

ABSTRACT
Tumor-specific T cells likely underpin effective immune checkpoint-blockade therapies. Yet, most studies focus on Treg cells and CD8+ tumor-infiltrating lymphocytes (TILs). Here, we study CD4+ TILs in human lung and colorectal cancers and observe that non-Treg CD4+ TILs average more than 70% of total CD4+ TILs in both cancer types. Leveraging high dimensional analyses including mass cytometry, we reveal that CD4+ TILs are phenotypically heterogeneous, within each tumor and across patients. Consistently, we find different subsets of CD4+ TILs showing characteristics of effectors, tissue resident memory (Trm) or exhausted cells (expressing PD-1, CTLA-4 and CD39). In both cancer types, the frequencies of CD39– non-Treg CD4+ TILs strongly correlate with frequencies of CD39– CD8+ TILs, which we and others have previously shown to be enriched for cells specific for cancer-unrelated antigens (bystanders). Ex-vivo, we demonstrate that CD39– CD4+ TILs can be specific for cancer-unrelated antigens, such as HCMV epitopes. Overall, our findings highlight that CD4+ TILs can also recognize cancer-unrelated antigens and suggest measuring CD39 expression as a straightforward way to quantify or isolate bystander CD4+ T cells.
KEYWORDS: CD4, CD8, CD39, TIL, HCMV, cancer, tumor, infiltrating, bystander
Introduction
Numerous studies have established the importance of T cells in controlling cancer.1 Nonetheless, tumors can escape this immune surveillance by diverse mechanisms.2 As various forms of cancer therapy stand, immunotherapy is rapidly evolving and has proved to effectively restore T cell-mediated immune responses. Strategies include immune checkpoint blocking receptors (i.e. anti-CTLA-4 or anti-PD1),3 autologous T cell transfer,4 as well as therapeutic cancer vaccines.5 However, the efficacy of these therapies is unpredictable and only some patients successfully respond.6,7 Therefore, a better understanding of T cell biology – CD8 and CD4 – in the tumor microenvironment is crucial to improve cancer therapies. Recently, we showed in the context of human colorectal and lung cancers that CD8+ tumor-infiltrating lymphocytes (TILs) are not only specific for tumor antigens but can also recognize a wide range of cancer-unrelated epitopes (called bystander CD8+ TILs).8 We suggested measuring CD39 expression as a straightforward way to quantify or isolate bystander CD8+ T cells as well as being a potential biomarker for immunotherapy.8–10 These observations are now confirmed in different cancer types.11–14
Although CD4+ TILs are also involved in tumor responses,15 many studies focused on the role of FoxP3-expressing regulatory T (Treg) cells in cancer.16–18 Treg cells suppress tumor immunity by various mechanisms including: 1) Disruption of the metabolic pathway (i.e. CD39 expression), 2) Modulation of dendritic cells function (i.e. CTLA-4 expression), 3) Production of anti-inflammatory molecules (i.e. IL-10, TGFβ), 4) Induction of apoptosis.19 Abundant Treg infiltration into tumors is strongly associated with poor prognosis in multiple cancer types.17,20 Because of their deleterious role, several molecules have been developed to specifically target these cells in human cancers (e.g. anti-CTLA-4, anti-CD25).21–25
Importantly, a large proportion of CD4+ TILs consist of non-Treg cells. Studies in mice have shown that these cells play a key role in anti-tumor responses.15,26 By producing IFNγ, they induce an up-regulation of MHC class I and II expression by tumor cells and dendritic cells (DC).27 Production of IFNγ by CD4+ TILs also induce expression of chemokines supporting homing of CD8+ T cells to the tumor site (e.g. CXCL10).27 Activated CD4+ T cells express CD40L by which they can activate DC, and support CD8+ T cells priming and memory formation.27 Furthermore, they can have a cytotoxic function and directly kill tumor cells as well.28 In humans, a number of case reports identified tumor-specific CD4+ T cells targeting neoantigens in various cancer types.29–31 Based on these observations, developing CD4-based therapeutic vaccination and/or adoptive cell therapies by targeting tumor-specific CD4+ T cells seems essential.26,32–35 The limited number of tools that allow studying CD4+ TILs (i.e. MHC class II tetramers, in-vitro assays) had so far made this population poorly characterized as compared to CD8+ TILs and Treg cells. Uncovering the role of these cells in the tumor microenvironment would thus help design new strategies to manipulate them and improve immunotherapy efficiency. A recent work studied antigen-specific CD4+ T cells in human papillomavirus (HPV)-induced cancers. These cancers express the tumor-specific proteins E6 and E7 from HPV, allowing identification of tumor-specific CD4+ T cells in a large cohort of patients. The authors showed that CD39+ CD4+ TILs are almost exclusively composed of tumor-specific T cells in these cancers.36 In our study, we comprehensively profiled CD4+ TILs in human colorectal and lung cancers using mass-cytometry and in-vitro antigens stimulation assay. Our findings highlight that non-Treg CD4+ TILs can be divided into two distinct populations. The first one expressing CD39 is exhausted and shows hallmarks of tumor-specific cells. The second one lacking CD39 expression is functional and can recognize cancer-unrelated antigens, just as observed for CD39– CD8+ TILs.8 Taken together, we hypothesize that measuring CD39 expression is a straightforward way to quantify or isolate bystander CD4+ TILs.
Results
Tumor infiltrating CD4+ T cells are composed of a majority of non-Treg cells
To study CD4+ T cells, we developed a mass cytometry panel consisting of 38 heavy metal-labeled antibodies that include markers of tissue residency, stimulatory and inhibitory receptors (Figure 1a and Table S1). We profiled two cohorts of patients, one with lung cancer and another with colorectal cancer. The composition of CD4+ T cells was assessed using a Uniform Manifold Approximation and Projection (UMAP) based on protein expression. UMAP is a dimension reduction algorithm that performs a pair-wise comparison of the cellular phenotypes to optimally plot similar cells close to each other.37 For our analysis, surface proteins, or dimensions, were reduced into two dimensions (UMAP1 and UMAP2). We first compared CD4+ T cells isolated from the PBMC and tumor of the same patient, which allowed for an accurate comparison between the two tissues. We observed that CD4+ T cells derived from blood and tumor were localized in different areas of the two-dimensional UMAP plot, indicating distinct phenotypes of CD4+ T cells between blood and tumor (Figure 1b and 1c). This difference was mainly driven by expression of tissues-resident markers (CD103, CD69)38,39 and inhibitory/exhaustion receptors (PD-1, CD39) in the tumor tissue (Figure 1b, 1c and 1d). Next, we focused our analysis on the tumor tissue. We observed that the majority of CD3+ TILs were composed of CD4+ TILs (Figure 1e), with only 35% and 24% of CD8+ TILs in lung cancer (n = 28) and colorectal cancer (n = 50), respectively. Of note, we also observed the presence of double negative (i.e. CD4 – CD8–) T cells infiltrating tumor tissue in both cohorts (Supplementary Figure 1a). Within CD4+ TILs, we distinguished Treg and non-Treg cells based on the expression of FoxP3 (figure 1f). Non-Treg CD4+ TILs accounted for a higher proportion of CD4+ TILs as compared to Treg cells, with a mean frequency of 78.8% vs. 19% in lung cancer and 66% vs. 35% in colorectal cancer (Figure 1g). In both cohorts, we did not observe any correlation between the frequencies of Treg or Non-Treg populations among CD4+ TILs with clinical parameters, such as overall survival, histological grade, pathological group, gender, smoking status or EGFR mutation status (Supplementary Figure 2).
Figure 1.
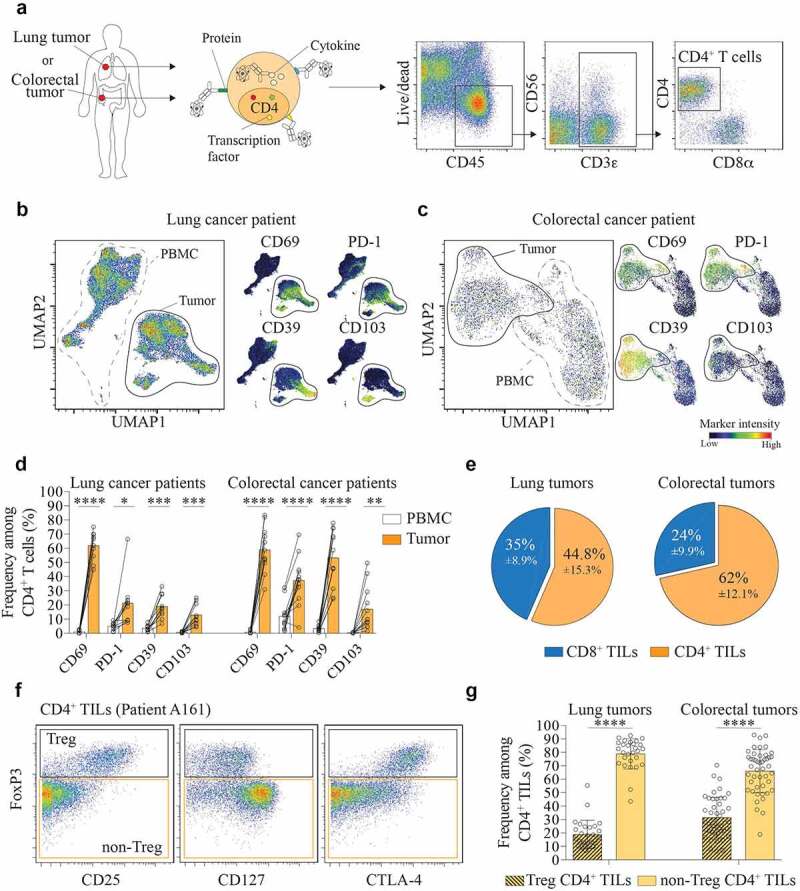
CD4+ TILs are composed of a majority of non-Treg cells.
(a) Identification of CD4+ T cells by mass-cytometry (Live CD45+CD3+CD4+). Representative dot plot from one colorectal tumor sample. (b) UMAP plot on CD4+ T cells from paired blood and tumor samples of a lung cancer patient (left panel). Normalized expression intensities of CD69, CD103, PD-1, and CD39 were calculated and overlaid on the UMAP plot (right panel). (c) UMAP plot on CD4+ T cells from paired blood and tumor samples of a colorectal cancer patient (left panel). Normalized expression intensities of CD69, CD103, PD-1, and CD39 were calculated and overlaid on the UMAP plot (right panel). (d) Expression of CD69, CD103, PD-1, and CD39 by CD4+ T cells in PBMC (white) and tumor (orange). Data are from paired samples of n = 10 (lung cancer) and n = 12 (colorectal cancer) biologically independent individuals. Means ± SD, Paired t test – two-tailed. *p ≤ 0.05, **p ≤ 0.01, ***p ≤ 0.001, ****p ≤ 0.0001. (e) Frequency of CD4+ and CD8+ TILs among total CD3+ TILs in lung tumors (left) and colorectal tumors (right). Data are from tumor samples of n = 28 (lung tumors) and n = 51 (colorectal tumors) biologically independent individuals. Means ± SD. (f) Gating strategy to distinguish between Treg (Live CD45+CD3+CD4+Foxp3+) and Non-Treg CD4+ TILs (Live CD45+CD3+CD4+FoxP3–). Representative dot plot from one colorectal tumor sample. (g) Frequency of Non-Treg and Treg CD4+ TILs in lung tumors (left) and colorectal tumors (right). Data are from tumor samples of n = 28 (lung tumors) and n = 51 (colorectal tumors) biologically independent individuals. Means ± SD, Paired t test – two-tailed. ****p ≤ 0.0001
Figure 2.
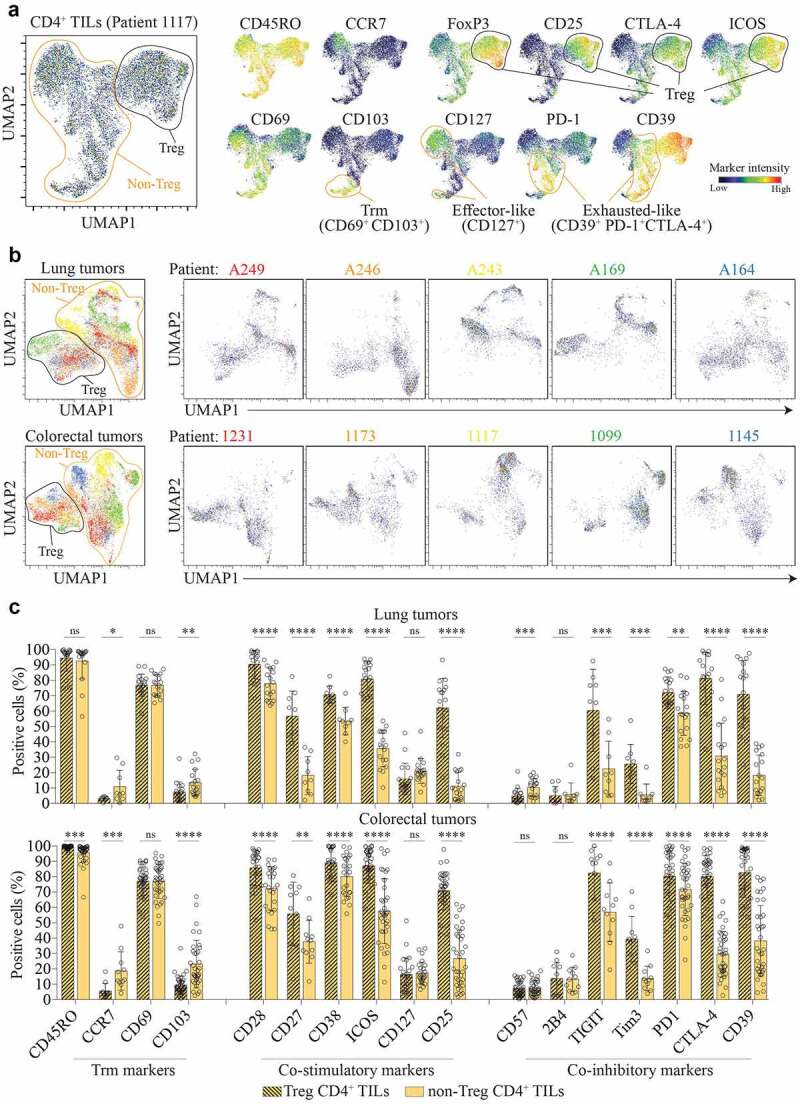
CD4+ TILs are heterogeneous within the tumor and across patients.
(a) UMAP plot on CD4+ TILs from the tumor of a colorectal cancer patient (1117) (left panel). Normalized expression intensities of CD45RO, CCR7, CD69, CD103, CD25, FoxP3, CTLA-4, CD127, ICOS, PD-1, and CD39 were calculated and overlaid on the UMAP plot (right panel). (b) UMAP plot on CD4+ TILs isolated from lung tumors (top panel) or colorectal tumors (bottom panel). Mass cytometry acquisition and UMAP analysis were performed simultaneously on five different patients. Patient identifiers refer to individual patients. n = 5 patients for each cancer type. (c) Expression of selected immune markers by Treg (black stripes) and non-Treg (yellow) CD4+ TILs. Data are from lung tumor samples of n = 28 (lung tumors) and n = 51 (colorectal tumors) biologically independent individuals. Means ± SD, Paired t test – two-tailed. *p ≤ 0.05, **p ≤ 0.01, ***p ≤ 0.001, ****p ≤ 0.0001
These results highlight that the CD4+ T cells infiltrating tumor tissue are phenotypically different from the CD4+ T cells in the blood. Furthermore, the majority of CD4+ T cells in tumor tissue are composed of non-Treg cells, supporting the importance of studying this population in tumor immune response.
Tumor infiltrating CD4+ T cells are heterogeneous with a contrasted phenotypic profile
We first performed an UMAP analysis to explore the heterogeneity of CD4+ TILs at the individual level. We distinguished several cell clusters, illustrating a broad phenotypic heterogeneity (Figure 2a). We first identified a cell population with Treg cells features (FoxP3+, CD25+, ICOS+, CD127–, CTLA-4+) (Figure 2a). Among the non-Treg CD4+ TILs, we observed presence of multiple cell clusters expressing co-stimulatory and co-inhibitory markers at variable intensities. For instance, these cells expressed CD69, excluding a blood contamination, but only a small fraction expressed the tissue resident memory (Trm) marker CD103. CD127 (IL-7R), which promotes survival of effector cells, is only found in some of the clusters. Within the cell clusters negative for CD127, we detected cells harboring hallmarks of exhaustion (called Exhausted-like) (Figure 2a). These cell clusters expressed differential levels of co-inhibitory receptors such as PD-1, CTLA-4 and CD39, suggesting an ongoing antigen exposure at the tumor site (Figure 2a and Supplementary Figure 1b). We next performed the UMAP analysis on two sets of data acquired in parallel to measure the heterogeneity between patients (one set of five lung tumors and another set of five colorectal tumors) (Figure 2b). We identified two main cell populations with or without Treg cell features (Figure 2b). Again, we observed several clusters of cells for each patient sample, corroborating the CD4+ TIL heterogeneity within individual tumor. Moreover, for both non-Treg and Treg CD4+ TILs, we noted that cluster positions differed for each patient, indicating a high degree of heterogeneity across patients as well. We then measured expressions of Trm markers, co-stimulatory and inhibitory receptors on Treg and Non-Treg cells in both cohorts. As extensively described in the literature, Treg cells expressed hallmarks of activated/exhausted cells in both cohorts with high expression levels of CD39, CTLA-4, ICOS, TIGIT, PD-1, CD25.40 All non-Treg CD4+ TILs expressed the memory marker CD45RO (>95% in both cohorts) and many of them expressed the activation/tissue residency marker CD69 (>75% in both cohorts). Interestingly, the other residency marker CD103, which is highly expressed by CD8+ TILs,8,41 was only found on a small fraction of CD4+ TILs (<23% in both cohorts) (Figure 2c). Expression of stimulatory markers and inhibitory receptors varied greatly between patients of the same cohort, which explain the important phenotypic diversity observed for CD4+ TILs between patients (Figure 2b). Non-Treg CD4+ TILs expressed co-stimulatory receptors, such as CD28, CD38, ICOS but only a small fraction expressed CD127 (21% in lung and 17.1% in colorectal cancer). Interestingly, some non-Treg CD4+ TILs expressed CD25 (11.1% in lung and 26.7% in colorectal cancer), suggesting that the use of CD25 and CD127 alone to identify Treg cells in the context of tumor infiltrates could lead to a contamination by non-Treg CD4+ TILs (i.e. Foxp3–). More interestingly, non-Treg CD4+ TILs also expressed hallmarks of exhausted cells at different levels across patients. Expression of inhibitory receptors associated with chronic antigen stimulation such as TIGIT (22.5% and 56.9%), PD-1 (58.5% and 71.6%) and CTLA-4 (30.7% and 29.6%) in both lung and colorectal cancers suggested a role for these cells in tumor immunity. Of note, frequencies of CD39+ non-Treg CD4+ TILs (38.2% and 20.2% for lung and colorectal cancer, respectively) were very heterogeneous, ranging from 4.6% to 70% across patients.
Overall, these data showed a high degree of phenotypic diversity among non-Treg CD4+ TILs within individual tumors and across patients. Phenotypic analysis showed that both effectors and exhausted cells were found in the same tumor.
CD39 expression defines two populations of non-Treg CD4+ TILs
As we and others have shown that cancer-unrelated bystander CD8+ TILs are abundant in cancer and phenotypically distinct (i.e. lack of CD39 expression),8,11,12 we explored whether this could be echoed on non-Treg CD4+ TILs. We first measured the expression of CD39 by non-Treg CD4+ TILs. Strikingly, we observed an important heterogeneity for CD39 expression across both cohorts, with patients showing up to 95% of CD39– non-Treg CD4+ TILs and others showing less than 10% (Figure 3a, 3b). We hypothesized that if CD39– non-Treg CD4+ TILs were bystander cells, they should express a different phenotypic and functional profile. By assessing inhibitory receptors associated with chronic antigen stimulation, we observed a significantly lower expression of TIGIT, Tim3, CTLA-4, and PD-1 on CD39– non-Treg CD4+ TILs as compared to theCD39+ counterparts (Figure 3c, 3d). These differences were also observed in markers intensity expression, with a significantly increased intensity of CTLA-4 and PD-1 by CD39+ non-Treg CD4+ TILs (Figure 3e). Furthermore, frequency of proliferating cells (i.e. Ki67+) was higher on CD39+ non-Treg CD4+ TILs, indicating a local T cell activation (Supplementary Figure 4a and 4b). To confirm these observations, we assessed functional properties of both populations. After in-vitro stimulation using PMA/Ionomycin, we measured production of IFNγ, IL-2 and TNFα. CD39– non-Treg CD4+ TILs produced more TNFα compared to their CD39+ counterparts, suggesting that these cells are more functional and less exhausted (Figure 3c and 3f and Supplementary Figure 4c). Finally, we performed a correlation analysis comparing frequencies of CD39– non-Treg CD4+ TILs with CD39– CD8+ TILs of the same patient (Figure 3g). In both tumor types, frequencies of bystander CD8+ TILs strongly correlate with frequencies of CD39– non-Treg CD4+ TILs.
Figure 3.

CD39– and CD39+ non-Treg CD4+ TILs are phenotypically and functionally distinct.
(a) Dot plots showing differential expression of CD39 vs. Foxp3 in CD4+ TILs of four colorectal cancer patients. (b) Frequency of CD39– and CD39+ Non-Treg CD4+ TILs in lung and colorectal cancers. Data are from tumor samples of n = 28 (lung) and n = 51 (colorectal) biologically independent individuals. Means ± SD, Paired t test – two-tailed. **p ≤ 0.01, ****p ≤ 0.0001. (c) Representative staining showing expression of CD39 vs. inhibitory receptors (CTLA-4 and PD-1) or cytokine TNFα on Non-Treg CD4+ TILs in colorectal cancer. (d) Expression of inhibitory receptors TIGIT, Tim3, CTLA-4 and PD-1 by CD39– (yellow) and CD39+ (orange) non-Treg CD4+ TILs. Data are from tumor samples of n = 18 (lung) and n = 36 (colorectal) biologically independent individuals. Means ± SD, Paired t test – two-tailed. ***p ≤ 0.001, ****p ≤ 0.0001. (e) Mean expression intensities of CTLA-4 and PD-1 by CD39–(yellow) and CD39+ (orange) non-Treg CD4+ TILs. Data are from tumor samples of n = 18 (lung) and n = 36 (colorectal) biologically independent individuals. Means ± SD, Paired t test – two-tailed. **p ≤ 0.01, ****p ≤ 0.0001. (f) Expression of TNFα by CD39– (yellow) and CD39+ (orange) non-Treg CD4+ TILs. Data are from tumor samples of n = 8 (lung) and n = 11 (colorectal) biologically independent individuals. Means ± SD, Paired t test – two-tailed. *p ≤ 0.05, **p ≤ 0.01. (g) Correlation between CD39– non-Treg CD4+ TILs and CD39– CD8+ TILs in lung cancer (left panel) and colorectal cancer (right panel). Data are from tumor samples of n = 23 (lung) and n = 35 (colorectal) biologically independent individuals. Pearson correlation test. ***p ≤ 0.001, ****p ≤ 0.0001
Figure 4.

Bystander CD4+ TILs infiltrate tumor and lack expression of CD39.
(a) Schematic of the in-vitro activation induced marker (AIM) assay to assess the reactivity of CD4+ TILs to HCMV pp65 epitopes as previously described (see methods,42 APC and CD4+ T cells were isolated from the same donor). (b) Representative staining showing expression of CD40L and CD69 by CD4+ T cells from PBMC or tumor sample after 18 h stimulation with HCMV pp65 peptides pool (see methods). (c) Frequency of HCMV (pp65)-specificCD4+ T cells in PBMC and tumor. Data are from n = 6 biologically independent individuals. Means ± SD, Wilcoxon signed-rank test. *p ≤ 0.05. (d) Fold change of HCMV (pp65)-specificCD4+ T cells frequency in PBMC and tumor compared to unstimulated controls. Data are from n = 6 biologically independent individuals. Means ± SD, Wilcoxon signed-rank test. *p ≤ 0.05. (e) Dot plots showing expression of CD39 by HCMV-specificCD4+ (orange) vs. total CD4+ (gray) TILs in lung cancer patients. Data are from n = 3 biologically independent individuals.
These data highlight that two distinct populations of non-Treg CD4+ T cells infiltrate tumors. The first one expressing CD39 is exhausted and shows hallmarks of tumor-specific cells (see discussion). The second one lacking CD39 expression is functional, and their infiltration to the tumor site correlates with the presence of CD39– CD8+ TILs.
Cancer-unrelated non-Treg CD4+ TILs lack CD39 expression
To confirm our hypothesis that bystander CD4+ TILs are present at the tumor site, we studied the antigen specificity of these cells. We optimized an activation-induced marker (AIM) assay to assess activation of CD4+ TILs stimulated with cancer-unrelated epitopes. From our previous observations showing the presence of HCMV-specific CD8+ TILs,8 we decided to use an HCMV pp65 peptide pool in our experiments (Figure 4a). By measuring the up-regulation of both CD40L and CD69 after peptide pool stimulation, we would be able to identify antigen-specific cells.42 As expected, we detected presence of HCMV-specific CD4+ T cells in PBMC (Figure 4b). Interestingly, we also detected presence of HCMV-specific CD4+ T cells in tumor tissues (Figure 4b). When compared with the negative control (PBMC incubated with DMSO), we observed a significant higher frequency and fold change of HCMV-specific cells in CD4+ TILs, confirming that similarly to CD8+ TILs, cancer-unrelated CD4+ T cells infiltrate tumor tissues (Figure 4c and d). Next, we measured the expression of CD39 on these bystander CD4+ TILs and found none (Figure 4e). Of note, the cells were stimulated during 18 hours and CD39 is considered as a late activation marker only up-regulated by CD4+ T cells after 48 h stimulation.43 In order to exclude any expression of CD39 by CD4+ T cells induced upon stimulation, cells were stained with CD39 prior to peptides stimulation (See Methods). As an internal control of this experiment, we observed CD39 expression for others populations of CD4+ TILs, confirming the validity of our assay (Figure 4e).
These observations demonstrate that cancer-unrelated CD4+ T cells infiltrate tumor tissues, and measuring CD39 expression could be a straightforward way to identify this population in tumor tissue (Supplementary Figure 5).
Discussion
Since the late 1990s, cancer research has highlighted the central role of T cells in antitumor immunity.44 Notably, because of their ability to directly kill tumor cells and our better understanding of MHC class I tumor antigens, the role of CD8+ T cells has come under scrutiny.45–47 In the meantime, many studies have also elucidated the detrimental role of CD4+ Treg cells in antitumor immunity and brought these cells at the center stage as immunotherapy targets.48 Our work brings to light that consistently, a major fraction of the T cells infiltrating the tumor are composed of non-Treg CD4+ T cells in both colorectal and lung cancers. Similar observation has been previously made in breast cancer.18 In lymph nodes, non-Treg CD4+ T cells support the priming of tumor-specific CD8+ T cells.49 These cells enhance the activity of CD8+ TILs by producing cytokines (i.e. TNFα, IFNγ) but can also act as effectors by eliminating tumor cells in a direct or indirect way.15,50 Contrary to the MHC class I molecule that is expressed by tumor cells and presents tumor antigens to CD8+ TILs, MHC class II is usually not expressed (or at low levels) by human tumor cells.51 However, we clearly observe an up-regulation of markers associated with chronic antigen exposure in non-Treg CD4+ TILs, such as PD-1 and CTLA-4, indicating that these cells can be activated at the tumor site as well.3 Of note, inhibitory receptors can be transiently expressed on functional T cells during activation (e.g. PD-1, TIGIT).52 However, in the context of cancer, accumulation of inhibitory receptors is associated with exhaustion of T cells due to chronic antigenic stimulation.52,53 We hypothesize that this activation could be mediated by antigen-presenting cells, such as macrophages and dendritic cells. The distinct phenotype of non-Treg CD4+ TILs observed across patients, especially regarding expression of inhibitory receptors, could be explained by tumor-intrinsic factors shaping the individual tumor immune microenvironment.54 Furthermore, we also observe heterogeneity of non-Treg CD4+ TILs within the same tumor, with cells showing an effector phenotype and others expressing hallmarks of chronic antigen stimulation, notably CD39.
CD39 is an enzyme that converts the extracellular ATP to AMP. In turn, CD73 converts AMP into adenosine, which is showed to possess immunosuppressive activity.55 Thus, conversion of extracellular ATP into adenosine by CD39 leads to inhibition of CD4, CD8, NK cell function, decreased phagocytosis and antigen presentation activities by macrophages and dendritic cells.56–58 Although we used in our study CD39 to stratify CD4+ TILs, it would be interesting to measure and study the role of CD73 on CD4+ TILs, as reported previously between effector CD73+ CD4+ T cells and CD39+ Treg in breast cancer.58 Widely reported in Treg-related literature, CD39 has also been described on HIV-, HBV- and tumor-specific CD8+ T cells as a marker expressed during chronic antigen stimulation.8,59–61 However, only a few groups have characterized this marker on non-Treg CD4+ TILs. In-vitro, CD39 is expressed on non-Treg CD4+ TILs after activation and on Listeria-specific CD4+ T cells after infection.43 Interestingly, a pioneer study reported an increased frequency of pathogenic CD39+ non-Treg CD4+ T cells in the peripheral blood of patients with renal allograft rejection.62 As previously observed for CD8+ TILs, CD39 could be a useful marker to identify tumor-specific CD4+ T cells within the tumor microenvironment as well. Similarly, although we did not identify any correlation between clinical features and frequencies of CD4+ TILs subsets (Supplementary Figures 2 and 3), it would be interesting to assess the presence of CD39– and CD39+ CD4+ TILs by immunohistochemistry on responder vs. non-responder patients treated with immunotherapy, as performed previously for CD39+ CD8+ TILs.9 Further studies will be needed to confirm this hypothesis and to better understand the regulation of CD39 on non-Treg CD4+ TILs.
By investigating the antigen specificity of CD4+ TILs, we detected cancer-unrelated bystander CD4+ TILs. These HCMV-specific cells lack CD39 expression, which mirrors our previous observations with CD39– CD8+ TILs specific for cancer-unrelated antigens (HCMV, EBV, and Flu).8 Future studies will be needed to confirm the reactivity of CD4+ TILs to EBV or Flu viruses. The observation of a possible correlated CD4 and CD8 T cell response in the tumors would be consistent with the notion that tumor-specific CD4 responses are also required for the induction of tumor-specific CD8 response as recently illustrated in mice.26 Of note, in some patients, up to 95% of non-Treg CD4+ TILs lack CD39 expression. Taken together, this observation could suggest that an important fraction of CD4+ (and CD8+) TILs is not tumor-specific in some patients. This hypothesis could explain, along with other factors, the absence of response in many patients treated with immunotherapy.63
In this study, we have been unable to assess the presence of tumor-specific CD4+ T cells. Although possible, studying tumor-specific CD4+ T cells remains technically challenging due to the difficulty to identify tumor epitopes for CD4+ T cells (i.e. neoantigens).64 Nevertheless, bystander CD4+ (and CD8+) TILs should not be considered as passive cells in the tumor microenvironment. Several reports have highlighted their role in modulating disease severity upon TCR-independent activation.65,66 Because of their TCR specificity for known viral epitopes, virus-specific bystander TILs could also be specifically targeted by therapeutic approaches to produce cytokines and enhance anti-tumor response.14
Overall, our findings highlight that non-Treg CD4+ TILs cells represent one of the main lymphocytes recruited at the tumor site as well as a potential target of interest for immunotherapy.
Methods
Human samples
PBMC and tumor samples were obtained from patients with colorectal cancer or lung cancer. The use of human tissues was approved by the appropriate institutional research boards, A*STAR and the Singapore Immunology Network, Singapore.
Cell isolation
Samples were prepared as previously described.67 In brief, tissues were mechanically dissociated into small pieces and incubated at 37°C for 15–40 min in DMEM + collagenase IV (1 mg/ml) + DNAse (15 μg/ml). Digestion was stopped by addition of RPMI containing 5% FBS. Dissociated tissues were filtered and washed in RPMI + 5% FBS + DNAse (15 μg/ml). All samples were cryopreserved in 90% FBS + 10% DMSO and stored in liquid nitrogen.
Mass-cytometry staining
Samples were stained as previously described.67,68 In brief, antibody conjugation was performed according to the protocol provided by Fluidigm (see Table S1 for clone list and metals). Prior to surface staining, cells were stained with Cisplatin (viability marker) at 5 µM in PBS for 5 min. Cells were then stained in PBS + 0.5% BSA buffer with surface antibodies at 4°C for 15 min. After two washing steps, cells were fixed in FoxP3 fixation buffer (eBioscience – 00-5521-00) for 30 min at 4°C. After washing in permeabilization buffer, cells were stained with biotinylated FoxP3 during 30 min at 4°C in permeabilization buffer. Cells were washed and stained with a heavy metal-labeled streptavidin for 30 min at 4°C in permeabilization buffer. After two washing steps, cells were fixed in 2% PFA overnight. Prior to CyTOF acquisition, cells were stained for DNA (Cell-ID intercalator-Ir, Fluidigm) for 10 min at room temperature and washed three times with dH20. For evaluation of cytokines production, samples were stimulated before staining with PMA at 50 ng/mL and Ionomycin at 1 μg/mL (Sigma) along with Brefeldin A for 4 h at 37°C.
Data analysis and UMAP
After mass cytometry (CyTOF) acquisition, any zero values were randomized using a uniform distribution of values between 0 and −1 using R. The signal of each parameter was normalized based on EQ beads (Fluidigm) as described previously.69 Samples were then used for UMAP analysis similar to that previously described using customized R scripts based on the ‘flowCore’ and ‘uwot’ R packages.37 In R, all data were transformed using the logicleTransform function (flowCore package) using parameters: w = 0.25, t = 16409, m = 4.5, a = 0 to roughly match scaling historically used in FlowJo. For heatmaps, median intensity corresponds to a logical data scale using formula previously described.70 The colors in the heat map represent the measured means intensity value of a given marker in a given sample. A seven-color scale is used with black–blue indicating low expression values, green–yellow indicating intermediately expressed markers, and orange-red representing highly expressed markers.
AIM (Activation-Induced Marker) assay
AIM assay was performed as described previously.42 Briefly, on day 1, frozen paired blood and tumor samples were thawed and prepared as stated above. CD4+ T cells (7AAD– CD45+ CD3+ CD4+) were sorted from blood or the tumors. Antigen presenting cells (APC – 7AAD– CD45+ CD3– CD56–) were sorted from paired PBMC using BD FACSAria II. Importantly, CD39 staining was performed with other surface antibodies before cell sorting. After cell sorting, cells were rested for 3 h at 37°C, incubated with a CD40 blocking antibody for 15 min and put in co-culture at a ratio CD4:APC of 1:5. CD40 blocking antibody prevents internalization and degradation of the CD40L receptor on CD4 T cells during peptide stimulation.42 Cells were then stimulated with either HCMV pp65 peptides pool (JPT PM-PP65-1, 86.25 μg/ml) or DMSO (negative control) for 18 h. On day 2, cells were washed, stained with surface antibodies (Table S2) and acquired on BD FACSCelesta. Activation was measured with CD69 and CD40L expression on total CD4+ T cells and bystander CD4+ T cells were analyzed for CD39 expression.
Supplementary Material
Acknowledgments
The authors thank all members of E.W.N lab, Fred Hutchinson Cancer Research Center, Seattle, the flow cytometry platform, all participating patients, the clinical research coordinators from NCCS.
Funding Statement
This work was supported by the Fred Hutchinson Cancer Research Center [New Development funding]; The Andy Hill Endowment Distinguished Researcher CARE fund to E.W.N. [NA].
Authors contribution
S.L. performed the experiments, analyzed the data and wrote the paper. S.Z. helped perform the experiments and reviewed the paper. S-L.K, A.T, I.B.T, D.T provided samples and discussed data and reviewed the paper. A.H, I-T.C and W.W.K discussed the data and gave insightful feedbacks. Y.S Led, initiated and designed the project, performed the experiments, analyzed the data and wrote the paper. E.W.N. Led, initiated and designed the project, developed scripts for CyTOF analysis, discussed the data and wrote the paper.
Disclosure statement
E.W.N is a co-founder, advisor and shareholder of ImmunoScape Pte. Ltd. E.W.N. is an advisor for Neogene Therapeutics and Nanostring Technologies. All other authors declare no potential conflicts of interest.
Data availability
Data are available on flow-repository: https://flowrepository.org/id/FR-FCM-Z4N2
Supplementary material
Supplemental data for this article can be accessed on the publisher’s website
References
- 1.Dunn GP, Bruce AT, Ikeda H, Old LJ, Schreiber RD.. Cancer immunoediting: from immunosurveillance to tumor escape. Nat Immunol. 2002;3: pp. 991–11. [DOI] [PubMed] [Google Scholar]
- 2.Schreiber RD, Old LJ, Smyth MJ.. Cancer immunoediting: integrating immunity’s roles in cancer suppression and promotion. Science. 2011;331: pp. 1565–1570. [DOI] [PubMed] [Google Scholar]
- 3.Pardoll DM. The blockade of immune checkpoints in cancer immunotherapy. Nat Rev Cancer. 2012;12: pp. 252–264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Rosenberg SA, Restifo NP. Adoptive cell transfer as personalized immunotherapy for human cancer. Science. 2015;348: pp. 62–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ott PA, Hu Z, Keskin DB, Shukla SA, et al. An immunogenic personal neoantigen vaccine for patients with melanoma. Nature. 2017;547(7662): pp. 217–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Maleki Vareki S, Garrigos C, Duran I. Biomarkers of response to PD-1/PD-L1 inhibition. Crit Rev Oncol Hematol. 2017;116: pp. 116–124. [DOI] [PubMed] [Google Scholar]
- 7.Li S, Simoni Y, Zhuang S, Gabel A, et al. Characterization of neoantigen-specific T cells in cancer resistant to immune checkpoint therapies. Proc Natl Acad Sci U S A. 2021;118(30): pp.1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Simoni Y, Becht E, Fehlings M, Loh CY, et al. Bystander CD8(+) T cells are abundant and phenotypically distinct in human tumour infiltrates. Nature. 2018;557(7706): pp. 575–579. [DOI] [PubMed] [Google Scholar]
- 9.Yeong J, Suteja L, Simoni Y, Lau KW, et al. Intratumoral CD39(+)CD8(+) T Cells Predict Response to Programmed Cell Death Protein-1 or Programmed Death Ligand-1 Blockade in Patients With NSCLC. J Thorac Oncol. 2021;16(8): pp. 1349–1358. [DOI] [PubMed] [Google Scholar]
- 10.Simoni Y, Becht E, Li S, Loh CY, et al. Partial absence of PD-1 expression by tumor-infiltrating EBV-specific CD8(+) T cells in EBV-driven lymphoepithelioma-like carcinoma. Clin Transl Immunology. 2020;9(9): p. e1175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Canale FP, Ramello MC, Nunez N, Araujo Furlan CL, et al. CD39 Expression Defines Cell Exhaustion in Tumor-Infiltrating CD8(+) T Cells. Cancer Res. 2018;78(1): pp. 115–128. [DOI] [PubMed] [Google Scholar]
- 12.Duhen T, Duhen R, Montler R, Moses J, et al. Co-expression of CD39 and CD103 identifies tumor-reactive CD8 T cells in human solid tumors. Nat Commun. 2018;9(1): p. 2724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Scheper W, Kelderman S, Fanchi LF, Linnemann C, et al. Low and variable tumor reactivity of the intratumoral TCR repertoire in human cancers. Nat Med. 2019;25(1): pp. 89–94. [DOI] [PubMed] [Google Scholar]
- 14.Rosato PC, Wijeyesinghe S, Stolley JM, Nelson CE, et al. Virus-specific memory T cells populate tumors and can be repurposed for tumor immunotherapy. Nat Commun. 2019;10(1): p. 567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Perez-Diez A, Joncker NT, Choi K, Chan WF, et al. CD4 cells can be more efficient at tumor rejection than CD8 cells. Blood. 2007;109(12): pp. 5346–5354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wing JB, Tanaka A, Sakaguchi S. Human FOXP3(+) Regulatory T Cell Heterogeneity and Function in Autoimmunity and Cancer. Immunity. 2019;50: pp. 302–316. [DOI] [PubMed] [Google Scholar]
- 17.Saito T, Nishikawa H, Wada H, Nagano Y, et al. Two FOXP3(+)CD4(+) T cell subpopulations distinctly control the prognosis of colorectal cancers. Nat Med. 2016;22(6): pp. 679–684. [DOI] [PubMed] [Google Scholar]
- 18.Plitas G, Konopacki C, Wu K, Bos PD, et al. Regulatory T Cells Exhibit Distinct Features in Human Breast Cancer. Immunity. 2016;45(5): pp. 1122–1134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Facciabene A, Motz GT, Coukos G. T-regulatory cells: key players in tumor immune escape and angiogenesis. Cancer Res. 2012;72: pp. 2162–2171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zou W. Regulatory T cells, tumour immunity and immunotherapy. Nat Rev Immunol. 2006;6: pp. 295–307. [DOI] [PubMed] [Google Scholar]
- 21.Tang F, Du X, Liu M, Zheng P, Liu Y. Anti-CTLA-4 antibodies in cancer immunotherapy: selective depletion of intratumoral regulatory T cells or checkpoint blockade? Cell Biosci. 2018;8: p. 30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Callahan MK, Wolchok JD, Allison JP. Anti-CTLA-4 antibody therapy: immune monitoring during clinical development of a novel immunotherapy. Semin Oncol. 2010;37: pp. 473–484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Seidel JA, Otsuka A, Kabashima K. Anti-PD-1 and Anti-CTLA-4 Therapies in Cancer: mechanisms of Action, Efficacy, and Limitations. Front Oncol. 2018;8: p. 86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Arce Vargas F, Furness AJS, Solomon I, Joshi K, et al. Fc-Optimized Anti-CD25 Depletes Tumor-Infiltrating Regulatory T Cells and Synergizes with PD-1 Blockade to Eradicate Established Tumors. Immunity. 2017;46(4): pp. 577–586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Arce Vargas F, Furness AJS, Litchfield K, Joshi K, et al. Fc Effector Function Contributes to the Activity of Human Anti-CTLA-4 Antibodies. Cancer Cell. 2018;33(4): pp. 649–63 e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Alspach E, Lussier DM, Miceli AP, et al. MHC-II neoantigens shape tumour immunity and response to immunotherapy. Nature. 2019;574(7780): pp. 696–701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Melssen M, Slingluff CL Jr. Vaccines targeting helper T cells for cancer immunotherapy. Curr Opin Immunol. 2017;47: pp. 85–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Quezada SA, Simpson TR, Peggs KS, Merghoub T, et al. Tumor-reactive CD4(+) T cells develop cytotoxic activity and eradicate large established melanoma after transfer into lymphopenic hosts. J Exp Med. 2010;207(3): pp. 637–650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Tran E, Turcotte S, Gros A, Robbins PF, et al. Cancer immunotherapy based on mutation-specific CD4+ T cells in a patient with epithelial cancer. Science. 2014;344(6184): pp. 641–645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Friedman KM, Prieto PA, Devillier LE, Gross CA, et al. Tumor-specific CD4+ melanoma tumor-infiltrating lymphocytes. J Immunother. 2012;35(5): pp. 400–408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kreiter S, Vormehr M, van de Roemer N, Diken M, et al. Mutant MHC class II epitopes drive therapeutic immune responses to cancer. Nature. 2015;520(7549): pp. 692–696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wong SB, Bos R, Sherman LA. Tumor-specific CD4+ T cells render the tumor environment permissive for infiltration by low-avidity CD8+ T cells. J Immunol. 2008;180: pp. 3122–3131. [DOI] [PubMed] [Google Scholar]
- 33.Church SE, Jensen SM, Antony PA, Restifo NP, Fox BA. Tumor-specific CD4+ T cells maintain effector and memory tumor-specific CD8+ T cells. Eur J Immunol. 2014;44: pp. 69–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Matsuzaki J, Tsuji T, Luescher IF, Shiku H, et al. Direct tumor recognition by a human CD4(+) T-cell subset potently mediates tumor growth inhibition and orchestrates anti-tumor immune responses. Sci Rep. 2015;5: p. 14896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Malandro N, Budhu S, Kuhn NF, Liu C, et al. Clonal Abundance of Tumor-Specific CD4(+) T Cells Potentiates Efficacy and Alters Susceptibility to Exhaustion. Immunity. 2016;44(1): pp. 179–193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kortekaas KE, Santegoets SJ, Sturm G, Ehsan I, et al. CD39 Identifies the CD4(+) Tumor-Specific T-cell Population in Human Cancer. Cancer Immunol Res. 2020;8(10): pp. 1311–1321. [DOI] [PubMed] [Google Scholar]
- 37.Becht E, McInnes L, Healy J, Dutertre CA, et al. Dimensionality reduction for visualizing single-cell data using UMAP. Nat Biotechnol. 2018;38–44. [DOI] [PubMed] [Google Scholar]
- 38.Walsh DA, Borges Da Silva H, Beura LK, Peng C, et al. The Functional Requirement for CD69 in Establishment of Resident Memory CD8(+) T Cells Varies with Tissue Location. J Immunol. 2019;203(4): pp. 946–955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.McNamara HA, Cockburn IA. Resident T cells seek the perfect place to work from home. Nat Immunol. 2021;22: pp. 1076–1078. [DOI] [PubMed] [Google Scholar]
- 40.Togashi Y, Shitara K, Nishikawa H. Regulatory T cells in cancer immunosuppression - implications for anticancer therapy. Nat Rev Clin Oncol. 2019;16: pp. 356–371. [DOI] [PubMed] [Google Scholar]
- 41.Oja AE, Piet B, van der Zwan D, Blaauwgeers H, et al. Functional Heterogeneity of CD4(+) Tumor-Infiltrating Lymphocytes With a Resident Memory Phenotype in NSCLC. Front Immunol. 2018;9: p. 2654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Reiss S, Baxter AE, Cirelli KM, Dan JM, et al. Comparative analysis of activation induced marker (AIM) assays for sensitive identification of antigen-specific CD4 T cells. PLoS One. 2017;12(10): p. e0186998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Raczkowski F, Rissiek A, Ricklefs I, Heiss K, et al. CD39 is upregulated during activation of mouse and human T cells and attenuates the immune response to Listeria monocytogenes. PLoS One. 2018;13(5): p. e0197151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Shankaran V, Ikeda H, Bruce AT, White JM, et al. IFNgamma and lymphocytes prevent primary tumour development and shape tumour immunogenicity. Nature. 2001;410(6832): pp. 1107–1111. [DOI] [PubMed] [Google Scholar]
- 45.Reading JL, Galvez-Cancino F, Swanton C, Lladser A, Peggs KS, Quezada SA. The function and dysfunction of memory CD8(+) T cells in tumor immunity. Immunol Rev. 2018;283: pp. 194–212. [DOI] [PubMed] [Google Scholar]
- 46.Farhood B, Najafi M, Mortezaee K. CD8(+) cytotoxic T lymphocytes in cancer immunotherapy: a review. J Cell Physiol. 2019;234: pp. 8509–8521. [DOI] [PubMed] [Google Scholar]
- 47.van der Leun Am, Thommen DS, Schumacher TN. CD8(+) T cell states in human cancer: insights from single-cell analysis. Nat Rev Cancer. 2020;20: pp. 218–232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Tanaka A, Sakaguchi S. Targeting Treg cells in cancer immunotherapy. Eur J Immunol. 2019;49: pp. 1140–1146. [DOI] [PubMed] [Google Scholar]
- 49.Ahrends T, Spanjaard A, Pilzecker B, Babala N, et al. CD4(+) T Cell Help Confers a Cytotoxic T Cell Effector Program Including Coinhibitory Receptor Downregulation and Increased Tissue Invasiveness. Immunity. 2017;47(5): pp. 848–61 e5. [DOI] [PubMed] [Google Scholar]
- 50.Mumberg D, Monach PA, Wanderling S, Philip M, et al. CD4(+) T cells eliminate MHC class II-negative cancer cells in vivo by indirect effects of IFN-gamma. Proc Natl Acad Sci U S A. 1999;96(15): pp. 8633–8638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Haabeth OA, Tveita AA, Fauskanger M, Schjesvold F, et al. How Do CD4(+) T Cells Detect and Eliminate Tumor Cells That Either Lack or Express MHC Class II Molecules? Front Immunol. 2014;5: p. 174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Wherry EJ, Kurachi M. Molecular and cellular insights into T cell exhaustion. Nat Rev Immunol. 2015;15: pp. 486–499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Pauken KE, Wherry EJ. Overcoming T cell exhaustion in infection and cancer. Trends Immunol. 2015;36: pp. 265–276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Li J, Byrne KT, Yan F, Yamazoe T, et al. Tumor Cell-Intrinsic Factors Underlie Heterogeneity of Immune Cell Infiltration and Response to Immunotherapy. Immunity. 2018;49(1): pp. 178–93 e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Bastid J, Cottalorda-Regairaz A, Alberici G, Bonnefoy N, Eliaou JF, Bensussan A. ENTPD1/CD39 is a promising therapeutic target in oncology. Oncogene. 2013;32: pp. 1743–1751. [DOI] [PubMed] [Google Scholar]
- 56.Elliott MR, Chekeni FB, Trampont PC, Lazarowski ER, et al. Nucleotides released by apoptotic cells act as a find-me signal to promote phagocytic clearance. Nature. 2009;461(7261): pp. 282–286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Ohta A, Gorelik E, Prasad SJ, Ronchese F, et al. A2A adenosine receptor protects tumors from antitumor T cells. Proc Natl Acad Sci U S A. 2006;103(35): pp. 13132–13137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Gourdin N, Bossennec M, Rodriguez C, Vigano S, et al. Autocrine Adenosine Regulates Tumor Polyfunctional CD73(+)CD4(+) Effector T Cells Devoid of Immune Checkpoints. Cancer Res. 2018;78(13): pp. 3604–3618. [DOI] [PubMed] [Google Scholar]
- 59.Gupta PK, Godec J, Wolski D, Adland E, et al. CD39 Expression Identifies Terminally Exhausted CD8+ T Cells. PLoS Pathog. 2015;11(10): p. e1005177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Deaglio S, Dwyer KM, Gao W, Friedman D, et al. Adenosine generation catalyzed by CD39 and CD73 expressed on regulatory T cells mediates immune suppression. J Exp Med. 2007;204(6): pp. 1257–1265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Borsellino G, Kleinewietfeld M, Di Mitri D, Sternjak A, et al. Expression of ectonucleotidase CD39 by Foxp3+ Treg cells: hydrolysis of extracellular ATP and immune suppression. Blood. 2007;110(4): pp. 1225–1232. [DOI] [PubMed] [Google Scholar]
- 62.Dwyer KM, Hanidziar D, Putheti P, Hill PA, et al. Expression of CD39 by human peripheral blood CD4+ CD25+ T cells denotes a regulatory memory phenotype. Am J Transplant. 2010;10(11): pp. 2410–2420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Chen DS, Mellman I. Elements of cancer immunity and the cancer-immune set point. Nature. 2017;541: pp. 321–330. [DOI] [PubMed] [Google Scholar]
- 64.Yossef R, Tran E, Deniger DC, Gros A, et al. Enhanced detection of neoantigen-reactive T cells targeting unique and shared oncogenes for personalized cancer immunotherapy. JCI Insight. 2018;3(19): pp. 1–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Whiteside SK, Snook JP, Williams MA, Weis JJ. Bystander T Cells: a Balancing Act of Friends and Foes. Trends Immunol. 2018;39: pp. 1021–1035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Christoffersson G, Chodaczek G, Ratliff SS, et al. Suppression of diabetes by accumulation of non-islet-specific CD8(+) effector T cells in pancreatic islets. Sci Immunol. 2018;3(21). [DOI] [PubMed] [Google Scholar]
- 67.Simoni Y, Fehlings M, Kloverpris HN, McGovern N, et al. Human Innate Lymphoid Cell Subsets Possess Tissue-Type Based Heterogeneity in Phenotype and Frequency. Immunity. 2017;46(1): pp. 148–161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Simoni Y, Fehlings M, Newell EW. Multiplex MHC Class I Tetramer Combined with Intranuclear Staining by Mass Cytometry. Methods Mol Biol. 2019;1989: pp. 147–158. [DOI] [PubMed] [Google Scholar]
- 69.Finck R, Simonds EF, Jager A, et al. Normalization of mass cytometry data with bead standards. Cytometry A. 2013;83(5): pp. 483–494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Moore WA, Parks DR. Update for the logicle data scale including operational code implementations. Cytometry A. 2012;81: pp. 273–277. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Data are available on flow-repository: https://flowrepository.org/id/FR-FCM-Z4N2