

Abstract
Rationale
Obstructive sleep apnea (OSA) is a common disorder associated with increased risk for cardiovascular disease, diabetes, and premature mortality. There is strong clinical and epidemiologic evidence supporting the importance of genetic factors influencing OSA but limited data implicating specific genes.
Objectives
To search for rare variants contributing to OSA severity.
Methods
Leveraging high-depth genomic sequencing data from the NHLBI Trans-Omics for Precision Medicine (TOPMed) program and imputed genotype data from multiple population-based studies, we performed linkage analysis in the CFS (Cleveland Family Study), followed by multistage gene-based association analyses in independent cohorts for apnea–hypopnea index (AHI) in a total of 7,708 individuals of European ancestry.
Measurements and Main Results
Linkage analysis in the CFS identified a suggestive linkage peak on chromosome 7q31 (LOD = 2.31). Gene-based analysis identified 21 noncoding rare variants in CAV1 (Caveolin-1) associated with lower AHI after accounting for multiple comparisons (P = 7.4 × 10−8). These noncoding variants together significantly contributed to the linkage evidence (P < 10−3). Follow-up analysis revealed significant associations between these variants and increased CAV1 expression, and increased CAV1 expression in peripheral monocytes was associated with lower AHI (P = 0.024) and higher minimum overnight oxygen saturation (P = 0.007).
Conclusions
Rare variants in CAV1, a membrane-scaffolding protein essential in multiple cellular and metabolic functions, are associated with higher CAV1 gene expression and lower OSA severity, suggesting a novel target for modulating OSA severity.
Keywords: obstructive sleep apnea, caveolin-1, apnea–hypopnea index, genetic association analysis, rare variants
At a Glance Commentary
Scientific Knowledge on the Subject
Obstructive sleep apnea (OSA) is a common heritable disorder. However, previous genome-wide association analysis using common genetic variants identified few genes. The “missing heritability” may be explained by rare variants.
What This Study Adds to the Field
We perform multistage gene-based analysis for apnea–hypopnea index using whole-genome sequencing data from the Trans-Omics for Precision Medicine program. We identify multiple rare variants at the regulatory region of Caveolin-1 for OSA. Cav-1 has been implicated in multiple pathophysiological processes and diseases relevant to OSA.
Obstructive sleep apnea (OSA) is a common disorder that affects 6% to more than 20% of adults (1–3). Characterized by repetitive episodes of upper airway obstruction during sleep, intermittent hypoxemia, sleep fragmentation, and excessive daytime sleepiness, OSA negatively affects a wide range of cardiovascular, neurological, and metabolic functions (1, 4), increasing risk for cardiovascular disease, diabetes, cancer, cognitive impairment, and premature mortality (1). OSA also is associated with important patient-reported outcomes, including quality of life, mood, and daytime functioning (4). Although positive airway pressure is an efficacious treatment for OSA, adherence to therapy often is poor, and the efficacy of common alternative treatments, including oral appliances and upper airway surgery, is quite variable. It is now generally recognized that OSA is a heterogeneous disease, which reflects variable contributions of anatomic and physiological factors that cause narrowing and collapse of the upper airway as well as instability of ventilation during sleep (5, 6). An understanding of the molecular mechanisms underlying OSA should provide insight into disease mechanisms and heterogeneity, facilitating personalized assessment of both OSA-associated risks and informing therapeutic decisions.
Family-based studies of OSA traits adjusted for obesity have yielded heritability estimates that range from 27% to 31%, with SNP-based heritability estimates from large-scale biobank studies for clinical traits ranging from 6% to 10% (7–9). Early genetic linkage studies reported multiple candidate regions contributing to OSA (10, 11), but recent large genome-wide association studies (GWASs) have identified fewer significant associations for polysomnographically defined OSA traits, likely reflecting modest sample sizes (12).
Advances in next-generation sequencing technology allow gene-based analysis to identify combined effects of multiple rare variants contributing to complex diseases (13). We and others have demonstrated that phenotype-associated rare variants can be enriched in families and are manifest by statistical evidence of linkage (14, 15). Recently, a strategy that integrates linkage and rare variant association analysis was shown to be powerful for identifying variants and genes contributing to complex traits such as blood pressure and sleep-related hypoxemia traits (16–20). In this study, we apply this strategy to search for rare variants underlying OSA using whole-genome sequencing data from the Trans-Omics in Precision Medicine (TOPMed) program. We focus on the apnea–hypopnea index (AHI), the primary measure of OSA severity that summarizes the number of apneas plus hypopneas per hour of sleep, because of its widespread clinical use and evidence of heritability. Some of the results of this study have been previously reported in the form of an abstract (21). Methods are detailed in the online supplement.
Results
Noncoding Rare Variants on Caveolin-1 Associated with AHI
The analysis flowchart is shown in Figure 1. Our analysis includes three stages. In stage I, we performed linkage analysis for AHI in 487 European American individuals from 118 families in the CFS (Cleveland Family Study) (see Table E1 in the online supplement) and identified the highest linkage peak on chromosome 7q31 (LOD = 2.31; Figure 2A). Applying the same procedure as in our previous study (20), we calculated a family-specific LOD score for each CFS family, identifying the top 12 families with family-specific LOD scores ⩾ 0.1 as those that potentially carry low-frequency or rare AHI variants. In the 20-cM genomic region centered at the linkage peak, there were 159,435 variants with minor allele frequency (MAF) ⩽ 0.05 that passed quality control filters. As rare variants in intergenic regions are less likely to be in linkage disequilibrium with variants in genes, we limited our search to the variants located within the 223 genes in the region and their corresponding 5 kb up- and downstream regions. We filtered out the variants that presented at most once in any of the 12 selected families because these variants did not contribute to the observed linkage evidence. This procedure identified 35,352 and 21,275 variants using thresholds of MAF ⩽ 0.05 and MAF ⩽ 0.01, respectively. When examining functional coding variants defined as missense, in-frame deletion or insertion, stop gained or lost, start gained or lost, splice acceptor or donor, or initiator or start codon, we observed 49 and 22 genes with at least two such functional variants for MAF ⩽ 0.05 and MAF ⩽ 0.01 thresholds, respectively. For the noncoding variants, we observed 210 and 198 genes with at least two variants for MAF ⩽ 0.05 and MAF ⩽ 0.01 thresholds, respectively. Gene-based burden tests and sequence kernel association tests (SKATs) (22, 23) were performed for these genes after stratifying by coding and noncoding variants with MAF thresholds of 0.05 and 0.01, respectively. Table E2 lists the number of genes with nominal empirical P values <0.05 for different categories of variants and analyses. We observed a 1.3- to 2.4-fold enrichment of genes showing association evidence with AHI in the categories we examined. In total, we observed 70 nonoverlapping genes with P < 0.05 in either the burden test or SKAT with MAF threshold 0.05 or 0.01, and these genes were carried forward to stage II analysis. The most significant gene identified was CAV1 (Caveolin-1), for analysis of 21 noncoding variants with MAF ⩽ 0.01 (Tables 1 and 2; burden test effect = −0.51; P = 1.1 × 10−5).
Figure 1.

Flowchart of discovery analysis. AHI = apnea–hypopnea index; ARIC = Atherosclerosis Risk in Communities; CAV1 = Caveolin-1; CFS = Cleveland Family Study; CHS = Cardiovascular Health Study; FHS = Framingham Heart Study; LOD = logarithm of the odds; MAF = minor allele frequency; MESA = Multi-Ethnic Study of Atherosclerosis; MrOS = Osteoporotic Fractures in Men Study; SKAT = sequence kernel association test; TOPMed = Trans-Omics in Precision Medicine; WASHS = Western Australian Sleep Health Study; WGS = whole-genome sequencing.
Figure 2.
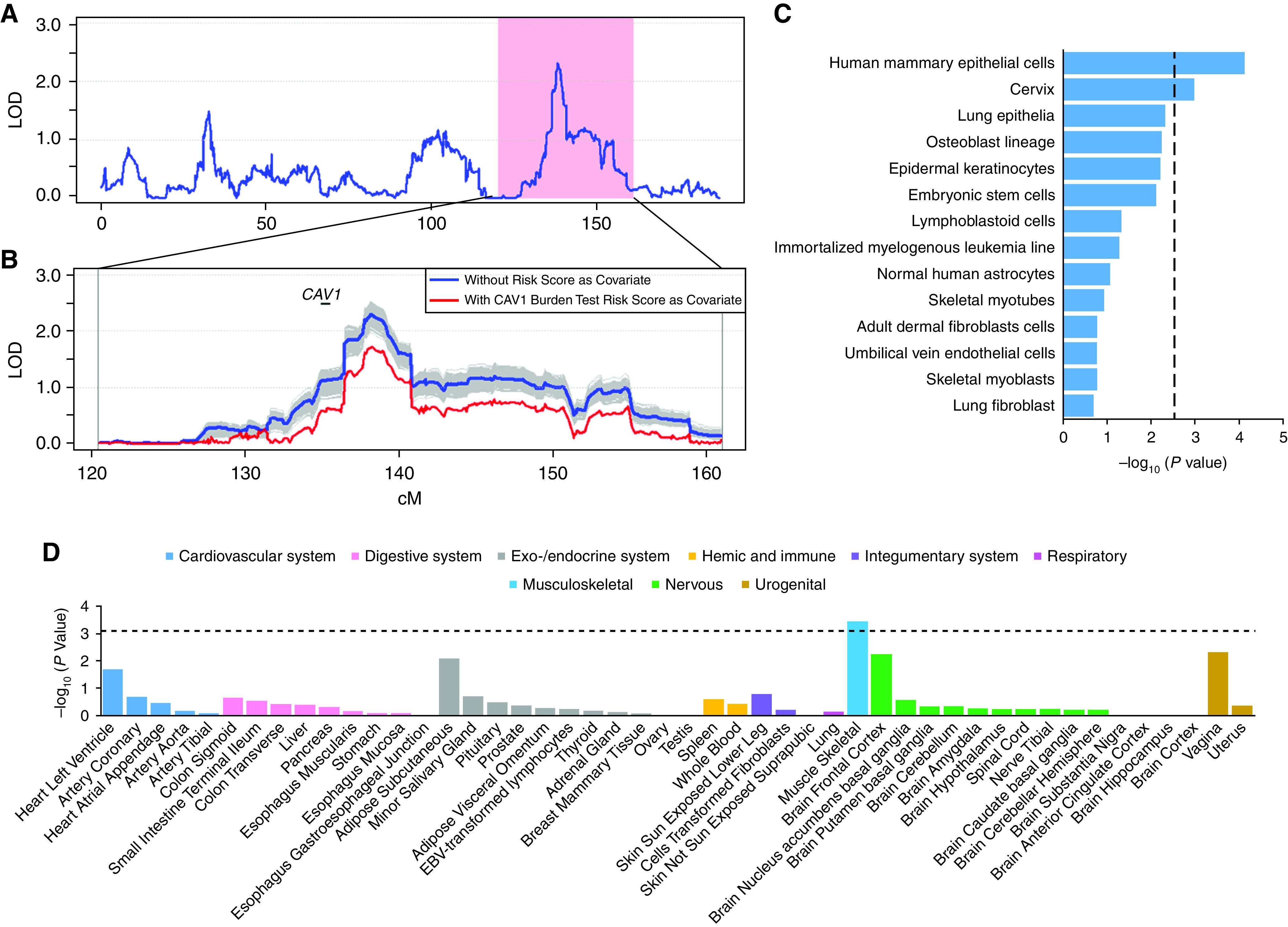
The 21 variants in CAV1 (Caveolin-1). (A) LOD score in 7q31 linked to apnea–hypopnea index in linkage analysis. The pink region is the 20-Mb target region in the sequencing analysis, and the protein coding genes are presented at the bottom. (B) LOD score in 7q31 when conditioning on the burden test risk score of the effect size weighted sum of the 21 identified rare variants in CAV1. The effect sizes were estimated in the CFS (Cleveland Family Study) cohort. The linkage curves are plotted with (red curve) and without (blue curve) adjusting for the burden test risk score. The location of CAV1 is marked by a solid bar. The shaded regions represent the 1,000 simulations when conditional on a randomly selected 21 frequency matched rare variants. (C) Cell type–specific regulatory annotation enrichment tests for the 21 noncoding variants in CAV1 in 14 cell lines defined in the Ensembl Regulatory Build. The vertical dotted line represents the degree of significance after adjusting for multiple tests. (D) Association of the 21 variants in CAV1 with CAV1 expression degree in 44 tissues from the Genotype-Tissue Expression Consortium. The horizontal dotted line represents the degree of significance after adjusting for multiple tests. LOD = logarithm of the odds.
Table 1.
Stage I, II, and III Gene-based Association P Values of Noncoding Variants for Apnea–Hypopnea Index
| MAF Threshold | Stage I (n = 487) |
Stage II (n = 2,772) |
Stage III (n = 4,449) |
Stages I, II, and III (n = 7,708) |
||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Gene | Number of Variants | Burden | SKAT | Burden | SKAT | Burden | SKAT | Burden | SKAT | |
| 0.01 | TES | 14 | 0.155 | 0.035 | 0.511 | 0.003 | 0.572 | 0.113 | 0.532 | 0.001 |
| CAV1 | 21 | 1.1 × 10−5 | 0.033 | 0.003 | 0.466 | 0.003 | 0.336 | 7.4 × 10−8 | 0.201 | |
| ASZ1 | 16 | 0.036 | 0.139 | 0.008 | 0.028 | 0.560 | 0.400 | 0.010 | 0.047 | |
| PTPRZ1 | 58 | 0.388 | 0.002 | 0.006 | 0.104 | 0.186 | 0.441 | 0.013 | 0.014 | |
| SND1 | 111 | 0.246 | 0.008 | 0.147 | 0.020 | 0.502 | 0.347 | 0.252 | 0.005 | |
| AKR1B1 | 5 | 0.414 | 0.005 | 0.206 | 0.008 | 0.012 | 0.146 | 0.131 | 7.9 × 10−5 | |
| 0.05 | CAV1 | 38 | 0.012 | 0.030 | 0.009 | 0.106 | 0.001 | 0.263 | 2.1 × 10−5 | 0.043 |
| POT1 | 93 | 0.351 | 0.036 | 0.007 | 0.513 | 0.300 | 0.461 | 0.021 | 0.287 | |
| SND1 | 319 | 0.010 | 0.038 | 0.061 | 0.041 | 0.311 | 0.637 | 0.016 | 0.045 | |
Definition of abbreviations: AKR1B1 = aldo-keto reductase family 1 member B; ASZ1 = ankyrin repeat, SAM and basic leucine zipper domain containing 1; CAV1 = Caveolin-1; MAF = minor allele frequency; POT1 = protection of telomeres 1; PTPRZ1 = protein tyrosine phosphatase receptor type Z1; SKAT = sequence kernel association test; SND1 = staphylococcal nuclease and Tudor domain containing 1; TES = testin LIM domain protein.
Only genes with either burden or SKAT P values <0.05 in both stages I and II are reported. The results for CAV1 are in boldface type.
Table 2.
Gene-based Association Analyses of 21 Noncoding Variants in Caveolin-1
| Analysis | Study | Trait | Sample Size | Number of Variants | Burden Effect | Burden P Value | SKAT P Value |
|---|---|---|---|---|---|---|---|
| Stage I | CFS | AHI | 487 | 21 | −0.51 | 1.1 × 10−5 | 0.03 |
| Stage II | FHS | AHI | 468 | 16 | −0.15 | 0.06 | 0.75 |
| ARIC | AHI | 1,006 | 17 | −0.08 | 0.17 | 0.73 | |
| CHS | AHI | 668 | 15 | −0.12 | 0.05 | 0.21 | |
| MESA | AHI | 630 | 19 | −0.33 | 0.02 | 0.19 | |
| Stages I and II | Meta-analysis | AHI | 3,259 | — | — | 1.2 × 10−6 | 0.15 |
| Stage III | ARIC | AHI | 583 | 14 | −0.07 | 0.24 | 0.48 |
| FHS | AHI | 181 | 18 | −0.18 | 0.03 | 0.58 | |
| MrOS | AHI | 2,178 | 17 | −0.29 | 0.01 | 0.10 | |
| WASHS | AHI | 1,507 | 14 | −0.04 | 0.18 | 0.39 | |
| Meta-analysis | AHI | 4,449 | — | — | 3.1 × 10−3 | 0.34 | |
| Stages I, II, and III | Meta-analysis | AHI | 7,708 | — | — | 7.4 × 10−8 | 0.20 |
| Generalization | UKB | OSA case-control | 16,052 | 19 | −57.8 | 0.19 | 0.283 |
| Gene expression | MESA | CAV1 expression in peripheral blood mononuclear cell | 922 | 16 | 13.6 | 0.39 | 0.08 |
| MESA | CAV1 expression in T cell | 404 | 15 | 27.4 | 0.04 | 0.09 | |
| MESA | CAV1 expression in monocyte | 401 | 15 | 6.0 | 0.66 | 0.86 |
Definition of abbreviations: AHI = apnea–hypopnea index; ARIC = Atherosclerosis Risk in Communities; CAV1 = Caveolin-1; CFS = Cleveland Family Study; CHS = Cardiovascular Health Study; FHS = Framingham Heart Study; MESA = Multi-Ethnic Study of Atherosclerosis; MrOS = Osteoporotic Fractures in Men Study; OSA = obstructive sleep apnea; SKAT = sequence kernel association test; UKB = UK Biobank; WASHS = Western Australian Sleep Health Study.
In stage II analysis, we used four independent cohorts from the TOPMed whole-genome sequencing project (ARIC [Atherosclerosis Risk in Communities] study, CHS [Cardiovascular Health Study], FHS [Framingham Heart Study], and MESA [Multi-Ethnic Study of Atherosclerosis]), consisting of 2,772 individuals of European ancestry whose genomes were sequenced and had AHI measured (see Table E1). To reduce the multiple-comparison penalty, we performed the same burden and SKAT analyses and restricted to the same variants in the 70 genes identified in stage I. For functional coding variants, we did not observe any genes reaching nominal significance in stage II. For the noncoding variants, we observed six and three genes with P < 0.05 in either the burden test or SKAT for variants with MAF thresholds of 0.01 and 0.05, respectively (Table 1). CAV1 was again significantly associated with lower AHI, with burden P values of 0.003 and 0.009 for MAF thresholds of 0.01 and 0.05, respectively, although it did not reach significance after correcting for multiple tests (70 genes, two statistical methods, and two MAF thresholds).
In stage III, we performed association analysis of the seven genes identified in stage II using imputed data from additional samples in ARIC and FHS that had been genotyped but not whole-genome sequenced, as well as MrOS (Osteoporotic Fractures in Men Study) and WASHS (Western Australian Sleep Health Study) (N = 4,449; see Table E1). We consistently observed association evidence of CAV1 with lower AHI (burden test P = 0.003 for MAF ⩽ 0.01 and P = 0.001 for MAF ⩽ 0.05, respectively; Table 1), which is significant after adjusting for multiple tests (seven genes, two statistical methods, and two MAF thresholds), although significance was reached by MAF ⩽ 0.05. When combining the association evidence from stage II and III samples, the P value of association evidence of CAV1 for both MAF ⩽ 0.05 and MAF ⩽ 0.01 is 1.14 × 10−4, which is significant after adjusting for multiple tests conducted in stage II analysis (70 genes, two statistical methods, and two MAF thresholds). When combining the association evidence from stages I, II, and III, the association evidence of CAV1 reached genome-wide significance for the MAF ⩽ 0.01 threshold (P = 7.4 × 10−8; Tables 1 and 2), which is significant after adjusting for the total number of tests: 223 genes, two statistical methods, and two MAF thresholds. We further examined the effect sizes of the 21 variants in stage II samples. We observed that 10 of 12 variants had protective effects (see Table E3), which is consistent with these variants’ demonstrating protective effects in the burden test using stage I, II, and III samples (Table 2). We also observed an association of AKR1B1 with AHI using the SKAT P value of 7.9 × 10−5, but this association did not reach significance after adjusting for multiple tests (Table 1).
Sensitivity and Generalization Analyses
We performed conditional linkage analysis by including the burden test risk score of 21 CAV1 noncoding variants as a covariate in further analysis of the CFS (see Methods in the online supplement). Individuals carrying any of the 21 noncoding variants have a mean AHI of 6.0 (SD, 11.3) events/h, approximately one-third the mean value of 19.6 (SD, 26.7) events/h observed in noncarriers, and have more favorable values for all nocturnal oxygen saturation parameters. Carriers also are slightly younger and have somewhat lower body mass index, neck circumference, and systolic and diastolic blood pressure, and they have a lower prevalence of cardiovascular disease than noncarriers but do not differ regarding waist-to-hip ratio, excessive daytime sleepiness, lung function, or diabetes (see Table E4).
The linkage LOD score dropped from 2.307 to 1.698 (by 26.4%) after conditioning on the burden test risk score of 21 CAV1 noncoding variants as a covariate. The decrease of LOD score is significant on the basis of 1,000 simulations with conditional analyses on the basis of 21 randomly selected rare variants (see Methods in the online supplement), allele frequency matched to the CAV1 variants (P < 0.001; Figure 2B), suggesting that those noncoding variants contribute to the linkage evidence on chromosome 7q31.
As CAV1 has been associated with type 2 diabetes (T2D) and pulmonary impairment, we next performed gene-based analyses also adjusting for lung function traits and T2D in a subset of CFS samples with available measures, and we did not observe attenuation in the degree of significance for the association with AHI (Table 3). We also performed gene-based analyses adjusting for OSA mechanistic endotypes in a subset of CFS samples with available data (n = 218). Burden test P values increased after adjusting for collapsibility and arousal threshold (from 0.01 to 0.07 and 0.099 for collapsibility and arousal threshold, respectively) but did not substantively change after adjusting for compensation or loop gain (Table 3). This result suggests that the association of CAV1 and AHI is mainly independent of lung function and T2D, although pharyngeal collapsibility and arousal threshold may contribute to the observed association.
Table 3.
Gene-based Association Analyses of 21 Noncoding Variants in Caveolin-1 with Apnea–Hypopnea Index Adjusting for Lung Function and Obstructive Sleep Apnea Physiological Endotypes
| Covariates | Number of Variants | Burden Effect | Burden P Value | SKAT P Value | Sample Size |
|---|---|---|---|---|---|
| No covariate | 21 | −0.49 | 3.31 × 10−5 | 0.038 | 454 |
| FEV1, predicted % | 21 | −0.49 | 2.92 × 10−5 | 0.032 | 454 |
| FVC, predicted % | 21 | −0.47 | 6.34 × 10−5 | 0.041 | 454 |
| No covariate | 21 | −0.40 | 0.012 | 0.054 | 350 |
| T2D | 21 | −0.40 | 0.016 | 0.065 | 350 |
| No covariate | 19 | −0.39 | 0.014 | 0.077 | 218 |
| Collapsibility, % eupnea | 19 | −0.25 | 0.070 | 0.119 | 218 |
| Compensation | 19 | −0.38 | 0.016 | 0.060 | 218 |
| Loop gain | 19 | −0.33 | 0.020 | 0.083 | 218 |
| Arousal threshold, % eupnea | 19 | −0.23 | 0.099 | 0.117 | 218 |
Definition of abbreviations: SKAT = sequence kernel association test; T2D = type 2 diabetes.
We performed gene-based analyses of the 21 noncoding variants in CAV1 using clinically defined OSA cases (n = 4,103) and control subjects (n = 16,052) (on the basis of International Classification of Diseases codes and self-reported daytime sleepiness; see Methods in the online supplement) in European individuals from the UK Biobank. The SKAT and burden test P values were 0.283 and 0.186, respectively. Although this analysis did not reach significance, these 21 variants consistently showed a protective effect on OSA (Table 2).
Bioinformatics Analysis
We investigated whether the 21 low-frequency noncoding variants in the CAV1 gene show gene-regulatory roles by performing gene-based association with the RNA sequencing data across the 44 tissues from the Genotype-Tissue Expression Consortium (24). We observed that these noncoding variants significantly contribute to the CAV1 expression degree in skeletal muscle (P = 3.8 × 10−4) in Genotype-Tissue Expression data after adjusting for 44 tests (Figure 2D). We also observed a positive association between the CAV1 variants with increased CAV1 expression degree in T cells (P = 0.042) in MESA (Table 2). In addition, we observed significant associations between higher CAV1 gene expression from peripheral white blood cells and lower AHI (P = 0.024 in the meta-analysis of MESA and FHS; Table 4). Higher CAV1 gene expression was also associated with more favorable values of oxygen saturation nadir during sleep (meta-analysis P = 0.007; Table 4), a marker of lower OSA severity.
Table 4.
Caveolin-1 Gene Expression Associated with Obstructive Sleep Apnea Traits
| Study | Average SpO2 |
Minimum SpO2 |
AHI |
|||
|---|---|---|---|---|---|---|
| Effect (SE) | P Value | Effect (SE) | P Value | Effect (SE) | P Value | |
| MESA | 0.015 (0.007) | 0.032 | 0.004 (0.002) | 0.019 | −0.0015 (0.0006) | 0.017 |
| FHS | 0.0004 (0.0051) | 0.940 | 0.002 (0.001) | 0.141 | −0.0005 (0.0007) | 0.434 |
| Meta-analysis | 0.005 (0.004) | 0.182 | 0.003 (0.001) | 0.007 | −0.001 (0.0005) | 0.024 |
Definition of abbreviations: AHI = apnea–hypopnea index; FHS = Framingham Heart Study; MESA = Multi-Ethnic Study of Atherosclerosis; SpO2 = nocturnal oxygen saturation as measured by pulse oximetry.
We next examined whether the 21 noncoding variants in CAV1 are enriched in the regulatory activity–predicted elements for the 14 cell lines defined in the Ensembl Regulatory Build (25) (which includes CTCF [CCCTC-binding factor] binding sites, enhancer, heterochromatin, promoter flank, and transcription start sites). We observed significant enrichment in cis-regulatory elements in human mammary epithelial cells and cervix after correcting for multiple tests (Figure 2C). Similar enrichment was observed in the genomic locations of the 21 variants with their corresponding cis-regulatory elements in the human mammary epithelial cells (see Figure E1).
Discussion
Genetic association studies for OSA have been limited by modest sample sizes with both quantitative phenotype and genomic data. To overcome this limitation, using data from 7,708 individuals, we performed an integrative search of rare variants associated with AHI, the main clinical quantitative metric for OSA, through linkage analysis followed by multiple-stage association analyses using whole-genome sequencing data from the TOPMed project and imputed genotype data from GWASs. This strategy, which reduced the multiple-comparison burden, has been successful for identifying rare variants associated with complex traits (16–20). We identified 21 noncoding rare variants in CAV1 that were associated with lower AHI with genome-wide significance when combining stage I, II, and III samples (P = 7.4 × 10−8). The biological plausibility that these variants play a protective role in OSA is supported by several lines of evidence: 1) the variants significantly contributed to the initially observed linkage peak for AHI; 2) the variants were associated with CAV1 expression degree in skeletal muscle and peripheral T cells, two sites that may contribute to the pathogenesis of OSA via effects on airway collapsibility and inflammation, respectively; 3) CAV1 expression in peripheral monocytes associated with both AHI and measures of oxygen saturation during sleep; and 4) associations were partially attenuated with adjustment for physiological mechanistic traits, namely, pharyngeal collapsibility and arousal threshold. All findings were consistent with effects of higher gene expression associated with less severe indices of disease, suggesting that future targeting of CAV1-associated pathways may have utility for the development of novel therapeutic strategies for OSA. Notably, a prior study showed that the burden of noncoding variants at regulatory regions may increase or decrease gene expression and are associated with disease outcomes either through loss or gain of function (26). The directional associations we observed between noncoding variants and both higher gene expression and protective effects on OSA severity are consistent with this literature, implicating Caveolin-1 deficiency with multiple physiological abnormalities relevant to OSA.
Although this is the first report implicating CAV1 with OSA, Cav-1 (Caveolin-1) protein has been implicated in multiple pathophysiological processes and diseases relevant to OSA (27). Cav-1 is a membrane-scaffolding protein that is critical for the formation of caveola, plasma membrane lipid rafts that harbor signaling complexes that respond to and transmit a variety of extracellular stimuli across the membrane. Cav-1 interacts and binds with GPCRs (G protein–coupled receptors), endothelial nitric oxide synthase, insulin receptors, TGF-β (transforming growth factor-β), the MAP (mitogen-activated protein) kinase family, the protein kinase C family, growth factor receptor tyrosine kinases, and others. As such, Cav-1 mediates a wide range of functions, which include transcytosis, lipid homeostasis, and mitochondrial function and modulation of reactive oxygen species. CAV1 gene expression is highest in lung and fat tissue but also is abundant in endothelial cells, fibroblasts, adipocytes, and the central nervous system (27). Cav-1 loss can drive inflammation, fibrosis, endothelial dysfunction, angiogenesis, cognitive impairment, and oncogenesis. Cav-1 deficiency is associated with OSA comorbidities, including obesity, diabetes, dyslipidemia, hypertension, pulmonary fibrosis, cardiac failure, Alzheimer’s disease, and cancer (see Figure E1). Depletion of Cav-1 is also implicated in impairment of the ability to maintain blood vessel homeostasis and promote vascular repair after endothelial injury, increasing risk for pulmonary arterial hypertension and lung injury (28, 29). CAV1 genetic variants may therefore be relevant for understanding both the mechanisms for OSA (e.g., pharyngeal collapsibility) as well as mechanisms contributing to OSA-related morbidities (e.g., pulmonary hypertension, diabetes).
Deficiency in Cav-1 may increase OSA severity through several pathways. CAV1 variants that increase gene expression may protect against OSA by effects on upper airway muscle function, as suggested by the findings that 1) the variants associated with OSA are expressed in skeletal muscle, and 2) associations were partially mediated by adjusting for a measure of pharyngeal collapsibility. Deficiencies in Cav-1 also might predispose to more severe OSA through effects on cardiopulmonary function that result in reduced oxygen reserves, thus predisposing to ventilatory instability or more severe desaturation with respiratory events (30). Cav-1 depletion reduces insulin sensitivity and enhances inflammation (31, 32), factors that recently were identified as antecedent risk factors for OSA (33, 34), in addition to reflecting outcomes of OSA. Although our analyses adjusted for body mass index and sensitivity analyses did not suggest pleiotropy or confounding by lung function or diabetes, analyses of larger samples and more precise phenotyping may identify mediating or pleiotropic associations of Cav-1 with OSA and cardiometabolic disease. Given the widespread expression and actions of Cav-1, it is also possible that there are more direct pathophysiological effects of Cav-1 on central or peripheral neuromuscular mechanisms regulating upper airway patency and breathing control.
Cav-1 is also a target for a number of stimuli, including pulmonary stretch, reactive oxygen species (35, 36), and leptin and insulin concentrations. Therefore, there is potential for CAV1 variants to interact with exposures and features common in OSA, modifying the clinical expression of OSA and its comorbidities. For example, Cav-1 can be induced by insulin to negatively regulate endothelial nitric oxide synthase, resulting in endothelial dysfunction (37), a feature of OSA-related cardiovascular disease (32, 38, 39).
Our prior genetic studies of sleep-related hypoxemia (a measure of OSA severity) support a role for genes that modulate lung responses to oxidative and other stressors, genes in inflammatory pathways, and genes influencing iron metabolism. These are pathways and disease processes that are also influenced by CAV1. A GWAS of 10 ethnically diverse cohorts identified significant associations between measures of overnight oxygenation with SNPs in the IL18R1 and HK1 regions (40), which are genes implicated in pulmonary inflammatory conditions (41). A combined linkage–association analysis identified associations between overnight hypoxemia and common variants in ANGPT2 (16), a gene also involved in lung injury syndromes, as well as with rare variants in DLC1 (20), a gene implicated in lung-related disease. Both of these genes have effects on pulmonary endothelial cell function, which is also a target of variants in CAV1. In an admixed population, a combined admixture and association analysis identified variants in FECH to associate with both AHI and hypoxemia (42), suggesting the importance of heme pathways in the etiology of OSA. Heme degradation and its products (e.g., carbon monoxide) play a role in carotid body oxygen sensing and influence redox balance, inflammatory response, and energy metabolism. Caveolae harbor and regulate heme degradation enzymes and negatively regulate heme oxygenase-1, with potential cascading effects on heme oxygenase and carbon monoxide signaling on antiinflammatory processes. Therefore, these distinct analyses, focusing on different OSA traits and using different analytical approaches, support a role for genetic variants in pathways implicated in lung inflammation and pulmonary vascular integrity and suggest the potential for these variants to be amplified with OSA-related exposures such as pulmonary stretch and oxidative stress.
Cav-1 deficiency also leads to sex hormone–dependent metabolic abnormalities in adipose tissue, insulin resistance, lipid metabolism, and hypertriglyceridemia (43). Although we did not have sufficient power to test for sex-specific associations, OSA has clear sexual dimorphism, with less severe disease in women.
A limitation of genetic studies of OSA is the lack of well-phenotyped samples with genotype data. The increasing availability of large samples from biobanks presents opportunities to further explore or validate findings using clinically available data. We analyzed a case-control sample from the UK Biobank in which OSA status was based on International Classification of Diseases codes and likely was markedly underestimated (i.e., only 1% prevalence), reducing statistical power for replication analysis. These analyses provided weak evidence of generalization (i.e., consistent directionality) despite differences in the phenotype and likely significant misclassification. Our analyses of rare variants and quantitative phenotypes suggest value in improving the phenotype data within large biobanks to address disorders such as OSA which are markedly underdiagnosed.
In addition to the associations with CAV1, we observed suggestive evidence of AKR1B1 (P = 7.9 × 10−5) with AHI, although this was not significant after correcting multiple tests. However, given the potential for type II error, this gene may be of interest in further studies.
There are several methodological issues that merit discussion. The P values at stage I may be inflated because of variant selection and because linkage and association were performed in the same pedigrees. Our previous simulation study suggested that such inflation is minimal (20). We used empirical P values when we selected genes for stage II analysis. Because of statistical power, we declared a significant gene either at stage II or III a priori analyses at reduced numbers of tests or using all three stage samples but adjusting for all test hypotheses. As noted, burden and SKAT can be correlated. Thus, the association evidence of CAV1 with AHI reported in this study represents a conservative result. In addition, our analysis is limited to individuals of European ancestry, which limits the generalizability to other populations, especially considering that OSA prevalence and severity as well as allele frequencies of rare variants vary by ancestry groups. Future genetic analysis in large non-European samples is needed to evaluate the generalizability of our results.
Study strengths include a three-stage analysis plan that allowed the assessment of rare variant associations across independent samples and pooled samples, analysis of a standardized quantitative phenotype, and the availability of high-quality genome-wide sequencing data. In stage I, we used efficient statistical approaches that weighted linkage evidence in association analysis to reduce multiple test burden, which differs from popular association methods that do not integrate family information in association testing. In evaluating association evidence, we used stage II and III samples that did not involve variant selection; therefore, our statistical analysis is robust against type I error. However, this approach will miss identifying genes located outside of linkage regions and is best suited for identifying rare variants that segregate within families. Therefore, our approach is complementary to current rare variant searching approaches.
Conclusions
We identified 21 noncoding rare variants in the gene CAV1 that associated with a clinically important and understudied phenotype, the AHI. Our study suggests a functional role of CAV1 in the pathogenesis of OSA. Although there is active research aimed at understanding the role of therapeutic interventions that target Cav-1–related pathways for a wide range of diseases (27), our study suggests that studies of these pathways may identify new approaches for treatment of individuals with OSA, a disease for which there are few existing molecular targets. Future omics analysis and model organism experiments in relevant tissues are needed to further understand the specific mechanisms for the effects of Cav-1 on OSA. More generally, the strategy we applied is a powerful approach in searching for rare variants of complex traits through a large genome sequencing study such as the TOPMed project.
Acknowledgments
Acknowledgment
The authors gratefully acknowledge the studies and participants who provided biological samples and data for TOPMed. The authors thank the staff and participants of the ARIC study for their important contributions. The authors also acknowledge the dedication of the FHS study participants, without whom this research would not be possible.
Footnotes
Supported by National Human Genome Research Institute grant R01HG011052 (X.Z.); NHLBI grants R35135818 (S.R.), R01HL153814 (H.W.), R01HL146697 (S.A.S.), K24HL159246 (P.L.L.), 75N92021D00002 (H.M.O.-B.), and R21HL145425 (T.S.); and American Academy of Sleep Medicine grant 228-SR-20 (S.A.S.). Molecular data for the Trans-Omics in Precision Medicine (TOPMed) program were supported by the NHLBI. Genome sequencing for NHLBI TOPMed: The Cleveland Family Study (phs000954.v2.p1) was performed at the University of Washington Northwest Genomics Center (grant 3R01HL098433-05S1). Genome sequencing for NHLBI TOPMed - NHGRI CCDG: Atherosclerosis Risk in Communities (phs001211.v1.p1) was performed at the Baylor College of Medicine Human Genome Sequencing Center (grants HHSN268201500015C and 3U54HG003273-12S2) and the Broad Institute Genomics Platform (grant 3R01HL092577-06S1). Genome sequencing for NHLBI TOPMed: Cardiovascular Health Study (phs001368.v1.p1) was performed at the Baylor College of Medicine Human Genome Sequencing Center (grant HHSN268201500015C). Genome sequencing for NHLBI TOPMed: Whole Genome Sequencing and Related Phenotypes in the Framingham Heart Study (phs000974.v3.p2) was performed at the Broad Institute Genomics Platform (grant 3R01HL092577-06S1). Genome sequencing for NHLBI TOPMed: MESA (phs001416.v1.p1) was performed at the Broad Institute Genomics Platform (grant 3U54HG003067-13S1). RNA sequencing for NHLBI TOPMed: MESA (phs001416.v1.p1) was performed at the Northwest Genomics Center (grant HHSN268201600032I). Core support including centralized genomic read mapping and genotype calling, along with variant quality metrics and filtering, was provided by the TOPMed Informatics Research Center (grant 3R01HL-117626-02S1, contract HHSN268201800002I). Core support including phenotype harmonization, data management, sample-identity quality control, and general program coordination was provided by the TOPMed Administrative Coordinating Center (grant R01HL-120393, grant U01HL-120393, contract HHSN268201800001I). The Genome Sequencing Program (GSP) was funded by the National Human Genome Research Institute (NHGRI), the National Heart, Lung, and Blood Institute (NHLBI), and the National Eye Institute (NEI). The GSP Coordinating Center (U24 HG008956) contributed to cross program scientific initiatives and provided logistical and general study coordination. The Centers for Common Disease Genomics (CCDG) program was supported by NHGRI and NHLBI, and whole genome sequencing was performed at the Baylor College of Medicine Human Genome Sequencing Center (UM1 HG008898). The Atherosclerosis Risk in Communities study has been funded in whole or in part with federal funds from the NHLBI, NIH, U.S. Department of Health and Human Services (contracts HHSN268201700001, HHSN268201700002, HHSN268201700003, HHSN268201700004, and HHSN268201700005). The Cleveland Family Study has been supported in part by NIH grants (R01-HL046380, KL2-RR024990, R35-HL135818, and R01-HL113338). The Cardiovascular Health Study was supported by NHLBI contracts HHSN268201200036C, HHSN268200800007C, HHSN268201800001C, N01HC55222, N01HC85079, N01HC85080, N01HC85081, N01HC85082, N01HC85083, N01HC85086, and 75N92021D00006 and NHLBI grants U01HL080295 and U01HL130114, with additional contribution from the National Institute of Neurological Disorders and Stroke. Additional support was provided by National Institute on Aging grant R01AG023629. A full list of principal CHS investigators and institutions can be found at CHS-NHLBI.org. The content is solely the responsibility of the authors and does not necessarily represent the official views of the NIH. The Framingham Heart Study (FHS) acknowledges the support of NHLBI contracts NO1-HC-25195, HHSN268201500001I, and 75N92019D00031 and grant supplement R01 HL092577-06S1 for this research. R.S.V. is supported in part by the Evans Medical Foundation and the Jay and Louis Coffman Endowment from the Department of Medicine, Boston University School of Medicine. MESA and the MESA SHARe project are conducted and supported by the NHLBI in collaboration with MESA investigators. Support for MESA is provided by contracts HHSN268201500003I, N01-HC-95159, N01-HC-95160, N01-HC-95161, N01-HC-95162, N01-HC-95163, N01-HC-95164, N01-HC-95165, N01-HC-95166, N01-HC-95167, N01-HC-95168, N01-HC-95169, UL1-TR-000040, UL1-TR-001079, and UL1-TR-001420. MESA Family is conducted and supported by the NHLBI in collaboration with MESA investigators. Support is provided by grants and contracts R01HL071051, R01HL071205, R01HL071250, R01HL071251, R01HL071258, and R01HL071259 and by National Center for Research Resources grant UL1RR033176. The provision of genotyping data was supported in part by the National Center for Advancing Translational Sciences, Clinical and Translational Sciences Institute grant UL1TR001881, and National Institute of Diabetes and Digestive and Kidney Disease Diabetes Research Center grant DK063491 to the Southern California Diabetes Endocrinology Research Center. The Osteoporotic Fractures in Men (MrOS) study is supported by NIH funding. Support is provided by the National Institute on Aging, the National Institute of Arthritis and Musculoskeletal and Skin Diseases (NIAMS), the National Center for Advancing Translational Sciences, and the NIH Roadmap for Medical Research under grants U01 AG027810, U01 AG042124, U01 AG042139, U01 AG042140, U01 AG042143, U01 AG042145, U01 AG042168, U01 AR066160, and UL1 TR000128. The NHLBI provides funding for the MrOS Sleep ancillary study Outcomes of Sleep Disorders in Older Men under grants R01 HL071194, R01 HL070848, R01 HL070847, R01 HL070842, R01 HL070841, R01 HL070837, R01 HL070838, and R01 HL070839. NIAMS provides funding for the MrOS ancillary study Replication of Candidate Gene Associations and Bone Strength Phenotype in MrOS under the grant R01 AR051124. NIAMS provides funding for the MrOS ancillary study GWAS in MrOS and SOF under grant RC2 AR058973. Funding for the Western Australian Sleep Health Study was obtained from the Sir Charles Gairdner and Hollywood Private Hospital Research Foundations, the Western Australian Sleep Disorders Research Institute, and the Centre for Genetic Epidemiology and Biostatistics at the University of Western Australia. Funding for the genome-wide association study genotyping was obtained from the Ontario Institute for Cancer Research and a McLaughlin Centre Accelerator Grant from the University of Toronto.
Author Contributions: J. Liang, H.W., T.S., S.R., and X.Z. designed this study. J. Liang, H.W., B.E.C., N.K., K.Y.H., J. Lee, T.S., S.R., and X.Z. participated in data acquisition and analysis and/or interpretation of data. J. Liang, H.W., T.S., S.R., and X.Z. drafted the manuscript. All authors reviewed and approved the final version of the paper that was submitted to the journal.
This article has an online supplement, which is accessible from this issue’s table of contents at www.atsjournals.org.
Originally Published in Press as DOI: 10.1164/rccm.202203-0618OC on July 13, 2022
Author disclosures are available with the text of this article at www.atsjournals.org.
References
- 1. Badran M, Yassin BA, Fox N, Laher I, Ayas N. Epidemiology of sleep disturbances and cardiovascular consequences. Can J Cardiol . 2015;31:873–879. doi: 10.1016/j.cjca.2015.03.011. [DOI] [PubMed] [Google Scholar]
- 2. Senaratna CV, Perret JL, Lodge CJ, Lowe AJ, Campbell BE, Matheson MC, et al. Prevalence of obstructive sleep apnea in the general population: a systematic review. Sleep Med Rev . 2017;34:70–81. doi: 10.1016/j.smrv.2016.07.002. [DOI] [PubMed] [Google Scholar]
- 3. Benjafield AV, Ayas NT, Eastwood PR, Heinzer R, Ip MSM, Morrell MJ, et al. Estimation of the global prevalence and burden of obstructive sleep apnoea: a literature-based analysis. Lancet Respir Med . 2019;7:687–698. doi: 10.1016/S2213-2600(19)30198-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Mbata G, Chukwuka J. Obstructive sleep apnea hypopnea syndrome. Ann Med Health Sci Res . 2012;2:74–77. doi: 10.4103/2141-9248.96943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Young T, Skatrud J, Peppard PE. Risk factors for obstructive sleep apnea in adults. JAMA . 2004;291:2013–2016. doi: 10.1001/jama.291.16.2013. [DOI] [PubMed] [Google Scholar]
- 6. Redline S, Tishler PV. The genetics of sleep apnea. Sleep Med Rev . 2000;4:583–602. doi: 10.1053/smrv.2000.0120. [DOI] [PubMed] [Google Scholar]
- 7. Liang J, Cade BE, Wang H, Chen H, Gleason KJ, Larkin EK, et al. Comparison of heritability estimation and linkage analysis for multiple traits using principal component analyses. Genet Epidemiol . 2016;40:222–232. doi: 10.1002/gepi.21957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Patel SR, Larkin EK, Redline S. Shared genetic basis for obstructive sleep apnea and adiposity measures. Int J Obes . 2008;32:795–800. doi: 10.1038/sj.ijo.0803803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Chen H, Cade BE, Gleason KJ, Bjonnes AC, Stilp AM, Sofer T, et al. Multiethnic meta-analysis identifies RAI1 as a possible obstructive sleep apnea-related quantitative trait locus in men. Am J Respir Cell Mol Biol . 2018;58:391–401. doi: 10.1165/rcmb.2017-0237OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Palmer LJ, Buxbaum SG, Larkin E, Patel SR, Elston RC, Tishler PV, et al. A whole-genome scan for obstructive sleep apnea and obesity. Am J Hum Genet . 2003;72:340–350. doi: 10.1086/346064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Larkin EK, Patel SR, Elston RC, Gray-McGuire C, Zhu X, Redline S. Using linkage analysis to identify quantitative trait loci for sleep apnea in relationship to body mass index. Ann Hum Genet . 2008;72:762–773. doi: 10.1111/j.1469-1809.2008.00472.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Cade BE, Chen H, Stilp AM, Gleason KJ, Sofer T, Ancoli-Israel S, et al. Genetic associations with obstructive sleep apnea traits in Hispanic/Latino Americans. Am J Respir Crit Care Med . 2016;194:886–897. doi: 10.1164/rccm.201512-2431OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Povysil G, Petrovski S, Hostyk J, Aggarwal V, Allen AS, Goldstein DB. Rare-variant collapsing analyses for complex traits: guidelines and applications. Nat Rev Genet . 2019;20:747–759. doi: 10.1038/s41576-019-0177-4. [DOI] [PubMed] [Google Scholar]
- 14. Zhu X, Feng T, Li Y, Lu Q, Elston RC. Detecting rare variants for complex traits using family and unrelated data. Genet Epidemiol . 2010;34:171–187. doi: 10.1002/gepi.20449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Jun G, Manning A, Almeida M, Zawistowski M, Wood AR, Teslovich TM, et al. Evaluating the contribution of rare variants to type 2 diabetes and related traits using pedigrees. Proc Natl Acad Sci USA . 2018;115:379–384. doi: 10.1073/pnas.1705859115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Wang H, Cade BE, Chen H, Gleason KJ, Saxena R, Feng T, et al. Variants in angiopoietin-2 (ANGPT2) contribute to variation in nocturnal oxyhaemoglobin saturation level. Hum Mol Genet . 2016;25:5244–5253. doi: 10.1093/hmg/ddw324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Wang H, Nandakumar P, Tekola-Ayele F, Tayo BO, Ware EB, Gu CC, et al. Combined linkage and association analysis identifies rare and low frequency variants for blood pressure at 1q31. Eur J Hum Genet . 2019;27:269–277. doi: 10.1038/s41431-018-0277-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. He KY, Wang H, Cade BE, Nandakumar P, Giri A, Ware EB, et al. Rare variants in fox-1 homolog A (RBFOX1) are associated with lower blood pressure. PLoS Genet . 2017;13:e1006678. doi: 10.1371/journal.pgen.1006678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. He KY, Li X, Kelly TN, Liang J, Cade BE, Assimes TL, et al. NHLBI Trans-Omics for Precision Medicine (TOPMed) Consortium, TOPMed Blood Pressure Working Group Leveraging linkage evidence to identify low-frequency and rare variants on 16p13 associated with blood pressure using TOPMed whole genome sequencing data. Hum Genet . 2019;138:199–210. doi: 10.1007/s00439-019-01975-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Liang J, Cade BE, He KY, Wang H, Lee J, Sofer T, et al. NHLBI Trans-Omics for Precision Medicine (TOPMed); TOPMed Sleep Working Group Sequencing analysis at 8p23 identifies multiple rare variants in DLC1 associated with sleep-related oxyhemoglobin saturation level. Am J Hum Genet . 2019;105:1057–1068. doi: 10.1016/j.ajhg.2019.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Wang H, Liang J, Cade B, Kurniansyah N, Sofer T, Redline S, et al. 0026 Targeted genome sequencing identifies multiple rare variants in Caveolin-1 associated with obstructive sleep apnea [abstract] Sleep (Basel) . 2022;45:A12. doi: 10.1164/rccm.202203-0618OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Chen H, Meigs JB, Dupuis J. Sequence kernel association test for quantitative traits in family samples. Genet Epidemiol . 2013;37:196–204. doi: 10.1002/gepi.21703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Wu MC, Lee S, Cai T, Li Y, Boehnke M, Lin X. Rare-variant association testing for sequencing data with the sequence kernel association test. Am J Hum Genet . 2011;89:82–93. doi: 10.1016/j.ajhg.2011.05.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. GTEx Consortium. The genotype-tissue expression (GTEx) project. Nat Genet . 2013;45:580–585. doi: 10.1038/ng.2653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Zerbino DR, Wilder SP, Johnson N, Juettemann T, Flicek PR. The ensembl regulatory build. Genome Biol . 2015;16:56. doi: 10.1186/s13059-015-0621-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Zhao J, Akinsanmi I, Arafat D, Cradick TJ, Lee CM, Banskota S, et al. A burden of rare variants associated with extremes of gene expression in human peripheral blood. Am J Hum Genet . 2016;98:299–309. doi: 10.1016/j.ajhg.2015.12.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Gokani S, Bhatt LK. Caveolin-1: a promising therapeutic target for diverse diseases. Curr Mol Pharmacol . 2022;15:701–715. doi: 10.2174/1874467214666211130155902. [DOI] [PubMed] [Google Scholar]
- 28. Jin Y, Lee SJ, Minshall RD, Choi AM. Caveolin-1: a critical regulator of lung injury. Am J Physiol Lung Cell Mol Physiol . 2011;300:L151–L160. doi: 10.1152/ajplung.00170.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Zhao YY, Liu Y, Stan RV, Fan L, Gu Y, Dalton N, et al. Defects in caveolin-1 cause dilated cardiomyopathy and pulmonary hypertension in knockout mice. Proc Natl Acad Sci USA . 2002;99:11375–11380. doi: 10.1073/pnas.172360799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Wang Y, Roche O, Xu C, Moriyama EH, Heir P, Chung J, et al. Hypoxia promotes ligand-independent EGF receptor signaling via hypoxia-inducible factor-mediated upregulation of caveolin-1. Proc Natl Acad Sci USA . 2012;109:4892–4897. doi: 10.1073/pnas.1112129109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Haddad D, Al Madhoun A, Nizam R, Al-Mulla F. Role of Caveolin-1 in diabetes and its complications. Oxid Med Cell Longev . 2020;2020:9761539. doi: 10.1155/2020/9761539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Pojoga LH, Underwood PC, Goodarzi MO, Williams JS, Adler GK, Jeunemaitre X, et al. Variants of the caveolin-1 gene: a translational investigation linking insulin resistance and hypertension. J Clin Endocrinol Metab . 2011;96:E1288–E1292. doi: 10.1210/jc.2010-2738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Huang T, Goodman M, Li X, Sands SA, Li J, Stampfer MJ, et al. C-reactive protein and risk of OSA in four US cohorts. Chest . 2021;159:2439–2448. doi: 10.1016/j.chest.2021.01.060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Jean-Louis G, Zizi F, Clark LT, Brown CD, McFarlane SI. Obstructive sleep apnea and cardiovascular disease: role of the metabolic syndrome and its components. J Clin Sleep Med . 2008;4:261–272. [PMC free article] [PubMed] [Google Scholar]
- 35. Budhiraja R, Parthasarathy S, Quan SF. Endothelial dysfunction in obstructive sleep apnea. J Clin Sleep Med . 2007;3:409–415. [PMC free article] [PubMed] [Google Scholar]
- 36. Hoyos CM, Melehan KL, Liu PY, Grunstein RR, Phillips CL. Does obstructive sleep apnea cause endothelial dysfunction? A critical review of the literature. Sleep Med Rev . 2015;20:15–26. doi: 10.1016/j.smrv.2014.06.003. [DOI] [PubMed] [Google Scholar]
- 37. Wang H, Wang AX, Liu Z, Chai W, Barrett EJ. The trafficking/interaction of eNOS and caveolin-1 induced by insulin modulates endothelial nitric oxide production. Mol Endocrinol . 2009;23:1613–1623. doi: 10.1210/me.2009-0115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Nieto FJ, Herrington DM, Redline S, Benjamin EJ, Robbins JA. Sleep apnea and markers of vascular endothelial function in a large community sample of older adults. Am J Respir Crit Care Med . 2004;169:354–360. doi: 10.1164/rccm.200306-756OC. [DOI] [PubMed] [Google Scholar]
- 39. Xiang AH, Azen SP, Raffel LJ, Tan S, Cheng LS, Diaz J, et al. Evidence for joint genetic control of insulin sensitivity and systolic blood pressure in Hispanic families with a hypertensive proband. Circulation . 2001;103:78–83. doi: 10.1161/01.cir.103.1.78. [DOI] [PubMed] [Google Scholar]
- 40. Cade BE, Chen H, Stilp AM, Louie T, Ancoli-Israel S, Arens R, et al. Associations of variants In the hexokinase 1 and interleukin 18 receptor regions with oxyhemoglobin saturation during sleep. PLoS Genet . 2019;15:e1007739. doi: 10.1371/journal.pgen.1007739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Stout-Delgado HW, Cho SJ, Chu SG, Mitzel DN, Villalba J, El-Chemaly S, et al. Age-dependent susceptibility to pulmonary fibrosis is associated with NLRP3 inflammasome activation. Am J Respir Cell Mol Biol . 2016;55:252–263. doi: 10.1165/rcmb.2015-0222OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Wang H, Cade BE, Sofer T, Sands SA, Chen H, Browning SR, et al. Admixture mapping identifies novel loci for obstructive sleep apnea in Hispanic/Latino Americans. Hum Mol Genet . 2019;28:675–687. doi: 10.1093/hmg/ddy387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Mukherjee R, Kim SW, Choi MS, Yun JW. Sex-dependent expression of caveolin 1 in response to sex steroid hormones is closely associated with development of obesity in rats. PLoS One . 2014;9:e90918. doi: 10.1371/journal.pone.0090918. [DOI] [PMC free article] [PubMed] [Google Scholar]