target antigen. CD19 is an antigen that is expressed on the surface of B cell-derived cancer cells. These CAR T-cells attach to the CD 19 expressing cells to stimulate CMs such as CD28 or 4-1BB, resulting in the activation and expansion of CAR T-cells that ultimately remove CD19-positive tumor cells.164 Tisagenlecleucel, axicabtagene ciloleucel, brexucabtagene autoleucel, and lisocabtagene maraleucel are CD19-directed autologous CAR T-cell therapies that have shown excellent efficacy and long-lasting benefit in patients with B-cell malignancies. They are now considered standard treatments for patients suffering from aggressive lymphomas and leukemia.165,166 Three CAR-T-cell products (tisagenlecleucel, axicabtagene ciloleucel, and lisocabtagene ciloleucel) are authorized to treat patients with R/R high-grade B-cell lymphoma (HGBCL). Four CAR T-cells (tisagenlecleucel, axicabtagene ciloleucel, lisocabtagene ciloleucel, and brexucabtagene autoleucel) have shown remarkable activity and are currently approved for R/R non-Hodgkin lymphoma (NHL), whereas brexucabtagene autoleucel and tisagenlecleucel are licensed for adults with R/R mantle cell lymphoma (MCL) and patients under the age of 25 years with R/R B-cell acute lymphoblastic leukemia (B-ALL), respectively.146
Tisagenlecleucel
It is also known as Kymriah™ or tisa-cel, which is an autologous second-generation CART-19 therapy that uses 4-1BB as CM. Tisa-cel, which was initially introduced by Novartis, is the first commercially authorized CAR T-cell therapy by the FDA as of 30 August 2017.147,148 This CAR-T therapy was first approved for the treatment of R/R B-ALL in pediatric and young adults aged between 3 and 25 years.137,147,149,150 As of 1 May 2018, tisa-cel has received approval for use in adult patients suffering from B-cell lymphomas, with an overall favorable success rate. Currently, it also serves as a therapeutic option for adults and children with several types of advanced-stage lymphomas. It is indicated for the treatment of R/R large B-cell lymphoma after two or more lines of systemic therapy, including diffuse large B-cell lymphoma (DLBCL) not otherwise specified or arising from follicular lymphoma (FL), or second-line DLBC, and HGBCL.150 A follow-up study involving 93 patients with R/R HGBCL who received tisa-cel showed an overall response rate of 52% with 40% of complete response. The FDA has also recently approved tisa-cel for R/R FL after two or more lines of systemic therapy, making it the third indication for the therapy. This was also approved by the European Commission in early May 2022 and suggested to offer long-term benefits in patients with R/R FL.151 Tisa-cel is associated with a greater incidence of cytokine release syndrome (CRS) in 22% of patients, as well as neurotoxicity in 12% of patients.138,146 Consistently, another longitudinal clinical study involving 115 patients that have taken this CAR T-cell therapy was observed to have a 53% overall response rate (ORR) and a 39% complete response (CR), with 27% developing CRS.167 However, it has limited use in treating patients with primary central nervous system (CNS) lymphoma and chronic lymphocytic leukemia (CLL) despite its hopeful efficacy and safety.168
Axicabtagene ciloleucel
Axicabtagene ciloleucel, also called YescartaTM or axi-cel, is another CART-19 agent with CD28 as CM. On 18 October 2017, the FDA authorized Axi-cel from Kite Pharma as the second-approved CAR T-cell for treating adult patients with large B-cell lymphoma that is refractory to first-line chemoimmunotherapy or relapses within 12 months of first-line chemoimmunotherapy, or relapsing or refractory after 2 or more lines of systemic therapy, including DLBCL not otherwise specified or arising from indolent lymphoma, primary mediastinal large B-cell lymphoma, and HGBCL.169,170 According to real-world patient data, patients with HGBCL who received axi-cel were documented to have an 82% ORR and a 64% CR rate.152 The Phase 3 ZUMA-7 trial compared the safety and efficacy of second-line axi-cel with that of the current standard-of-care (SOC) as second-line treatment in patients with R/R LBCL. The most updated findings from the phase 3 ZUMA-7 trial showed that at a median follow-up of 24.9 months, the estimated median event-free survival (EFS) using axi-cel was 8.3 months, the ORR was 83% and the CR rate was 65%, which were generally better than SOC treatment.171 The most recent report by Neelapu et al. indicated that axi-cel is highly effective as part of first-line therapy for high-risk DLBCL, with 78% CR, 89% ORR, and a manageable safety profile.172 Besides, this product has been authorized recently for adult patients with R/R FL after two or more lines of systemic therapy, with 92% and 76% ORR and CR rate, respectively. The median duration of remission was found to be 18 months or more, but 8% and 21% of patients developed CRS and neurotoxicity, respectively.153 However, axi-cel is still being evaluated and is not yet approved for the treatment of patients with primary CNS lymphoma and MCL.
Brexucabtagene autoleucel
Kite Pharma’s brexucabtagene autoleucel, also referred to as TecartusTM or Brexu-cel, was the third-approved CAR T-cell for the treatment of subsets of patients with leukemia and lymphoma. It is an autologous anti-CD19-CAR T product that was authorized by the FDA on 24 July 2020, for treating adult patients with R/R MCL.154 It was licensed under accelerated approval following favorable responses in clinical studies, with an ORR of 93% and a CR rate of 67%. Besides, the durability of response in that patients has shown incredible outcomes after 12 months of follow-up, with 61% progression-free survival (PFS) and 83% of overall survival (OS).141 As of 1 October 2021, brexu-cel was also approved as the first CAR T-cell therapy for adults suffering from R/R B cell-ALL. It showed a 71% CR rate at nearly 16 months of follow-up, 12.8 months median duration of remission, and 18.2 months of OS.155,156 CRS, neurotoxicity syndrome, cytopenia, and infection were among the major side events linked to brexu-cel treatment.141
Lisocabtagene maraleucel
Lisocabtagene maraleucel, the so-called BreyanziTM or liso-cel, is a CART-19 agent containing a CD3ζ and 4-1BB. On 5 February 2021, liso-cel from Juno Therapeutics was first approved by the FDA for the therapeutic use in adult patients suffering from large B cell lymphoma after two or more lines of systemic therapy, involving DLBCL non-specified or arising from indolent lymphoma, HGBCL, primary mediastinal large B-cell lymphoma, and grade 3b FL.157 Moreover, though it is not yet approved by the FDA, liso-cel is showing an impressive response for MCL, CLL, and primary CNS lymphoma.141,158 About two-thirds of patients with large B cell lymphoma treated with liso-cel had remissions lasting at least 6–9 months. In contrast, liso-cel is linked with adverse effects such as CRS, neurotoxicity, cytopenia, and infection, but generally at a lower incidence rate than axi-cel or tisa-cel.159 One of the key factors for effective immunotherapy is the cellular composition and phenotype of the adoptively transferred T-cells, including T-cell subtypes and subpopulations. Liso-cel is administered in a defined composition with a specific ratio of CD4+ CAR T-cells and CD8+ CAR T-cells.159 A balanced ratio of CD4+ T-cells and CD8+ T-cells can have a positive effect on the ability of products to eradicate the tumor. This is supported by a report indicating that the treatment of B-ALL patients with a 1:1 ratio of CD4+ and CD8+ (constant CD4:CD8 ratio) CAR T-cells could achieve high remission rates. Immunotherapy with a CAR-T-cell product of defined composition enables the identification of factors that are associated with CAR-T-cell expansion, persistence, and toxicity. It also facilitates the development of lymphodepletion and CAR-T-cell dosing strategies that reduce toxicity and improve disease-free survival.173 Therefore, the subsets must be isolated at the start of the production of liso-cel and separately modified to gain a defined CD4:CD8 ratio.159
BCMA-directed CAR T-cell therapy
BCMA-directed CAR T-cell therapy is manufactured by employing BCMA as an antigen detected by the CAR T-cells. BCMA is a cell surface receptor under the tumor necrosis factor (TNF) receptor superfamily that detects B-cell activating factor (BAFF).112,133 This antigen can be expressed by normal B cells and, more excessively, by MM.122,124,125 Based on this concept, BCMA targeted CAR T-cell therapy was developed as a therapeutic option for patients with R/R MM. So far, two CAR T-cell products directed at BCMA, namely idecabtagene vicleucel and ciltacabtagene autoleucel, have been approved by the FDA to treat advanced MM.
Idecabtagene vicleucel
Idecabtagene vicleucel, also named as AbecmaTM or ide-cel, is a BCMA-targeted CAR T-cell therapy. It was first introduced by the Bluebird Bio and approved by the FDA on 26 March 2021 based on the findings from the KarMMa trial. Ide-cel was the first CAR T-cell product used for treating patients suffering from R/R MM after four or more prior lines of therapy, such as a proteasome inhibitor, an immunomodulatory agent, and an anti-CD38 monoclonal antibody.160,161 Patients with R/R MM who were heavily treated using ide-cel showed significantly improved responses, with a 73% ORR and a 33% CR rate. These patients have also shown improved survival, with 8.8 and 19.4 months of PFS and OS, respectively. Side effects such as CRS and neurotoxicity were low, accounting for 5% and 3%, respectively.160,161
Ciltacabtagene autoleucel
On 28 February 2022, another BCMA-directed agent, known as ciltacabtagene autoleucel (cilta-cel or Carvykti™) has been licensed by the FDA as the sixth-approved CAR T-cell therapy. It is the most recently approved CAR T-cell product that was initially announced by the Janssen Pharmaceutical Companies of Johnson & Johnson and Legend Biotech. This product was approved for subsets of patients suffering from R/R MM after four or more prior lines of therapy.162 The approval was based on the findings from phase 1b/2 CARTITUDE-1. The reports from this trial indicated that patients with R/R MM who treated cita-cel had 98% ORR and 80% of CR rate, with PFS of 18 months in 66% of patients.163
Ongoing clinical trials on CAR T-cell therapy
Many years ago, the first CAR T-cell clinical trial began by targeting the folate receptor to treat patients with advanced epithelial ovarian cancer.143 The breakthrough, however, came in the subsequent years with CD19-targeted CAR T-cells used to treat B cell malignancies. The field of CAR-T-cell therapy is then continually developing, with an explosion of over a thousand clinical trials on CAR-T therapies globally to cure advanced cancers that are resistant or refractory to the conventional therapies.174 While some of these clinical trials have shown promising outcomes in cancer patients, many other CAR T-cells targeting various antigens are currently under investigation.175,176 The ongoing clinical trials around CAR T-cell therapy generally cover a wide range of production techniques, antigen targets, and cancer types, with a special focus on a variety of areas, including deeper investigation of current FDA-licensed CAR T-cell products, research into existing or new target antigens, and exploration of CAR T-cell-based combination therapies to improve efficacy and safety.146
Although they have been approved by the FDA and other agencies, current FDA-approved CAR T-cell products are still active areas of experiment in various settings. They are being tested for new indications and evaluated to find ways that improve clinical responses and the safety of patients with different types of malignancies.146 For instance, licensed CD19-targeted CAR T-cell therapy in the treatment of CLL is recently under clinical trials and early reports showed impressive outcomes.177 Besides, in light of the successful results obtained using CAR T-cell therapies targeting CD19 and BCMA in blood cancers in the past few years, a large number of studies have been initiated in search of more effective tumor antigens closer to the ideal targets. Ideal targets are highly specific tumor antigens with extensive tumor coverage and stability to ensure safe and successful use of CAR T-cells in tumor clearance, but it is hard to find them.178 More than half of the current clinical trials employing CAR-T therapy are focusing on hematological malignancies.124 CARs targeting several novel blood cancer antigens such as CD30 in R/R Hodgkin’s lymphoma (HL) and CD33, CD123, and FLT3 in acute myeloid leukemia (AML) are in the pipeline of clinical development.118,179 Other studies have discovered phenomenal target antigens, such as CD20 and CD22 that are abundantly expressed in B cell lineages.181–183 Other ongoing clinical studies are also evaluating the safety and efficacy of CAR T-cell therapy targeting CD5, CD7, and CD4 in R/R T-cell lymphoma and T-cell ALL.183
Apart from blood cancers, advancements in CAR-T-cell therapy have led to their potential use in the treatment of solid tumors. Multiple clinical trials on the use of CAR T-cell therapy in solid cancers by targeting various antigens, including TnMUC1, mesothelin, CEA, ROR1, GD2, PSCA, glypican 3 (GPC3), carbonic anhydrase IX (CAIX), HER2, B7H3, GUCY2C, CD19, and epidermal growth factor (EGFR), are currently underway.184 Solid tumors such as breast cancer, hepatocellular carcinoma, pancreatic cancer, renal cancer, colorectal cancer, prostate cancer, ovarian cancer, lung cancer, and brain cancers are under investigation using existing or novel antigens.185 Despite numerous clinical trials on CAR-T-cell treatment in solid cancers, most of them have been less successful, and none of these targets has yet been approved.
Moreover, clinical trials regarding CAR T-cell therapy combined with other conventional treatments, such as immune checkpoint inhibitors, are now underway to find a cure for blood cancer. Prior findings from clinical studies revealed that concurrent use of these two varieties of immunotherapies is safe in treating various types of blood cancers, such as R/R CLL and R/R NHL.186 Combination therapies have also been demonstrated to have a higher in vivo CAR-T-cell proliferation and exciting response rates for specific hematological malignancies, showing that combination therapy is more efficacious than CAR T-cell therapy alone.118,183,186,187 Despite these early promising results, many more clinical trials on various CAR T-cell-based combination therapies are still in progress to examine their efficacy and safety in cancer patients. Table 4 summarizes the major selected ongoing clinical trials on CAR T-cell therapy for various types of blood and solid malignancies (clinicalTrials.gov).
Table 4.
Some major ongoing clinical trials on CAR T-cell therapy in blood and solid cancers.
| Clinical trial | Target | Cancer type | Phase | Updated result/status |
|---|---|---|---|---|
| Hematological malignancies | ||||
| NCT03277729 | CD20 | R/R B-NHL | Phase 1/2 | High overall and complete response with an extremely favorable safety profile in B-NHL.188 |
| NCT03262298 | CD22 | R/R B-ALL | Phase 1/2 | Dose-dependent antileukemic activity was observed by targeting CD22 in B-ALL.182 |
| NCT02690545, NCT02917083) | CD30 | R/R HL | Phase 1/2 | It shows an ORR of 72% and a CR rate of 59% in R/R HL with an excellent safety profile.118 |
| NCT02203825 | NKG2D | R/R AML | Phase 1 | Targeting NKG2D-Ligands observed to result in high efficacy against AML.189 |
| NCT03971799 | CD33 | R/R AML | phase 1/2 | Recruiting |
| NCT04014881 | CD123 | R/R AML | Phase 1 | Recruiting |
| NCT04010877 | CLL-1/CD123/CD33 | R/R AML | Phase 1/2 | Recruiting |
| NCT05023707 | FLT3 | R/R AML | Phase 1/2 | The potent In vitro antitumor activities of Flt3-CAR T-cells, combined with their low off-target cytotoxicity, offer hope in the treatment of AML.190 |
| NCT02842320 | IL-1RAP | CML | CAR T-cell therapy exhibited cytotoxicity against leukemic cells expressing IL-1RAP.191 | |
| NCT04689659 | CD7 | R/R T- ALL | Phase 1/2 | Phase 1 trial indicated CD7-targeted CAR T-cells result in efficient expansion and achieved a high CR rate with manageable safety profiles.183 |
| NCT02203825 | NKG2D | R/R T-ALL, AML, MDS, MM | Phase 1 | Targeting NKG2D-Ligands shows robust efficacy against T-ALL.189 |
| NCT04351022 | CD38 | R/R AML | Phase 1/2 | 66.7% patient achieved CR with OS of 7.9 months.192 |
| NCT03081910 | CD5 | R/R T-NHL | Phase 1 | CD5 CAR T-cells are safe and can induce clinical responses in heavily treated patients with R/R CD5+ T-NHL.193 |
| NCT04712864 | CD4 | R/R T-cell lymphoma | Phase 1 | CD4CART-cells have potent cytotoxic effects on T-cell lymphoma.194 |
| NCT04594135 | CD5 | T-ALL | Phase 1 | CD5 CAR T-cells eradicate T-ALL blasts In vitro and control disease progression in xenograft mouse models of T-ALL.195 |
| Solid malignancies | ||||
| NCT03500991 | HER2 | CNS tumor | Phase 1 | HER2-specific CAR T-cells is well tolerated and activate a localized immune response in pediatric and young adult patients.196 |
| NCT03618381 | EGFR806 | Advanced solid cancers | Phase 1 | Recruiting |
| NCT03525782 | MUC1/PD-1 | NSCLC | Phase 1/2 | Recruiting |
| NCT03198052 | PSCA/MUC1/TGFβ, HER2/Mesothelin, Lewis-Y/GPC3/AXL/EGFR, Claudin18.2/B7H3 | Lung cancer | Phase 1 | Recruiting |
| NCT04099797 | GD2 | Brain cancer | Phase 1 | Recruiting |
| NCT02932956 | GPC3 | Liver cancer | Phase 1 | Active but not recruiting |
| NCT01583686 | Mesothelin | Ovarian, cervical, pancreatic, lung cancer | Phase 1/2 | Recruiting |
| NCT02349724 | CEA | Lung, colorectal, gastric, breast, pancreatic cancer | Phase 1 | Recruiting |
| NCT04483778 | B7H3, CD19 | Advanced solid tumors | Phase 1 | Recruiting |
| NCT04025216 | TnMUC1 | R/R solid cancers | Phase 1 | Active but not recruiting |
Abbreviations: ALL, Acute lymphoblastic leukemia; AML, Acute Myeloid Leukemia; CEA, carcinoembryonic antigen; CLL-1, C-type lectin-like molecule-1; CML, Chronic Myeloid Leukemia; CNS, central nervous system; EGFR, Epidermal growth factor receptor; GPC3, Glypican-3 IL1RAP, HL, Hodgkin’s Lymphoma; IL1 receptor-associated protein; MDS, myelodysplastic syndrome; MM, multiple myeloma; NHL, non-Hodgkin’s Lymphoma; NKG2D, Natural killer group 2D; NSCLC, Non-small cell lung cancer; PSCA, Prostate stem cell antigen
Challenges of CAR T-cell therapy and the possible solutions
CAR T-cell therapy is a more versatile ACT that can generally generate long-term remission in blood cancer patients and spark excitement among patients and oncologists.117 However, this therapy has inherent limitations as modified T-cells can only detect antigens that are only naturally expressed on the cell surface, narrowing the ranges of potential target antigens. Generally, CAR T-cell therapeutic approach is not as effective as expected due to several challenges. Limited therapeutic efficacy, adverse effects, high cost, and feasibility issues are the main challenges associated with CAR T-cell therapy, hindering it from becoming the first-line treatment and having broader implications.11
Limited CAR T-cell therapeutic efficacy
One of the significant challenges associated with CAR T-cell therapy is its limited therapeutic efficacy, with generally less than a 50% cure rate.197 According to the accumulated body of data, the therapeutic response of patients receiving CAR-T-cell therapy is unpredictable and varies by patient, with some patients getting cure while others do not respond to the treatment.138,198–200 While the outcome of successful treatment is associated with producing multivalent and long-term clinical responses, loss of therapeutic response is caused by treatment resistance (refractory) and relapse of cancer for various reasons.159
CAR-T treatment resistance of cancer may result from the complete loss (called antigen-null cancer cells) or partial loss of target antigen expression by tumor cells (called antigen-dim cancer cells), known as antigen escape.202–205 Antigen escape is thought to be caused by the presentation of a more heterogeneous or challenging target by tumor cells that leads to resistance and/or relapse. This is evident from a relapse associated with loss or down-regulation of CD19 expression in 30–70% of R/R ALL patients treated with CD19-directed CAR-T-cells.198,199,206 Likewise, a complete or partial loss of the BCMA antigen has been observed in patients suffering from advanced MM and under BCMA-directed CAR-T-cell therapy.207–209 Besides, CAR T-cell therapeutic failure could be the outcome of T-cell exhaustion that leads to limited T-cell proliferation, activity, and persistence.138,199,210 Additionally, in vivo tonic signaling, T-cell exhaustion, and poor performance can occur as a result of oligomerization of the ScFv of CARs, such as dibody, tribody, and tetrabody.15
The efficacy of CAR T-cell therapy is more markedly reduced in solid tumors due to antigen escape, inefficient trafficking, or infiltration of CAR-T-cells into the center of the tumor, dense extracellular matrix, and a hostile or immunosuppressive tumor microenvironment characterized by a lack of proinflammatory stimulants, excess inhibitory immune checkpoint molecules, and suppression of CAR T-cell activities. Moreover, it is challenging to specifically direct CAR T-cells against solid cancer cells and spare healthy ones because the majority of the target antigens present on solid are also expressed in low quantities on normal cells.40,76,180,201,211,212,213
In order to overcome the relapse and resistance in cancer patients under CAR-T-cell therapy, a number of novel strategies are being explored in a variety of cancer types. One strategy is combining CAR T-cell therapy with other traditional therapeutic approaches to cure malignancies, which show encouraging results. For instance, using immune checkpoint blockade with monoclonal antibodies (mAbs), such as PD-1, PD-L1, and CTLA-4 antibodies, to restore CAR-T-cell function has been shown to overcome resistance, diminish suppression of antitumor immunity or T-cell exhaustion, and thereby reduce the burden of malignancy.209,213 In addition, inhibition of immunosuppressive cells, such as regulatory T (Treg) cells, tumor-associated macrophages (TAMs), and myeloid-derived suppressor cells (MDSCs), is a helpful strategy to address CAR T-cell exhaustion and enhance the therapeutic efficacy of the therapy.214 Blocking of immunosuppressive factors like TGF-β receptor II (TGFBR2), IL-10, and IL-35 with mAbs or small molecular inhibitors is also important to transform suppressive signals into stimulating signals, reverse the T-cell exhaustion, and improve the efficacy of CAR T-cell therapy in cancer patients such as CLL.215 Moreover, inhibition transcription factors such as Nr4a, TOX, TOX2, and hematopoietic progenitor kinase 1 (HPK1) are another promising strategy to avert T-cell exhaustion and improve immune therapy responses.216 Improving the metabolism of T-cells and modification of gene integration sites also provide a new avenue for reducing T-cell exhaustion and increasing CAR-T-cell efficacy.217
Another novel approach to improve the therapeutic efficacy is reengineering the structure of CAR-T-cells, such as the ectodomain, TMD, and endodomain, to design T-cells with improved clinical impacts, such as efficiency, persistence, infiltration, and anti-apoptosis capacity.218,219 This includes developing CARs with two scFv targeting more than one tumor antigen, which is useful for achieving long-term remission.220 The generation of artificial non-immunoglobulin-based alternatives to ScFv such as engineered binding scaffolds through protein technologies can eliminate T-cell exhaustion, and poor performance as a result of ScFv oligomerization.15 Besides, another potential strategy for dealing with resistance is developing CAR T-cells that are resistant to the hostile tumor milieu.221 Moreover, additional techniques, including the use of innate-like T-cells and the development of new TCR, are also currently under consideration. More recently, the use of NK cells equipped with tumor-targeting CARs is being explored in clinical settings.11,222 However, further long-lasting follow-up studies with a larger sample size are still required to learn more about the clinical impacts of these different approaches to regaining CAR T-cell efficacy.
Adverse effects
Ideally, CAR T-cells are modified to be specific to certain antigens expressed on cancer cells that are not present on healthy cells to ensure safety while curing cancer patients. But in practice, despite its potential to successfully treat certain types of malignancies, it is associated with a number of adverse effects.223 Adverse effects are prevalent enough to be one of the greatest challenges during CAR T-cell therapy due to the nonspecific nature of the CAR T-cells attacking normal cells and resulting in systemic toxicities. The occurrence of side effects generally depends on the overall patient’s health status, the cancer type, location, and severity, the molecular target of the therapy, and the therapeutic dose.224 Overall, CAR T-cell treatment may result in adverse events ranging from flu-like symptoms, like fevers, chills, fatigue, headache, loss of appetite, nausea, vomiting, and diarrhea, to more serious and potentially life-threatening clinical conditions, such as CRS, neurotoxicity, B-cell aplasia, hypogammaglobulinemia, cardiovascular toxicity, hemophagocytic lymphohistiocytosis/macrophage activation syndrome (HLH/MAS), bleeding disorders, and infections.117,126,225,226
Fortunately, most of the adverse clinical syndromes associated with CAR T-cell therapy are safely manageable if they are diagnosed and addressed early. For instance, CRS, which is the most frequent serious adverse effect, can be treated with either tocilizumab or steroids. On the other hand, neurotoxicity can be managed with corticosteroids and HLH/MAS with tocilizumab, corticosteroids, or anti-cytokine therapy.225 Recently, clinical use of anakinra for adjunctive treatment of CRS, ICANS, and/or CAR T-cell-associated HLH/MAS has shown promising outcomes. Anakinra is a recombinant IL-1 receptor antagonist since IL-1 is involved in the development of CRS and ICANS and IL-1 blockade may be useful in preventing and treating these adverse events by suppressing additional inflammatory pathways. Although anakinra is still under prospective clinical trial, early results indicate that its early use reduces the rates of both severe CRS and ICANS, and it appears to be safe and feasible.227,228 According to the Parker et al. report, the overall rate of severe CRS was observed in 6% of patients who received anakinra, while ICANS was seen in 13% of them.228 Currently, several prospective studies evaluating anakinra in adults for prevention or treatment of CRS and ICANS are underway.228,229 Moreover, exogenous immunoglobulin replacement therapy is used to get relief from hypogammaglobulinemia.230 But better toxicity management approaches are still required to lower the occurrence of adverse toxicities and achieve safe CAR T-cell therapy by understanding the possible underlying causes and mechanisms.
CAR-T-cell therapy is still under continuous advancement to search for novel strategies to mitigate side effects by specifically targeting malignant T-cells while sparing healthy cells.11 In recent years, the structure of CAR-T-cells has evolved quickly to reduce systemic toxicity and boost antigen-specific activation. A synthetic control technique for CAR T-cells using a separate drug to regulate T-cells is currently being developed. It is essential to minimize adverse events while parallelly allowing for control of T-cell activity and persistence.6 Another interesting development in this field to enhance the activity and safety of tumor-specific cytotoxic T-cells is switch receptors. Switch receptors are designed by adding a drug-dependent one or more genes that induce apoptosis and CAR depletion upon stimulation by secondary inducing agents, known as ‘off-switch’ or suicide gene strategies. Suicide genes, including herpes simplex virus thymidine kinase and inducible caspase 9 (iCas9), have recently been incorporated into CAR-T-cells to mitigate adverse toxicities.6,231,232 ‘On-switch’ or split CAR techniques, on the other hand, use CAR T-cells possessing CARs split into two distinct receptor proteins that lead to activation and tumor-killing only when the two proteins come into contact and dimerize.233 Alternatively, CAR T-cells are modified to contain switches using bispecific molecules that target both antigens on the surface of tumor cells and CD3 on the surface of T-cells and regulate their activities, decreasing toxicity.234 Engineering CAR cells to have dual-antigen receptors could also be another potential avenue to ameliorate adverse effects by lowering the likelihood of T-cells attacking healthy cells.40
Remote-controlled CAR T-cells, known as SNIP CAR-T, are developed currently to ensure the safety of the therapy as they are activated only when an oral drug is administered. The medication is useful to control the cells’ activity after they have been infused back into the patient. They are modified cells that are tunable for each patient, making them not only safer but also more potent and more versatile than the original CAR T-cells.235 To control CAR T-cell function, permanent degradation of the CAR protein itself rather than the entire cell by targeting with PROteolysis Targeting Chimeras (PROTACs) has also been attempted. This selective destruction of the protein without harming the T-cell is achieved by ubiquitin tagging for proteasomal degradation to decrease on-target off-tumor toxicity.236
High cost and feasibility issues
CAR T-cells are difficult to develop and manufacture cost-effectively. The technique is time-consuming and costly, requiring intensive labor and laboratory expertise. Besides, CAR T-cell therapy is also criticized for its expensive cost.237,238 However, these challenges of CAR-T-cell therapy could be reduced by improving the manufacturing technology and the production of more advanced generations such as uniCAR-T-cells, next generations, and CAR NK cell therapy.11 Moreover, the new strategies for rapid manufacturing approaches such as FasTCAR and T-Charge systems may reduce the production time as well as the cost of the therapy. FasTCAR was first introduced by Gracell Biotechnologies to shorten the production time to just 2 days, hence known as ‘next day manufacturing.’239 It is demonstrated to be feasible with a manageable toxicity profile and has received approval currently by the NMPA in China for adult patients with R/R B-ALL.240 T-Charge is a novel strategy for CAR-T-cell expansion, which has also been developed recently. It is a next-generation platform that involves CAR T-cell expansion within the patient’s body or in-vivo by eliminating the need for an extended culture time outside of the body or ex-vivo. T-Charge maintains T-cells’ ability to self-renew and differentiate, resulting in a product containing more proliferative capacity and fewer exhausted T-cells. Such unique characteristics of the T-Charge platform may revolutionize CAR-T-cell therapy and lead to improved patient outcomes with a reduced risk of severe adverse events. The Novartis T-Charge platform is currently being studied in first-in-human clinical trials.241
Another technology that provides a manufacturing protocol for producing CAR T-cells rapidly at a clinical scale, known as DNA nano-vectors, has also recently been developed. DNA nano-vectors, which are nonviral and nonintegrating small-sized vector platforms capable of replicating extra chromosomally in the nucleus of dividing cells, improve the capacity of generating recombinant human T-cells efficiently.242 Thus, it is a safe, quick, and persistent manufacturing procedure of modified T-cells that may reduce the cost of CAR T-cell therapy. Nonetheless, further technological innovation is required to improve the accessibility of these therapeutic modalities.
Growing autologous types of CAR T-cells in the laboratory can take 2 to 8 weeks, which may not be afforded by the patients as well as creates large variations in the product. It is a highly personalized treatment that does not easily fit into standard oncological practice and results in another big challenge in the application of CAR T-cell therapy. The quality of the CAR T-cells created from autologous patient T-cells might also vary greatly, making it difficult to compare dose and efficacy. This challenge can partly be overcome by reconsidering other T-cell sources, such as allogeneic CAR T-cells that could be manufactured in advance from highly potent T-cell sources, such as healthy donors and induced pluripotent stem cells (iPSCs), which will then be administered off the shelf.243 Using iPSCs is found to be a limitless source of off-the-shelf CAR T-cells that help to treat patients in a timelier fashion.244 This means CAR T-cells can be produced in bulk and can be stored in a biobank for prompt utilization to treat patients. Cryopreservation using cryoprotective agents is an effective method of preserving cells to remain viable and functional until shipped to clinics for infusion into patients. The most common cryo-storing agent for CAR T-cells is DMSO, which is a cell membrane–permeating cryoprotective agent that helps the cells survive the stresses of freezing and thawing. However, DMSO may harm organelles, cytoskeletons, and cell membranes, thus negatively affecting cell viability and functionality, with roughly 50% of cells undergoing thawing and cryopreservation, with DMSO remaining viable.245 While using fresh cells may be favorable and contribute to improved clinical outcomes, it can also increase the complexity and cost of manufacturing and logistics. Hence, DMSO-based cryostorage is essential to make cell manufacturing and therapy more efficient.246 Biobanking allogeneic CAR T products reduce the time it takes to make autologous CAR T-cells and allow patients to receive their therapy immediately rather than waiting for weeks. Additionally, allogeneic CAR cells can be produced on a larger industrial scale, resulting in a more affordable production pipeline and becoming more available for larger numbers of patients.247 This offshore production also permits patients to be treated simultaneously with multiple CAR T-cells that target several antigens, prevent tumor resistance, and improve efficacy.107 Moreover, biobanking of allogeneic CAR T-cell products is essential to ensure potent and predictable anti-tumor activity in all patients. But allogeneic CAR T-cells may induce an autoimmune reaction that risks the patient to potentially fatal graft-versus-host disease (GvHD) or kills off the transferred T-cells before they have brought the desired effect.248 In this regard, however, the autologous CAR T-cells are preferred to minimize the patient’s risk of immune reactions to the cells, and thus currently available CAR T-cell therapies autologous types that are customized for each patient.
Discussion
The field of CAR-T-cell therapy has significantly improved since its first discovery three decades ago by reprogramming T-cells with a synthetic CAR receptor to the most recent and advanced CAR T-cell therapy. It has become a breakthrough in contemporary medicine that acts as a game changer to revolutionize the treatment of advanced-stage malignancies. Over the past years, CAR-T-cell therapy has gone through a series of steps in its development in an attempt to improve therapeutic effects and reduce potential adverse effects. It has progressed through five generations since its inception to the most recent and advanced fifth generation, with great improvement in T-cell activation, growth, and perseverance. Despite these advances, innovative CAR designs in recent generations have only been demonstrated in vitro or in the stage of preclinical animal model studies, with no confirmed clinical or patient data. In addition, there is no currently available clinical study that clearly compares different generations of CARs, making it difficult to determine which CAR design will deliver the most effective clinical benefit for patients and warranting clinical studies to fill these knowledge gaps.
Despite the fact that so many clinical trials around CAR T-cell therapy have been ongoing for the last few years, only some of them have shown promising outcomes, with just six of them having received FDA approval for hematological cancer and none having been licensed for therapeutic use in solid tumors. Approved CAR-T therapies are either CD19 or BCMA-targeting and have a high overall response rate and long-lasting remissions in patients with aggressive malignancies. As studies regarding CAR T-cell therapy proceed, many of the cancer community have increasing optimism for the therapy’s future. Multiple clinical studies have shown remarkable results from cancer patients treated with CAR T-cell treatment, raising hopes for a new approval soon. Although CAR T-cell therapy is showing an exciting outcome in cancer, less than half of individuals who have been treated have only been cured and have long-term survival. The success rate of CAR T-cell therapy is reduced due to the relapsing and refractory nature of cancer, with solid cancer posing a greater challenge. The mechanisms underlying the resistance and relapses in cancer patients treated with CAR-T-cells are yet elusive, pending more research to learn more. CAR T-cell technology is also not criticized regarding its adverse effects and cost, prompting a search for a safer CAR T-cell treatment at a reasonable cost.
Concluding remarks
In summary, CAR T-cell therapy, which is manufactured by ex vivo genetic modification and proliferation of T-cells, is a personalized therapeutic technology in managing cancer patients. It is considered a living, self-replicating immune-boosting drug that destroys cancer cells. CAR-T-cell therapies have evolved significantly in recent years, progressing from the earliest first generation to the most advanced fifth generation in an attempt to improve safety and efficacy.
Numerous clinical studies regarding CAR T-cell therapy are now underway, and several of them have shown incredible success in treating cancer patients. Although CAR T-cell therapy has demonstrated impressive clinical outcomes in some advanced hematological malignancies, only few of them have recently received approval for treating blood cancer. But approvals around CAR T-cell therapy are rapidly changing, and several other CAR T-cell products are expected to be licensed in the near future. However, several hurdles, such as limited efficacy, adverse effects, greater wait times for treatment, and the expensive price of the therapy, are prohibiting it from becoming the first-line treatment and being utilized broadly in cancer patients. This warrants further research to overcome these hurdles, improve the anti-tumor activity, and reduce the adverse effects of CAR T-cell therapy, which may offer novel insights into the success and future advances of this therapeutic technology.
Funding Statement
The author(s) reported that there is no funding associated with the work featured in this article.
Abbreviations
| ACT | Adoptive cell therapy |
| AML | acute myeloid leukemia |
| BAFF | B-cell activating factor |
| B-cell-ALL | B cell acute lymphoblastic leukemia |
| BCMA | B cell maturation antigen |
| CAR | Chimeric antigen receptor |
| CART-19 | CD19-targeted CAR T-cell therapies |
| CLL | chronic lymphocytic leukemia |
| CM | costimulatory molecule |
| CRS | cytokine release syndrome |
| DLBCL | diffuse large B-cell lymphoma |
| FasL FDA | Food and Drug Administration |
| FL | follicular lymphoma |
| FL, HGBCL | high-grade B cell lymphoma |
| HL | Hodgkin lymphoma |
| HLA | human leukocyte antigen |
| HLH/MAS | hemophagocytic lymphohistiocytosis/macrophage activation syndrome |
| ICOS | inducible T-cell co-stimulator |
| ITAMs | immunoreceptor tyrosine-based activation motifs |
| JAK | Janus kinase |
| MCL | mantle cell lymphoma |
| MM | multiple myeloma |
| NFAT | nuclear factor of the activated T-cell |
| scFv | single chain variable fragment |
| STAT | Signal transducer and activator of transcription |
| TCR | T-cell receptor |
| TMD | transmembrane domain |
| TRAIL | TNF-related apoptosis-inducing ligand |
| TRUCK | T-cell redirected for universal cytokine-mediated killing |
Author contributions
All authors made a significant contribution to the work reported, whether that is in the conception, study design, execution, acquisition of data, analysis and interpretation, or in all these areas; took part in drafting, revising, or critically reviewing the article; gave final approval of the version to be published; have agreed on the journal to which the article has been submitted; and agreed to be accountable for all aspects of the work.
Disclosure statement
No potential conflict of interest was reported by the author(s).
References
- 1.Perica K, Varela JC, Oelke M, Schneck J.. Adoptive T-cell immunotherapy for cancer. Rambam Maimonides Med J. 2015;6(1):1. doi: 10.5041/RMMJ.10179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Pardoll D. Does the immune system see tumors as foreign or self? Annu Rev Immunol. 2003;21(1):807–25. doi: 10.1146/annurev.immunol.21.120601.141135. [DOI] [PubMed] [Google Scholar]
- 3.Park JH, Brentjens RJ. Adoptive immunotherapy for B-cell malignancies with autologous chimeric antigen receptor modified tumor targeted T-cells. Discov Med. 2010;9:277. [PMC free article] [PubMed] [Google Scholar]
- 4.Robbins PF, Kawakami Y. Human tumor antigens recognized by T-cells. Curr Opin Immunol. 1996;8(5):628–636. doi: 10.1016/S0952-7915(96)80078-1. [DOI] [PubMed] [Google Scholar]
- 5.Strati P, Neelapu SS. Chimeric antigen receptor–engineered T-cell therapy in lymphoma. Curr Oncol Rep. 2019;21(5):1–7. doi: 10.1007/s11912-019-0789-z. [DOI] [PubMed] [Google Scholar]
- 6.Zhang E, Xu H. A new insight in chimeric antigen receptor-engineered T-cells for cancer immunotherapy. J Hematol Oncol. 2017;10(1):1–11. doi: 10.1186/s13045-016-0379-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sommermeyer D, Hudecek M, Kosasih PL, Gogishvili T, Maloney DG, Turtle CJ, Riddell SR. Chimeric antigen receptor-modified T cells derived from defined CD8+ and CD4+ subsets confer superior antitumor reactivity in vivo. Leukemia. 2016;30(2):492–500. doi: 10.1038/leu.2015.247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kuwana Y, Asakura Y, Utsunomiya N, Nakanishi M, Arata Y, Itoh S, Nagase F, Kurosawa Y. Expression of chimeric receptor composed of immunoglobulin-derived V regions and T-cell receptor-derived C regions. Biochem Biophys Res Commun. 1987;149(3):960–968. doi: 10.1016/0006-291X(87)90502-X. [DOI] [PubMed] [Google Scholar]
- 9.Gross G, Gorochov G, Waks T, Eshhar Z, editors. Generation of effector T-cells expressing chimeric T-cell receptor with antibody type-specificity. Transplant Proc. Israel. 1989;21(1 Pt 1):127–130. [PubMed] [Google Scholar]
- 10.Gross G, Waks T, Eshhar Z. Expression of immunoglobulin-T-cell receptor chimeric molecules as functional receptors with antibody-type specificity. Proc Nat Acad Sci. 1989;86(24):10024–10028. doi: 10.1073/pnas.86.24.10024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Huang R, Li X, He Y, Zhu W, Gao L, Liu Y, Gao L, Wen Q, Zhong JF, Zhang C, et al. Recent advances in CAR-T-cell engineering. J Hematol Oncol. 2020;13(1):1–19. doi: 10.1186/s13045-020-00910-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Dotti G, Gottschalk S, Savoldo B, Brenner MK. Design and development of therapies using chimeric antigen receptor‐expressing T-cells. Immunol Rev. 2014;257(1):107–126. doi: 10.1111/imr.12131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Guedan S, Calderon H, Posey JA, Maus MV. Engineering and design of chimeric antigen receptors. Mol Ther-Met Clin Dev. 2019;12:145–156. doi: 10.1016/j.omtm.2018.12.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Jayaraman J, Mellody MP, Hou AJ, Desai RP, Fung AW, Pham AHT, Chen YY, Zhao W. Car-T design: elements and their synergistic function. EBioMedicine. 2020;58:102931. doi: 10.1016/j.ebiom.2020.102931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zajc CU, Salzer B, Taft JM, Reddy ST, Lehner M, Traxlmayr MW. Driving CARs with alternative navigation tools–the potential of engineered binding scaffolds. Febs J. 2021;288(7):2103–2118. doi: 10.1111/febs.15523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Whilding LM, Parente-Pereira AC, Zabinski T, Davies DM, Petrovic RM, Kao YV, Saxena SA, Romain A, Costa-Guerra JA, Violette S, et al. Targeting of aberrant αvβ6 integrin expression in solid tumors using chimeric antigen receptor-engineered T Cells. Mol Ther. 2017;25(1):259–273. doi: 10.1016/j.ymthe.2016.10.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zhang T, Barber A, Sentman CL. Generation of antitumor responses by genetic modification of primary human T-cells with a chimeric NKG2D receptor. Cancer Res. 2006;66(11):5927–5933. doi: 10.1158/0008-5472.CAN-06-0130. [DOI] [PubMed] [Google Scholar]
- 18.Hudecek M, Sommermeyer D, Kosasih PL, Silva-Benedict A, Liu L, Rader C, Jensen MC, Riddell SR. The nonsignaling extracellular spacer domain of chimeric antigen receptors is decisive for in vivo antitumor activity. Cancer Immunol Res. 2015;3(2):125–135. doi: 10.1158/2326-6066.CIR-14-0127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Guest RD, Hawkins RE, Kirillova N, Cheadle EJ, Arnold J, O’Neill A, Irlam J, Chester KA, Kemshead JT, Shaw DM. The role of extracellular spacer regions in the optimal design of chimeric immune receptors: evaluation of four different scFvs and antigens. J Immunother. 2005;28(3):203–211. doi: 10.1097/01.cji.0000161397.96582.59. [DOI] [PubMed] [Google Scholar]
- 20.James SE, Greenberg PD, Jensen MC, Lin Y, Wang J, Till BG, Raubitschek AA, Forman SJ, Press OW. Antigen sensitivity of CD22-specific chimeric TCR is modulated by target epitope distance from the cell membrane. J Immunol. 2008;180(10):7028–7038. doi: 10.4049/jimmunol.180.10.7028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wilkie S, Picco G, Foster J, Davies DM, Julien S, Cooper L, Arif S, Mather SJ, Taylor-Papadimitriou J, Burchell JM, et al. Retargeting of human T-cells to tumor-associated MUC1: the evolution of a chimeric antigen receptor. J Immunol. 2008;180(7):4901–4909. doi: 10.4049/jimmunol.180.7.4901. [DOI] [PubMed] [Google Scholar]
- 22.Zhang C, Liu J, Zhong JF, Zhang X. Engineering car-T-cells. Biomarker Res. 2017;5(1):1–6. doi: 10.1186/s40364-017-0081-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Elazar A, Chandler NJ, Davey AS, Weinstein JY, Nguyen JV, Trenker R, Cross RS, Jenkins MR, Call MJ, Call ME. De novo-designed transmembrane domains tune engineered receptor functions. Elife. 2022;11:e75660. doi: 10.7554/eLife.75660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Chandran SS, Klebanoff CA. T-Cell receptor‐based cancer immunotherapy: emerging efficacy and pathways of resistance. Immunol Rev. 2019;290(1):127–147. doi: 10.1111/imr.12772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Salter AI, Rajan A, Kennedy JJ, Ivey RG, Shelby SA, Leung I, Templeton ML, Muhunthan V, Voillet V, Sommermeyer D. Comparative analysis of TCR and CAR signaling informs CAR designs with superior antigen sensitivity and in vivo function. Sci Signal. 2021;14(697):eabe2606. doi: 10.1126/scisignal.abe2606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wu L, Wei Q, Brzostek J, Gascoigne NR. Signaling from T-cell receptors (TCRs) and chimeric antigen receptors (CARs) on T-cells. Cell Mol Immunol. 2020;17(6):600–612. doi: 10.1038/s41423-020-0470-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Rudolph MG, Stanfield RL, Wilson IA. How TCRs bind MHCs, peptides, and coreceptors. Annu Rev Immunol. 2006;24(1):419–466. doi: 10.1146/annurev.immunol.23.021704.115658. [DOI] [PubMed] [Google Scholar]
- 28.Becker ML, Near R, Mudgett-Hunter M, Margolies MN, Kubo RT, Kaye J, Hedrick SM. Expression of a hybrid immunoglobulin-T-cell receptor protein in transgenic mice. Cell. 1989;58(5):911–921. doi: 10.1016/0092-8674(89)90943-4. [DOI] [PubMed] [Google Scholar]
- 29.Hogquist KA, Jameson SC. The self-obsession of T-cells: how TCR signaling thresholds affect fate ’decisions’ and effector function. Nat Immunol. 2014;15(9):815–823. doi: 10.1038/ni.2938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hudecek M, Lupo-Stanghellini M-T, Kosasih PL, Sommermeyer D, Jensen MC, Rader C, Riddell SR. Receptor affinity and extracellular domain modifications affect tumor recognition by ROR1-specific chimeric antigen receptor T-cells tumor recognition by ROR1-specific CARs. Clin Cancer Res. 2013;19(12):3153–3164. doi: 10.1158/1078-0432.CCR-13-0330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Huang J, Brameshuber M, Zeng X, Xie J, Li Q-J, Chien Y-H, Valitutti S, Davis M. A single peptide-major histocompatibility complex ligand triggers digital cytokine secretion in CD4+ T-cells. Immunity. 2013;39(5):846–857. doi: 10.1016/j.immuni.2013.08.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Watanabe K, Terakura S, Martens AC, Van Meerten T, Uchiyama S, Imai M, Sakemura R, Goto T, Hanajiri R, Imahashi N, et al. Target antigen density governs the efficacy of anti–CD20-CD28-CD3 ζ chimeric antigen receptor–modified effector CD8 + T cells. J Immunol. 2015;194(3):911–920. doi: 10.4049/jimmunol.1402346. [DOI] [PubMed] [Google Scholar]
- 33.Mansilla-Soto J, Eyquem J, Haubner S, Hamieh M, Feucht J, Paillon N, Zucchetti AE, Li Z, Sjöstrand M, Lindenbergh PL, et al. HLA-Independent T-cell receptors for targeting tumors with low antigen density. Nat Med. 2022;28(2):345–352. doi: 10.1038/s41591-021-01621-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Brameshuber M, Kellner F, Rossboth BK, Ta H, Alge K, Sevcsik E, Göhring J, Axmann M, Baumgart F, Gascoigne NRJ, et al. Monomeric TCRs drive T-cell antigen recognition. Nat Immunol. 2018;19(5):487–496. doi: 10.1038/s41590-018-0092-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Chang ZL, Lorenzini MH, Chen X, Tran U, Bangayan NJ, Chen YY. Rewiring T-cell responses to soluble factors with chimeric antigen receptors. Nat Chem Biol. 2018;14(3):317–324. doi: 10.1038/nchembio.2565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Davenport A, Cross R, Watson K, Liao Y, Shi W, Prince H, Beavis PA, Trapani JA, Kershaw MH, Ritchie DS, et al. Chimeric antigen receptor T-cells form nonclassical and potent immune synapses driving rapid cytotoxicity. Proc Nat Acad Sci. 2018;115(9):E2068–E76. doi: 10.1073/pnas.1716266115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Gudipati V, Rydzek J, Doel-Perez I, Gonçalves VDR, Scharf L, Königsberger S, Lobner E, Kunert R, Einsele H, Stockinger H. Inefficient CAR-proximal signaling blunts antigen sensitivity. Nat Immunol. 2020;21(8):848–856. doi: 10.1038/s41590-020-0719-0. [DOI] [PubMed] [Google Scholar]
- 38.Brocker T. Chimeric Fv-ζ or Fv-ε receptors are not sufficient to induce activation or cytokine production in peripheral T-cells. Blood. 2000;96:1999–2001. [PubMed] [Google Scholar]
- 39.Weinkove R, George P, Dasyam N, McLellan AD. Selecting costimulatory domains for chimeric antigen receptors: functional and clinical considerations. Clin Transl Immunol. 2019;8(5):e1049. doi: 10.1002/cti2.1049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Srivastava S, Riddell SR. Engineering CAR-T-cells: design concepts. Trends Immunol. 2015;36(8):494–502. doi: 10.1016/j.it.2015.06.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Eshhar Z, Waks T, Gross G, Schindler DG. Specific activation and targeting of cytotoxic lymphocytes through chimeric single chains consisting of antibody-binding domains and the gamma or zeta subunits of the immunoglobulin and T-cell receptors. Proc Nat Acad Sci. 1993;90(2):720–724. doi: 10.1073/pnas.90.2.720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Firor AE, Jares A, Ma Y. From humble beginnings to success in the clinic: chimeric antigen receptor-modified T-cells and implications for immunotherapy. Exp Biol Med. 2015;240(8):1087–1098. doi: 10.1177/1535370215584936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Smith AJ, Oertle J, Warren D, Prato D. Chimeric antigen receptor (CAR) T-cell therapy for malignant cancers: summary and perspective. J Cell Immunother. 2016;2(2):59–68. doi: 10.1016/j.jocit.2016.08.001. [DOI] [Google Scholar]
- 44.Imai C, Mihara K, Andreansky M, Nicholson I, Pui C, Geiger T, Campana D. Chimeric receptors with 4-1BB signaling capacity provoke potent cytotoxicity against acute lymphoblastic leukemia. Leukemia. 2004;18(4):676–684. doi: 10.1038/sj.leu.2403302. [DOI] [PubMed] [Google Scholar]
- 45.Maher J, Brentjens RJ, Gunset G, Rivière I, Sadelain M. Human T-lymphocyte cytotoxicity and proliferation directed by a single chimeric TCRζ/CD28 receptor. Nat Biotechnol. 2002;20(1):70–75. doi: 10.1038/nbt0102-70. [DOI] [PubMed] [Google Scholar]
- 46.Finney HM, Lawson AD, Bebbington CR, Weir ANC. Chimeric receptors providing both primary and costimulatory signaling in T-cells from a single gene product. J Immunol. 1998;161:2791–2797. [PubMed] [Google Scholar]
- 47.Acuto O, Michel F. CD28-Mediated co-stimulation: a quantitative support for TCR signalling. Nat Rev Immunol. 2003;3(12):939–951. doi: 10.1038/nri1248. [DOI] [PubMed] [Google Scholar]
- 48.Hombach A, Wieczarkowiecz A, Marquardt T, Heuser C, Usai L, Pohl C, Seliger B, Abken H. Tumor-specific T cell activation by recombinant immunoreceptors: cD3ζ signaling and CD28 costimulation are simultaneously required for efficient IL-2 secretion and can be integrated into one combined CD28/CD3ζ signaling receptor molecule. J Immunol. 2001;167(11):6123–6131. doi: 10.4049/jimmunol.167.11.6123. [DOI] [PubMed] [Google Scholar]
- 49.Jenkins M, Burrell E, Ashwell JD. Antigen presentation by resting B cells. Effectiveness at inducing T-cell proliferation is determined by costimulatory signals, not T-cell receptor occupancy. J Immunol. 1990;144:1585–1590. [PubMed] [Google Scholar]
- 50.Krause A, Guo H-F, Latouche J-B, Tan C, Cheung N-K, Sadelain M. Antigen-Dependent CD28 signaling selectively enhances survival and proliferation in genetically modified activated human primary T lymphocytes. J Exp Med. 1998;188(4):619–626. doi: 10.1084/jem.188.4.619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Finney HM, Akbar AN, Lawson AD. Activation of resting human primary T-cells with chimeric receptors: costimulation from CD28, inducible costimulator, CD134, and CD137 in series with signals from the TCRζ chain. J Immunol. 2004;172(1):104–113. doi: 10.4049/jimmunol.172.1.104. [DOI] [PubMed] [Google Scholar]
- 52.Lu P, Lu X-A, Zhang X, Xiong M, Zhang J, Zhou X, Qi F, Yang J, He T. Which is better in CD19 CAR-T treatment of r/r B-ALL, CD28 or 4-1BB? A parallel trial under the same manufacturing process. Am Soc Clin Oncol. 2018;36(15):3041–3041 [Google Scholar]
- 53.Kawalekar OU, O’Connor RS, Fraietta JA, Guo L, McGettigan SE, Posey JA, Patel PR, Guedan S, Scholler J, Keith B, et al. Distinct signaling of coreceptors regulates specific metabolism pathways and impacts memory development in CAR T-cells. Immunity. 2016;44(2):380–390. doi: 10.1016/j.immuni.2016.01.021. [DOI] [PubMed] [Google Scholar]
- 54.Long AH, Haso WM, Shern JF, Wanhainen KM, Murgai M, Ingaramo M, Smith JP, Walker AJ, Kohler ME, Venkateshwara VR, et al. 4-1BB costimulation ameliorates T-cell exhaustion induced by tonic signaling of chimeric antigen receptors. Nat Med. 2015;21(6):581–590. doi: 10.1038/nm.3838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Quintarelli C, Orlando D, Boffa I, Guercio M, Polito VA, Petretto A, Lavarello C, Sinibaldi M, Weber G, Del Bufalo F, et al. Choice of costimulatory domains and of cytokines determines CAR T-cell activity in neuroblastoma. Oncoimmunology. 2018;7(6):e1433518. doi: 10.1080/2162402X.2018.1433518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Ramos CA, Rouce R, Robertson CS, Reyna A, Narala N, Vyas G, Mehta B, Zhang H, Dakhova O, Carrum G, et al. In vivo fate and activity of second- versus third-generation CD19-specific CAR-T cells in B cell non-Hodgkin’s lymphomas. Mol Ther. 2018;26(12):2727–2737. doi: 10.1016/j.ymthe.2018.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Doherty K. Obe-Cel may mark additional treatment option for R/R B-ALL. 2022.
- 58.Abate-Daga D, Davila ML. Car models: next-generation car modifications for enhanced T-cell function. Mol Ther-Oncolytics. 2016;3:16014. doi: 10.1038/mto.2016.14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Eggermont LJ, Paulis LE, Tel J, Figdor CG. Towards efficient cancer immunotherapy: advances in developing artificial antigen-presenting cells. Trends Biotechnol. 2014;32(9):456–465. doi: 10.1016/j.tibtech.2014.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Sadelain M, Brentjens R, Rivière I. The basic principles of chimeric antigen receptor design. Cancer Discov. 2013;3(4):388–398. doi: 10.1158/2159-8290.CD-12-0548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Song D-G, Ye Q, Santoro S, Fang C, Best A, Powell JD. Chimeric NKG2D CAR-expressing T-cell-mediated attack of human ovarian cancer is enhanced by histone deacetylase inhibition. Hum Gene Ther. 2013;24(3):295–305. doi: 10.1089/hum.2012.143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Song D-G, Ye Q, Poussin M, Harms GM, Figini M, Powell JD. CD27 costimulation augments the survival and antitumor activity of redirected human T-cells In vivo. Blood. 2012;119:696–706. [DOI] [PubMed] [Google Scholar]
- 63.Schubert M-L, Schmitt A, Neuber B, Hückelhoven-Krauss A, Kunz A, Wang L, Gern U, Michels B, Sellner L, Hofmann S, et al. Third-generation CAR T-cells targeting CD19 are associated with an excellent safety profile and might improve persistence of CAR T-cells in treated patients. Blood. 2019;134:51. doi: 10.1182/blood-2019-125423. [DOI] [Google Scholar]
- 64.Künkele A, Johnson AJ, Rolczynski LS, Chang CA, Hoglund V, Kelly-Spratt KS, Jensen MC. Functional tuning of CARs reveals signaling threshold above which CD8+ CTL antitumor potency is attenuated due to cell Fas–FasL-dependent AICD. Cancer Immunol Res. 2015;3(4):368–379. doi: 10.1158/2326-6066.CIR-14-0200. [DOI] [PubMed] [Google Scholar]
- 65.Hombach AA, Rappl G, Abken H. Arming cytokine-induced killer cells with chimeric antigen receptors: cD28 outperforms combined CD28–OX40 “super-stimulation”. Mol Ther. 2013;21(12):2268–2277. doi: 10.1038/mt.2013.192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Stephan MT, Ponomarev V, Brentjens RJ, Chang AH, Dobrenkov KV, Heller G, Sadelain M. T cell–encoded cd80 and 4-1bbl induce auto- and transcostimulation, resulting in potent tumor rejection. Nat Med. 2007;13(12):1440–1449. doi: 10.1038/nm1676. [DOI] [PubMed] [Google Scholar]
- 67.Pegram HJ, Lee JC, Hayman EG, Imperato GH, Tedder TF, Sadelain M, Brentjens RJ. Tumor-Targeted T-cells modified to secrete IL-12 eradicate systemic tumors without need for prior conditioning. Blood. 2012;119:4133–4141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Kueberuwa G, Kalaitsidou M, Cheadle E, Hawkins RE, Gilham DE. CD19 CAR T-cells expressing IL-12 eradicate lymphoma in fully lymphoreplete mice through induction of host immunity. Mol Ther-Oncolytics. 2018;8:41–51. doi: 10.1016/j.omto.2017.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Chmielewski M, Abken H. Trucks: the fourth generation of CARs. Expert Opin Biol Ther. 2015;15(8):1145–1154. doi: 10.1517/14712598.2015.1046430. [DOI] [PubMed] [Google Scholar]
- 70.Martínez-Lostao L, Anel A, Pardo J. How do cytotoxic lymphocytes kill cancer cells? Clin Cancer Res. 2015;21(22):5047–5056. doi: 10.1158/1078-0432.CCR-15-0685. [DOI] [PubMed] [Google Scholar]
- 71.Li J, Li W, Huang K, Zhang Y, Kupfer G, Zhao Q. Chimeric antigen receptor T-cell (CAR-T) immunotherapy for solid tumors: lessons learned and strategies for moving forward. J Hematol Oncol. 2018;11(1):1–18. doi: 10.1186/s13045-018-0568-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Tokarew N, Ogonek J, Endres S, von Bergwelt-Baildon M, Kobold S. Teaching an old dog new tricks: next-generation CAR T-cells. Br J Cancer. 2019;120(1):26–37. doi: 10.1038/s41416-018-0325-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Kagoya Y, Tanaka S, Guo T, Anczurowski M, Wang C-H, Saso K, Butler MO, Minden MD, Hirano N. A novel chimeric antigen receptor containing a jak–stat signaling domain mediates superior antitumor effects. Nat Med. 2018;24(3):352–359. doi: 10.1038/nm.4478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Lyman GH, Nguyen A, Snyder S, Gitlin M, Chung KC. Economic evaluation of chimeric antigen receptor T-cell therapy by site of care among patients with relapsed or refractory large B-cell lymphoma. JAMA network open. 2020;3(4):e202072-e. doi: 10.1001/jamanetworkopen.2020.2072 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Brentjens RJ, Santos E, Nikhamin Y, Yeh R, Matsushita M, La Perle K, Quintás-Cardama A, Larson SM, Sadelain M. Genetically targeted T-cells eradicate systemic acute lymphoblastic leukemia xenografts. Clin Cancer Res. 2007;13(18):5426–5435. doi: 10.1158/1078-0432.CCR-07-0674. [DOI] [PubMed] [Google Scholar]
- 76.Lim WA, June CH. The principles of engineering immune cells to treat cancer. Cell. 2017;168(4):724–740. doi: 10.1016/j.cell.2017.01.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Moreno C, Haynie C, Johnson A, Weber KS. Alternative CAR Therapies: recent Approaches in Engineering Chimeric Antigen Receptor Immune Cells to Combat Cancer. Biomedicines. 2022;10(7):1493. doi: 10.3390/biomedicines10071493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Savanur MA, Weinstein-Marom H, Gross G. Implementing logic gates for safer immunotherapy of cancer. Front Immunol. 2021;12:4678. doi: 10.3389/fimmu.2021.780399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Lanitis E, Poussin M, Klattenhoff AW, Song D, Sandaltzopoulos R, June CH, Powell DJ. Chimeric Antigen Receptor T Cells with Dissociated Signaling Domains Exhibit Focused Antitumor Activity with Reduced Potential for Toxicity In vivo. Cancer Immunol Res. 2013;1(1):43–53. doi: 10.1158/2326-6066.CIR-13-0008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Kloss CC, Condomines M, Cartellieri M, Bachmann M, Sadelain M. Combinatorial antigen recognition with balanced signaling promotes selective tumor eradication by engineered T-cells. Nat Biotechnol. 2013;31(1):71–75. doi: 10.1038/nbt.2459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Qin H, Ramakrishna S, Nguyen S, Fountaine TJ, Ponduri A, Stetler-Stevenson M, Yuan CM, Haso W, Shern JF, Shah NN, et al. Preclinical development of bivalent chimeric antigen receptors targeting both CD19 and CD22. Mol Ther-Oncolytics. 2018;11:127–137. doi: 10.1016/j.omto.2018.10.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Grada Z, Hegde M, Byrd T, Shaffer DR, Ghazi A, Brawley VS, Corder A, Schönfeld K, Koch J, Dotti G, et al. TanCAR: a novel bispecific chimeric antigen receptor for cancer immunotherapy. Mol Ther-Nucleic Acids. 2013;2:e105. doi: 10.1038/mtna.2013.32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Fedorov VD, Themeli M, Sadelain M. PD-1–and CTLA-4–based inhibitory chimeric antigen receptors (iCars) divert off-target immunotherapy responses. Sci Transl Med. 2013;5(215):215ra172. doi: 10.1126/scitranslmed.3006597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Haubner S, Mansilla-Soto J, Nataraj S, He X, Park JH, Wang X, Rivière I, Sadelain M. IF-Better Gating: combinatorial Targeting and Synergistic Signaling for Enhanced CAR T-cell Efficacy. Blood. 2021;138(Supplement 1):2774. doi: 10.1182/blood-2021-149263. [DOI] [Google Scholar]
- 85.Cho JH, Collins JJ, Wong WW. Universal chimeric antigen receptors for multiplexed and logical control of T-cell responses. Cell. 2018;173(6):1426–38. e11. doi: 10.1016/j.cell.2018.03.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Cho JH, Okuma A, Sofjan K, Lee S, Collins JJ, Wong WW. Engineering advanced logic and distributed computing in human CAR immune cells. Nat Commun. 2021;12(1):1–14. doi: 10.1038/s41467-021-21078-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Roybal KT, Rupp LJ, Morsut L, Walker WJ, McNally KA, Park JS, Lim W. Precision tumor recognition by T-cells with combinatorial antigen-sensing circuits. Cell. 2016;164(4):770–779. doi: 10.1016/j.cell.2016.01.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Morsut L, Roybal KT, Xiong X, Gordley RM, Coyle SM, Thomson M, Lim WA. Engineering customized cell sensing and response behaviors using synthetic notch receptors. Cell. 2016;164(4):780–791. doi: 10.1016/j.cell.2016.01.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Choe JH, Watchmaker PB, Simic MS, Gilbert RD, Li AW, Krasnow NA, Downey KM, Yu W, Carrera DA, Celli A, et al. SynNotch-CAR T-cells overcome challenges of specificity, heterogeneity, and persistence in treating glioblastoma. Sci Transl Med. 2021;13(591):eabe7378. doi: 10.1126/scitranslmed.abe7378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Feldmann A, Hoffmann A, Bergmann R, Koristka S, Berndt N, Arndt C, Rodrigues Loureiro L, Kittel-Boselli E, Mitwasi N, Kegler A, et al. Versatile chimeric antigen receptor platform for controllable and combinatorial T-cell therapy. Oncoimmunology. 2020;9(1):1785608. doi: 10.1080/2162402X.2020.1785608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Feldmann A, Hoffmann A, Kittel-Boselli E, Bergmann R, Koristka S, Berndt N, Arndt C, Loureiro LR, Mitwasi N, Jureczek J, et al. A Novel Revcar Platform for Switchable and Gated Tumor Targeting. Blood. 2019;134:5611. doi: 10.1182/blood-2019-128436. [DOI] [Google Scholar]
- 92.Salzer B, Schueller CM, Zajc CU, Peters T, Schoeber MA, Kovacic B, Buri MC, Lobner E, Dushek O, Huppa JB, et al. Engineering AvidCars for combinatorial antigen recognition and reversible control of CAR function. Nat Commun. 2020;11(1):1–16. doi: 10.1038/s41467-020-17970-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Liu Y, Liu G, Wang J, Zheng Z-Y, Jia L, Rui W, Huang D, Zhou Z-X, Zhou L, Wu X, et al. Chimeric STAR receptors using TCR machinery mediate robust responses against solid tumors. Sci Transl Med. 2021;13(586):eabb5191. doi: 10.1126/scitranslmed.abb5191. [DOI] [PubMed] [Google Scholar]
- 94.Wang J, Zhang X, Zhou Z, Liu Y, Yu L, Jia L, Yang J, Li J, Yu H, Li W, et al. A novel adoptive synthetic tcr and antigen receptor (star) T-Cell therapy for B-Cell acute lymphoblastic leukemia. Am J Hematol. 2022;97(8):992–1004. doi: 10.1002/ajh.26586. [DOI] [PubMed] [Google Scholar]
- 95.Yee C. The use of endogenous T-cells for adoptive transfer. Immunol Rev. 2014;257(1):250–263. doi: 10.1111/imr.12134. [DOI] [PubMed] [Google Scholar]
- 96.Miliotou AN, Papadopoulou LC. Car T-cell therapy: a new era in cancer immunotherapy. Curr Pharm Biotechnol. 2018;19(1):5–18. doi: 10.2174/1389201019666180418095526. [DOI] [PubMed] [Google Scholar]
- 97.Jin C, Yu D, Hillerdal V, Wallgren A, Karlsson-Parra A, Essand M. Allogeneic lymphocyte-licensed DCs expand T-cells with improved antitumor activity and resistance to oxidative stress and immunosuppressive factors. Mol Ther-Met Clin Dev. 2014;1:14001. doi: 10.1038/mtm.2014.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Powell JD, Brennan AL, Zheng Z, Huynh H, Cotte J, Levine BL. Efficient clinical-scale enrichment of lymphocytes for use in adoptive immunotherapy using a modified counterflow centrifugal elutriation program. Cytotherapy. 2009;11(7):923–935. doi: 10.3109/14653240903188921. [DOI] [PubMed] [Google Scholar]
- 99.Riddell SR, Sommermeyer D, Berger C, Liu LS, Balakrishnan A, Salter A, Hudecek M, Maloney DG, Turtle CJ. Adoptive therapy with chimeric antigen receptor–modified T cells of defined subset composition. Cancer J. 2014;20(2):141. doi: 10.1097/PPO.0000000000000036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Cordes N, Kolbe C, Lock D, Holzer T, Althoff D, Schäfer D, Blaeschke F, Kotter B, Karitzky S, Rossig C, et al. Anti-CD19 CARs displayed at the surface of lentiviral vector particles promote transduction of target-expressing cells. Mol Ther-Met Clin Dev. 2021;21:42–53. doi: 10.1016/j.omtm.2021.02.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Jin C, Fotaki G, Ramachandran M, Nilsson B, Essand M, Yu D. Safe engineering of CAR T-cells for adoptive cell therapy of cancer using long‐term episomal gene transfer. Embo Mol Med. 2016;8(7):702–711. doi: 10.15252/emmm.201505869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Jensen TI, Axelgaard E, Bak RO. Therapeutic gene editing in haematological disorders with CRISPR/Cas9. Br J Haematol. 2019;185(5):821–835. doi: 10.1111/bjh.15851. [DOI] [PubMed] [Google Scholar]
- 103.DeWitt MA, Corn JE, Carroll D. Genome editing via delivery of Cas9 ribonucleoprotein. Methods. 2017;121:9–15. doi: 10.1016/j.ymeth.2017.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Frank AM, Braun AH, Scheib L, Agarwal S, Schneider IC, Fusil F, Perian S, Sahin U, Thalheimer FB, Verhoeyen E, et al. Combining T-cell-specific activation and In vivo gene delivery through CD3-targeted lentiviral vectors. Blood Adv. 2020;4(22):5702–5715. doi: 10.1182/bloodadvances.2020002229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Agarwal S, Hanauer JD, Frank AM, Riechert V, Thalheimer FB, Buchholz CJ. In vivo generation of CAR T-cells selectively in human CD4+ lymphocytes. Mol Ther. 2020;28(8):1783–1794. doi: 10.1016/j.ymthe.2020.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Pfeiffer A, Thalheimer FB, Hartmann S, Frank AM, Bender RR, Danisch S, Costa C, Wels WS, Modlich U, Stripecke R, et al. In vivo generation of human CD 19- CAR T cells results in B-cell depletion and signs of cytokine release syndrome. Embo Mol Med. 2018;10(11):e9158. doi: 10.15252/emmm.201809158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Levine BL. Personalized cell-based medicine: activated and expanded T-cells for adoptive immunotherapy. Bioprocess J. 2007;6(2):14. doi: 10.12665/J62.Levine. [DOI] [Google Scholar]
- 108.Levine BL, Miskin J, Wonnacott K, Keir C. Global manufacturing of CAR T-cell therapy. Mol Ther-Met Clin Dev. 2017;4:92–101. doi: 10.1016/j.omtm.2016.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Mock U, Nickolay L, Cheung G-K, Zhan H, Peggs K, Johnston IC, Kaiser A, Pule M, Thrasher A, Qasim W, et al. Automated lentiviral transduction of T-cells with CARs using the CliniMACS Prodigy. Blood. 2015;126(23):2043. doi: 10.1182/blood.V126.23.2043.2043. [DOI] [Google Scholar]
- 110.Abou-el-Enein M, Elsallab M, Feldman SA, Fesnak AD, Heslop HE, Marks P, Till BG, Bauer G, Savoldo B. Scalable manufacturing of CAR T-cells for cancer ImmunotherapyClinical production of CAR T-cells. Blood Cancer Discovery. 2021;2(5):408–422. doi: 10.1158/2643-3230.BCD-21-0084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Alzubi J, Lock D, Rhiel M, Schmitz S, Wild S, Mussolino C, Hildenbeutel M, Brandes C, Rositzka J, Lennartz S, et al. Automated generation of gene-edited CAR T cells at clinical scale. Mol Ther-Met Clin Dev. 2021;20:379–388. doi: 10.1016/j.omtm.2020.12.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Makita S, Imaizumi K, Kurosawa S, Tobinai K. Chimeric antigen receptor T-cell therapy for B-cell non-Hodgkin lymphoma: opportunities and challenges. Drugs Context. 2019;8:1–14. doi: 10.7573/dic.212567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Hartmann J, Schüßler‐lenz M, Bondanza A, Buchholz CJ. Clinical development of CAR T-cells—challenges and opportunities in translating innovative treatment concepts. Embo Mol Med. 2017;9(9):1183–1197. doi: 10.15252/emmm.201607485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Wall D, Krueger J. Chimeric antigen receptor T-cell therapy comes to clinical practice. Current Oncol. 2020;27(s2):115–123. doi: 10.3747/co.27.5283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Muranski P, Boni A, Wrzesinski C, Citrin DE, Rosenberg SA, Childs R, Restifo NP. Increased intensity lymphodepletion and adoptive immunotherapy—how far can we go? Nat Clin Pract Oncol. 2006;3(12):668–681. doi: 10.1038/ncponc0666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Turtle CJ, Hanafi L-A, Berger C, Sommermeyer D, Pender B, Robinson EM, Melville K, Budiarto TM, Steevens NN, Chaney C, et al. Addition of fludarabine to cyclophosphamide lymphodepletion improves in vivo expansion of CD19 chimeric antigen receptor-modified T cells and clinical outcome in adults with B cell acute lymphoblastic leukemia. Blood. 2015;126(23):3773. doi: 10.1182/blood.V126.23.3773.3773. [DOI] [Google Scholar]
- 117.Ghilardi G, Chong E, Svoboda J, Wohlfarth P, Nasta S, Williamson S, Landsburg JD, Gerson JN, Barta SK, Pajarillo R. Bendamustine is safe and effective for lymphodepletion before tisagenlecleucel in patients with refractory or relapsed large B-cell lymphomas. Ann Oncol. 2022. doi: 10.1016/j.annonc.2022.05.521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Maude SL, Frey N, Shaw PA, Aplenc R, Barrett DM, Bunin NJ, Chew A, Gonzalez VE, Zheng Z, Lacey SF, et al. Chimeric antigen receptor T-cells for sustained remissions in leukemia. N Engl J Med. 2014;371(16):1507–1517. doi: 10.1056/NEJMoa1407222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Ramos CA, Grover NS, Beaven AW, Lulla PD, Wu M-F, Ivanova A, Wang T, Shea TC, Rooney CM, Dittus C, et al. Anti-CD30 CAR-T-cell therapy in relapsed and refractory Hodgkin lymphoma. J Clin Oncol. 2020;38(32):3794. doi: 10.1200/JCO.20.01342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Dawicki W, Allen KJ, Garg R, Geoghegan EM, Berger MS, Ludwig DL, Dadachova E. Targeted lymphodepletion with a CD45-directed antibody radioconjugate as a novel conditioning regimen prior to adoptive cell therapy. Oncotarget. 2020;11(39):3571. doi: 10.18632/oncotarget.27731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Nath R, Geoghegan EM, Ulrickson ML, Spross JA, Lichtenstein RH, Konerth S, Fisher DR, Liang Q, Ludwig D, Reddy V, et al. Sierra clinical trial dosimetry results support low dose anti-CD45 Iodine (131I) Apamistamab [Iomab-B] for targeted lymphodepletion prior to adoptive cell therapy. Blood. 2019;134:1958. doi: 10.1182/blood-2019-128838. [DOI] [Google Scholar]
- 122.Mahadeo KM, Khazal SJ, Abdel-Azim H, Fitzgerald JC, Taraseviciute A, Bollard CM, Tewari P, Duncan C, Traube C, McCall D, et al. Management guidelines for paediatric patients receiving chimeric antigen receptor T-cell therapy. Nat Rev Clin Oncol. 2019;16(1):45–63. doi: 10.1038/s41571-018-0075-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Brudno JN, Kochenderfer JN. Chimeric antigen receptor T-cell therapies for lymphoma. Nat Rev Clin Oncol. 2018;15(1):31–46. doi: 10.1038/nrclinonc.2017.128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Su X, Vale R. Mechanisms of chimeric antigen receptor (CAR) signaling during T-cell activation. Biophys J. 2018;114(3):107a–108a. doi: 10.1016/j.bpj.2017.11.625. [DOI] [Google Scholar]
- 125.Yu JX, Hubbard-Lucey VM, Tang J. The global pipeline of cell therapies for cancer. Nat Rev Drug Discov. 2019;18(11):821–822. doi: 10.1038/d41573-019-00090-z. [DOI] [PubMed] [Google Scholar]
- 126.Mikkilineni L, Kochenderfer JN. Chimeric antigen receptor T-cell therapies for multiple myeloma. Blood. 2017;130:2594–2602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Abbasi MH, Riaz A, Khawar MB, Farooq A, Majid A, Sheikh N. CAR-T-Cell therapy: present progress and future strategies. Biomed Res Ther. 2022;9(2):4920–4929. doi: 10.15419/bmrat.v9i2.726. [DOI] [Google Scholar]
- 128.Cantrell DA. T-Cell antigen receptor signal transduction. Immunology. 2002;105(4):369. doi: 10.1046/j.1365-2567.2002.01391.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Cullen S, Martin S. Mechanisms of granule-dependent killing. Cell Death Differ. 2008;15(2):251–262. doi: 10.1038/sj.cdd.4402244. [DOI] [PubMed] [Google Scholar]
- 130.de Saint Basile G, Ménasché G, Fischer A. Molecular mechanisms of biogenesis and exocytosis of cytotoxic granules. Nat Rev Immunol. 2010;10(8):568–579. doi: 10.1038/nri2803. [DOI] [PubMed] [Google Scholar]
- 131.Nagata S, Tanaka M. Programmed cell death and the immune system. Nat Rev Immunol. 2017;17(5):333–340. doi: 10.1038/nri.2016.153. [DOI] [PubMed] [Google Scholar]
- 132.Davenport AJ, Jenkins MR, Cross RS, Yong CS, Prince HM, Ritchie DS, Trapani JA, Kershaw MH, Darcy PK, Neeson PJ, et al. CAR-T-Cells inflict sequential killing of multiple tumor targeT-cells. Cancer Immunol Res. 2015;3(5):483–494. doi: 10.1158/2326-6066.CIR-15-0048. [DOI] [PubMed] [Google Scholar]
- 133.Nai Y, Du L, Shen M, Li T, Huang J, Han X, Luo F, Wang W, Pang D, Jin A, et al. TRAIL-R1-Targeted CAR-T-cells exhibit dual antitumor efficacy. Front Mol Biosci. 2021;8. doi: 10.3389/fmolb.2021.756599 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Tang X-J, Sun X-Y, Huang K-M, Zhang L, Yang Z-S, Zou D-D, Wang B, Warnock GL, Dai L-J, Luo J, et al. Therapeutic potential of CAR-T-cell-derived exosomes: a cell-free modality for targeted cancer therapy. Oncotarget. 2015;6(42):44179. doi: 10.18632/oncotarget.6175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Priceman SJ, Forman SJ, Brown CE. Smart CARs engineered for cancer immunotherapy. Curr Opin Oncol. 2015;27(6):466. doi: 10.1097/CCO.0000000000000232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Rohaan MW, Wilgenhof S, Haanen JB. Adoptive cellular therapies: the current landscape. Virchows Archiv. 2019;474(4):449–461. doi: 10.1007/s00428-018-2484-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Kochenderfer JN, Wilson WH, Janik JE, Dudley ME, Stetler-Stevenson M, Feldman SA, Maric I, Raffeld M, Nathan DA, Lanier BJ, et al. Eradication of B-lineage cells and regression of lymphoma in a patient treated with autologous T-cells genetically engineered to recognize CD19. Blood. 2010;116(20):4099–4102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Maude SL, Laetsch TW, Buechner J, Rives S, Boyer M, Bittencourt H, Bader P, Verneris MR, Stefanski HE, Myers GD. Tisagenlecleucel in children and young adults with B-cell lymphoblastic leukemia. N Engl J Med. 2018;378(5):439–448. doi: 10.1056/NEJMoa1709866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Schuster SJ, Bishop MR, Tam CS, Waller EK, Borchmann P, McGuirk JP, Jäger U, Jaglowski S, Andreadis C, Westin JR, et al. Tisagenlecleucel in adult relapsed or refractory diffuse large B-cell lymphoma. N Engl J Med. 2019;380(1):45–56. doi: 10.1056/NEJMoa1804980. [DOI] [PubMed] [Google Scholar]
- 140.Neelapu SS, Locke FL, Bartlett NL, Lekakis LJ, Miklos DB, Jacobson CA, Braunschweig I, Oluwole OO, Siddiqi T, Lin Y. Axicabtagene ciloleucel CAR T-cell therapy in refractory large B-cell lymphoma. N Engl J Med. 2017;377(26):2531–2544. doi: 10.1056/NEJMoa1707447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Locke FL, Ghobadi A, Jacobson CA, Miklos DB, Lekakis LJ, Oluwole OO, Lin Y, Braunschweig I, Hill BT, Timmerman JM. Long-Term safety and activity of axicabtagene ciloleucel in refractory large B-cell lymphoma (ZUMA-1): a single-arm, multicentre, phase 1–2 trial. Lancet Oncol. 2019;20(1):31–42. doi: 10.1016/S1470-2045(18)30864-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Wang M, Munoz J, Goy A, Locke FL, Jacobson CA, Hill BT, Timmerman JM, Holmes H, Jaglowski S, Flinn IW. KTE-X19 CAR T-cell therapy in relapsed or refractory mantle-cell lymphoma. N Engl J Med. 2020;382(14):1331–1342. doi: 10.1056/NEJMoa1914347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Waldman AD, Fritz JM, Lenardo MJ. A guide to cancer immunotherapy: from T-cell basic science to clinical practice. Nat Rev Immunol. 2020;20(11):651–668. doi: 10.1038/s41577-020-0306-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Kershaw MH, Westwood JA, Parker LL, Wang G, Eshhar Z, Mavroukakis SA, White DE, Wunderlich JR, Canevari S, Rogers-Freezer L, et al. A phase I study on adoptive immunotherapy using gene-modified T-cells for ovarian cancer. Clin Cancer Res. 2006;12(20):6106–6115. doi: 10.1158/1078-0432.CCR-06-1183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Lamers C, Sleijfer S, Vulto AG, Kruit W, Kliffen M, Debets R, Gratama JW, Stoter G, Oosterwijk E. Treatment of metastatic renal cell carcinoma with autologous T-lymphocytes genetically retargeted against carbonic anhydrase IX: first clinical experience. J Clin Oncol. 2006;24(13):e20–2. doi: 10.1200/JCO.2006.05.9964. [DOI] [PubMed] [Google Scholar]
- 146.Brown CE, Alizadeh D, Starr R, Weng L, Wagner JR, Naranjo A, Ostberg JR, Blanchard MS, Kilpatrick J, Simpson J. Regression of glioblastoma after chimeric antigen receptor T-cell therapy. N Engl J Med. 2016;375(26):2561–2569. doi: 10.1056/NEJMoa1610497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Sengsayadeth S, Savani BN, Oluwole O, Dholaria B. Overview of approved CAR‐T therapies, ongoing clinical trials, and its impact on clinical practice. Ejhaem. 2022;3:6–10. doi: 10.1002/jha2.338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.FDA . Novel Drugs Approved by the FDA, 2011–2017. 2018.
- 149.Braendstrup P, Levine BL, Ruella M. The long road to the first FDA-approved gene therapy: chimeric antigen receptor T-cells targeting CD19. Cytotherapy. 2020;22(2):57–69. doi: 10.1016/j.jcyt.2019.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Prasad V. Tisagenlecleucel—the first approved CAR-T-cell therapy: implications for payers and policy makers. Nat Rev Clin Oncol. 2018;15(1):11–12. doi: 10.1038/nrclinonc.2017.156. [DOI] [PubMed] [Google Scholar]
- 151.Ahmad A, Uddin S, Steinhoff M. Car-T-Cell therapies: an overview of clinical studies supporting their approved use against acute lymphoblastic leukemia and large b-cell lymphomas. Int J Mol Sci. 2020;21(11):3906. doi: 10.3390/ijms21113906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Laura Joszt M. FDA Approves Tisa-Cel for Third Indication, r/r Follicular Lymphoma. Innovation Multi Care. May 2022. [Google Scholar]
- 153.Nastoupil LJ, Jain MD, Feng L, Spiegel JY, Ghobadi A, Lin Y, Dahiya S, Lunning M, Lekakis L, Reagan P. Standard-Of-Care axicabtagene ciloleucel for relapsed or refractory large B-cell lymphoma: results from the US Lymphoma CAR T Consortium. J Clin Oncol. 2020;38(27):3119. doi: 10.1200/JCO.19.02104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Jacobson CA, Chavez JC, Sehgal A, William BM, Munoz J, Salles GA, Casulo C, Munshi PN, Maloney DG, De Vos S, et al. Outcomes in ZUMA-5 with axicabtagene ciloleucel (axi-cel) in patients (pts) with relapsed/refractory (R/R) indolent non-Hodgkin lymphoma (iNHL) who had the high-risk feature of progression within 24 months from initiation of first anti-CD20–containing chemoimmunotherapy (POD24). USA: American Society of Clinical Oncology; 2021. [Google Scholar]
- 155.FDA . FDA approves FirsT-cell-based gene therapy for adult patients with relapsed or refractory mcl. 2020.
- 156.FDA . FDA approval of Tecartus (brexucabtagene autoleucel) for adult patients with relapsed or refractory B-cell precursor acute lymphoblastic leukemia. DISCO Burst Edition. 2021.
- 157.Shah BD, Ghobadi A, Oluwole OO, Logan AC, Boissel N, Cassaday RD, Leguay T, Bishop MR, Topp MS, Tzachanis D. KTE-X19 for relapsed or refractory adult B-cell acute lymphoblastic leukaemia: phase 2 results of the single-arm, open-label, multicentre ZUMA-3 study. The Lancet. 2021;398(10299):491–502. doi: 10.1016/S0140-6736(21)01222-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.FDA . FDA approves lisocabtagene maraleucel for relapsed or refractory large B-cell lymphoma. 2021.
- 159.Kharfan-Dabaja MA, Yassine F, Moustafa MA, Iqbal M, Murthy H. Lisocabtagene maraleucel in relapsed or refractory diffuse large B cell lymphoma: what is the evidence? Hematol Oncol Stem Cell Ther. 2021. doi: 10.1016/j.hemonc.2021.09.004. [DOI] [PubMed] [Google Scholar]
- 160.Abramson JS, Palomba ML, Gordon LI, Lunning MA, Wang M, Arnason J, Mehta A, Purev E, Maloney DG, Andreadis C. Lisocabtagene maraleucel for patients with relapsed or refractory large B-cell lymphomas (TRANSCEND NHL 001): a multicentre seamless design study. The Lancet. 2020;396(10254):839–852. doi: 10.1016/S0140-6736(20)31366-0. [DOI] [PubMed] [Google Scholar]
- 161.Munshi NC, Anderson JL, Shah N, Madduri D, Berdeja J, Lonial S, Raje N, Lin Y, Siegel D, Oriol A. Idecabtagene vicleucel in relapsed and refractory multiple myeloma. N Engl J Med. 2021;384(8):705–716. doi: 10.1056/NEJMoa2024850. [DOI] [PubMed] [Google Scholar]
- 162.Jagannath S, Lin Y, Goldschmidt H, Reece D, Nooka A, Senin A, Rodriguez-Otero P, Powles R, Matsue K, Shah N. KarMma-RW: comparison of idecabtagene vicleucel with real-world outcomes in relapsed and refractory multiple myeloma. Blood Cancer J. 2021;11(6):1–9. doi: 10.1038/s41408-021-00507-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.FDA FDA approval of carvykti (ciltacabtagene autoleucel) for the treatment of adult patients with relapsed or refractory multiple myeloma after four or more prior lines of therapy, including a proteasome inhibitor, an immunomodulatory agent, and an anti-cd38 monoclonal antibody. FDA DISCO Burst Edition. 2022.
- 164.Madduri D, Berdeja JG, Usmani SZ, Jakubowiak A, Agha M, Cohen AD, Stewart AK, Hari P, Htut M, O’Donnell E, et al. CARTITUDE-1: phase 1b/2 study of ciltacabtagene autoleucel, a B-cell maturation antigen-directed chimeric antigen receptor T-cell therapy, in relapsed/refractory multiple myeloma. Blood. 2020;136:22–25. doi: 10.1182/blood-2020-136307. [DOI] [Google Scholar]
- 165.King AC, Orozco JS. Axicabtagene ciloleucel: the first FDA-approved CAR T-cell therapy for relapsed/refractory large B-cell lymphoma. J Adv Pract Oncol. 2019;10(8):878. doi: 10.6004/jadpro.2019.10.8.9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Grupp SA, Maude SL, Shaw PA, Aplenc R, Barrett DM, Callahan C, Lacey SF, Levine BL, Melenhorst JJ, Motley L, et al. Durable remissions in children with relapsed/refractory all treated with T-cells engineered with a CD19-targeted chimeric antigen receptor (CTL019). Blood. 2015;126(23):681. doi: 10.1182/blood.V126.23.681.681. [DOI] [Google Scholar]
- 167.Kochenderfer JN, Dudley ME, Kassim SH, Somerville RP, Carpenter RO, Stetler-Stevenson M, Yang JC, Phan GQ, Hughes MS, Sherry RM, et al. Chemotherapy-Refractory diffuse large B-cell lymphoma and indolent B-cell malignancies can be effectively treated with autologous T-cells expressing an anti-CD19 chimeric antigen receptor. J Clin Oncol. 2015;33(6):540. doi: 10.1200/JCO.2014.56.2025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Schuster SJ, Tam CS, Borchmann P, Worel N, McGuirk JP, Holte H, Waller EK, Jaglowski S, Bishop MR, Damon LE, et al. Long-Term clinical outcomes of tisagenlecleucel in patients with relapsed or refractory aggressive B-cell lymphomas (JULIET): a multicentre, open-label, single-arm, phase 2 study. Lancet Oncol. 2021;22(10):1403–1415. doi: 10.1016/S1470-2045(21)00375-2. [DOI] [PubMed] [Google Scholar]
- 169.Lemal R, Tournilhac O. State-Of-The-Art for CAR T-cell therapy for chronic lymphocytic leukemia in 2019. J Immunother Cancer. 2019;7(1):1–6. doi: 10.1186/s40425-019-0686-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.FDA FDA approves axicabtagene ciloleucel for second-line treatment of large B-cell lymphoma. 2022.
- 171.FDA FDA approves axicabtagene ciloleucel for large B-cell lymphoma. 2017.
- 172.Elsawy M, Chavez JC, Avivi I, Larouche J-F, Wannesson L, Cwynarski K, Osman K, Davison K, Rudzki JD, Dahiya S, et al., editors. Patient-Reported Outcomes (PROs) in Zuma-7, a phase 3, randomized, open-label study evaluating the efficacy of Axicabtagene Ciloleucel (Axi-Cel) versus Standard-of-Care (SOC) therapy in patients with relapsed/refractory Large B-Cell Lymphoma (LBCL) 2022 Tandem Meetings|Transplantation & Cellular Therapy Meetings of ASTCT and CIBMTR; 2022: Tandem Meetings; United States. [Google Scholar]
- 173.Neelapu SS, Dickinson M, Munoz J, Ulrickson ML, Thieblemont C, Oluwole OO, Herrera AF, Ujjani CS, Lin Y, Riedell PA. Axicabtagene ciloleucel as first-line therapy in high-risk large B-cell lymphoma: the phase 2 ZUMA-12 trial. Nat Med. 2022;28(4):735–742. doi: 10.1038/s41591-022-01731-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Turtle CJ, Hanafi L-A, Berger C, Gooley TA, Cherian S, Hudecek M, Sommermeyer D, Melville K, Pender B, Budiarto TM, et al. CD19 CAR–T cells of defined CD4+:CD8+ composition in adult B cell ALL patients. J Clin Invest. 2016;126(6):2123–2138. doi: 10.1172/JCI85309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.De Bousser E, Callewaert N, Festjens N. T-cell engaging immunotherapies, highlighting chimeric antigen receptor (CAR) T-cell therapy. Cancers. 2021;13(23):6067. doi: 10.3390/cancers13236067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Maldini CR, Ellis GI, Riley JL. Car T-cells for infection, autoimmunity and allotransplantation. Nat Rev Immunol. 2018;18(10):605–616. doi: 10.1038/s41577-018-0042-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Tian Y, Li Y, Shao Y, Zhang Y. Gene modification strategies for next-generation CAR T-cells against solid cancers. J Hematol Oncol. 2020;13(1):1–16. doi: 10.1186/s13045-020-00890-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Singh N, Frey NV, Grupp SA, Maude SL. Car T-cell therapy in acute lymphoblastic leukemia and potential for chronic lymphocytic leukemia. Curr Treat Options Oncol. 2016;17(6):1–11. doi: 10.1007/s11864-016-0406-4. [DOI] [PubMed] [Google Scholar]
- 179.Wei J, Han X, Bo J, Han W. Target selection for CAR-T therapy. J Hematol Oncol. 2019;12(1):1–9. doi: 10.1186/s13045-019-0758-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Schultz L, Mackall C. Driving CAR T-cell translation forward. Sci Transl Med. 2019;11(481):eaaw2127. doi: 10.1126/scitranslmed.aaw2127. [DOI] [PubMed] [Google Scholar]
- 181.Almåsbak H, Aarvak T, Vemuri MC. Car T-cell therapy: a game changer in cancer treatment. J Immunol Res. 2016;2016:1–10. doi: 10.1155/2016/5474602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Rennert P, Su L, Dufort F, Birt A, Sanford T, Wu L, Ambrose C, Lobb R. A novel CD19-anti-CD20 bridging protein prevents and reverses CD19-negative relapse from CAR19 T cell treatment in vivo. Blood. 2019;134:252. doi: 10.1182/blood-2019-130654.31118164 [DOI] [Google Scholar]
- 183.Fry TJ, Shah NN, Orentas RJ, Stetler-Stevenson M, Yuan CM, Ramakrishna S, Wolters P, Martin S, Delbrook C, Yates B, et al. CD22-Targeted CAR T-cells induce remission in B-ALL that is naive or resistant to CD19-targeted CAR immunotherapy. Nat Med. 2018;24(1):20–28. doi: 10.1038/nm.4441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Pan J, Tan Y, Wang G, Deng B, Ling Z, Song W, Seery S, Zhang Y, Peng S, Xu J, et al. Donor-Derived CD7 chimeric antigen receptor T-cells for T-cell acute lymphoblastic leukemia: first-in-human, phase I trial. J Clin Oncol. 2021;39(30):3340–3351. doi: 10.1200/JCO.21.00389. [DOI] [PubMed] [Google Scholar]
- 185.Kirtane K, Elmariah H, Chung CH, Abate-Daga D. Adoptive cellular therapy in solid tumor malignancies: review of the literature and challenges ahead. J Immunother Cancer. 2021;9(7):e002723. doi: 10.1136/jitc-2021-002723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Marofi F, Motavalli R, Safonov VA, Thangavelu L, Yumashev AV, Alexander M, Shomali N, Chartrand MS, Pathak Y, Jarahian M, et al. Car T-cells in solid tumors: challenges and opportunities. Stem Cell Res Ther. 2021;12(1):1–16. doi: 10.1186/s13287-020-02128-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Gauthier J, Hirayama AV, Purushe J, Hay KA, Lymp J, Li DH, Yeung CCS, Sheih A, Pender BS, Hawkins RM, et al. Feasibility and efficacy of CD19-targeted CAR T-cells with concurrent ibrutinib for CLL after ibrutinib failure. Blood. 2020;135(19):1650–1660. doi: 10.1182/blood.2019002936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Sallman DA, Brayer J, Sagatys EM, Lonez C, Breman E, Agaugué S, Verma B, Gilham DE, Lehmann FF, Davila ML. NKG2D-Based chimeric antigen receptor therapy induced remission in a relapsed/refractory acute myeloid leukemia patient. Haematologica. 2018;103(9):e424. doi: 10.3324/haematol.2017.186742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Shadman M, Yeung C, Redman MW, Lee SY, Lee DH, Ramachandran A, Ra S, Marzbani EA, Graf SA, Warren EH, et al. Third generation CD20 targeted CAR T-cell therapy (MB-106) for treatment of patients with relapsed/refractory B-cell non-Hodgkin lymphoma. Blood. 2020;136(Supplement 1):38–39. doi: 10.1182/blood-2020-136440. [DOI] [Google Scholar]
- 190.Driouk L, Gicobi JK, Kamihara Y, Rutherford K, Dranoff G, Ritz J, Baumeister SHC. Chimeric antigen receptor T-cells targeting NKG2D-ligands show robust efficacy against acute myeloid leukemia and T-cell acute lymphoblastic leukemia. Front Immunol. 2020;11:580328. doi: 10.3389/fimmu.2020.580328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Maiorova V, Mollaev MD, Vikhreva P, Kulakovskaya E, Pershin D, Chudakov DM, Kibardin A, Maschan MA, Larin S. Natural Flt3Lg-Based Chimeric Antigen Receptor (Flt3-CAR) T-cells Successfully Target Flt3 on AML Cell Lines. Vaccines. 2021;9(11):1238. doi: 10.3390/vaccines9111238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.Warda W, Larosa F, Neto Da Rocha M, Trad R, Deconinck E, Fajloun Z, Faure C, Caillot D, Moldovan M, Valmary-Degano S, et al. Cml hematopoietic stem cells expressing il1rap can be targeted by chimeric antigen receptor–engineered T cells. Cancer Res. 2019;79(3):663–675. doi: 10.1158/0008-5472.CAN-18-1078. [DOI] [PubMed] [Google Scholar]
- 193.Cui Q, Qian C, Xu N, Kang L, Dai H, Cui W, Song B, Yin J, Li Z, Zhu X, et al. CD38-Directed CAR-T-cell therapy: a novel immunotherapy strategy for relapsed acute myeloid leukemia after allogeneic hematopoietic stem cell transplantation. J Hematol Oncol. 2021;14(1):1–5. doi: 10.1186/s13045-021-01092-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194.Hill L, Rouce RH, Smith TS, Yang L, Srinivasan M, Zhang H, Perconti S, Mehta B, Dakhova O, Randall J, et al. CD5 CAR T-cells for treatment of patients with relapsed/refractory CD5 expressing T-cell lymphoma demonstrates safety and anti-tumor activity. Biol Blood Marrow Transplant. 2020;26(3):S237. doi: 10.1016/j.bbmt.2019.12.482. [DOI] [Google Scholar]
- 195.Cheng J, Chen G, Lv H, Xu L, Liu H, Chen T, Qu L, Wang J, Cheng L, Hu S, et al. CD4-Targeted T-cells rapidly induce remissions in mice with T-cell Lymphoma. Biomed Res Int. 2021;2021. doi: 10.1155/2021/6614784 [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 196.Mamonkin M, Rouce RH, Tashiro H, Brenner MK. A T-cell–directed chimeric antigen receptor for the selective treatment of T-cell malignancies. Blood. 2015;126:983–992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197.Vitanza NA, Johnson AJ, Wilson AL, Brown C, Yokoyama JK, Künkele A, Chang CA, Rawlings-Rhea S, Huang W, Seidel K, et al. Locoregional infusion of HER2-specific CAR T-cells in children and young adults with recurrent or refractory CNS tumors: an interim analysis. Nat Med. 2021;27(9):1544–1552. doi: 10.1038/s41591-021-01404-8. [DOI] [PubMed] [Google Scholar]
- 198.Shah NN, Fry TJ. Mechanisms of resistance to CAR T-cell therapy. Nat Rev Clin Oncol. 2019;16(6):372–385. doi: 10.1038/s41571-019-0184-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199.Fousek K, Watanabe J, Joseph SK, George A, An X, Byrd TT, Morris JS, Luong A, Martínez-Paniagua MA, Sanber K, et al. Car T-cells that target acute B-lineage leukemia irrespective of CD19 expression. Leukemia. 2021;35(1):75–89. doi: 10.1038/s41375-020-0792-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200.Maude SL, Barrett DM, Rheingold SR, Aplenc R, Teachey DT, Callahan C, Baniewicz D, White C, Talekar MK, Shaw PA, et al. Efficacy of humanized CD19-targeted chimeric antigen receptor (CAR)-modified T-cells in children and young adults with relapsed/refractory acute lymphoblastic leukemia. Blood. 2016;128(22):217. doi: 10.1182/blood.V128.22.217.217.27207794 [DOI] [Google Scholar]
- 201.Kalos M, Levine BL, Porter DL, Katz S, Grupp SA, Bagg A, June CH. T-Cells with chimeric antigen receptors have potent antitumor effects and can establish memory in patients with advanced leukemia. Sci Transl Med. 2011;3(95):95ra73. doi: 10.1126/scitranslmed.3002842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202.Orlando EJ, Han X, Tribouley C, Wood PA, Leary RJ, Riester M, Levine JE, Qayed M, Grupp SA, Boyer M. Genetic mechanisms of target antigen loss in CAR19 therapy of acute lymphoblastic leukemia. Nat Med. 2018;24(10):1504–1506. doi: 10.1038/s41591-018-0146-z. [DOI] [PubMed] [Google Scholar]
- 203.Fischer J, Paret C, El Malki K, Alt F, Wingerter A, Neu MA, Kron B, Russo A, Lehmann N, Roth L, et al. CD19 isoforms enabling resistance to CART-19 immunotherapy are expressed in B-ALL patients at initial diagnosis. J Immunother. 2017;40(5):187. doi: 10.1097/CJI.0000000000000169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204.Sotillo E, Barrett DM, Black KL, Bagashev A, Oldridge D, Wu G, Sussman R, Lanauze C, Ruella M, Gazzara MR. Convergence of Acquired Mutations and Alternative Splicing of CD19 Enables Resistance to CART-19 Immunotherapy. Cancer Discov. 2015;5(12):1282–1295. doi: 10.1158/2159-8290.CD-15-1020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205.Gardner R, Wu D, Cherian S, Fang M, Hanafi L-A, Finney O, Smithers H, Jensen MC, Riddell SR, Maloney DG, et al. Acquisition of a CD19-negative myeloid phenotype allows immune escape of MLL-rearranged B-ALL from CD19 CAR-T-cell therapy. Blood. 2016;127(20):2406–2410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 206.Majzner RG, Mackall CL. Tumor antigen escape from CAR T-cell therapy. Cancer Discov. 2018;8(10):1219–1226. doi: 10.1158/2159-8290.CD-18-0442. [DOI] [PubMed] [Google Scholar]
- 207.Green DJ, Pont M, Sather BD, Cowan AJ, Turtle CJ, Till BG, Nagengast AM, Libby EN, Becker PS, Coffey DG, et al. Fully human BCMA targeted chimeric antigen receptor T-cells administered in a defined composition demonstrate potency at low doses in advanced stage high risk multiple myeloma. Blood. 2018;132(Supplement 1):1011. doi: 10.1182/blood-2018-99-117729. [DOI] [Google Scholar]
- 208.Brudno JN, Maric I, Hartman SD, Rose JJ, Wang M, Lam N, Stetler-Stevenson M, Salem D, Yuan C, Pavletic S, et al. T Cells genetically modified to express an Anti–B-Cell maturation antigen chimeric antigen receptor cause remissions of poor-prognosis relapsed multiple myeloma. J Clin Oncol. 2018;36(22):2267. doi: 10.1200/JCO.2018.77.8084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209.Cohen AD, Garfall AL, Stadtmauer EA, Melenhorst JJ, Lacey SF, Lancaster E, Vogl DT, Weiss BM, Dengel K, Nelson A, et al. B cell maturation antigen–specific car T cells are clinically active in multiple myeloma. J Clin Invest. 2019;129(6):2210–2221. doi: 10.1172/JCI126397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210.Jafarzadeh L, Masoumi E, Fallah-Mehrjardi K, Mirzaei HR, Hadjati J. Prolonged persistence of chimeric antigen receptor (CAR) T-cell in adoptive cancer immunotherapy: challenges and ways forward. Front Immunol. 2020;11:702. doi: 10.3389/fimmu.2020.00702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 211.Li D, Li N, Zhang Y-F, Fu H, Feng M, Schneider D, Su L, Wu X, Zhou J, Mackay S, et al. Persistent polyfunctional chimeric antigen receptor T-cells that target glypican 3 eliminate orthotopic hepatocellular carcinomas in mice. Gastroenterology. 2020;158(8):2250–65. e20. doi: 10.1053/j.gastro.2020.02.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 212.Jensen MC, Riddell SR. Designing chimeric antigen receptors to effectively and safely target tumors. Curr Opin Immunol. 2015;33:9–15. doi: 10.1016/j.coi.2015.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 213.Caruso HG, Hurton LV, Najjar A, Rushworth D, Ang S, Olivares S, Mi T, Switzer K, Singh H, Huls H, et al. Tuning sensitivity of CAR to EGFR density limits recognition of normal tissue while maintaining potent antitumor activity. Cancer Res. 2015;75(17):3505–3518. doi: 10.1158/0008-5472.CAN-15-0139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 214.McGowan E, Lin Q, Ma G, Yin H, Chen S, Lin Y. PD-1 disrupted CAR-T-cells in the treatment of solid tumors: promises and challenges. Biomed Pharmacother. 2020;121:109625. doi: 10.1016/j.biopha.2019.109625. [DOI] [PubMed] [Google Scholar]
- 215.Liu Y, Wang L, Predina J, Han R, Beier UH, Wang LCS, Kapoor V, Bhatti TR, Akimova T, Singhal S. Inhibition of p300 impairs Foxp3+ T regulatory cell function and promotes antitumor immunity. Nat Med. 2013;19(9):1173–1177. doi: 10.1038/nm.3286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 216.Sawant DV, Yano H, Chikina M, Zhang Q, Liao M, Liu C, Callahan DJ, Sun Z, Sun T, Tabib T, et al. Adaptive plasticity of IL-10+ and IL-35+ Treg cells cooperatively promotes tumor T-cell exhaustion. Nat Immunol. 2019;20(6):724–735. doi: 10.1038/s41590-019-0346-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 217.Seo H, Chen J, González-Avalos E, Samaniego-Castruita D, Das A, Wang YH, López-Moyado IF, Georges RO, Zhang W, Onodera A, et al. TOX and TOX2 transcription factors cooperate with NR4A transcription factors to impose CD8 + T cell exhaustion. Proc Nat Acad Sci. 2019;116(25):12410–12415. doi: 10.1073/pnas.1905675116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 218.Nabe S, Yamada T, Suzuki J, Toriyama K, Yasuoka T, Kuwahara M, Shiraishi A, Takenaka K, Yasukawa M, Yamashita M, et al. Reinforce the antitumor activity of CD 8 + T cells via glutamine restriction. Cancer Sci. 2018;109(12):3737–3750. doi: 10.1111/cas.13827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 219.Zhao J, Lin Q, Song Y, Liu D. Universal CARs, universal T-cells, and universal CAR T-cells. J Hematol Oncol. 2018;11(1):1–9. doi: 10.1186/s13045-018-0677-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 220.Liu D, Zhao J, Song Y. Engineering switchable and programmable universal CARs for CAR T therapy. J Hematol Oncol. 2019;12(1):1–9. doi: 10.1186/s13045-019-0763-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 221.Rafiq S, Hackett CS, Brentjens RJ. Engineering strategies to overcome the current roadblocks in CAR T-cell therapy. Nat Rev Clin Oncol. 2020;17(3):147–167. doi: 10.1038/s41571-019-0297-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 222.Kloss CC, Lee J, Zhang A, Chen F, Melenhorst JJ, Lacey SF, Maus MV, Fraietta JA, Zhao Y, June CH, et al. Dominant-Negative TGF-β receptor enhances PSMA-targeted human CAR T cell proliferation and augments prostate cancer eradication. Mol Ther. 2018;26(7):1855–1866. doi: 10.1016/j.ymthe.2018.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 223.Albinger N, Hartmann J, Ullrich E. Current status and perspective of CAR-T and CAR-NK cell therapy trials in Germany. Gene Ther. 2021;28(9):513–527. doi: 10.1038/s41434-021-00246-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 224.Abreu TR, Fonseca NA, Gonçalves N, Moreira JN. Current challenges and emerging opportunities of CAR-T-cell therapies. J Controlled Release. 2020;319:246–261. doi: 10.1016/j.jconrel.2019.12.047. [DOI] [PubMed] [Google Scholar]
- 225.Zhao L, Cao YJ. Engineered T-cell therapy for cancer in the clinic. Front Immunol. 2019;10:2250. doi: 10.3389/fimmu.2019.02250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 226.Brudno JN, Kochenderfer JN. Recent advances in CAR T-cell toxicity: mechanisms, manifestations and management. Blood Rev. 2019;34:45–55. doi: 10.1016/j.blre.2018.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 227.Si S, Teachey DT. Spotlight on tocilizumab in the treatment of CAR-T-cell-induced cytokine release syndrome: clinical evidence to date. Ther Clin Risk Manag. 2020;16:705. doi: 10.2147/TCRM.S223468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 228.Diorio C, Vatsayan A, Talleur AC, Annesley C, Jaroscak JJ, Shalabi H, Ombrello AK, Hudspeth M, Maude SL, Gardner RA, et al. Anakinra utilization in refractory pediatric CAR T-cell associated toxicities. Blood Adv. 2022;6(11):3398–3403. doi: 10.1182/bloodadvances.2022006983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 229.Park JH, Sauter CS, Palomba ML, Shah GL, Dahi PB, Lin RJ, Scordo M, Batlevi CL, Perales M-A, Kane P, et al. A phase ii study of prophylactic anakinra to prevent CRS and neurotoxicity in patients receiving CD19 CAR T-cell therapy for relapsed or refractory lymphoma. Blood. 2021;138:96. doi: 10.1182/blood-2021-150431. [DOI] [Google Scholar]
- 230.Oliai C, Crosetti A, De Vos S, Eradat H, Mead MD, Larson SM, Tsai S, Liu A, Khachatrian G, Hannigan C, et al. IL-1 receptor antagonist for prevention of severe immune effector cell-associated neurotoxicity syndrome. United States: Wolters Kluwer Health; 2021. [Google Scholar]
- 231.Bupha-Intr O, Haeusler G, Chee L, Thursky K, Slavin M, Teh B. CAR-T-Cell therapy and infection: a review. Expert Rev Anti Infect Ther. 2021;19(6):749–758. doi: 10.1080/14787210.2021.1855143. [DOI] [PubMed] [Google Scholar]
- 232.Bonini C, Ferrari G, Verzeletti S, Servida P, Zappone E, Ruggieri L, Ponzoni M, Rossini S, Mavilio F, Traversari C. HSV-Tk gene transfer into donor lymphocytes for control of allogeneic graft-versus-leukemia. Science. 1997;276(5319):1719–1724. doi: 10.1126/science.276.5319.1719. [DOI] [PubMed] [Google Scholar]
- 233.Quintarelli C, Vera JF, Savoldo B, Giordano Attianese GM, Pule M, Foster AE, Heslop HE, Rooney CM, Brenner MK, Dotti G. Co-Expression of cytokine and suicide genes to enhance the activity and safety of tumor-specific cytotoxic T lymphocytes. Blood. 2007;110:2793–2802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 234.C-Y W, Roybal KT, Puchner EM, Onuffer J, Lim WA. Remote control of therapeutic T-cells through a small molecule–gated chimeric receptor. Science. 2015;350(6258):aab4077. doi: 10.1126/science.aab4077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 235.Frankel SR, Baeuerle PA. Targeting T-cells to tumor cells using bispecific antibodies. Curr Opin Chem Biol. 2013;17(3):385–392. doi: 10.1016/j.cbpa.2013.03.029. [DOI] [PubMed] [Google Scholar]
- 236.Labanieh L, Majzner RG, Klysz D, Sotillo E, Fisher CJ, Vilches-Moure JG, Pacheco KZB, Malipatlolla M, Xu P, Hui JH, et al. Enhanced safety and efficacy of protease-regulated CAR-T-cell receptors. Cell. 2022;185(10):1745–63. e22. doi: 10.1016/j.cell.2022.03.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 237.Lee SM, Kang CH, Choi SU, Kim Y, Hwang JY, Jeong HG, Park CH. A chemical switch system to modulate chimeric antigen receptor T-cell activity through proteolysis-targeting chimaera technology. Acs Synth Biol. 2020;9(5):987–992. doi: 10.1021/acssynbio.9b00476. [DOI] [PubMed] [Google Scholar]
- 238.Fiorenza S, Ritchie DS, Ramsey SD, Turtle CJ, Roth JA. Value and affordability of CAR T-cell therapy in the United States. Bone Marrow Transplant. 2020;55(9):1706–1715. doi: 10.1038/s41409-020-0956-8. [DOI] [PubMed] [Google Scholar]
- 239.Zhang C, He J, Liu L, Wang J, Wang S, Liu L, Ge J, Gao L, Gao L, Kong P, et al. Novel CD19 chimeric antigen receptor T-cells manufactured next-day for acute lymphoblastic leukemia. Blood Cancer J. 2022;12(6):1–9. doi: 10.1038/s41408-022-00688-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 240.Ardigen 2021. CAR-T-Cell therapies: limitations and improvement strategies. Poland: Ardigen BLOG. [Google Scholar]
- 241.Novartis . Charging towards the next-generation of CAR-T. 2022.
- 242.Bozza M, De Roia A, Correia MP, Berger A, Tuch A, Schmidt A, Zörnig I, Jäger D, Schmidt P, Harbottle RP, et al. A nonviral, nonintegrating DNA nanovector platform for the safe, rapid, and persistent manufacture of recombinant T-cells. Science Adv. 2021;7(16):eabf1333. doi: 10.1126/sciadv.abf1333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 243.Depil S, Duchateau P, Grupp S, Mufti G, Poirot L. ‘Off-The-Shelf’allogeneic CAR T-cells: development and challenges. Nat Rev Drug Discov. 2020;19(3):185–199. doi: 10.1038/s41573-019-0051-2. [DOI] [PubMed] [Google Scholar]
- 244.Cutmore LC, Marshall JF. Current perspectives on the use of off the shelf CAR-T/NK cells for the treatment of cancer. Cancers. 2021;13(8):1926. doi: 10.3390/cancers13081926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 245.Elavia N, McManus A, Highfill SL, Ren J, Shah NN, Fry TJ, Brudno J, Kochenderfer JN, Stroncek D, Panch SR. The post-thaw recovery of cryopreserved chimeric antigen receptor (CAR) T-cells during manufacture is better than that of cryopreserved peripheral blood CD3+ cells. Blood. 2017;130:4475. [Google Scholar]
- 246.Tay A. Cryopreservation to Improve Cell Manufacturing and Biobanking: as adoptive cell therapies and stem cell therapies come to be applied more generally, cryopreservation technology must cope with quality and supply challenges. Genet Eng Biotechnol News. 2020;40(S5):S10–S2. doi: 10.1089/gen.40.S5.05. [DOI] [Google Scholar]
- 247.Harrison RP, Zylberberg E, Ellison S, Levine BL. Chimeric antigen receptor–t-cell therapy manufacturing: modelling the effect of offshore production on aggregate cost of goods. Cytotherapy. 2019;21(2):224–233. doi: 10.1016/j.jcyt.2019.01.003. [DOI] [PubMed] [Google Scholar]
- 248.Sanber K, Savani B, Jain T. Graft‐versus‐host disease risk after chimeric antigen receptor T‐cell therapy: the diametric opposition of T-cells. Br J Haematol. 2021;195(5):660–668. doi: 10.1111/bjh.17544. [DOI] [PubMed] [Google Scholar]