

Abstract
Background and Objectives
Friedreich ataxia (FRDA) is an autosomal recessive ataxia with no approved treatments. Leriglitazone is a selective peroxisome proliferator–activated receptor γ agonist that crosses the blood-brain barrier and, in preclinical models, improved mitochondrial function and energy production. We assessed effects of leriglitazone in patients with FRDA in a proof-of-concept study.
Methods
In this double-blind, randomized controlled trial, eligible participants (age 12–60 years) had genetically confirmed FRDA, a Scale for the Assessment and Rating of Ataxia (SARA) total score <25, and a SARA item 1 score of 2–6, inclusive. Key exclusion criteria were age at FRDA onset ≥25 years and history of cardiac dysfunction. Participants were randomly assigned (2:1) to receive a daily, oral, individualized dose of leriglitazone or placebo for 48 weeks. The primary endpoint was the change from baseline to week 48 in spinal cord area (C2-C3) (measured by MRI). Secondary endpoints included the change from baseline to week 48 in iron accumulation in the dentate nucleus (quantitative susceptibility mapping) and total N-acetylaspartate to myo-inositol (tNAA/mIns) ratio.
Results
Overall, 39 patients were enrolled (mean age 24 years; 43.6% women; mean time since symptom onset 10.5 years): 26 patients received leriglitazone (20 completed) and 13 received placebo (12 completed). There was no difference between groups in spinal cord area from baseline to week 48 (least-squares [LS] mean change [standard error (SE)]: leriglitazone, −0.39 [0.55] mm2; placebo, 0.08 [0.72] mm2; p = 0.61). Iron accumulation in the dentate nucleus was greater with placebo (LS mean change [SE]: leriglitazone, 0.10 [1.33] ppb; placebo, 4.86 [1.84] ppb; p = 0.05), and a numerical difference was seen in tNAA/mIns ratio (LS mean change [SE]: leriglitazone, 0.03 [0.02]; placebo, −0.02 [0.03]; p = 0.25). The most frequent adverse event was peripheral edema (leriglitazone 73.1%, placebo 0%).
Discussion
The primary endpoint of change in spinal cord area was not met. Secondary endpoints provide evidence supporting proof of concept for leriglitazone mode of action and, with acceptable safety data, support larger studies in patients with FRDA.
Trial Registration Information
ClinicalTrials.gov: NCT03917225; EudraCT: 2018-004405-64; submitted April 17, 2019; first patient enrolled April 2, 2019. clinicaltrials.gov/ct2/show/NCT03917225?term=NCT03917225&draw=2&rank=1.
Classification of Evidence
This study provides Class I evidence that individualized dosing of leriglitazone, compared with placebo, is not associated with changes in spinal cord area in patients with FRDA.
Friedreich ataxia (FRDA) is an autosomal recessive multisystem disorder characterized by neurologic impairment, hypertrophic cardiomyopathy, skeletal abnormalities, and carbohydrate intolerance.1 Most patients are homozygous for a pathologic expansion of an alpha glucosidase (GAA) repeat in the first intron of the frataxin (FXN) gene.1 This expansion results in the formation of repressive chromatin, inhibiting transcription of FXN mRNA.2 FXN is needed for the synthesis of iron-sulfur (Fe-S) clusters in mitochondria,3 and its deficiency results in mitochondrial dysfunction,4 oxidative stress,5 and altered iron metabolism.6 Longer pathologic GAA repeats are associated with more severe disease and earlier disease onset.7,8
FRDA neuropathology is characterized by marked differences in the vulnerability of different neuronal systems in the course of disease. Loss of large primary sensory neurons in the dorsal root ganglia (sensory information) occurs very early and may be partly developmental,9 whereas cerebellar pathology (namely, dentate nucleus atrophy)10 is only detected at symptom onset. Pyramidal degeneration worsens with increased disease duration,11 and patients with FRDA generally lose the ability to walk, sit, or stand without support within 15–20 years of disease onset.12 In advanced disease, auditory and visual impairment become significant,13 and in some patients, hypertrophic cardiomyopathy progresses, resulting in heart failure, arrhythmias, and premature death.14 There are currently no approved treatments that have been shown to slow progression of FRDA, with therapies focusing on symptom management.15,16 Therefore, effective treatment represents a substantial unmet medical need.
Peroxisome proliferator–activated receptor γ (PPARγ) is a transcription factor involved in the regulation of fundamental cellular processes, such as differentiation, development, and metabolism, and a key regulator of mitochondrial biogenesis and function.17 Dysregulation of the PPARγ and PPARγ coactivator 1 α pathway has been reported in tissues from a mouse model of FRDA, in FXN-deficient cell models, and in fibroblasts and lymphoblasts from patients with FRDA.18-21 Leriglitazone is a novel, selective PPARγ agonist.17 It is one of the active metabolites of pioglitazone (Actos; Takeda Chemical Industries, Ltd., Osaka, Japan), an approved treatment for type 2 diabetes.22 Leriglitazone has high oral bioavailability and crosses the blood-brain barrier, reaching CNS concentrations significantly above the level safely achieved with pioglitazone and other glitazones.23
Preclinical and clinical studies support leriglitazone as a possible treatment for FRDA. A recent study found that leriglitazone protected dorsal root ganglia neurons from apoptosis in vitro and improved motor function in a mouse model of FRDA.17 Lipid droplet accumulation in FXN-deficient cardiomyocytes was also reduced, owing to improvements in fatty acid β-oxidation.17 A good safety and tolerability profile of leriglitazone, as well as sufficient CNS activation of PPARγ receptors, was demonstrated in a phase 1, randomized, double-blind, placebo-controlled study in 33 healthy male volunteers.24
Various parameters can be used to assess progression and severity of disease in patients with FRDA. Natural history studies indicate that progression is best assessed in ambulatory patients25-29 possibly because rating scales are most sensitive to impairments in gait and stance and reach a ceiling effect in nonambulatory patients.12,25 The annual rate of clinical decline can be estimated using linear regression of disease duration vs scores in rating scales such as the Scale for the Assessment and Rating of Ataxia (SARA).28,30 This was one of the primary endpoints in the European FRDA Consortium for Translational Studies natural history study.27-29 MRI can also be used to detect morphometric, microstructural, and biochemical changes in the CNS of patients with FRDA. For example, structural MRI showed a decrease in spinal cord area and an increase in eccentricity in patients relative to controls, consistent with atrophy of dorsal and lateral columns of the spinal cord and correlating with measures of clinical decline.31,32 In addition, structural MRI showed a progressive decrease in spinal cord area, and magnetic resonance spectroscopy (MRS) showed a progressive decrease in the total N-acetylaspartate concentration/myo-inositol (tNAA/mIns) ratio over time in an early-stage patient cohort.33 MRI quantitative susceptibility mapping (QSM) showed a progressive increase in dentate nucleus iron concentration in a longitudinal study in patients with FRDA.34
Here, we report efficacy and safety results from a phase 2, randomized, double-blind, placebo-controlled, proof-of-concept study on the effects of leriglitazone on biochemical, imaging, neurophysiologic, and clinical markers in patients with FRDA.
Methods
Study Design
This was a 48-week, double-blind, placebo-controlled study conducted at 5 centers in Europe (Belgium, France, Germany, and 2 in Spain) between April 2019 and September 2020. Participants were randomly assigned in a 2:1 ratio (using interactive response technology) to receive leriglitazone or placebo orally once per day for 48 weeks. Participants received an individualized starting dose of leriglitazone based on sex and age (150 mg in adult male patients, 130 mg in adult female patients, and 2.2 mg/kg in adolescents [12–17 years old]) and informed by physiologic pharmacokinetic (PK) modeling and a phase 1 study.24 Dose was modified based on PK parameters at week 4 to achieve a target exposure of 170 μg ⋅ h/mL and remained stable thereafter except for adjustments for safety or tolerability. To preserve blinding, an unblinded PK expert calculated the necessary dose adjustments and communicated dose changes to the investigator. Random dose adjustments were also made in the placebo group. Plasma concentrations of leriglitazone and its main metabolite M3 were assessed at all postbaseline visits. Values were extrapolated using an algorithm developed from PK data to determine area under the plasma concentration-time curve over the past 24-hour dosing interval at steady state (AUC0–24) values and inform dose adjustments.
Standard Protocol Approvals, Registrations, and Patient Consents
The study (FRAMES; ClinicalTrials.gov NCT03917225; EudraCT: 2018-004405-64) was conducted in accordance with the Declaration of Helsinki and the International Conference on Harmonisation guidelines for Good Clinical Practice. All patients provided written informed consent, and all sites obtained independent ethics committee and institutional review board approval before study initiation.
Patients
Male and female patients aged 12–60 years at screening were eligible for study enrollment if they had a genetically confirmed diagnosis of FRDA (the individual FXN genetic status of participants was not obtained in this study), a total SARA score of less than 25, and a score of between 2 and 6, inclusive, on SARA item 1 (gait). The main exclusion criteria were age at FRDA onset of 25 years or older and a history of heart failure or left ventricular ejection fraction (LVEF) below 55% (on echocardiogram). Full inclusion and exclusion criteria are given in the eMethods, links.lww.com/NXG/A553.
Efficacy Variables and Assessments
The primary efficacy endpoint was the change from baseline to week 48 in spinal cord area cervical segment C2-C3, as assessed by morphometric T1-weighted brain image MRI measurements, in the leriglitazone group vs the placebo group. MRI and MRS acquisition and processing methods are described in the eMethods, links.lww.com/NXG/A553.
Secondary efficacy endpoints assessed change from baseline at 48 weeks vs placebo in MRI and MRS imaging parameters, biochemical magnetic resonance parameters, and clinical and patient questionnaires. Cervical spinal cord fractional anisotropy and mean, axial, and radial diffusivity at cervical segments C2 to C7 (10−3 mm2/s) were assessed by diffusion tensor imaging (DTI). In the brain, FreeSurfer was used for brain morphometry to obtain the volumes of the medulla, pons, midbrain, thalamus, third and fourth ventricles, caudate, and putamen. Brain DTI was used for fractional anisotropy and mean, axial, and radial diffusivity in the superior and inferior peduncles, and the posterior limb of the internal capsule. Fixel-based analysis (FBA) (which, unlike DTI, can identify structural changes in individual fiber populations within voxels containing crossing fibers) was used for fiber density (FD), fiber cross-section (FC), and FD and cross-section (FDC) analyses of the corticospinal tract; the superior and inferior cerebellar peduncles; the posterior limb of the internal capsule; the superior corona radiata; and the medial lemniscus.35 Biochemical magnetic resonance parameters included the assessment of the volume of the dentate nucleus (mm3) and iron concentration in the dentate nucleus (ppb shift) by MRI QSM,36 and cervical spinal cord tNAA/mIns ratio by MRS. Cerebellar dysfunction of the upper limbs was measured using the cerebellar composite functional scale (CCFS) score, a quantitative assessment of 2 functional tests (9-hole peg test and clicking) corrected for age.37 Clinician-reported and patient-reported outcomes included total score on the SARA; quality of life assessed by the 5-level, 5-dimension EuroQol questionnaire (EQ-5D-5L); clinicians' and patients' global impressions of improvement, including Clinical Global Impression-Severity, Clinical Global Impression-Improvement (CGI-I), Patient Global Impression-Improvement (PGI-I); Fatigue Severity Score; and Activities of Daily Living (ADL) (subscale from the Friedreich Ataxia Rating Scale).
Exploratory efficacy endpoints included adiponectin concentrations in plasma, as a well-known marker of PPARγ engagement, and gene expression of FXN in whole blood.38
Baseline evaluations consisted of imaging (MRI and MRS), clinical status using SARA, CCFS, ADL, global clinical rating scales, patient questionnaires, and assessment of biochemical markers. Postbaseline evaluations at 24 and 48 weeks consisted of imaging evaluations, evaluations of clinical status using SARA, CCFS, global clinical rating scales, and patient questionnaires. Assessment of biochemical markers in plasma took place at weeks 12, 24, and 48. Compliance was summarized overall for the entire study by assessment of dose taken and leriglitazone levels in plasma.
Safety Outcomes and Assessments
Safety and tolerability of leriglitazone were assessed by adverse events (AEs), vital signs, 12-lead ECG in triplicate, echocardiogram (with assessment of LVEF), clinical laboratory tests, and palatability.
Statistical Methods
Clinically, meaningful effects were assessed based on point estimates and 95% confidence intervals (CI); p values were used only as a guide to activity. Sample size was based on the expected change in the primary outcome measure over 1 year, and study feasibility assured by available sites and time needed for completion. SAS software version 9.4 was used for statistical analyses.
The modified intent-to-treat (mITT) population included all patients who took at least 1 dose (partial or complete) of the study drug and had at least 1 postbaseline spinal cord area cervical segment C2-C3 measurement and SARA assessment at the same visit. The per-protocol population included all patients in the mITT population who did not have a major protocol deviation. The safety population included all patients who took at least 1 dose (partial or complete) of the study drug. All efficacy analyses are presented for the mITT population.
For analysis of the primary efficacy endpoint, treatment comparison of mean change from baseline at 24 and 48 weeks was performed using an analysis of covariance (ANCOVA) model, with treatment arm as a fixed effect and baseline value as a covariate. This methodology was also used for most secondary efficacy endpoints, as well as post hoc subpopulation analyses. For the CGI-I and PGI-I endpoints, descriptive statistics and t tests were used to compare the 2 treatment arms at 24 and 48 weeks. All safety data were summarized descriptively by treatment received.
Post hoc analyses included an O'Brien composite variable analysis, with change in iron concentration (assessed by QSM), cervical spinal cord tNAA/mIns ratio (assessed by MRS), and CCFS score as variables (eMethods, links.lww.com/NXG/A553).
Data Availability
All data and related documentation underlying the reported results will be made available after anonymization of patient information up to 4 years after publication of this article. In the interim, the authors will share the data with qualified investigators whose proposal of data use has been approved by an independent review committee.
Protocol and Statistical Analysis Plan
The study protocol and statistical analysis plan are available in eSAP 1 and eSAP 2, respectively, links.lww.com/NXG/A553.
Results
Patients
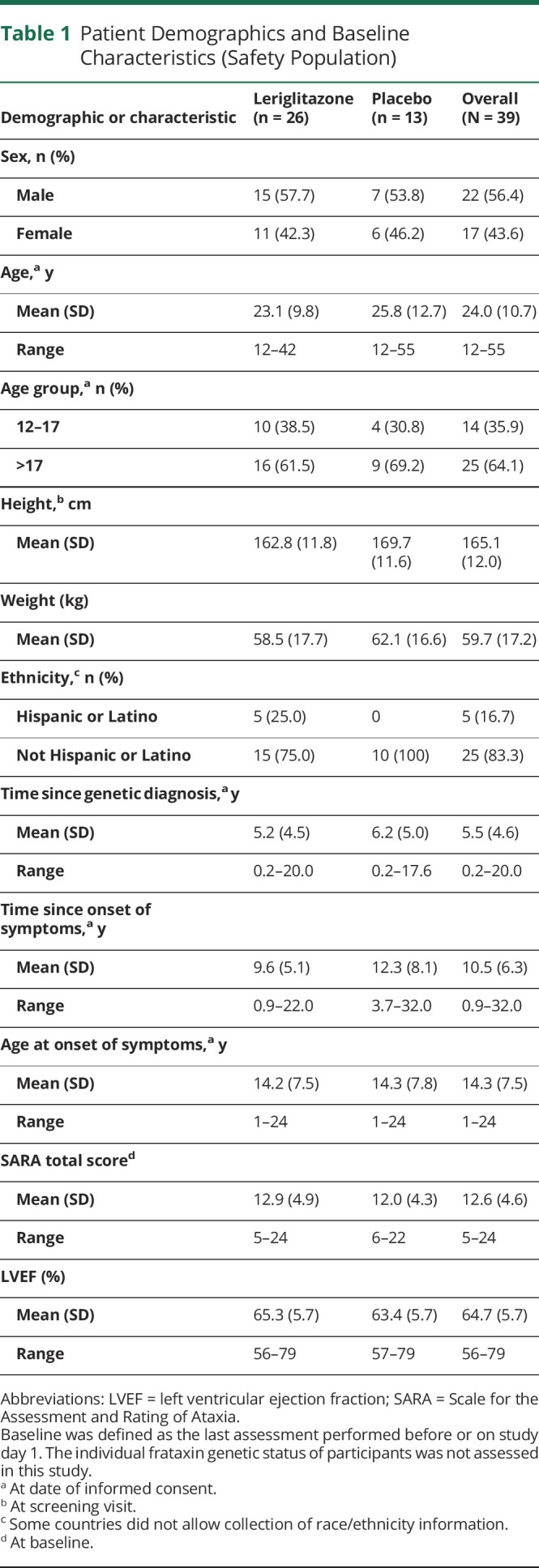
Between April 2019 and September 2020, 50 patients were screened for enrollment, 26 were randomly assigned to receive leriglitazone and 13 to receive placebo (Figure 1). In the leriglitazone group, 20 (76.9%) patients completed the treatment period; in the placebo group, 12 (92.3%) completed the treatment period. All patients had at least 1 protocol deviation, and for most patients (n = 28 [71.8%]), this was due to travel or hospital restrictions related to the COVID-19 pandemic. Patient demographics and baseline characteristics were similar, across treatment groups in age, sex, height, weight, SARA total score and LVEF (Table 1). There were 14 (35.9%) adolescents and 25 (64.1%) adults. The mean (SD) time since the onset of symptoms was shorter for the leriglitazone group (9.6 [5.1] years) than for the placebo group (12.3 [8.1] years). Overall, 2 (5.1%) patients had a medical history of at least 1 cardiac disorder, and 3 (7.7%) patients reported hypertrophic cardiomyopathy. At baseline, 20.5% of patients had a normal ECG result, 76.9% had a not clinically significant, abnormal ECG result, and 35.9% had a not clinically significant, abnormal echocardiogram result (eTable 1, links.lww.com/NXG/A553).
Figure 1. CONSORT Diagram.

The safety analysis set included all randomized patients who received at least 1 dose (partial or complete) of the study drug. The mITT analysis set included all patients who took at least 1 dose (partial or complete) of the study drug and had at least 1 postbaseline spinal cord area segment C2-C3 measurement and SARA assessment at the same visit. The PP analysis set included all patients in the mITT analysis set who did not have a major protocol deviation. CONSORT = Consolidated Standards of Reporting Trials; mITT = modified intent-to-treat; PP = per protocol; SARA = Scale for the Assessment and Rating of Ataxia.
Table 1.
Patient Demographics and Baseline Characteristics (Safety Population)
After initial dose adjustments to achieve a target exposure of 170 µg ⋅ h/mL, additional adjustments due to safety or tolerability issues were reported in 12 (46.2%) patients in the leriglitazone group and 1 (7.7%) patient in the placebo group. The overall mean extrapolated AUC0–24 for all patients was approximately 166 μg ⋅ h/mL. Exposure and PPARγ engagement were within target range in all patients (eFigure 1, links.lww.com/NXG/A553). The mean overall compliance was 99.4% (100.0%, leriglitazone; 98.4%, placebo).
Efficacy Assessments
MRI Morphometry and Microstructure
The mean spinal cord area (SD) at baseline was 41.09 (4.95) mm2 in the leriglitazone group and 40.20 (5.44) mm2 in the placebo group. Figure 2 shows the mean (standard error [SE]) change in spinal cord area from baseline at weeks 24 and 48 (individual patient data are shown in eFigure 2, links.lww.com/NXG/A553). The least-squares (LS) mean change (SE) (95% CI) from baseline at week 48 was −0.39 (0.55) (95% CI −1.53 to 0.74) mm2 in the leriglitazone group and 0.08 (0.72) (95% CI −1.39 to 1.54) mm2 in the placebo group (p = 0.61). There was a smaller LS mean change (SE) (95% CI) in dentate nuclei volume (normalized by total intracranial volume) at week 48 in the leriglitazone group (0.0002) (0.0001) (95% CI 0.0001–0.0003) than in the placebo group (0.0005) (0.0001) (95% CI 0.0003–0.0006) (p = 0.03) (eTable 2, links.lww.com/NXG/A553). The mean changes from baseline to week 48 in brain FD, FC, and FDC, as assessed by FBA, were minimal (data not shown; example MR images are shown in eFigure 3, links.lww.com/NXG/A553). The only nominally significant difference between treatment groups was in FD in the superior cerebellar peduncle, which was higher in the leriglitazone group (data not shown). No difference was seen between treatment groups in spinal cord DTI, brain DTI, or brain morphometry from baseline to week 48 (data not shown).
Figure 2. Change From Baseline at Week 24 and Week 48 in Spinal Cord Cervical Area Cervical Segment C2-C3 (mm2), Estimated From MRI T1-Weighted Brain Images (mITT Population) (Primary Endpoint).

Data shown are mean ± SE. The LS mean (SE), 95% CI and p value for the difference between treatment arms, as assessed by ANCOVA (with treatment arm as a fixed effect and the baseline value as a covariate) are shown in the table. Baseline was defined as the last assessment performed before or on study day 1. N values in the table show the number of patients included in the ANCOVA analyses. ANCOVA = analysis of covariance; CI = confidence interval; LS = least-squares; mITT = modified intent-to-treat; SE = standard error.
Biochemical Magnetic Resonance Markers
Figure 3 shows the mean (SE) change in iron accumulation in the dentate nucleus (as assessed by QSM) from baseline at week 48 (individual patient data shown in eFigure 4, links.lww.com/NXG/A553). There was a nominally significant difference between treatment groups at weeks 24 and 48, with patients in the leriglitazone group showing no further accumulation of iron and patients in the placebo group showing an increase in iron levels (eTable 2, links.lww.com/NXG/A553). The LS mean change (SE) (95% CI) from baseline to week 24 was −1.66 (1.58) (95% CI −4.94 to 1.62) ppb in the leriglitazone group and 6.24 (2.58) (95% CI 0.88–11.60) ppb in the placebo group (p = 0.02). The LS mean change (SE) (95% CI) from baseline to week 48 was 0.10 (1.33) (95% CI −2.65 to 2.85) ppb in the leriglitazone group compared with 4.86 (1.84) (95% CI 1.05–8.67) ppb in the placebo group (p = 0.05). Example MR images are shown in eFigure 3, links.lww.com/NXG/A553.
Figure 3. Change From Baseline at Week 48 in (A) QSM Signal in the Dentate Nucleus (ppb), (B) Cervical Spinal Cord tNAA/mIns Ratio (as Assessed by MRS), (C) CCFS (Total Score), and (D) Patient Ranking for the CCFS, QSM, and MRS Endpoints, Showing Magnitude of Improvement or Worsening at Week 48 (mITT Population).

Data shown in A to C are mean ± SE, with the LS mean (SE), 95% CI and p value for the difference between treatment arms, as assessed by ANCOVA (with treatment arm as a fixed effect and the baseline as a covariate) shown in the tables. Baseline was defined as the last assessment performed before or on study day 1. n values show the number of patients included in the ANCOVA analyses. Dotted lines in panels (A–C) show the expected changes from baseline per year from the literature (panel A34; panel B33; panel C42). Panel D shows ranking only for those patients who did not require imputation for the O'Brien composite sum of ranks test. aCCFS total score = log10 (7 + Z pegboard dominant hand/10 + 4 *Z click dominant hand/10). ANCOVA = analysis of covariance; CCFS = cerebellar composite functional scale; CI = confidence interval; LS = least-squares; mITT = modified intent-to-treat; MRS = magnetic resonance spectroscopy; QSM = quantitative susceptibility mapping; SE = standard error; tNAA/mIns = total N-acetylaspartate concentration/myo-inositol.
There were a decline in cervical spinal cord tNAA/mIns ratio in the placebo group over the treatment period and an increase in the leriglitazone group (Figure 3, eTable 2; individual patient data shown in eFigure 4, links.lww.com/NXG/A553). The LS mean change (SE) (95% CI) from baseline to week 24 was 0.03 (0.02) (95% CI −0.02 to 0.08) in the leriglitazone group and −0.06 (0.04) (95% CI −0.14 to 0.02) in the placebo group (p = 0.04). The LS mean change (SE) (95% CI) from baseline to week 48 was 0.03 (0.02) (95% CI −0.02 to 0.08) in the leriglitazone group and −0.02 (0.03) (95% CI −0.08 to 0.05) in the placebo group (p = 0.25).
Quantitative Score
From baseline to week 48, the CCFS total score increased (worsened) in the placebo group as expected but decreased in the leriglitazone group, although the difference between groups was not significant (Figure 3; eTable 2; individual patient data shown in eFigure 4, links.lww.com/NXG/A553).
Clinician-Reported and Patient-Reported Outcomes
There was no difference between treatment groups in change of total SARA score from baseline to 48 weeks (eTable 2, links.lww.com/NXG/A553). At baseline, the EQ-5D-5L scores were similar between groups, although a greater proportion of patients in the leriglitazone group had problems at baseline with “usual activities” (leriglitazone, n = 5 [22.7%]; placebo, n = 1 [8.3%]). There were no changes in EQ-5D-5L scores in either group at week 48 (eTable 2, links.lww.com/NXG/A553) and no differences between groups in any other clinician-reported and patient-reported outcomes (data not shown).
Exploratory Endpoints
There was an increase of approximately 700% in the mean concentration of plasma adiponectin in the leriglitazone group between baseline and week 48, compared with approximately 10% in the placebo group. The LS mean change (SE) (95% CI) from baseline in adiponectin levels was 43,309.3 (3,619.9) (95% CI 35,905.8–50,712.8) ng/mL in the leriglitazone group and 1,596.2 (4,683.0) (95% CI −7,981.6 to 11,173.9) ng/mL in the placebo group (p < 0.001) (eFigure 1, links.lww.com/NXG/A553). There were little change from baseline to week 48 in FXN level in either treatment group and no difference between groups (eTable 2, links.lww.com/NXG/A553).
Post Hoc Analyses
The O'Brien nonparametric rank sum post hoc analysis was performed to assess whether the same patients were improving in the 3 variables showing the most difference between groups (QSM, CCFS, and tNAA/mIns ratio). Figure 3D shows patient rankings for these endpoints at week 48. The mean average rank was numerically lower in the leriglitazone group than the placebo group for all 3 variables (QSM: 14.8 vs 19.3; tNAA/mIns ratio: 15.3 vs 18.6; CCFS total score: 14.6 vs 19.7). At week 48, the mean (SD) O'Brien composite value was lower in the leriglitazone group than the placebo group (44.7 [18.7] vs 57.5 [14.2]; p = 0.04).
Subpopulation Analysis
In the leriglitazone group, 73% of patients showed faster disease progression than the average patient, compared with 42% in the placebo group (eFigure 5, links.lww.com/NXG/A553). When patients in each group were matched based on progression, greater differences in favor of leriglitazone were seen in the SARA and ADL endpoints, compared with the mITT population analyses (eFigure 6, links.lww.com/NXG/A553).
Safety Assessments
All patients experienced at least 1 AE (Table 2). Most events (88.5%) experienced by patients treated with leriglitazone were mild or moderate in severity. One serious AE (SAE) was reported (atrial dilatation in a patient receiving leriglitazone). There were no deaths reported in this study.
Table 2.
Overall Summary of AEs (Safety Population)
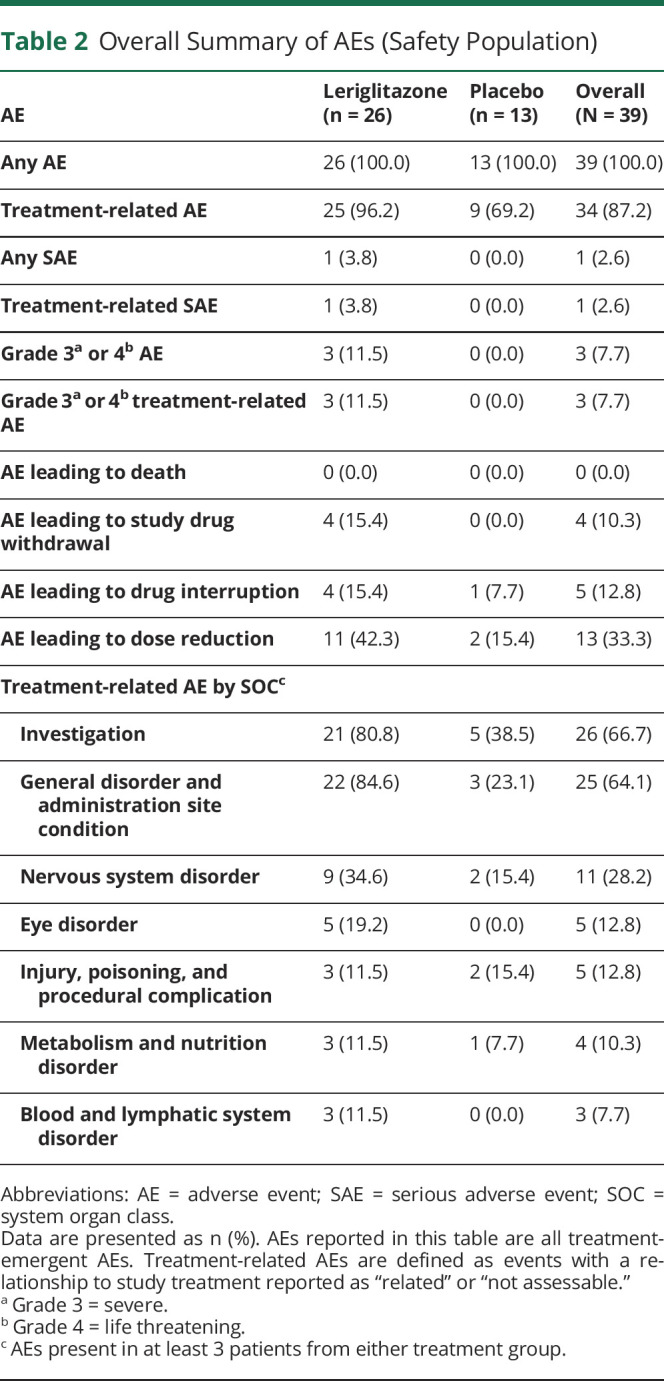
Treatment-related AEs were reported in 25 (96.2%) patients in the leriglitazone group and 9 (69.2%) patients in the placebo group. Dose reductions due to AEs were reported in 11 (42.3%) patients in the leriglitazone group and 2 (15.4%) patients in the placebo group. Treatment interruption due to AEs was reported in 4 (15.4%) patients in the leriglitazone group and 1 (7.7%) patient in the placebo group. Four (15.4%) patients in the leriglitazone group discontinued treatment owing to an AE. The most frequently reported AE was peripheral edema, reported in 19 (73.1%) patients in the leriglitazone group (18 cases were considered related to treatment) and no patients in the placebo group (Table 3). The median (range) duration of these events was 3.1 (0.1–52.4) weeks. There were additional reports of unspecified edema, face edema, localized edema, and generalized edema and swelling. Overall, 10 (38.5%) patients required diuretic treatment in the leriglitazone group.
Table 3.
AEs of Special Interest by Preferred Term (Safety Population)
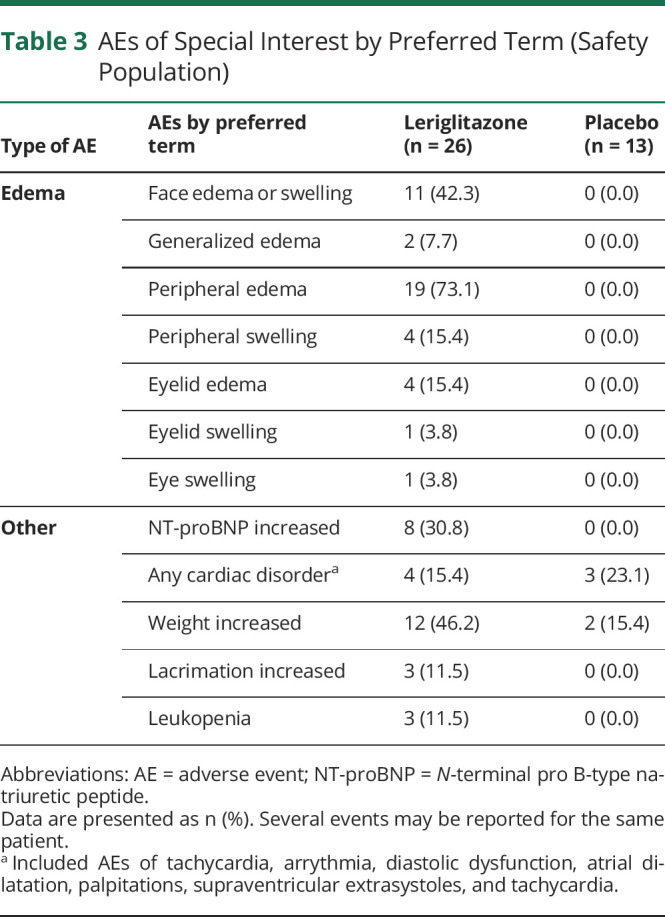
Cardiac disorders were reported in 4 (15.4%) patients in the leriglitazone group and 3 (23.1%) patients in the placebo group (Table 3). The SAE of atrial dilatation (grade 3) reported in the leriglitazone group was considered by the investigator to be related to study medication. However, the event occurred 28 days after stopping treatment and concomitantly with vomiting and dehydration due to viral gastroenteritis. Toxicology findings showed no evidence of myocardial damage by leriglitazone in this patient, who also had a medical history of nonobstructive hypertrophic cardiomyopathy.
No safety signals were identified in laboratory results. Increases in N-terminal pro B-type natriuretic peptide (NT-proBNP) concentration were reported as AEs in 8 (30.8%) patients taking leriglitazone (Table 3). All were considered related to treatment, but none were associated with significant clinical symptoms, ECG or echocardiographic changes. Of the 8 patients who reported an increase in NT-proBNP, 7 also reported edema. However, of the 23 patients who reported edema, 16 did not report an increase in NT-proBNP. Assessment of vital signs showed no clinically meaningful difference between groups in blood pressure, pulse rate, or temperature. The weight increase was reported in 12 (46.2%) patients in the leriglitazone group and 2 (15.4%) patients in the placebo group and led to a dose reduction in 4 patients (Table 3). The mean change in weight (SD) from baseline to follow-up was greater in the leriglitazone group than the placebo group (5.2 [3.8] kg; 10.2% and 3.9 [2.8] kg; 6.3%, respectively) (eFigure 7, links.lww.com/NXG/A553).
Leriglitazone treatment was not associated with any clinically significant changes in ECG results or echocardiograms (clinical interpretations by treatment group shown in eTable 1, links.lww.com/NXG/A553; individual patient data for septal wall thickness and left ventricular mass shown in eTable 3, links.lww.com/NXG/A553). LVEF remained stable over time without significant differences between groups (eFigure 8, links.lww.com/NXG/A553).
Classification of Evidence Statement
Primary Question
Is leriglitazone well tolerated and effective for the treatment of patients with FRDA?
Class
I.
Results
The primary endpoint of change from baseline to week 48 in spinal cord area (C2-C3) (assessed by MRI) vs placebo was not met in this study, likely owing to the absence of disease progression in either treatment group. Secondary endpoint results provided evidence supporting proof of concept for leriglitazone mode of action, and treatment was generally well tolerated.
Discussion
In this double-blind, randomized, placebo-controlled, phase 2 study of leriglitazone in patients with FRDA, there was no difference between treatment groups in the primary endpoint of change from baseline in cervical spinal cord cross-sectional area after 48 weeks of treatment. This result needs to be mitigated by the fact that there was no evidence of disease progression in either group. There was a nominally significant difference between patient groups in QSM measures of iron accumulation in the dentate nucleus, and a numerical difference in favor of leriglitazone was seen for the tNAA/mIns ratio.
Data from a Friedreich's Ataxia Research Alliance (FARA) natural history early-stage cohort (mean [SD] time since diagnosis and disease onset of 2.3 [2.2] years and 5.5 [4.0] years, respectively) have shown that patients with FRDA show a progressive decrease in spinal cord area over time, with a mean (SD) annual rate of atrophy at C2-C3 of −1.03 (1.09) mm2 (−2.4%; p < 0.01) (similar results obtained with manual and automated segmentation).33 Although the same methodology was used to scan and measure spinal cord area in the FRAMES study, our cohort did not show similar longitudinal atrophy. As the FRAMES study had an average time from disease onset of 10.5 years, one possible explanation is that the rate of atrophy slows as the disease progresses, as seen with clinical measures such as the SARA. Of interest, the mean (SD) change from baseline in SARA total score was also slower in the FRAMES study (leriglitazone, week 48: 1.8 [3.6]) compared with the FARA study (leriglitazone, year 1: 2.0 [1.1]).33 The more advanced disease stage of our cohort could have prevented the observation of any effects of leriglitazone on this aspect of disease progression. Likely for the same reason, minimal progression was also seen in measures of cervical spinal cord mean axial and radial diffusivity (as assessed by DTI) and brain FD and FC (as assessed by FBA), regardless of treatment group.
Biochemical magnetic resonance parameters suggest a metabolic improvement in the treated group. Iron concentration in the dentate nucleus as assessed by QSM showed an increase at week 48 in the placebo arm only, although this result should be interpreted with caution considering the small placebo group size and the weak statistical significance. An increase in iron concentration in the DN has been shown previously in a longitudinal study in patients with FRDA.34 The increase in our study (4.3 ppb over 48 weeks) was less than that seen previously (6.7–7.4 ppb per year),34 most likely due to the sensitivity of this measure to disease duration and differences in QSM methodology between the 2 studies. The role of iron accumulation in FRDA pathogenesis is supported by both in vitro and in vivo studies.39,40 Yeast FXN-deficient variants and patient fibroblasts accumulate iron in the mitochondrial matrix, causing oxidative damage and adversely affecting mitochondrial function. Increased cellular and mitochondrial iron uptake is the consequence of impaired Fe-S cluster biogenesis, a direct consequence of FXN deficiency.41 Therefore, lack of iron accumulation in the leriglitazone group likely reflects more efficient Fe-S cluster synthesis, due to increased mitochondrial biogenesis and possibly increased FXN levels, as observed in preclinical models.17
In our study, a numerical decline in the tNAA/mIns ratio (as assessed by MRS) in the spinal cord was observed in the placebo group, while a slight increase was seen in the leriglitazone group. Decline in the placebo group was consistent with longitudinal data from the FARA natural history cohort (mean [SD] decrease in tNAA/mIns ratio: FRAMES, −0.025 [0.11]; FARA cohort, −0.032 [0.06]).33 Improvement in the treated group is consistent with QSM results, suggesting that leriglitazone at least partially corrects metabolic deficits, as shown in preclinical FRDA models.17
Progressive cerebellar impairment is the major cause of worsening of ataxia in FRDA. A recent review recommended CCFS as the best assessment for upper limb coordination in patients with FRDA.42 In this study, CCFS scores increased from baseline to week 48 in the placebo but not the leriglitazone group, although the difference between groups was not statistically significant. In this study, variability was lower in CCFS than in SARA scores, perhaps owing to the simpler structure of the test and its fully quantitative computerized assessment.
Although there was a nominally significant difference between groups in the change in dentate nuclei volume from baseline to week 48, it is not clear whether this represents a true efficacy signal because the increase in dentate nuclei volume seen in both treatment groups was not expected. This result may be due to the “blooming effect,” in which iron-rich areas of the dentate nuclei appear larger than in histologic measurements.43
The O'Brien nonparametric rank sum post hoc analysis showed an improvement in the leriglitazone group compared with the placebo group in average ranks for the 3 composite variables. Matching pairs of patients in the subpopulation analysis also showed an increase in the difference between treatment groups in the SARA and ADL endpoints. Although these post hoc analyses were performed after data lock and should be interpreted with caution, their results are supportive of a clinical benefit of leriglitazone on these outcomes.
Treatment with leriglitazone was generally well tolerated. The PK and adiponectin plasma concentration results show that even in patients requiring a dose reduction, exposure to study drug and PPARγ activation were within the target range for CNS efficacy.24 The most frequently reported AE was peripheral edema, reported in 19 (73.1%) patients in the leriglitazone group. Most events of edema occurred during the first 12 weeks and were managed by dose reduction and/or administration of diuretics. These events were not unexpected and are commonly observed with PPARγ-targeted treatments in patients with type 2 diabetes.44 The weight increase in the leriglitazone group was also not unexpected and may result from fluid retention and an increase in adipocytes.45-48
Patients with FRDA are at increased risk of cardiac dysfunction due to cardiomyopathy.49 Although only 7.7% of patients in the FRAMES cohort reported a clinical history of heart disease, 79.5% had an abnormal ECG at baseline, indicating that most patients had subclinical cardiomyopathy. During the study, we observed a relatively low (∼15%) occurrence of cardiac AEs, which was similar between treatment groups, suggesting that these events were most likely related to underlying heart disease rather than directly due to leriglitazone. The increase in NT-proBNP concentration reported as AEs in 8 patients taking leriglitazone was not unexpected and is likely to be a class effect in response to increased intravascular fluid; none of these patients showed any clinical signs of cardiac failure.
There was no evidence of an increased risk of liver damage or bladder cancer in this study, as has been reported after pioglitazone use.50
Limitations of the study include the fact that a proportion of patients had advanced disease at baseline and therefore had possibly already reached a plateau of spinal cord atrophy. The sample size was small, and the observation time was short; this limits somewhat the power of the analyses and the conclusions that can be drawn regarding clinician-reported and patient-reported outcomes. Potential unblinding due to edema (not formally assessed) and an effect on subjective ratings measured in the study cannot be excluded. The process used for adjusting the dose of leriglitazone in this study was complex; a simpler dosing algorithm is under consideration for future studies.
In conclusion, the FRAMES study assessed the effects of leriglitazone on biochemical, imaging, neurophysiologic, and clinical parameters in patients with FRDA. Although the primary endpoint was not met in this study, results from secondary endpoints provide evidence for clinical proof of concept for use of leriglitazone in patients with FRDA and support assessment in larger studies.
Acknowledgment
The authors thank all the patients and investigators involved in the study. Medical writing support (for nonintellectual content) was provided by Emily Manktelow, PhD, of Oxford PharmaGenesis, Melbourne, Australia, and was funded by Minoryx, in accordance with the Good Publication Practice 3 (GPP3) guidelines (ismpp.org/gpp3).
Glossary
- ADL
Activities of Daily Living
- AE
adverse event
- ANCOVA
analysis of covariance
- AUC
area under the curve
- CCFS
cerebellar composite functional scale
- CGI-I
Clinical Global Impression-Improvement
- CI
confidence interval
- DTI
diffusion tensor imaging
- EQ-5D-5L
5-level, 5-dimension EuroQol questionnaire
- FARA
Friedreich's Ataxia Research Alliance
- FBA
fixel-based analysis
- FC
fiber cross-section
- FD
fiber density
- FDC
FD and cross-section
- FRDA
Friedreich ataxia
- FXN
frataxin
- GAA
alpha glucosidase
- LS
least-squares
- LVEF
left ventricular ejection fraction
- mITT
modified intent-to-treat
- MRS
magnetic resonance spectroscopy
- NTproBNP
N-terminal pro B-type natriuretic peptide
- PGI-I
Patient Global Impression-Improvement
- PK
pharmacokinetic
- PP
per protocol
- PPARγ
peroxisome proliferator–activated receptor γ
- QSM
quantitative susceptibility mapping
- SAE
serious AE
- SARA
Scale for the Assessment and Rating of Ataxia
- SE
standard error
- SOC
system organ class
- tNAA/mIns
total N-acetylaspartate concentration/myo-inositol
Appendix. Authors

Footnotes
Class of Evidence: NPub.org/coe
Study Funding
The study was sponsored by Minoryx Therapeutics BE, the Belgian subsidiary of Minoryx Therapeutics SL, and received funding from Region Wallonne (SPW-EER/DRDT/DPjR/DEMO/ML/Déf-7939).
Disclosure
M. Pandolfo reports research support outside of this study from the Friedreich's Ataxia Research Alliance, Fonds National de la Recherche Scientifique, Belgium, and has received consulting fees from Voyager Therapeutics, Chiesi Canada, Minoryx, Exicure, and Design Therapeutics. K. Reetz has received grants from the German Federal Ministry of Education and Research (BMBF 01GQ1402, 01DN18022), the German Research Foundation (IRTG 2150, ZUK32/1), Alzheimer Forschung Initiative e.V. (AFI 13812, NL-18002CB), Friedreich's Ataxia Research Alliance, and honoraria for presentations or advisory boards from Biogen and Roche. F.J. Rodriguez de Rivera reports no disclosures relevant to the manuscript. P.-G. Henry reports support by research grants from Minoryx for this study and acknowledges research grants from the Friedreich's Ataxia Research Alliance, GoFAR, Ataxia UK, the Bob Allison Ataxia Research Center, and NIH grants P41 EB027061 and P30 NS076408. J. Joers reports research support from Minoryx for this study through a research grant to C. Lenglet and P.-G. Henry. C. Lenglet reports support by research grants from Minoryx for this study; acknowledges research grants from the Friedreich's Ataxia Research Alliance, GoFAR, Ataxia UK, the Bob Allison Ataxia Research Center, NIH grants P41 EB027061 and P30 NS076408; and reports support by a research grant from Biogen Inc. for activities outside this study. I. Adanyeguh reports research support from Minoryx for this study through a research grant to C. Lenglet and P.-G. Henry. D. Deelchand reports research support from Minoryx for this study through a research grant to C.Lenglet and P.-G. Henry. F. Mochel reports support by a research grant from Minoryx outside this study, consulting fees from Minoryx, support by research grant from Metafora Biosystems and the Agence Nationale de la Recherche (ANR) in France outside this study. F. Pousset reports support by research grants from Minoryx for this study, support from The French Association of Friedreich's Ataxia, and support by research grants from Novartis and Pfizer. S. Pascual, A. Vilà, A. Mantilla, and M. Pascual are employees of Minoryx and have stock options. D. Van den Eede and I. Martín-Ugarte are previous employees of Minoryx and have stock options. M. Martinell is CEO of Minoryx and has shares and stock options. U. Meya is a previous employee and now consultant for Minoryx with stock options. A. Durr reports research support outside of this study from the National Institute of Health, Hospital Clinical Research Program, Biogen/Ionis, French National Research Agency and The French Association of Friedreich's Ataxia. A. Darling reports no disclosures relevant to the manuscript. Full disclosure form information provided by the authors is available with the full text of this article at Neurology.org/NG.
References
- 1.Campuzano V, Montermini L, Molto MD, et al. Friedreich's ataxia: autosomal recessive disease caused by an intronic GAA triplet repeat expansion. Science. 1996;271(5254):1423-1427. [DOI] [PubMed] [Google Scholar]
- 2.Gottesfeld JM. Molecular mechanisms and therapeutics for the GAA. TTC expansion disease Friedreich ataxia. Neurotherapeutics. 2019;16(4):1032-1049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Tsai CL, Barondeau DP. Human frataxin is an allosteric switch that activates the Fe-S cluster biosynthetic complex. Biochemistry. 2010;49(43):9132-9139. [DOI] [PubMed] [Google Scholar]
- 4.Stepanova A, Magrane J. Mitochondrial dysfunction in neurons in Friedreich's ataxia. Mol Cell Neurosci. 2020;102:103419. [DOI] [PubMed] [Google Scholar]
- 5.Chiang S, Huang MLH, Park KC, Richardson DR. Antioxidant defense mechanisms and its dysfunctional regulation in the mitochondrial disease, Friedreich's ataxia. Free Radic Biol Med. 2020;159:177-188. [DOI] [PubMed] [Google Scholar]
- 6.Schmucker S, Puccio H. Understanding the molecular mechanisms of Friedreich's ataxia to develop therapeutic approaches. Hum Mol Genet. 2010;19(R1):R103-R110. [DOI] [PubMed] [Google Scholar]
- 7.Durr A, Cossee M, Agid Y, et al. Clinical and genetic abnormalities in patients with Friedreich's ataxia. N Engl J Med. 1996;335(16):1169-1175. [DOI] [PubMed] [Google Scholar]
- 8.Montermini L, Richter A, Morgan K, et al. Phenotypic variability in Friedreich ataxia: role of the associated GAA triplet repeat expansion. Ann Neurol. 1997;41(5):675-682. [DOI] [PubMed] [Google Scholar]
- 9.Koeppen AH, Morral JA, Davis AN, et al. The dorsal root ganglion in Friedreich's ataxia. Acta Neuropathol. 2009;118(6):763-776. [DOI] [PubMed] [Google Scholar]
- 10.Koeppen AH, Davis AN, Morral JA. The cerebellar component of Friedreich's ataxia. Acta Neuropathol. 2011;122(3):323-330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Koeppen AH, Mazurkiewicz JE. Friedreich ataxia: neuropathology revised. J Neuropathol Exp Neurol. 2013;72(2):78-90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Rummey C, Corben LA, Delatycki MB, et al. Psychometric properties of the Friedreich Ataxia Rating Scale. Neurol Genet. 2019;5(6):371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Reetz K, Dogan I, Hohenfeld C, et al. , EFACTS Study Group. Nonataxia symptoms in Friedreich ataxia: report from the registry of the European Friedreich's Ataxia Consortium for Translational Studies (EFACTS). Neurology. 2018;91(10):e917–e930. [DOI] [PubMed] [Google Scholar]
- 14.Pousset F, Legrand L, Monin ML, et al. A 22-year follow-up study of long-term cardiac outcome and predictors of survival in Friedreich ataxia. JAMA Neurol. 2015;72(11):1334-1341. [DOI] [PubMed] [Google Scholar]
- 15.Clay A, Hearle P, Schadt K, Lynch DR. New developments in pharmacotherapy for Friedreich ataxia. Expert Opin Pharmacother. 2019;20(15):1855-1867. [DOI] [PubMed] [Google Scholar]
- 16.Pandolfo M. Treatment of Friedreich's ataxia. Expert Opin Orphan Drugs. 2013;1(3):221-234. [Google Scholar]
- 17.Rodríguez-Pascau L, Britti E, Calap-Quintana P, et al. PPAR gamma agonist leriglitazone improves frataxin-loss impairments in cellular and animal models of Friedreich ataxia. Neurobiol Dis. 2021;148:105162. [DOI] [PubMed] [Google Scholar]
- 18.Jasoliya MJ, McMackin MZ, Henderson CK, Perlman SL, Cortopassi GA. Frataxin deficiency impairs mitochondrial biogenesis in cells, mice and humans. Hum Mol Genet. 2017;26(14):2627-2633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lin H, Magrane J, Rattelle A, et al. Early cerebellar deficits in mitochondrial biogenesis and respiratory chain complexes in the KIKO mouse model of Friedreich ataxia. Dis Model Mech. 2017;10(11):1343-1352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Marmolino D, Manto M, Acquaviva F, et al. PGC-1alpha down-regulation affects the antioxidant response in Friedreich's ataxia. PLoS One. 2010;5(4):e10025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Coppola G, Marmolino D, Lu D, et al. Functional genomic analysis of frataxin deficiency reveals tissue-specific alterations and identifies the PPARgamma pathway as a therapeutic target in Friedreich's ataxia. Hum Mol Genet. 2009;18(13):2452-2461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Desouza CV, Shivaswamy V. Pioglitazone in the treatment of type 2 diabetes: safety and efficacy review. Clin Med Insights Endocrinol Diabetes. 2010;3:43-51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Poli S, Rodriguez-Pascau L, Britti E, et al. MIN102 (leriglitazone), a brain penetrant PPAR gamma agonist for the treatment of Friedreich's ataxia (4147). Neurology. 2020;94(15):4147. [Google Scholar]
- 24.Meya U, Pina G, Pascual S, et al. A phase 1 study to assess the safety, tolerability, pharmacokinetics, and effects on biomarkers of MIN-102 (leriglitazone) (4149). Neurology. 2020;94(15 suppl):4149. [Google Scholar]
- 25.Pandolfo M. Neurologic outcomes in Friedreich ataxia: study of a single-site cohort. Neurol Genet. 2020;6(3):e415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Patel M, Isaacs CJ, Seyer L, et al. Progression of Friedreich ataxia: quantitative characterization over 5 years. Ann Clin Transl Neurol. 2016;3(9):684-694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Reetz K, Dogan I, Costa AS, et al. Biological and clinical characteristics of the European Friedreich's Ataxia Consortium for Translational Studies (EFACTS) cohort: a cross-sectional analysis of baseline data. Lancet Neurol. 2015;14(2):174-182. [DOI] [PubMed] [Google Scholar]
- 28.Reetz K, Dogan I, Hilgers RD, et al. Progression characteristics of the European Friedreich's Ataxia Consortium for translational studies (EFACTS): a 2 year cohort study. Lancet Neurol. 2016;15(13):1346-1354. [DOI] [PubMed] [Google Scholar]
- 29.Reetz K, Dogan I, Hilgers RD, et al. , EFACTS study group. Progression characteristics of the European Friedreich's Ataxia Consortium for translational studies (EFACTS): a 4-year cohort study. Lancet Neurol. 2021;20(5):362-372. [DOI] [PubMed] [Google Scholar]
- 30.Schmitz-Hubsch T, du Montcel ST, Baliko L, et al. Scale for the assessment and rating of ataxia: development of a new clinical scale. Neurology. 2006;66(11):1717-1720. [DOI] [PubMed] [Google Scholar]
- 31.Dogan I, Romanzetti S, Didszun C, et al. Structural characteristics of the central nervous system in Friedreich ataxia: an in vivo spinal cord and brain MRI study. J Neurol Neurosurg Psychiatry. 2019;90(5):615-617. [DOI] [PubMed] [Google Scholar]
- 32.Chevis CF, da Silva CB, D'Abreu A, et al. Spinal cord atrophy correlates with disability in Friedreich's ataxia. Cerebellum. 2013;12(1):43-47. [DOI] [PubMed] [Google Scholar]
- 33.Joers JM, Adanyeguh IM, Deelchand DK, et al. Spinal cord magnetic resonance imaging and spectroscopy detect early-stage alterations and disease progression in Friedreich ataxia. Brain Commun. 2022;6(5):fcac246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ward PGD, Harding IH, Close TG, et al. Longitudinal evaluation of iron concentration and atrophy in the dentate nuclei in Friedreich ataxia. Mov Disord. 2019;34(3):335-343. [DOI] [PubMed] [Google Scholar]
- 35.Raffelt DA, Tournier JD, Smith RE, et al. Investigating white matter fibre density and morphology using fixel-based analysis. Neuroimage. 2017;144(pt A):58-73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wang Y, Liu T. Quantitative susceptibility mapping (QSM): decoding MRI data for a tissue magnetic biomarker. Magn Reson Med. 2015;73(1):82-101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.du Montcel ST, Charles P, Ribai P, et al. Composite cerebellar functional severity score: validation of a quantitative score of cerebellar impairment. Brain. 2008;131(pt 5):1352-1361. [DOI] [PubMed] [Google Scholar]
- 38.Miyazaki Y, Mahankali A, Wajcberg E, Bajaj M, Mandarino LJ, DeFronzo RA. Effect of pioglitazone on circulating adipocytokine levels and insulin sensitivity in type 2 diabetic patients. J Clin Endocrinol Metab. 2004;89(9):4312-4319. [DOI] [PubMed] [Google Scholar]
- 39.Llorens JV, Soriano S, Calap-Quintana P, Gonzalez-Cabo P, Molto MD. The role of iron in Friedreich's ataxia: insights from studies in human tissues and cellular and animal models. Front Neurosci. 2019;13:75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Palau F. Friedreich's ataxia and frataxin: molecular genetics, evolution and pathogenesis (Review). Int J Mol Med. 2001;7(6):581-589. [DOI] [PubMed] [Google Scholar]
- 41.Terzi EM, Sviderskiy VO, Alvarez SW, Whiten GC, Possemato R. Iron-sulfur cluster deficiency can be sensed by IRP2 and regulates iron homeostasis and sensitivity to ferroptosis independent of IRP1 and FBXL5. Sci Adv. 2021;7(22):eabg4302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Tanguy Melac A, Mariotti C, Filipovic Pierucci A, et al. , EFACTS group. Friedreich and dominant ataxias: quantitative differences in cerebellar dysfunction measurements. J Neurol Neurosurg Psychiatry. 2018;89(6):559-565. [DOI] [PubMed] [Google Scholar]
- 43.Solbach K, Kraff O, Minnerop M, et al. Cerebellar pathology in Friedreich's ataxia: atrophied dentate nuclei with normal iron content. Neuroimage Clin. 2014;6:93-99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Nesto RW, Bell D, Bonow RO, et al. Thiazolidinedione use, fluid retention, and congestive heart failure: a consensus statement from the American Heart Association and American Diabetes Association. October 7, 2003. Circulation. 2003;108(23):2941-2948. [DOI] [PubMed] [Google Scholar]
- 45.Kelly IE, Han TS, Walsh K, Lean ME. Effects of a thiazolidinedione compound on body fat and fat distribution of patients with type 2 diabetes. Diabetes Care. 1999;22(2):288-293. [DOI] [PubMed] [Google Scholar]
- 46.Miyazaki Y, Mahankali A, Matsuda M, et al. Effect of pioglitazone on abdominal fat distribution and insulin sensitivity in type 2 diabetic patients. J Clin Endocrinol Metab. 2002;87(6):2784-2791. [DOI] [PubMed] [Google Scholar]
- 47.Nakamura T, Funahashi T, Yamashita S, et al. Thiazolidinedione derivative improves fat distribution and multiple risk factors in subjects with visceral fat accumulation: double-blind placebo-controlled trial. Diabetes Res Clin Pract. 2001;54(3):181-190. [DOI] [PubMed] [Google Scholar]
- 48.Takamura T, Nohara E, Nagai Y, Kobayashi K. Stage-specific effects of a thiazolidinedione on proliferation, differentiation and PPARgamma mRNA expression in 3T3-L1 adipocytes. Eur J Pharmacol. 2001;422(1-3):23-29. [DOI] [PubMed] [Google Scholar]
- 49.Tsou AY, Paulsen EK, Lagedrost SJ, et al. Mortality in Friedreich ataxia. J Neurol Sci. 2011;307(1-2):46-49. [DOI] [PubMed] [Google Scholar]
- 50.ACTOS (Pioglitazone Hydrochloride) Prescribing Information. U.S. Food and Drug Administration, 2011. Accessed October 14, 2021. accessdata.fda.gov/drugsatfda_docs/label/2011/021073s043s044lbl.pdf. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
All data and related documentation underlying the reported results will be made available after anonymization of patient information up to 4 years after publication of this article. In the interim, the authors will share the data with qualified investigators whose proposal of data use has been approved by an independent review committee.