

Abstract
Neurodegenerative diseases are a major cause of death and disability worldwide and are influenced by many factors including age, genetics, and injuries. While these diseases are often thought to result from the accumulation and spread of aberrant proteins, recent studies have demonstrated that they can be shaped by the innate and adaptive immune system. Resident myeloid cells typically mount a sustained response to the degenerating central nervous system (CNS) but peripheral leukocytes such as T and B cells can also alter disease trajectories. Here, we review the sometimes-dichotomous roles played by immune cells during neurodegenerative diseases and explore how brain trauma can serve as a disease initiator or accelerant. We also offer insights into how the failure to properly resolve a CNS injury might promote development of a neurodegenerative disease.
Keywords: neurodegeneration, brain injury, innate immunity, adaptive immunity
The central nervous system (CNS) is susceptible to disease and injury
The CNS is a composed of many different cell types including neurons (see glossary), endothelial cells, macrophages, and glial cells like oligodendrocytes, astrocytes, and microglia[1]. While the concept of CNS immune privilege has been greatly revised in recent years[2–7], CNS communication with the periphery is tightly regulated under homeostatic conditions, and immune residents are maintained in a relatively quiescent state[8]. For example, microglia are the main innate immune sentinel within the CNS parenchyma[9]. During steady state, microglia possess a small cell body with long processes and continually scan their surroundings for pathogens, damage, cellular missteps, etc.[10,11]. The parenchymal network of microglia is very sensitive to inflammatory queues and will respond almost immediately to changes in their surroundings, such as those that occur following injury or degeneration.
The hallmarks of neurodegenerative diseases have long been described as marked neuronal loss, reactive gliosis, and aberrant protein accumulation in disease-specific regions of the CNS. Recent studies, however, have highlighted the importance of the innate and adaptive immune system in the pathophysiology of these diseases[12–15]. In fact, immune activation and reactivity is known to increase with the deposition aberrant protein during states of neurodegeneration[16,17]. Immune activation also occurs after brain trauma, and emerging data have linked repetitive head injury to the etiology of specific neurodegenerative diseases[18–25]. Here, we explore how the immune system responds to and shapes neurodegenerative diseases and discuss how a failure to engage proper immune-mediated repair mechanisms after brain injury might contribute to degenerative processes in the CNS.
Innate immune activation during neurodegenerative diseases
Microglia acquire a reactive signature in the degenerating CNS
In neurodegenerative diseases, microglia transition from homeostatic to reactive. Early studies demonstrated the presence of HLA-DR+ or CD68+ reactive microglia in brain regions with aberrant protein deposits[26,27], and for many decades the observations were limited to descriptive histology, because the tools to elucidate the molecular signature and function of these cells did not exist. However, with the advent of single cell sequencing, it became possible to further define microglia and other cell populations in greater detail, which led to the identification of disease associated microglia (DAM) in Alzheimer’s disease (AD)[12]. Only a limited discussion of these cells will be provided here because they have been reviewed extensively elsewhere[28,29]. DAM-like cells likely arise in most, if not all neurodegenerative diseases (Figure 1)[30–33]. Conversion into a DAM requires microglia to undergo a two-step process in which they down-regulate homeostatic markers (e.g., P2ry12, Cx3cr1, and Tmem119) and then upregulate genes associated with phagocytosis, antigen presentation, and lipid metabolism (e.g., Apoe, Trem2, B2m, Itgax, Lpl) in a triggering receptor molecule expressed on myeloid cells 2 (TREM2) dependent manner [12,29,34]. TREM2 is a transmembrane protein found on microglia and other myeloid cells that facilitates phagocytosis and suppresses inflammation[35], and mutations in this protein are considered a risk factor for human AD (AD)[15,36]. Manipulation of Trem2 expression in different murine models of neurodegeneration has produced mixed results. Using the 5xFAD model of AD, TREM2 deficiency impeded the formation of DAMs and exacerbated disease[37]. Similarly, elevating Trem2 expression in 5xFAD mice by crossing them with a human TREM2 BAC transgenic revealed a beneficial role for TREM2 in AD. These mice had reduced amyloid beta (Aβ) plaque loads, improved memory performance, and more DAMs[38]. However, manipulation of TREM2 in other models of neurodegeneration has yielded opposite conclusions. In the PS19 murine tauopathy model, TREM2 deficiency protected against brain atrophy and reduced microglial activation but did not change the load of phosphorylated tau (pTau) in the brain[39]. The authors of this study concluded that TREM2 signaling promotes inflammation and neurodegeneration in the context of a primary tauopathy. Similarly, studies in the APP/PS1 mouse model of AD as well as the super oxide dismutase (SOD-1) model of amyotropic lateral sclerosis (ALS) revealed that TREM2 deficiency preserved microglial homeostasis in both models and reduced Aβ plaque loads in the APP/PS1 AD mouse model [32]. While it is clear that TREM2 signaling can promote microglial activation, the impact of this activation on neurodegenerative diseases can vary depending on the disease model, genetic background, and environmental factors like the microbiome or pathogen exposure[40], or the specific stage of disease[41]. A recent study, for example, demonstrated in APP/PS1 mice that TREM2 deficiency ameliorates amyloid pathology early but exacerbates it late[41]. These data indicate that TREM2 signaling can have both positive and negative effects on a single disease paradigm depending on disease stage.
Figure 1. The innate and adaptive immune reaction neurodegeneration.

Neurodegenerative diseases are marked by cortical atrophy and the accumulation of aberrant proteins inside and outside CNS cells. Aberrant proteins distress neurons, contributing to their death and subsequent cortical atrophy. This initially triggers and innate immune reaction in the CNS that can lead to engagement of adaptive immune cells such as CD8+ T cells, CD4+ T cells, and B cells. Within the CNS parenchyma microglia and astrocytes become reactive and acquire new functional properties. These cells are sometimes referred to as disease-associated microglia (DAM) and disease-associated astrocytes (DAA), respectively. The reactive microglia and astrocytes often attempt to contain the spread of aberrant proteins and aid distressed neurons. They also release cytokines and chemokines that facilitate recruitment of T and B cells from circulation. CD8+ T cells have been observed engaging both neurons and microglia during neurodegenerative diseases, suggesting that the participate in removing these two cell populations. CD4+ T cells also interact with microglia and possibly neurons during neurodegenerative diseases. The role B cells is not entirely clear, but these cells have the potential to influence the course of disease by producing antibodies against aberrant proteins and/or releasing immunoregulatory cytokines.
Another approach used to investigate the role of microglia in neurodegenerative diseases is depletion. These studies have also demonstrated positive and negative roles for microglia. Microglia depletion with the colony stimulating factor 1 receptor (CSF1R) inhibitor, PLX3397, at the early stage of disease progression in PS19 mouse model of tauopathy reduced the amount of pTau protein[42]. It was concluded in this study that microglia played a detrimental role at the early stage of a tauopathy by phagocytosing pTau from neurons and later secreting this protein in exosomes, thus supporting a prion-like seeding mechanism for pTau propagation[42]. By contrast, a complex study in 5xFAD mice injected intracerebrally with pTau from human AD patients found that mice chronically depleted of microglia with PLX3397 had increased pTau deposition[43]. The authors concluded that TREM2 activated DAM were required to slow the propagation of pTau during AD. Depletion studies in ALS mice also showed a beneficial role for microglia. The rNLS8 mouse model of ALS is a reversibly inducible line in which administration of doxycycline (DOX) suppresses the expression of human TDP-43 (hTDP-43) protein. Following removal of DOX, mice develop a progressive motor neuron disease within 6 weeks that can be reversed by re-feeding DOX, after which mice regain motor function[44]. Importantly, when microglia in rNLS8 mice were depleted with PLX3397 during this recovery phase, they did not recover motor function and retained high levels of hTDP-43[45]. A beneficial role for microglia in ALS was confirmed in a separate study that used both inducible transgenic and viral transduction models of hTDP-43 mediated neurodegeneration[46,47]. In this study, hTDP-43 was found to promote TREM2 signaling in a subset of microglia that upregulated CD11c and became phagocytic. In addition, Trem2 deficiency impeded microglia clearance of hTDP-43, resulting in enhanced neuronal damage and motor deficits. While these data considered collectively demonstrate that microglia can acquire different roles in neurodegenerative diseases, the emerging consensus is that microglia, and more specifically DAM, play a beneficial role in the late stage of these diseases.
At present, Trem2 is the most heavily studied gene in DAM regulation, but other factors also contribute to this microglia signature and the progression of neurodegenerative diseases. Genes related to lipid metabolism (e.g., Lpl) are also upregulated in DAM[28]. Lipid droplet formation is linked to Grn, a gene that encodes for the protein progranulin and whose mutation causes some forms of frontotemporal dementia (FTD)[48]. A recent study expanded upon the DAM phenotype by using electron microscopy and RNA sequencing to identify and characterize lipid-droplet accumulating microglia (LDAM) in the brains of aged mice and humans[49]. These cells contained large adipose vacuoles that mirrored what Alois Alzheimer described in the first patient diagnosed with AD[49,50]. While LDAM appeared in the aged brain, they were distinct from DAM in that they downregulated some DAM signature genes including Axl[49] – a receptor tyrosine kinase that facilitates clearance of apoptotic cells. In fact, DAM are known to have an enhanced phagocytic capacity, whereas LDAM show impairment in acquiring dead cells and instead produce elevated levels of reactive oxygen species (ROS) and inflammatory cytokines[49]. While both microglia subsets appear with ageing and in neurodegenerative diseases, these data suggest that LDAM represent dysfunctional microglia that may contribute to CNS deterioration. By contrast, DAM are a beneficial microglia subset that possess the functional capacity to slow the process of CNS degeneration. Additional studies are required to prove this hypothesis definitively and determine the relationship between DAM and LDAM in the degenerating brain. Differential depletion of these two subsets could partly explain the variable outcomes observed in microglia depletion studies. If DAM are indeed a beneficial microglia subset, it will also be important in future studies to develop strategies to enhance their functions while discouraging the generation of LDAM.
Another factor that contributes to microglia regulation in ageing and neurodegeneration is CD22[51]. CD22 is a member of the Siglec family that is typically found on B cells but is also upregulated on ageing microglia[51]. A recent study showed that inhibition of CD22 in aged mice increased the clearance of myelin protein, Aβ oligomers, or α-synuclein fibrils injected into the brain[51], demonstrating that CD22 impedes microglia phagocytosis. In mice, long term inhibition of CD22 also helped restore the homeostatic signature of microglia in the aged brain and improved cognitive function. These data help support the concept that decay of microglia phagocytic capacity is an important contributor to neurodegeneration and the aging process. Further studies are needed to determine what factors in addition to CD22 contribute to microglial decay and how best to reverse or slow this trajectory in the context of neurodegeneration.
While the precise contribution of each microglial subset over time to aging and neurodegeneration is unclear, recent studies demonstrate convincingly that these innate immune CNS residents play an active role in both processes. Microglial homeostasis is a key feature of a healthy CNS, and the discovery of factors that help preserve or promote this state is imperative. Nimble microglia in an undifferentiated state offer the greatest potential to deal with CNS challenges. Everything from antimicrobial immunity to CNS repair and the slowing of neurodegeneration requires a healthy pool of microglia.
Microglial activation of astrocytes
In addition to clearing aberrant peptide / proteins and apoptotic cells, microglia subsets including DAM play a role in activating neighboring astrocytes (Figure 1). Similar to the discovery of DAM, single nuclei sequencing of cells extracted from the brains of 5xFAD mice has uncovered a disease-associated astrocyte (DAA) signature characterized by increased expression of genes associated with the complement cascade, ageing, and endocytosis[13]. In terms of gene expression, DAA were similar to reactive A1 astrocytes identified in an independent study[52]. The A1 astrocytes were induced by direct exposure to the TLR4 agonist, lipopolysaccharide (LPS), or to LPS-activated microglia, to secrete inflammatory mediators like C1q, TNF, and IL-1α[52]. A1 astrocytes lose their homeostatic functions such as phagocytosis and synapse maintenance and instead promote the death of co-cultured neurons[52]. While the A1/A2 nomenclature developed from this study remain controversial, it is important to note astrocytes with the A1 signature were identified in CNS tissue from AD, PD, ALS, Huntington’s disease, and multiple sclerosis patients and likely represent an inflammatory phenotype [52]. The astrocytes were closely associated CD68+ reactive microglia. These data support the concept that alteration of astrocyte homeostasis by inflammatory queues or microglia-derived factors has the potential to facilitate neurodegeneration.
Astrocyte studies often focus on their role in maintaining the blood brain barrier (BBB) (the glia limitans perivascularis) or neuronal homeostasis; however, a population of surface associated astrocytes also comprise the glia limitans superficialis, which is the barrier beneath that pia mater that separates the CNS parenchyma from cerebral spinal fluid (CSF) containing spaces. Damage to the glia limitans superficialis generates a disequilibrium between CSF and interstitial fluids within the parenchyma, which can be harmful to the CNS. For example, mild traumatic brain injury (mTBI) can damage surface associated astrocytes and breach the glia limitans superficialis, allowing toxic mediators like ROS and leukocytes to move from the meningeal space into the CNS parenchyma (Figure 2A,B)[53,54]. A leaky glia limitans superficialis results in parenchyma cell death, but it is not known how conversion of astrocytes to an inflammatory phenotype influences the integrity or tolerance of this important barrier. Future studies will need to investigate whether surface associated astrocytes are converted to an inflammatory phenotype during states of neurodegeneration and the impact this has on the integrity of the glia limitans.
Figure 2. Immune Response to Brain Injury.
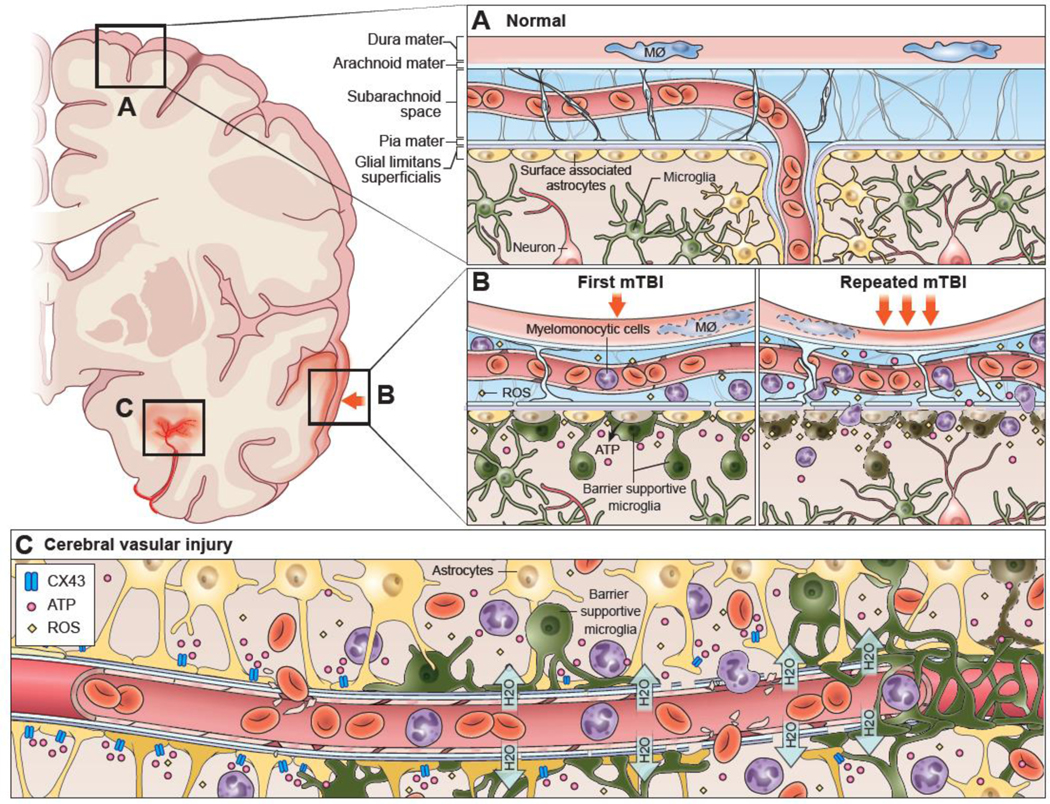
(A) The normal anatomy of the meninges (dura mater, arachnoid mater, pia mater) and superficial neocortex is depicted. During steady state, the meninges are surveyed primarily by meningeal macrophages. These cells are dynamic and reside mostly in the dura mater. Beneath the glia limitans superficialis, which is comprised of surface associated astrocytes, the brain parenchyma is surveyed by microglia. (B) Mild traumatic brain injuries (mTBI) often cause meningeal damage, resulting in vascular leakage and death of meningeal macrophages, endothelial cells, and other meningeal residents. Astrocytes along the glia limitans superficialis can also become distressed or damaged. Part of their damage response includes release of ATP via connexin hemichannels into the underlying brain parenchyma. This causes microglia to extend processes in a purinergic receptor dependent manner to the glial limitans. These reactive microglia help seal the glia limitans barrier and limit the degree of leak into the brain parenchyma. When astrocytes die, microglia transform into a jellyfish-like, amoeboid morphology and begin clearing debris. A major source of cellular and barrier damage following a single mTBI are reactive oxygen species (ROS), which are released into the meningeal space. A second mTBI can significantly enhance the amount of meningeal and parenchymal damage by increasing ROS concentrations. For example, a second injury encountered within a day of the first causes rapid death of barrier supportive microglia, resulting in increased leakage of the glia limitans superficialis. Myelomonocytic cells from circulation also massively invade the damaged meninges after a second injury and even enter the brain parenchyma by traversing the damaged glia limitans superficialis. Importantly, transcranial or intravenous delivery of the free radical scavenger, glutathione, can be used therapeutically to lessen cellular damage (both in the meninges and parenchyma), glia limitans breakdown, and inflammation following single or repetitive mTBI (not depicted). (C) Cerebrovascular injuries resulting from a stroke, intracerebral hemorrhage or TBI are often associated with the leakage of water (edema) and / or red blood cells into the brain parenchyma. This causes distressed astrocytes comprising the BBB to release ATP via connexin hemichannels. Microglia respond to this ATP within minutes using purinergic receptors and project barrier sealing processes that wrap the damaged blood vessel(s). Failure microglia to wrap these blood vessels results in more extensive vascular leakage and secondary parenchyma damage. Myelomonocytic cells can also be massively recruited from circulation following cerebrovascular injury. These cells are required at later stages for vascular repair but can acutely increase brain swelling (within the first 12 hours of injury) by promoting the entry of water into the brain while extravasating.
The contribution of adaptive immunity to neurodegenerative diseases
Immune targeting of Aβ peptides during AD
Before the advent of single cell sequencing and the identification of DAMs, the field of neurodegenerative disease research focused primarily on aberrant proteins (i.e., Aβ and pTau) that accumulated in the brain, which within the AD field, led to the development of the amyloid hypothesis[55–57]. According to this theory, Aβ plays a causative role in AD, initiating neurodegeneration and a cascade of neuroinflammation, such as the transformation of homeostatic microglia into DAMs (Figure 1)[58]. This led to a series of studies in animal models of AD that involved immunization with Aβ peptides to initiate an immune response and slow disease progression. Importantly, this approach markedly reduced the deposition of amyloid plaques in the brain as well as cognitive decline when Aβ peptide immunization was performed early in disease progression[59,60]. While these studies did not demonstrate the type of immunity responsible for slowing disease, it was subsequently shown that injection of Aβ specific antibodies into the periphery of mice entered the CNS, bound to amyloid plaques, and reduced their number by triggering Fc receptor mediated uptake by microglia[61]. In addition, a study in humans observed that there were naturally occurring antibodies to Aβ in the blood and CSF of healthy adults that decreased with ageing and advancing AD[62]. These findings collectively led to numerous clinical trials targeting Aβ, but to date, none of them have proven efficacious[63]. However, in 2021 the U.S. Food and Drug Administration (FDA) approved Aducanumab (a monoclonal Aβ antibody) for use in AD patients despite conflicting results[64–66]. While Aducanumab was effective in reducing amyloid plaques, no anti-Aβ therapy to date has improved cognitive outcomes in humans with AD[67]. These failures suggest that either there are other mechanisms involved in the progression of AD besides Aβ or that the clinical trials targeting Aβ intervened too late in the disease process to be effective.
The contribution of B cells to neurodegeneration
While B cells participate in antigen presentation and production of antibodies, few studies focused on AD or other neurodegenerative diseases have addressed their role in disease progression. One study disrupted in the entire adaptive immune system in the 5xFAD model of AD by generating Rag2−/− IL2rγ−/− mice. These mice showed a two-fold increase in amyloid plaques and signs of microglia distress, including an enhanced pro-inflammatory phenotype and decreased phagocytic activity[68]. In addition, treatment of these mice with preimmune IgG reduced the Aβ plaque burden by ~50% despite the absence of T cells, B cells, or NK cells. This study demonstrated an important role for antibodies in restraining the spread of amyloid plaques. Because B cells have other functions besides antibody production, a recent study genetically and therapeutically removed B cells in three different murine models of AD and revealed an unexpected slowing of disease progression[69]. B cell depleted mice had reduced amyloid plaques and improved performance on behavioral tests. This benefit after B cell depletion was attributed to enhanced microglia function, although the actual mechanism remains unclear. Interestingly, therapeutic depletion of B cells in 5xFAD mice with anti-CD20 at the onset of clinical symptoms slowed disease progression without affecting serum titers of Aβspecific antibodies[69], demonstrating that it is possible to eliminate the disease-enhancing features of B cells without affecting antibody-producing plasma cells. These data suggest a more nuanced view of B lymphocytes in the development of neurogenerative diseases like AD. This makes sense given that B cells can take on many different roles in inflammatory reactions, including immune regulation[70]. Thus, it is conceivable that B cells could simultaneously produce beneficial antibodies while also dampening microglia activity during AD.
Infiltration of the degenerating CNS by CD8+ T cells
T cells have long been linked to neurodegenerative diseases, with early reports demonstrating both CD4+ and CD8+ T cells in the brains of AD patients (Figure 1)[71–73]. Studies in Rag2−/− IL2rγ−/− 5xFAD mice suggested a role for the adaptive immune system in AD but did not specifically evaluate T cells[68]. CD8+ T cells have been found in the cortex of patients with frontotemporal dementia (caused by a P301L mutation in the tau protein) as well as in the hippocampus of THY-Tau22 mice – a murine tauopathy model[74]. Depletion of CD4+ and CD8+ T cells in these mice by administering anti-CD3 from 4 to 9 months of age reduced behavioral deficits without affecting the deposition of pTau[74]. These data suggested that T cells contributed to neurocognitive decline independent of pTau deposition, even though the latter is typically linked to the neural pathology observed in mice and humans with tauopathies[75,76]. Another recent post-mortem study of AD brain tissue detected CD8+ T cells in the perivascular spaces along Aβ+ blood vessels as well as in the leptomeninges adjacent to hippocampal Aβ+ plaques[14]. A population of CD3+CD8+CD27− T effector memory CD45RA+ (TEMRA) cells was also found in the blood of patients with AD and mild cognitive impairment[14]. These cells were associated with reduced cognition and were clonally expanded within the CSF of AD patients. The specificity of at least two CD8+ T cell clones was determined to be against Epstein-barr virus (EBV) antigens; however, it is important to note that these data do not establish a causal relationship between EBV-specific T cells and AD, nor do they demonstrate that CD8+ T cells are responsible for a decline in cognition during this disease. Effector / memory T cells directed against previous pathogens are known to survey inflamed tissues and may be doing so innocuously during AD. However, the presence of clonal CD8+ T cells along CNS borders of AD patients is an important finding that warrants further investigation. Additional mapping of T cell specificities may shed light on how these cells participate in disease progression. Within the blood of Parkinson’s Disease (PD) patients, CD4+ and CD8+ T cells specific for alpha-synuclein (α-syn) have been found, indicating that a peripheral T cell response can be mounted against an aberrantly deposited CNS protein during a human neurodegenerative disease[77,78]. It remains to be determined whether T cell responses against Aβ or tau are generated in humans with AD.
While the specificity of CNS-infiltrating cytotoxic lymphocytes (CTL) has not been determined in many different neurodegenerative diseases, several studies have focused instead on the targets of these cells. Healthy neurons typically express very little MHC I[79,80] but can sometimes upregulate antigen presenting machinery when distressed or following changes in neural activity[81,82]. Aberrant protein accumulation in neurons during neurodegenerative diseases has the potential to enhance expression of antigen presenting molecules like β2-microglobulin and MHC I. A recent study demonstrated that, in addition to displaying peptides, MHC I itself can facilitate aberrant pTau accumulation in neurons[83]. This process occurs intracellularly, but once on the cell surface, MHC I can render neurons susceptible to CD8+ T cell engagement. CD8+ T cells juxtaposed to neurons have been observed in animal models of AD (APP-PS1 mice)[84] and ALS (SOD1G93A mutant)[85] as well as in the brains of patients with PD (Figure 1)[86]. Depletion of CD8+ T cells reduced neuronal loss in SOD1G93A mutant mice[85] and altered neuronal gene expression in APP-PS1 mice[84], suggesting that CTL play a pathogenic role in these models of neurodegenerative disease. Conversely, genetic deletion of CD8+ T cells in the MPTP mouse model of PD demonstrated no role for these cells in the loss of dopaminergic neurons in the substantia nigra[87]. The study concluded instead that CD4+ T cells were responsible for neuronal damage in this model. Collectively, these data demonstrate that distressed neurons, a common occurrence in the degenerating CNS, can display antigen presenting molecules and sometimes become targets of CD8+ T cells. It is worth considering, however, whether selective removal of such neurons by adaptive immune cells is necessarily maladaptive or is instead an attempt to eliminate defective cells.
Another potential target to consider during the development of neurodegenerative diseases is microglia[9]. Microglia are known to participate in neurodegenerative diseases and are capable of presenting antigens[88,89]. For example, a recent study demonstrated in a model of nasal virus infection that microglia can cross-present viral antigen acquired from adjacently infected neurons in the olfactory bulb[89]. The microglia never become infected but nevertheless display viral peptides to infiltrating virus-specific CTL that then release antiviral cytokines to purge virus from infected neurons non-cytolytically. This mechanism of antigen cross-presentation by microglia is likely to become operational during neurodegenerative diseases, as neurons shed aberrant proteins and other cellular components that are viewed by the immune system as damage-associated molecular patterns (DAMPs)[90]. pTau aggregates were recently shown to contain RNA in degenerating P301S mice and humans with AD and FTD[91]. We postulate that these complexes of RNA and aberrant protein elicit a response resembling antiviral immunity when acquired by microglia. Early post mortem studies in AD patient brain tissue showed T cells in apposition to cells resembling microglia (Figure 1)[72]. CD8+ T cells were also observed engaging microglia in the brains of APP-PS1 mice[92]. Microglia during states of neurodegeneration possess the cardinal features of antigen presenting cells (APCs). The key now is to determine what these cells are presenting and how they influence the functions of infiltrating T cells[93].
CD4+ T cell subsets differentially impact neurodegeneration
Like CD8+ T cells, CD4+ T cells are elevated in the blood and CNS during a variety of different neurodegenerative diseases (Figure 1)[73,77,78,87,94]. CD4+ T cells are often viewed as “helpers” that facilitate the activities of CD8+ T cells and B cells, but they can also engage effector functions and cause tissue pathology. Genetic removal of CD4+ T cells in the MPTP mouse model of PD resulted in preservation of dopaminergic neurons in the substantial nigra[87]. It is unclear why CD4+ T cells engage and damage neurons during PD, as neurons do not typically express MHC II. Nevertheless, it was suggested in the MPTP model that CD4+ T cells damaged dopaminergic neurons in a Fas ligand (FasL)-dependent manner. FasL is an effector molecule used by T cells to induce death of infected target cells. Assuming that neurons do not express MHC II during states of neurodegeneration, it is conceivable that CD4+ T cells cause bystander (i.e., peptide-MHC independent) damage to neurons.
Because CD4+ T cells can differentiate into many different subtypes, their contribution to neurodegeneration is likely to vary depending on the specific disease and T cell subtype under investigation. CD4+ CD25+ Foxp3+ regulatory T cells (Tregs) are commonly studied in many different diseases due to their regulatory properties. Studies in humans with neurodegenerative disorders have shown that Tregs are increased[95,96] and decreased[97–99]. In RAG2 deficient SOD1 mutant mice with ALS, reconstitution with CD4+ CD25+ Tregs enhanced survival[98]. In addition, a decreased number Tregs were found in the blood of patients with rapidly progressing ALS, suggesting in combination with the mouse data that these cells slowed disease progression[98]. By contrast, transient depletion of Foxp3+ Tregs in the 5xFAD mouse model of AD decreased plaque load and reversed cognitive decline while enhancing peripheral immune cell recruitment to sites of cerebral plaques[100]. Pharmacological inhibition of Tregs also reduced AD pathology, whereas Treg activation enhanced Aβ+ plaques and worsened cognitive deficits[100]. Additional studies are required to determine the mechanisms by which Tregs influence different neurodegenerative diseases and whether therapeutic manipulation of these cells is warranted.
Accelerating neurodegeneration: brain injury as a risk factor
While the etiology of neurodegenerative diseases is not entirely understood, it is clear that traumatic brain injuries (TBI) increase the risk of developing AD, PD, ALS, and other neurodegenerative diseases[101–103]. Repetitive head injuries such as those experienced when playing contact sports like American football can give rise to a neurodegenerative disease often referred to as chronic traumatic encephalopathy (CTE)[18]. Postmortem analysis of brain sections from CTE patients has revealed aberrant deposition of pTau, Aβ, and α-synuclein – the amounts of which depend on disease stage[18,104]. Other types of head trauma also lead to aberrant protein accumulation in the CNS, resembling that observed during development of neurodegenerative diseases[105]. These studies establish a clear link between head trauma (especially repetitive) and neurodegenerative processes.
Brain injury triggers an immediate local innate immune reaction
To understand how brain injuries might give rise to a neurodegenerative disease, it is important to examine how the CNS responds to damage. Like neurodegenerative diseases, brain injuries elicit an immune reaction that develops in stages and can even become chronic if the injury is not repaired properly. The initial phase of brain injury is usually mechanical or biophysical. For example, the brain can receive a physical impact such as a TBI or it can experience an internal injury like an occluded vessel or vascular breach (e.g., stroke or intracerebral hemorrhage)[106–108]. Following injury, cells within the meninges (meningeal macrophages, peripheral nerves, fibroblasts, endothelium), glia limitans superficialis (astrocytes) and brain parenchyma (neurons, microglia, oligodendrocytes, astrocytes, endothelium) are all vulnerable to immediate destruction, and physical barriers such as the meninges and vasculature begin to leak, creating neural disequilibrium (Figure 2A,B). As cells die, they release alarmins and DAMPs into the extracellular space that initiate inflammatory and stress responses in neighboring cells[106,108]. The release of these mediators causes innate immune cells like microglia to engage in barrier preserving activities.
Following mild TBI (mTBI), meningeal vasculature and the glia limitans superficialis are routinely disrupted in both mice and humans, triggering surface associated astrocytes to release ATP via connexin hemichannels into the underlying brain parenchyma (Figure 2A,B)[53]. In mice, detection of ATP via the purinergic receptor, P2RY12, causes microglia to extend processes within minutes to the glia limitans superficialis where they seal gaps between individual astrocytes (Figure 2B). When glia limitans astrocytes die after murine mTBI, microglia become further activated in response to P2RY6 signaling and transform into a jellyfish-like morphology, allowing them to fill holes left by dead astroctyes and to phagocytose debris (Figure 2B)[53]. These two purinergic receptor dependent responses represent important mechanisms by which microglia help preserve glia limitans integrity after mTBI. Inhibition of these responses results in an extensive leak of fluids from the subarachnoid space into the brain parenchyma[53].
Similar to their activity along the glia limitans superficialis after TBI, microglia were also shown to respond rapidly and preserve BBB integrity following cerebral vascular injury (CVI)[109,110]. In response to murine CVI, microglia in the brain extend processes and wrap damaged blood vessels within minutes of injury (Figure 2C)[109,110]. They do this in a P2RY12-dependent manner by responding to ATP released via connexin hemichannels expressed by BBB astrocytes. Microglia depletion or inhibition of this purinergic response during CVI results in extensive vascular leakage and secondary damage in the brain parenchyma[110]. These data demonstrate that microglia play an important role in forming a quasi-barrier after vascular injury, and a failure to perform this function can result in significant secondary brain damage. It is also important to note that microglia must be in a homeostatic P2RY12+ state to project processes and seal barriers. Thus, any scenario that causes microglia to become reactive and downregulate P2RY12 (e.g., infection, neurodegeneration, prior injury, etc.) will impede their barrier sealing capacity after injury. This may explain in part why infections have such a profoundly negative impact on clinical outcome in patients with brain injuries and strokes[111–114].
Because microglia have such an important role in CNS homeostasis[9], return of these sentinels to a naïve state is an important step in the recovery process after injury. The molecular signature of microglia (as a surrogate for functional potential) in response to brain injury has been examined in several different animal models from days to months post-injury. As expected, based on imaging studies, microglia become reactive quickly after injury (within 24 hrs). They downregulate homeostatic genes (e.g., P2ry12, Siglec H, Tmem119, Sall1, Tgfbr1) and upregulate genes associated with RNA / protein synthesis, inflammation, phagocytosis, lipid metabolism, and cytokine production, among others[115–117]. The gene expression pattern to some degree resembles that observed during neurodegenerative diseases[116], although this may simply reflect a common microglial response to different types of brain damage (i.e., trauma versus degeneration). Changes in microglial gene expression can persist for months following TBI in rodents, some of which are linked to their ability to modulate neuronal synapses[117]. It is unclear how these changes influence actual neural function, but a sustained shift of microglia programming away from homeostasis after TBI could reflect an unresolved anatomical disturbance and / or an inability of these cells to regain homeostatic properties.
We postulate that it is quite important for microglia to regain some form of naivety following CNS injury. One reason for this is that the CNS will eventually face new challenges that require a nimble microglial response. An example of such a challenge is a secondary injury. A recent study demonstrated that the neuroprotective, barrier-sealing microglia response observed along the glia limitans superficialis after a single mTBI is lost when a second injury is experienced one day later (Figure 2B)[54]. High parameter flow cytometry revealed that microglia, one day following a single mTBI, were large (high FSC), granulated (high SSC), and clearly activated (P2RY12lo CX3CR1hi F480hi). Morphologically, these cells were non-ramified and contained inclusions, supporting their conversion into phagocytes. Following exposure to a second mTBI, many of these cells died within a few hours, which combined with their lack of naivete, contributed to extensive leak of the glia limitans superficialis and cell death in the brain parenchyma. Importantly, it was possible therapeutically preserve the glia limitans, reduce cortical cell death, and prevent microglia death by therapeutically administering the ROS scavenger, glutathione, immediately following reinjury[54]. Repetitive head injuries are known to trigger neurodegenerative diseases[118–121], and we postulate that restoration of microglia homeostasis is an important variable to consider when assessing the ability of the brain to withstand a secondary injury. Microglia cannot perform their barrier-sealing functions when they are in a reactive state.
Infiltrating myelomonocytic cells contribute to damage and repair after injury
Another important component of the innate immune reaction is the recruitment of peripherally derived myelomonocytic cells (monocytes and neutrophils) (Figure 2). While CNS injury studies typically conclude that myelomonocytic cells influence outcomes in a positive or negative manner[108,122,123], it is important to note that these cells can make dichotomous contributions to the same injury paradigm that depend on time, location, and function[124]. CVI, TBI, spinal cord injury, and stroke can trigger a massive recruitment of myelomonocytic cells into the CNS, which is often associated with brain swelling or edema[107,110]. A recent study in a model of CVI demonstrated a causal relationship between myelomonocytic cell extravasation and cerebral edema[110]. As myelomonocytic cell extravasate across cerebrovasculature, they breach BBB integrity and cause water to enter the brain, which can be fatal. This extravasation in response to CVI can be completely blocked by therapeutic administration of antibodies against the adhesion molecules LFA-1 and VLA-4. Importantly, LFA1/VLA4 blockade was able to prevent fatal brain swelling if given within 6 hours of a CVI in mice[110]. These data demonstrate that myelomonocytic cells can cause unintended damage to the CNS as they traverse the BBB, which might lead to the conclusion these cells are pathogenic following CVI. However, examination of cerebrovascular repair over the ensuing week in the same model of CVI revealed that myelomonocytic cells (specifically, CCR2+ monocytes) played an essential role in angiogenesis[110]. This was accomplished by endowing microglia with pro-angiogenic properties, such as the ability to produce VEGF-A. Interference with monocyte recruitment during CNS repair blocks development of repair-associated microglia (RAM) and angiogenesis. Thus, myelomonocytic cells can damage blood vessels in the initial hours following a CVI but then contribute to their repair. These studies have important implications for when therapeutics targeting peripherally-derived cells, like the LFA1/VLA4 blockade, should be administered.
In addition to facilitating parenchymal angiogenesis and microglia crosstalk, peripheral monocytes are also known to promote repair of damaged blood vessels in the meninges following mTBI[125]. TBI in mice and humans often causes damage to meningeal vasculature that needs to be repaired[53,125]. In mice, meningeal vascular repair after mTBI was shown to depend on recruitment of nonclassical monocytes that promote angiogenic programming and release of metalloproteinases like MMP-2 that break down provisional matrix[125]. Successful immune-mediated repair is a necessary step toward restoring CNS homeostasis after injury, and many studies have focused on how immune cells help remodel the damaged CNS[108,126–128]. However, it is also known there are factors that can alter the CNS repair trajectory. For example, secondary mTBI and peripheral infection were both shown to impede meningeal vascular repair[111,125]. In fact, a broad range of microbes block repair of damaged blood vessels in the meninges and brain parenchyma through induction of type I interferon signaling that deviates pro-angiogenic programming in myeloid cells[111]. Infections and repetitive brain injuries represent two factors among many that have the potential to exacerbate faulty wound-healing in the CNS and foster development of neurodegenerative diseases.
Faulty CNS repair may promote neurodegeneration
With a detailed understanding of mechanisms underlying breakdown and repair of the injured CNS, it becomes possible to speculate about how repetitive head injuries trigger neurodegeneration. When CTE was first described in the 1920’s, the disease was characterized by disruptions of barrier surfaces, meningeal and cortical hemorrhages, and behavioral changes[129]. Our knowledge about CTE has grown considerably over the past century, and the disease is now defined in stages, with the most severe form consisting of glial activation and diffuse pTau staining throughout the brain[23,130]. As discussed, both TBI and CVI breach vascular and non-vascular (glia limitans) CNS barriers, resulting in the activation of microglia and astrocytes. A single non-severe CNS injury has a reasonable chance of repairing if immune-mediated repair mechanisms can engage undisturbed. However, CNS injuries do not always resolve acutely[125], and factors such as age, genetics, prior injury, infection, etc. all have the potential to impede the repair trajectory. Sustained leakage of CNS barriers places the parenchyma in a state of chronic disequilibrium and immune activation. We postulate that this failure to repair in a timely manner fosters the development of neurodegenerative processes. CTE provides an example of how repetitive head trauma can lead to a chronic barrier disruption, profound changes in glial cells, and an aberrant deposition of pTau (Figure 3)[20,129,130]. Once initiated via this cycle of repeat trauma and inadequate repair, aberrant proteins such as pTau may begin to spread from neuron to neuron in a prion-like manner[25] and/or in exosomes secreted into the extracellular space[42], fostering a neurodegenerative disease. Other types of injuries, including CVI’s, may similarly strain the CNS, promoting spreadable aberrant protein deposition if unresolved. We propose that rapid restoration of injured CNS barriers is essential for the maintenance of CNS homeostasis and the deterrence of neurodegenerative diseases.
Figure 3. Induction of neurodegeneration by repetitive brain Injury.

(A) The normal anatomy of the meninges and superficial neocortex is depicted with a blood vessel traversing the pia mater and entering the brain. (B) Repeated brain injuries can induce neurodegenerative processes by promoting leaky CNS barriers. Exposure of the meninges and brain to multiple injuries can lead to chronic vascular leakage and disruption of the glia limitans superficialis and perivascularis. Continual leakage of materials from the blood and CSF into the parenchyma can contribute to neural disequilibrium and dysfunction. Microglia and astrocytes in areas of repetitive brain damage often assume a reactive state, and astrocytes are sometimes observed forming a scar along the damaged glia limitans. Repetitive brain injuries can also lead to the deposition of aberrant proteins like pTau both inside and outside of neurons. This can trigger a microglial response and recruitment of peripheral immune cells.
Concluding Remarks
Neurodegenerative diseases have a major negative impact on human health, and there are few treatment options available. Microglia play an important role in influencing the development and progression of neurodegenerative diseases, as they are the main innate immune occupant of the CNS parenchyma and constantly survey their surroundings. While microglia can slow the progression of neurodegenerative diseases by acquiring aberrant proteins, confining plaques, and phagocytosing dead cells, they can become strained over time and lose their neuroprotective functionality. A sustained loss of microglial naivete almost certainly impedes the important homeostatic functions that microglia perform, such as the maintenance of neuronal synapses. Reactive microglia are also unable to perform their barrier-sealing functions in response to injuries, rendering the CNS more susceptible to secondary damage.
Adaptive immune cells such as CD4+ T cells, CD8+ T cells, and B cells can also enter the degenerating CNS and survey its barriers as well as the parenchyma. How these cells influence disease progression varies widely based on the specific type of neurodegeneration under investigation. For T cells it will be important in future studies to further map the peptides they recognize and determine the direct consequence of them engaging distinct CNS targets (microglia, neurons, etc.) displaying those peptides. B cells require similar investigative scrutiny to distinguish their antibody production capacity from their regulatory and antigen presenting cell functions during neurodegenerative diseases. Additional knowledge about the innate and adaptive immune responses mounted during different neurodegenerative diseases will aid in the development of immunomodulatory therapeutics to slow disease progression. It will also be important to gain further insights into how CNS injuries facilitate development of neurodegenerative diseases. It is clear that repetitive brain injuries can initiate degenerative processes in the CNS, and we propose that a sustained breach of CNS barriers due to faulty or incomplete immune-mediated repair leads to chronic glial activation and neuronal distress that can progress to CTE and other related neurodegenerative diseases. More studies are required to evaluate this theory and to determine how best to introduce therapeutics (see Outstanding Questions). Given the heavy involvement of the immune system in both neurodegenerative diseases and CNS injuries, immunomodulatory therapies offer great potential for repairing damage and slowing degeneration.
Outstanding Questions.
How do specific microglia subtypes contribute to neurodegenerative diseases?
What is the specificity of adaptive immune cells generated during neurodegenerative diseases, and how should we manipulate these cells therapeutically?
Do microglia and astrocytes regain their homeostatic properties following CNS injury?
Are repaired brain regions more susceptible to future injury and degeneration?
Could treating individual head injuries prevent the development of neurodegenerative diseases like CTE?
Highlights.
Studies have demonstrated an important role for the innate and adaptive immune systems in shaping neurodegenerative disease progression.
Microglia become reactive during neurodegenerative diseases and along with astrocytes can slow disease progression by cleaning up aberrant proteins and impeding their spread, but these functions can decay with time.
An adaptive immune response consisting of CD8+ T cells, CD4+ T cells, and B cells is sometimes generated during neurodegenerative diseases contributing to divergent outcomes depending on their functional programming.
Traumatic brain and cerebrovascular injuries trigger rapid innate immune reactions that help seal and repair damaged CNS barriers.
Repetitive head injuries can cause sustained barrier leakage, immune activation, and aberrant protein deposition, leading to development of a neurodegenerative disease.
Acknowledgements
This work was supported by the intramural program at the National Institute of Neurological Disorders & Stroke (NINDS), National Institutes of Health (NIH). We thank Erina He in the NIH Medical Arts Design Section for her help with the illustrations shown in Figures 1 to 3.
Glossary
- Endothelium
cells that comprise the lining of the vasculature.
- Macrophage
a type of immune cell derived from monocytes that participates in phagocytosis and sometimes antigen presentation.
- Glia
non-neuronal CNS resident cells that include oligodendrocytes, astrocytes, and microglia.
- Oligodendrocyte
a type of glial cell that produces the myelin, which insulates neuronal axons.
- Astrocyte
a type of glial cell with many functions that include creating important CNS barriers like the glia limitans superficialis and perivascularis and maintaining neuronal homeostasis.
- Microglia
a brain resident myeloid cell that surveils the CNS parenchyma and responds to damage and other perturbations.
- HLA-DR+ reactive microglia
activated microglia that express the human leukocyte antigen complex-DR (HLA-DR) (also known as major histocompatibility complex II) on their cell surface with the capacity to present antigen. MHC II+ microglia also exist in mice.
- CD68+ microglia
microglia that express a high level of CD68, a lysosomal protein indicative of activation and phagocytic activity
- Disease associated microglia
a microglia subset associated with neurodegenerative diseases that arises in a TREM2 dependent manner and downregulates expression of genes related to homeostasis.
- Triggering receptor expressed on myeloid cells 2 (TREM2)
a transmembrane protein found on myeloid cells linked to phagocytosis and inflammation whose dysfunction is a risk factor for Alzheimer’s disease
- Alzheimer’s disease
a human neurodegenerative disease characterized by cortical atrophy, aberrant accumulation of extracellular amyloid beta plaques and neurofibrillary phosphorylated tau tangles as well as glial cell activation and cognitive changes such as memory deficits.
- Amyloid beta (Aβ)
peptide that aggregates in Alzheimer’s disease to form extracellular plaques.
- Phosphorylated tau (pTau)
peptide found in mainly axons of neurons that becomes phosphorylated in neurodegenerative diseases and disrupts cellular function.
- Human TAR DNA-binding protein 43 (hTDP-43)
a protein that binds RNA and DNA but can form oligomers and accumulate when phosphorylated, leading to neurodegenerative diseases like ALS.
- Glia limitans perivascularis
barrier consisting of astrocytic endfeet that wrap blood vessels in CNS parenchyma, forming part of the BBB.
- Glia limitans superficialis
barrier of surface associated astrocytes that reside beneath the pia mater and separate the CNS parenchyma from the CSF-containing meninges.
- Cerebral spinal fluid (CSF)
fluid produced by the choroid plexus in the CNS ventricular system that flows through the subarachnoid space and ventricles of the CNS.
- B cells
adaptive immune cells responsible for antibody production, antigen presentation, and immunoregulatory functions.
- CD4+ T cells
adaptive immune cells that possess different regulatory, effector, and helper functions whose T cell receptors interact with cells displaying cognate peptide-MHC II complexes.
- CD8+ T cells
adaptive immune cells, often referred to as cytotoxic lymphocytes, that can engage and kill cells containing intracellular pathogens after T cell receptor mediated recognition of cognate peptide-MHC I complexes on the target cell surface.
- Parkinson’s disease
neurodegenerative disease characterized by a loss of dopaminergic neurons in the substantia nigra and the accumulation of alpha-synuclein in Lewy bodies and Lewy neurites, resulting in movement impairments including tremors and rigidity.
- Alpha-synuclein (α-syn)
a protein responsible for vesicular transport between synapses that accumulates during Parkinson’s disease.
- T effector memory cell (TEMRA)
An effector or memory CD8+ T cell that has been exposed to an antigen previously and is capable of patrolling blood and tissues throughout the body in search of that antigen.
- Antigen presenting cell
a cell capable of processing and presenting cells on the cell surface in major histocompatibility complexes to T cells; dendritic cells, B cells, and macrophages are often considered professional antigen presenting cells.
- T regulatory cell
subset of T lymphocytes that suppress other cells in the immune system.
- Amyotrophic lateral sclerosis (ALS)
neurodegenerative disease characterized by accumulation of the aberrant protein TDP-43 in the spinal cord motor neurons, leading to paralysis and eventually death.
- Chronic traumatic encephalopathy
neurodegenerative disease caused by repeated head injuries that results in the accumulation of pTau in cortical neurons.
- Meninges
barrier tissue consisting of the dura mater, arachnoid mater, and pia mater that encases the brain and spinal cord.
- Frontotemporal dementia
a heterogenous neurodegenerative disease characterized by accumulation of aberrant phosphorylated tau and / or TDP-43 in predominantly neurons of the frontal and temporal regions of the brain
- Reactive oxygen species (ROS)
highly reactive byproducts of oxygen metabolism that can cause irreversible damage to nucleic acids, proteins, membranes, and other cellular structures.
- Damage-associated molecular pattern (DAMPs)
molecules released by damaged and dying cells that activate the innate immune system by binding to pattern recognition receptors.
- Cytokine
proteins secreted by cells that facilitate communication and inflammatory reactions.
- Chemokine
type of protein that directs the movement of another cell like the movement of leukocytes from the blood into an inflamed tissue.
- Edema
swelling caused by excess fluid accumulation in a tissue
- Myelomonocytic cell
peripherally derived immune cells of a myeloid origin that include neutrophils or monocytes.
- LFA1/VLA4 blockade
administration of antibodies against the adhesion molecules LFA-1 and VLA-4 that prevents extravasation of peripheral cells from the vasculature.
Footnotes
Declaration of Interests
The authors declare no competing interests.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Mastorakos P. and McGavern D. (2019) The anatomy and immunology of vasculature in the central nervous system. Sci Immunol 4. 10.1126/sciimmunol.aav0492 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Louveau A. et al. (2015) Revisiting the Mechanisms of CNS Immune Privilege. Trends Immunol 36, 569–577. 10.1016/j.it.2015.08.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Louveau A. et al. (2015) Structural and functional features of central nervous system lymphatic vessels. Nature 523, 337–341. 10.1038/nature14432 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Aspelund A. et al. (2015) A dural lymphatic vascular system that drains brain interstitial fluid and macromolecules. J Exp Med 212, 991–999. 10.1084/jem.20142290 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Cugurra A. et al. (2021) Skull and vertebral bone marrow are myeloid cell reservoirs for the meninges and CNS parenchyma. Science. 10.1126/science.abf7844 [DOI] [PMC free article] [PubMed]
- 6.Fitzpatrick Z. et al. (2020) Gut-educated IgA plasma cells defend the meningeal venous sinuses. Nature 587, 472–476. 10.1038/s41586-020-2886-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ahn JH et al. (2019) Meningeal lymphatic vessels at the skull base drain cerebrospinal fluid. Nature 572, 62–66. 10.1038/s41586-019-1419-5 [DOI] [PubMed] [Google Scholar]
- 8.Buckley MW and McGavern DB (2022) Immune dynamics in the CNS and its barriers during homeostasis and disease. Immunol Rev. 10.1111/imr.13066 [DOI] [PMC free article] [PubMed]
- 9.Nayak D. et al. (2014) Microglia development and function. Annu Rev Immunol 32, 367–402. 10.1146/annurev-immunol-032713-120240 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Nimmerjahn A. et al. (2005) Resting microglial cells are highly dynamic surveillants of brain parenchyma in vivo. Science 308, 1314–1318. 10.1126/science.1110647 [DOI] [PubMed] [Google Scholar]
- 11.Li Q. and Barres BA (2018) Microglia and macrophages in brain homeostasis and disease. Nat Rev Immunol 18, 225–242. 10.1038/nri.2017.125 [DOI] [PubMed] [Google Scholar]
- 12.Keren-Shaul H. et al. (2017) A Unique Microglia Type Associated with Restricting Development of Alzheimer’s Disease. Cell 169, 1276–1290.e1217. 10.1016/j.cell.2017.05.018 [DOI] [PubMed] [Google Scholar]
- 13.Habib N. et al. (2020) Disease-associated astrocytes in Alzheimer’s disease and aging. Nat Neurosci 23, 701–706. 10.1038/s41593-020-0624-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gate D. et al. (2020) Clonally expanded CD8 T cells patrol the cerebrospinal fluid in Alzheimer’s disease. Nature 577, 399–404. 10.1038/s41586-019-1895-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Jonsson T. et al. (2013) Variant of TREM2 associated with the risk of Alzheimer’s disease. N Engl J Med 368, 107–116. 10.1056/NEJMoa1211103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Serrano-Pozo A. et al. (2011) Reactive glia not only associates with plaques but also parallels tangles in Alzheimer’s disease. Am J Pathol 179, 1373–1384. 10.1016/j.ajpath.2011.05.047 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Pascoal TA et al. (2021) Microglial activation and tau propagate jointly across Braak stages. Nat Med 27, 1592–1599. 10.1038/s41591-021-01456-w [DOI] [PubMed] [Google Scholar]
- 18.Mez J. et al. (2017) Clinicopathological Evaluation of Chronic Traumatic Encephalopathy in Players of American Football. Jama 318, 360–370. 10.1001/jama.2017.8334 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Adams JW et al. (2018) Lewy Body Pathology and Chronic Traumatic Encephalopathy Associated With Contact Sports. J Neuropathol Exp Neurol 77, 757–768. 10.1093/jnen/nly065 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Chancellor KB et al. (2021) Altered oligodendroglia and astroglia in chronic traumatic encephalopathy. Acta Neuropathol 142, 295–321. 10.1007/s00401-021-02322-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Cherry JD et al. (2020) CCL2 is associated with microglia and macrophage recruitment in chronic traumatic encephalopathy. J Neuroinflammation 17, 370. 10.1186/s12974020-02036-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kenney K. et al. (2018) Dementia After Moderate-Severe Traumatic Brain Injury: Coexistence of Multiple Proteinopathies. J Neuropathol Exp Neurol 77, 50–63. 10.1093/jnen/nlx101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.McKee AC et al. (2013) The spectrum of disease in chronic traumatic encephalopathy. Brain 136, 43–64. 10.1093/brain/aws307 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Tagge CA et al. (2018) Concussion, microvascular injury, and early tauopathy in young athletes after impact head injury and an impact concussion mouse model. Brain 141, 422–458. 10.1093/brain/awx350 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Woerman AL et al. (2016) Tau prions from Alzheimer’s disease and chronic traumatic encephalopathy patients propagate in cultured cells. Proc Natl Acad Sci U S A 113, E8187–e8196. 10.1073/pnas.1616344113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.McGeer PL et al. (1987) Reactive microglia in patients with senile dementia of the Alzheimer type are positive for the histocompatibility glycoprotein HLA-DR. Neurosci Lett 79, 195–200. 10.1016/0304-3940(87)90696-3 [DOI] [PubMed] [Google Scholar]
- 27.Croisier E. et al. (2005) Microglial inflammation in the parkinsonian substantia nigra: relationship to alpha-synuclein deposition. J Neuroinflammation 2, 14. 10.1186/17422094-2-14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Deczkowska A. et al. (2018) Disease-Associated Microglia: A Universal Immune Sensor of Neurodegeneration. Cell 173, 1073–1081. 10.1016/j.cell.2018.05.003 [DOI] [PubMed] [Google Scholar]
- 29.Butovsky O. and Weiner HL (2018) Microglial signatures and their role in health and disease. Nat Rev Neurosci 19, 622–635. 10.1038/s41583-018-0057-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lewis ND et al. (2014) RNA sequencing of microglia and monocyte-derived macrophages from mice with experimental autoimmune encephalomyelitis illustrates a changing phenotype with disease course. J Neuroimmunol 277, 26–38. 10.1016/j.jneuroim.2014.09.014 [DOI] [PubMed] [Google Scholar]
- 31.Chiu IM et al. (2013) A neurodegeneration-specific gene-expression signature of acutely isolated microglia from an amyotrophic lateral sclerosis mouse model. Cell Rep 4, 385–401. 10.1016/j.celrep.2013.06.018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Krasemann S. et al. (2017) The TREM2-APOE Pathway Drives the Transcriptional Phenotype of Dysfunctional Microglia in Neurodegenerative Diseases. Immunity 47, 566-581.e569. 10.1016/j.immuni.2017.08.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Mrdjen D. et al. (2018) High-Dimensional Single-Cell Mapping of Central Nervous System Immune Cells Reveals Distinct Myeloid Subsets in Health, Aging, and Disease. Immunity 48, 380–395.e386. 10.1016/j.immuni.2018.01.011 [DOI] [PubMed] [Google Scholar]
- 34.Zhou Y. et al. (2020) Human and mouse single-nucleus transcriptomics reveal TREM2-dependent and TREM2-independent cellular responses in Alzheimer’s disease. Nat Med 26, 131–142. 10.1038/s41591-019-0695-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Takahashi K. et al. (2005) Clearance of apoptotic neurons without inflammation by microglial triggering receptor expressed on myeloid cells-2. J Exp Med 201, 647–657. 10.1084/jem.20041611 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Guerreiro R. et al. (2013) TREM2 variants in Alzheimer’s disease. N Engl J Med 368, 117–127. 10.1056/NEJMoa1211851 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wang Y. et al. (2015) TREM2 lipid sensing sustains the microglial response in an Alzheimer’s disease model. Cell 160, 1061–1071. 10.1016/j.cell.2015.01.049 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lee CYD et al. (2018) Elevated TREM2 Gene Dosage Reprograms Microglia Responsivity and Ameliorates Pathological Phenotypes in Alzheimer’s Disease Models. Neuron 97, 1032–1048.e1035. 10.1016/j.neuron.2018.02.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Leyns CEG et al. (2017) TREM2 deficiency attenuates neuroinflammation and protects against neurodegeneration in a mouse model of tauopathy. Proc Natl Acad Sci U S A 114, 11524–11529. 10.1073/pnas.1710311114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Montalvo V. et al. (2013) Environmental factors determine DAP12 deficiency to either enhance or suppress immunopathogenic processes. Immunology 140, 475–482. 10.1111/imm.12158 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Jay TR et al. (2017) Disease Progression-Dependent Effects of TREM2 Deficiency in a Mouse Model of Alzheimer’s Disease. J Neurosci 37, 637–647. 10.1523/jneurosci.2110-16.2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Asai H. et al. (2015) Depletion of microglia and inhibition of exosome synthesis halt tau propagation. Nat Neurosci 18, 1584–1593. 10.1038/nn.4132 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Gratuze M. et al. (2021) Activated microglia mitigate Aβ-associated tau seeding and spreading. J Exp Med 218. 10.1084/jem.20210542 [DOI] [PMC free article] [PubMed]
- 44.Walker AK et al. (2015) Functional recovery in new mouse models of ALS/FTLD after clearance of pathological cytoplasmic TDP-43. Acta Neuropathol 130, 643–660. 10.1007/s00401-015-1460-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Spiller KJ et al. (2018) Microglia-mediated recovery from ALS-relevant motor neuron degeneration in a mouse model of TDP-43 proteinopathy. Nat Neurosci 21, 329–340. 10.1038/s41593-018-0083-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Xie M. et al. (2022) TREM2 interacts with TDP-43 and mediates microglial neuroprotection against TDP-43-related neurodegeneration. Nat Neurosci 25, 26–38. 10.1038/s41593-021-00975-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Xie M. et al. (2022) Microglial TREM2 in amyotrophic lateral sclerosis. Dev Neurobiol 82, 125–137. 10.1002/dneu.22864 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Baker M. et al. (2006) Mutations in progranulin cause tau-negative frontotemporal dementia linked to chromosome 17. Nature 442, 916–919. 10.1038/nature05016 [DOI] [PubMed] [Google Scholar]
- 49.Marschallinger J. et al. (2020) Lipid-droplet-accumulating microglia represent a dysfunctional and proinflammatory state in the aging brain. Nat Neurosci 23, 194–208. 10.1038/s41593-019-0566-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Alzheimer A. et al. (1995) An English translation of Alzheimer’s 1907 paper, “Uber eine eigenartige Erkankung der Hirnrinde”. Clin Anat 8, 429–431. 10.1002/ca.980080612 [DOI] [PubMed] [Google Scholar]
- 51.Pluvinage JV et al. (2019) CD22 blockade restores homeostatic microglial phagocytosis in ageing brains. Nature 568, 187–192. 10.1038/s41586-019-1088-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Liddelow SA et al. (2017) Neurotoxic reactive astrocytes are induced by activated microglia. Nature 541, 481–487. 10.1038/nature21029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Roth TL et al. (2014) Transcranial amelioration of inflammation and cell death after brain injury. Nature 505, 223–228. 10.1038/nature12808 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Mason HD et al. (2021) Glia limitans superficialis oxidation and breakdown promotes cortical cell death after repeat head injury. JCI Insight. 10.1172/jci.insight.149229 [DOI] [PMC free article] [PubMed]
- 55.Selkoe DJ (1991) The molecular pathology of Alzheimer’s disease. Neuron 6, 487–498. 10.1016/0896-6273(91)90052-2 [DOI] [PubMed] [Google Scholar]
- 56.Hardy J. and Allsop D. (1991) Amyloid deposition as the central event in the aetiology of Alzheimer’s disease. Trends Pharmacol Sci 12, 383–388. 10.1016/01656147(91)90609-v [DOI] [PubMed] [Google Scholar]
- 57.Beyreuther K. and Masters CL (1991) Amyloid precursor protein (APP) and beta A4 amyloid in the etiology of Alzheimer’s disease: precursor-product relationships in the derangement of neuronal function. Brain Pathol 1, 241–251. 10.1111/j.17503639.1991.tb00667.x [DOI] [PubMed] [Google Scholar]
- 58.Selkoe DJ and Hardy J. (2016) The amyloid hypothesis of Alzheimer’s disease at 25 years. EMBO Mol Med 8, 595–608. 10.15252/emmm.201606210 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Morgan D. et al. (2000) A beta peptide vaccination prevents memory loss in an animal model of Alzheimer’s disease. Nature 408, 982–985. 10.1038/35050116 [DOI] [PubMed] [Google Scholar]
- 60.Schenk D. et al. (1999) Immunization with amyloid-beta attenuates Alzheimer-disease-like pathology in the PDAPP mouse. Nature 400, 173–177. 10.1038/22124 [DOI] [PubMed] [Google Scholar]
- 61.Bard F. et al. (2000) Peripherally administered antibodies against amyloid beta-peptide enter the central nervous system and reduce pathology in a mouse model of Alzheimer disease. Nat Med 6, 916–919. 10.1038/78682 [DOI] [PubMed] [Google Scholar]
- 62.Britschgi M. et al. (2009) Neuroprotective natural antibodies to assemblies of amyloidogenic peptides decrease with normal aging and advancing Alzheimer’s disease. Proc Natl Acad Sci U S A 106, 12145–12150. 10.1073/pnas.0904866106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Long JM and Holtzman DM (2019) Alzheimer Disease: An Update on Pathobiology and Treatment Strategies. Cell 179, 312–339. 10.1016/j.cell.2019.09.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Rubin R. (2021) Recently Approved Alzheimer Drug Raises Questions That Might Never Be Answered. Jama 326, 469–472. 10.1001/jama.2021.11558 [DOI] [PubMed] [Google Scholar]
- 65.Liu KY and Howard R. (2021) Can we learn lessons from the FDA’s approval of aducanumab? Nat Rev Neurol 17, 715–722. 10.1038/s41582-021-00557-x [DOI] [PubMed] [Google Scholar]
- 66.Howard R. and Liu KY (2020) Questions EMERGE as Biogen claims aducanumab turnaround. Nat Rev Neurol 16, 63–64. 10.1038/s41582-019-0295-9 [DOI] [PubMed] [Google Scholar]
- 67.Alexander GC et al. (2021) Revisiting FDA Approval of Aducanumab. N Engl J Med 385, 769–771. 10.1056/NEJMp2110468 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Marsh SE et al. (2016) The adaptive immune system restrains Alzheimer’s disease pathogenesis by modulating microglial function. Proc Natl Acad Sci U S A 113, E13161325. 10.1073/pnas.1525466113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Kim K. et al. (2021) Therapeutic B-cell depletion reverses progression of Alzheimer’s disease. Nat Commun 12, 2185. 10.1038/s41467-021-22479-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Rojas OL et al. (2019) Recirculating Intestinal IgA-Producing Cells Regulate Neuroinflammation via IL-10. Cell 176, 610–624 e618. 10.1016/j.cell.2018.11.035 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Itagaki S. et al. (1988) Presence of T-cytotoxic suppressor and leucocyte common antigen positive cells in Alzheimer’s disease brain tissue. Neurosci Lett 91, 259–264. 10.1016/0304-3940(88)90690-8 [DOI] [PubMed] [Google Scholar]
- 72.Rogers J. et al. (1988) Expression of immune system-associated antigens by cells of the human central nervous system: relationship to the pathology of Alzheimer’s disease. Neurobiol Aging 9, 339–349. 10.1016/s0197-4580(88)80079-4 [DOI] [PubMed] [Google Scholar]
- 73.Togo T. et al. (2002) Occurrence of T cells in the brain of Alzheimer’s disease and other neurological diseases. J Neuroimmunol 124, 83–92. 10.1016/s0165-5728(01)00496-9 [DOI] [PubMed] [Google Scholar]
- 74.Laurent C. et al. (2017) Hippocampal T cell infiltration promotes neuroinflammation and cognitive decline in a mouse model of tauopathy. Brain 140, 184–200. 10.1093/brain/aww270 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Braak H. and Braak E. (1991) Neuropathological stageing of Alzheimer-related changes. Acta Neuropathol 82, 239–259. 10.1007/bf00308809 [DOI] [PubMed] [Google Scholar]
- 76.Xu H. et al. (2014) Memory deficits correlate with tau and spine pathology in P301S MAPT transgenic mice. Neuropathol Appl Neurobiol 40, 833–843. 10.1111/nan.12160 [DOI] [PubMed] [Google Scholar]
- 77.Sulzer D. et al. (2017) T cells from patients with Parkinson’s disease recognize α-synuclein peptides. Nature 546, 656–661. 10.1038/nature22815 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Lindestam Arlehamn CS et al. (2020) alpha-Synuclein-specific T cell reactivity is associated with preclinical and early Parkinson’s disease. Nat Commun 11, 1875. 10.1038/s41467-020-15626-w [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Joly E. et al. (1991) Viral persistence in neurons explained by lack of major histocompatibility class I expression. Science 253, 1283–1285. 10.1126/science.1891717 [DOI] [PubMed] [Google Scholar]
- 80.Joly E. and Oldstone MB (1992) Neuronal cells are deficient in loading peptides onto MHC class I molecules. Neuron 8, 1185–1190. 10.1016/0896-6273(92)90138-4 [DOI] [PubMed] [Google Scholar]
- 81.Corriveau RA et al. (1998) Regulation of class I MHC gene expression in the developing and mature CNS by neural activity. Neuron 21, 505–520. 10.1016/s08966273(00)80562-0 [DOI] [PubMed] [Google Scholar]
- 82.Neumann H. et al. (1995) Induction of MHC class I genes in neurons. Science 269, 549–552. 10.1126/science.7624779 [DOI] [PubMed] [Google Scholar]
- 83.Zalocusky KA et al. (2021) Neuronal ApoE upregulates MHC-I expression to drive selective neurodegeneration in Alzheimer’s disease. Nat Neurosci 24, 786–798. 10.1038/s41593-021-00851-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Unger MS et al. (2020) CD8(+) T-cells infiltrate Alzheimer’s disease brains and regulate neuronal- and synapse-related gene expression in APP-PS1 transgenic mice. Brain Behav Immun 89, 67–86. 10.1016/j.bbi.2020.05.070 [DOI] [PubMed] [Google Scholar]
- 85.Coque E. et al. (2019) Cytotoxic CD8(+) T lymphocytes expressing ALS-causing SOD1 mutant selectively trigger death of spinal motoneurons. Proc Natl Acad Sci U S A 116, 2312–2317. 10.1073/pnas.1815961116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Galiano-Landeira J. et al. (2020) CD8 T cell nigral infiltration precedes synucleinopathy in early stages of Parkinson’s disease. Brain 143, 3717–3733. 10.1093/brain/awaa269 [DOI] [PubMed] [Google Scholar]
- 87.Brochard V. et al. (2009) Infiltration of CD4+ lymphocytes into the brain contributes to neurodegeneration in a mouse model of Parkinson disease. J Clin Invest 119, 182–192. 10.1172/jci36470 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Herz J. et al. (2015) Therapeutic antiviral T cells noncytopathically clear persistently infected microglia after conversion into antigen-presenting cells. J Exp Med 212, 1153-1169. 10.1084/jem.20142047 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Moseman EA et al. (2020) T cell engagement of cross-presenting microglia protects the brain from a nasal virus infection. Sci Immunol 5. 10.1126/sciimmunol.abb1817 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Matzinger P. (2002) The danger model: a renewed sense of self. Science 296, 301–305. 10.1126/science.1071059 [DOI] [PubMed] [Google Scholar]
- 91.Lester E. et al. (2021) Tau aggregates are RNA-protein assemblies that mislocalize multiple nuclear speckle components. Neuron 109, 1675–1691.e1679. 10.1016/j.neuron.2021.03.026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Unger MS et al. (2018) Doublecortin expression in CD8+ T-cells and microglia at sites of amyloid-β plaques: A potential role in shaping plaque pathology? Alzheimers Dement 14, 1022–1037. 10.1016/j.jalz.2018.02.017 [DOI] [PubMed] [Google Scholar]
- 93.Schetters STT et al. (2017) Neuroinflammation: Microglia and T Cells Get Ready to Tango. Front Immunol 8, 1905. 10.3389/fimmu.2017.01905 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Engelhardt JI et al. (1993) Lymphocytic infiltrates in the spinal cord in amyotrophic lateral sclerosis. Arch Neurol 50, 30–36. 10.1001/archneur.1993.00540010026013 [DOI] [PubMed] [Google Scholar]
- 95.Rosenkranz D. et al. (2007) Higher frequency of regulatory T cells in the elderly and increased suppressive activity in neurodegeneration. J Neuroimmunol 188, 117–127. 10.1016/j.jneuroim.2007.05.011 [DOI] [PubMed] [Google Scholar]
- 96.Saresella M. et al. (2010) PD1 negative and PD1 positive CD4+ T regulatory cells in mild cognitive impairment and Alzheimer’s disease. J Alzheimers Dis 21, 927–938. 10.3233/jad-2010-091696 [DOI] [PubMed] [Google Scholar]
- 97.Ciccocioppo F. et al. (2019) The Characterization of Regulatory T-Cell Profiles in Alzheimer’s Disease and Multiple Sclerosis. Sci Rep 9, 8788. 10.1038/s41598-01945433-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Beers DR et al. (2011) Endogenous regulatory T lymphocytes ameliorate amyotrophic lateral sclerosis in mice and correlate with disease progression in patients with amyotrophic lateral sclerosis. Brain 134, 1293–1314. 10.1093/brain/awr074 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Larbi A. et al. (2009) Dramatic shifts in circulating CD4 but not CD8 T cell subsets in mild Alzheimer’s disease. J Alzheimers Dis 17, 91–103. 10.3233/jad-2009-1015 [DOI] [PubMed] [Google Scholar]
- 100.Baruch K. et al. (2015) Breaking immune tolerance by targeting Foxp3(+) regulatory T cells mitigates Alzheimer’s disease pathology. Nat Commun 6, 7967. 10.1038/ncomms8967 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Brett BL et al. (2022) Traumatic Brain Injury and Risk of Neurodegenerative Disorder. Biol Psychiatry 91, 498–507. 10.1016/j.biopsych.2021.05.025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Crane PK et al. (2016) Association of Traumatic Brain Injury With Late-Life Neurodegenerative Conditions and Neuropathologic Findings. JAMA Neurol 73, 1062-1069. 10.1001/jamaneurol.2016.1948 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Mackay DF et al. (2019) Neurodegenerative Disease Mortality among Former Professional Soccer Players. N Engl J Med 381, 1801–1808. 10.1056/NEJMoa1908483 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Stein TD et al. (2015) Beta-amyloid deposition in chronic traumatic encephalopathy. Acta Neuropathol 130, 21–34. 10.1007/s00401-015-1435-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Uryu K. et al. (2007) Multiple proteins implicated in neurodegenerative diseases accumulate in axons after brain trauma in humans. Exp Neurol 208, 185–192. 10.1016/j.expneurol.2007.06.018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Corps KN et al. (2015) Inflammation and neuroprotection in traumatic brain injury. JAMA Neurol 72, 355–362. 10.1001/jamaneurol.2014.3558 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Simard JM et al. (2007) Brain oedema in focal ischaemia: molecular pathophysiology and theoretical implications. Lancet Neurol 6, 258–268. 10.1016/s1474-4422(07)70055-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Russo MV and McGavern DB (2016) Inflammatory neuroprotection following traumatic brain injury. Science 353, 783–785. 10.1126/science.aaf6260 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Lou N. et al. (2016) Purinergic receptor P2RY12-dependent microglial closure of the injured blood-brain barrier. Proc Natl Acad Sci U S A 113, 1074–1079. 10.1073/pnas.1520398113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Mastorakos P. et al. (2021) Temporally distinct myeloid cell responses mediate damage and repair after cerebrovascular injury. Nat Neurosci 24, 245–258. 10.1038/s41593-02000773-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Mastorakos P. et al. (2021) Antimicrobial immunity impedes CNS vascular repair following brain injury. Nat Immunol 22, 1280–1293. 10.1038/s41590-021-01012-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Kesinger MR et al. (2015) Hospital-acquired pneumonia is an independent predictor of poor global outcome in severe traumatic brain injury up to 5 years after discharge. J Trauma Acute Care Surg 78, 396–402. 10.1097/ta.0000000000000526 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Kourbeti IS et al. (2012) Infections in traumatic brain injury patients. Clin Microbiol Infect 18, 359–364. 10.1111/j.1469-0691.2011.03625.x [DOI] [PubMed] [Google Scholar]
- 114.Vermeij FH et al. (2009) Stroke-associated infection is an independent risk factor for poor outcome after acute ischemic stroke: data from the Netherlands Stroke Survey. Cerebrovasc Dis 27, 465–471. 10.1159/000210093 [DOI] [PubMed] [Google Scholar]
- 115.Arneson D. et al. (2018) Single cell molecular alterations reveal target cells and pathways of concussive brain injury. Nat Commun 9, 3894. 10.1038/s41467-018-062220 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Liu C. et al. (2021) Transcriptional profiling of microglia in the injured brain reveals distinct molecular features underlying neurodegeneration. Glia 69, 1292–1306. 10.1002/glia.23966 [DOI] [PubMed] [Google Scholar]
- 117.Makinde HM et al. (2020) Microglia Adopt Longitudinal Transcriptional Changes After Traumatic Brain Injury. J Surg Res 246, 113–122. 10.1016/j.jss.2019.08.024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Omalu BI et al. (2005) Chronic traumatic encephalopathy in a National Football League player. Neurosurgery 57, 128–134; discussion 128–134. 10.1227/01.neu.0000163407.92769.ed [DOI] [PubMed] [Google Scholar]
- 119.Keene CD et al. (2018) First confirmed case of chronic traumatic encephalopathy in a professional bull rider. Acta Neuropathol 135, 303–305. 10.1007/s00401-017-1801-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.McMillan TM et al. (2017) Long-term health outcomes after exposure to repeated concussion in elite level: rugby union players. J Neurol Neurosurg Psychiatry 88, 505-511. 10.1136/jnnp-2016-314279 [DOI] [PubMed] [Google Scholar]
- 121.Shively SB et al. (2016) Characterisation of interface astroglial scarring in the human brain after blast exposure: a post-mortem case series. Lancet Neurol 15, 944–953. 10.1016/s1474-4422(16)30057-6 [DOI] [PubMed] [Google Scholar]
- 122.Hsieh CL et al. (2014) CCR2 deficiency impairs macrophage infiltration and improves cognitive function after traumatic brain injury. J Neurotrauma 31, 1677–1688. 10.1089/neu.2013.3252 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Stirling DP et al. (2009) Depletion of Ly6G/Gr-1 leukocytes after spinal cord injury in mice alters wound healing and worsens neurological outcome. J Neurosci 29, 753–764. 10.1523/jneurosci.4918-08.2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Jassam YN et al. (2017) Neuroimmunology of Traumatic Brain Injury: Time for a Paradigm Shift. Neuron 95, 1246–1265. 10.1016/j.neuron.2017.07.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Russo MV et al. (2018) Distinct myeloid cell subsets promote meningeal remodeling and vascular repair after mild traumatic brain injury. Nat Immunol 19, 442–452. 10.1038/s41590-018-0086-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Wattananit S. et al. (2016) Monocyte-Derived Macrophages Contribute to Spontaneous Long-Term Functional Recovery after Stroke in Mice. J Neurosci 36, 4182–4195. 10.1523/JNEUROSCI.4317-15.2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Liu C. et al. (2016) Macrophages Mediate the Repair of Brain Vascular Rupture through Direct Physical Adhesion and Mechanical Traction. Immunity 44, 1162–1176. 10.1016/j.immuni.2016.03.008 [DOI] [PubMed] [Google Scholar]
- 128.Shechter R. et al. (2009) Infiltrating blood-derived macrophages are vital cells playing an anti-inflammatory role in recovery from spinal cord injury in mice. PLoS Med 6, e1000113. 10.1371/journal.pmed.1000113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.MARTLAND HS (1928) PUNCH DRUNK. Journal of the American Medical Association 91, 1103–1107. 10.1001/jama.1928.02700150029009 [DOI] [Google Scholar]
- 130.McKee AC et al. (2016) The first NINDS/NIBIB consensus meeting to define neuropathological criteria for the diagnosis of chronic traumatic encephalopathy. Acta Neuropathol 131, 75–86. 10.1007/s00401-015-1515-z [DOI] [PMC free article] [PubMed] [Google Scholar]