

Abstract
Evobrutinib, a Bruton's tyrosine kinase (BTK) inhibitor, has shown therapeutic potential in relapsing multiple sclerosis. This analysis aimed to develop pharmacokinetic (PK) and pharmacodynamic (PD; BTK occupancy [BTKO]) models of evobrutinib and simulate PK and BTKO profiles under alternative dosing regimens. Data were obtained from two phase I evobrutinib studies in healthy adult participants (Japanese and non‐Japanese). Overall, 2326 observations were available from 76 participants; n = 42 from Study MS200527_0017 Part A received evobrutinib 25, 75, or 200 mg once‐daily oral doses for 6 days while fasted; n = 18 from Study MS200527_0019 and n = 16 from Study MS200527_0017 Part B received single evobrutinib 75 mg oral doses with food (low‐fat meal) and while fasted. Population PK/PD modeling for evobrutinib concentrations and BTKO (fraction unbound) were performed using nonlinear mixed‐effects modeling. The effect of once‐daily/twice‐daily regimens and doses of 10–200 mg on BTKO were simulated. A two‐compartment model with sequential zero‐first order absorption and first‐order elimination adequately described the data. Bioavailability increased by 49% with food compared with when fasted. There was no difference in PK parameters between Japanese and non‐Japanese participants. The BTKO profile of evobrutinib was described by the irreversible binding population model. The simulated percentage of participants with minimum BTKO increased in a dose‐dependent manner across the BTKO thresholds of interest (70%, 80%, 90%, and 95% occupancy). Evobrutinib doses of 25 mg once‐daily, 50 mg twice‐daily, or 75 mg twice‐daily while fasted are possible choices for further development, assuming BTKO ≥70% at trough is needed to achieve efficacy.
Study Highlights.
WHAT IS THE CURRENT KNOWLEDGE ON THE TOPIC?
Evobrutinib is a Bruton's tyrosine kinase (BTK) inhibitor that impacts a range of key cell types involved in the pathogenesis of multiple sclerosis, such as B cells and myeloid cells, including macrophages and microglia. As evobrutinib binds BTK irreversibly, it provides prolonged target inhibition and, therefore, may be suitable for the chronic treatment of autoimmune diseases.
WHAT QUESTION DID THIS STUDY ADDRESS?
The aim of this analysis was to develop a population pharmacokinetic model using data from clinical studies, to describe evobrutinib plasma concentration–time data in participants under different conditions and dosing regimens. In addition, the current analysis aimed to develop a pharmacodynamic model to describe the BTK occupancy (BTKO) profile of evobrutinib, to determine dosing regimens achieving the target occupancy necessary for efficacy, based on preclinical experiments.
WHAT DOES THIS STUDY ADD TO OUR KNOWLEDGE?
A two‐compartment model with sequential zero‐first order absorption and first‐order elimination was able to describe the observations obtained from participants treated under different conditions and at different dose levels. The bioavailability of evobrutinib increased by 49% with food compared with when fasted. The simulated percentage of participants with minimum BTKO increased in a dose‐dependent manner across the BTKO thresholds of interest (70%, 80%, 90%, and 95% occupancy).
HOW MIGHT THIS CHANGE CLINICAL PHARMACOLOGY OR TRANSLATIONAL SCIENCE?
The simulated percentage of participants with minimum BTKO increased in a dose‐dependent manner across the BTKO thresholds. Assuming BTKO ≥70% at trough is needed to achieve efficacy in the different indications considered, the results suggest that daily doses of evobrutinib 25 mg once‐daily, 50 mg twice‐daily, or 75 mg twice‐daily while fasted are plausible choices for further development.
INTRODUCTION
Bruton's tyrosine kinase (BTK) participates in both adaptive and innate immune responses. 1 , 2 It has been considered a promising target for the treatment of multiple sclerosis (MS) 3 , 4 , 5 due to its role in mediating both B cell receptor (BCR) and Fc receptor signaling in B cells and myeloid cells. 6
Evobrutinib is a novel, highly selective, central nervous system (CNS)‐penetrant, orally administered, covalent BTK inhibitor. 6 , 7 Evobrutinib‐mediated BTK inhibition prevents BCR and Fc receptor‐mediated signaling, thereby inhibiting subsequent activation and function of human B cells and innate immune cells, including myeloid cells and basophils. 6 As evobrutinib binds BTK covalently, it provides prolonged target inhibition long after the compound has been cleared from the circulation, and therefore may be suitable for the chronic treatment of autoimmune diseases like MS. 6
BTK occupancy (BTKO) assays can be used to determine the selection of doses and systemic drug levels required to achieve maximal occupancy of BTK, with increasing occupancy levels inferring increasing inhibition of BCR signaling. 8 The clinical efficacy of BTK inhibition has been tested in a murine experimental autoimmune encephalomyelitis model, where a high degree of BTKO was achieved in peripheral blood 2 h after the first dose of evobrutinib, coinciding with maximum evobrutinib exposure. 7 In the CNS, BTKO was delayed, reaching 75%–100% occupancy at 24 h after the last dose when plasma levels had declined. 7
BTKO was also studied in a first‐in‐human, phase I trial of evobrutinib in healthy participants at doses up to 500 mg. 9 In this trial, BTKO was dose‐dependent, with maximum occupancy of >90% within 4 h of single doses ≥200 mg while fasted. 9 Following administration of single evobrutinib doses ≥100 mg while fasted, BTKO was long‐lasting, with >50% occupancy at 96 h postdose. 9 After 14 days administration of evobrutinib 25 mg once‐daily (q.d.) while fasted, very high BTKO was achieved (88% median at trough), in contrast to serum evobrutinib concentrations that were all below the limit of quantification (BLQ) at trough, demonstrating gradual turnover with half‐life of almost 1 week of BTK protein in vivo. 9
The aim of the current analysis was to develop a population pharmacokinetic (PK) model using data from clinical studies, in which evobrutinib was administered as a tablet, to describe evobrutinib plasma concentration–time data in healthy participants under different conditions and dosing regimens. A secondary objective was to develop a pharmacodynamic (PD) model to describe the BTKO profile of evobrutinib, to determine dosing regimens achieving the target occupancy necessary for efficacy, based on preclinical modeling.
METHODS
Ethical considerations
An independent ethics committee or institutional review board at each study site approved the protocols of the studies included in this analysis. The studies were conducted in accordance with the Declaration of Helsinki and International Conference on Harmonization Good Clinical Practice Guidelines. Written informed consent was obtained from each participant before any study‐related activities were performed.
Study population
For the population PK modeling, population PD modeling, and simulation exercises, data were obtained from two phase I studies conducted in healthy adult volunteers. Data from the safety analysis sets were used.
Study MS200527_0019
This was a randomized, open‐label, three‐period, three‐formulation, crossover study in which participants received a single evobrutinib 75 mg dose, administered orally either as powder in a capsule formulation under fasted conditions (data not used), or tablet formulation (3 × 25 mg tablets) with food (low‐fat meal) or under fasted conditions. For each treatment period, PK samples were collected on Day 1 predose and 0.25, 0.5, 1, 1.5, 2, 2.5, 3, 4, 5, 6, 8, 12, 24 h (Day 2), 36 h, and 48 h (Day 2) postdose. At 72 h, the treatment for the subsequent period was administered. PD sampling for BTKO was not performed in this study.
Study MS200527_0017
This study was performed in two sequential parts in healthy adult participants.
Part A was an open‐label, multiple‐dose escalation study, in which three cohorts of Japanese and non‐Japanese participants (7 Japanese and 7 non‐Japanese participants per cohort) received evobrutinib 25 mg, 75 mg, or 200 mg q.d. doses, administered orally in a tablet formulation (1, 3, or 8 × 25 mg tablets, respectively) for 6 days. All participants fasted for 8 h predose and 2 h postdose. Serial blood samples for PK and PD assessments were collected at scheduled timepoints throughout the study. PK samples were collected on Day 1 predose and at 0.25, 0.5, 1, 1.5, 2, 3, 4, 6, 8, 10, 12, 16, 24 h (Day 1 24 h = Day 2 predose), 48 h (Day 3 predose), 72 h (Day 4 predose), 96 h (Day 5 predose), and 120 h (Day 6 predose). On the last day (Day 6) of the study, samples were collected at 0.25, 0.5, 1, 1.5, 2, 3, 4, 6, 8, 10, 12, 16, and 24 h postdose. Blood samples for the PD analysis (BTKO) were collected on Day 1 predose and at 2, 24 h (Day 2), 120 h (Day 6), 122, 124, 126, 144 h (Day 7), 168 h (Day 8), 216 h (Day 10), and 264 h (Day 12) after the first dose.
Part B was an open‐label, two‐period, two‐sequence, randomized, crossover study, in which 8 Japanese and 8 non‐Japanese participants received a single evobrutinib 75 mg dose, administered orally in a tablet formulation both with food (fasted for 8 h followed by a low‐fat meal; evobrutinib was administered 30 min after starting the meal, then participants fasted for a further 4 h) and while fasted (fasted for 8 h predose and 4 h postdose), with a washout period of 3 days between evobrutinib doses. PK samples were collected on Day 1 and Day 4 predose and at 0.25, 0.5, 1, 1.5, 2, 3, 4, 6, 8, 10, 12, 16 h, and 24 h. PD sampling for BTKO was not performed for Part B.
Bioanalysis
Measurement of evobrutinib concentration in plasma was performed by liquid chromatography coupled to tandem mass spectrometry. Plasma samples were extracted using a liquid–liquid extraction with ethyl acetate and subsequently analyzed. The lower limit of quantification for evobrutinib concentrations was 0.600 ng/ml.
Total BTK (BTKT) was measured using a sandwich enzyme‐linked immunosorbent assay with mouse monoclonal anti‐BTK (catalog number 611117; BD Biosciences) as capture antibody and rabbit anti‐BTK (catalog number 8547BF; Cell Signaling Technologies) with goat anti‐rabbit horseradish peroxidase (catalog number AP132P; Millipore) as detection antibody. Free BTK (BTKF) was measured using a biotinylated probe (Merck Patent GmbH; June 2016) that irreversibly binds to the active site of BTK. Labeled BTK was captured from the lysate using streptavidin‐coated enzyme‐linked immunosorbent assay plates and detected using a different anti‐BTK antibody (catalog number PA5‐20085; Pierce) than in the BTKT assay. Total protein was measured and lysates were adjusted to 0.5 ng/ml total protein before measuring BTKT and BTKF. A standard curve was constructed for both assays using recombinant human BTK protein (catalog number PR5442A; Invitrogen). The lower limit of quantification of the assays for BTKT and BTKF were 1.9 and 0.6 ng/ml, respectively, with the upper limit of quantification of 450 and 200 ng/ml, respectively, in peripheral blood mononuclear cell lysate. 9 The fraction of unbound BTK was calculated using the following formula: fraction unbound = (BTKF/BTKT)/(BTKF predose/BTKT predose).
Population PK model development and analysis
The population PK model was developed first for the MS200527_0019 study (tablet formulation) and then for the combined MS200527_0019 and MS200527_0017 studies (tablet formulations).
Model development focused on alternative model structures (with emphasis on alternative absorption model structures, i.e., exploring the possibility that absorption‐related parameters depended on formulation/food state), the exploration of alternative statistical structures, as well as the usual covariate testing. The high proportion of BLQ observations were incorporated in the model using the M3 method. 10
Identification of covariates was implemented by the Stepwise Covariate Model (SCM) building tool of Perl‐speaks‐NONMEM (PsN; v4.4.8, http://psn.sourceforge.net/index.php) 11 using delta objective function value. Analyses that explored covariates as potential predictors of PK were performed.
The following covariates were tested: sex, weight, body mass index, body surface area, Japanese versus non‐Japanese ethnicity, age, evobrutinib dose, and food state (with food or while fasted). The resultant final model only contained covariates that met the predefined statistical inclusion and exclusion criteria. In addition, covariates were only retained on basis of their relevance in view of the purpose of the model.
The goodness‐of‐fit and appropriateness of the model was assessed by means of standard residual‐based diagnostic plots and visual predictive checks.
The final model was a two‐compartment PK model (Figure 1), with sequential zero‐first order absorption and first‐order elimination. The model was fitted to the plasma concentration–time data of evobrutinib, using the NONMEM® program (version 7.3). 12 The estimation method used in NONMEM was importance sampling with the Laplacian option. NONMEM was used to simulate PK profiles.
FIGURE 1.
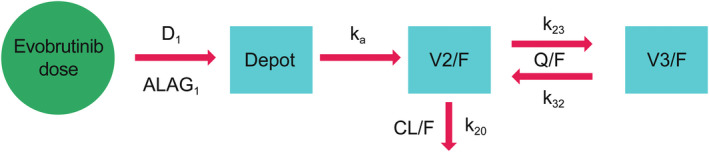
Evobrutinib population pharmacokinetic model structure. ALAG, absorption lag time; CL/F, apparent clearance; D 1, duration of zero‐order input; F, oral bioavailability; k 20, elimination rate constant; k 23, transfer rate from central to peripheral compartment; k 32, transfer rate from peripheral to central compartment; k a , first‐order absorption rate constant; Q/F, apparent intercompartmental clearance; V2/F, apparent central volume of distribution; V3/F, apparent peripheral volume of distribution.
BTKO model development and analysis
The structure of the population PD model for BTKO (fraction unbound) used in this study is the same as one used previously to characterize BTKO after oral administration of evobrutinib in mouse models of rheumatoid arthritis and systemic lupus erythematosus. 6 The irreversible binding model was described as:
where C p is the evobrutinib plasma concentration; k in and k out are the zero‐order synthesis and first‐order elimination rates of BTK protein, respectively; k irrev is the second‐order irreversible binding rate constant, and BTKFU is the time‐dependent fraction of unoccupied BTK.
At steady‐state,
where BTKFU,0 is the baseline fraction unoccupied BTK at time = 0, which is assumed to equal 1 (an assumption already included in the data, since they are baseline corrected). Consequently, the zero‐order synthesis and first‐order elimination rates of BTK protein are assumed equal, numerically, to each other at steady‐state.
Identification of covariates was performed with the SCM. The goodness‐of‐fit and appropriateness of the model was assessed by means of standard residual‐based diagnostic plots and visual predictive checks. The first‐order conditional with interaction estimation method in NONMEM was used. NONMEM was also used to simulate BTKO profiles.
Simulation of alternative regimens
The developed population PK and PD models were used as a basis to simulate BTKO profiles at steady‐state under alternative dosing regimens. The simulations aimed to determine dose levels/regimens to be used in future studies or to support dose levels/regimens being used in current studies of the evobrutinib development program.
Simulated profiles were obtained for the tablet formulation with food and while fasted. q.d. and twice‐daily (b.i.d.) regimens were simulated to reflect doses ranging from 10 mg q.d. to 200 mg b.i.d., given for 14 days (steady‐state). Correlations ≥30% between the random effects of the PK and the BTKO model were used in performing the simulations. There were five in total, with the maximum correlation being 42%.
In total, 1000 participant profiles per dosing regimen were simulated. The percentage of participants above BTKO thresholds of interest (70%, 80%, 90%, and 95%) were calculated. The simulations did not include residual error and parameter uncertainty was not used in the simulation.
RESULTS
Data availability for population PK model
The available datasets from studies MS200527_0019 and MS200527_0017 are outlined in Table 1. BLQ observations (total of 675 of 2326, i.e., 29%) were used in the modeling data set.
TABLE 1.
Available datasets from Study MS200527_0019 and Study MS200527_0017
| ID | Study MS200527_0019 | Study MS200527_0017 | Total | |||
|---|---|---|---|---|---|---|
| N | % | N | % | N | % | |
| 18 | 24 | 58 | 76 | 76 | 100 | |
| Observations | ||||||
| All | 576 | 100 | 1750 | 100 | 2326 | 100 |
| 25 mg q.d. | – | – | 434 | 25 | 434 | 19 |
| 75 mg q.d. | 576 | 100 | 882 | 50 | 1458 | 63 |
| 200 mg q.d. | – | – | 434 | 25 | 434 | 19 |
| BLQ observations | ||||||
| All | 166 | 29 | 509 | 29 | 675 | 29 |
| 25 mg q.d. | – | – | 195 | 11 | 195 | 8 |
| 75 mg q.d. | 166 | 29 | 265 | 15 | 431 | 19 |
| 200 mg q.d. | – | – | 49 | 3 | 49 | 2 |
| Predose | 36 | 6 | 74 | 4 | 110 | 5 |
| Postdose | 130 | 23 | 435 | 25 | 565 | 24 |
| Fed/fasted observations | ||||||
| Fed | 288 | 50 | 224 | 13 | 512 | 22 |
| Fasted | 288 | 50 | 1526 | 87 | 1814 | 78 |
Abbreviations: BLQ, below the limit of quantification; N, number of participants/observations; q.d., once‐daily.
Population PK model
Population PK model parameter estimates are shown in Table 2. The model included between‐participant variability (BPV) on apparent clearance (CL/F), apparent central volume of distribution (V2/F), apparent intercompartmental clearance (Q/F), apparent peripheral volume of distribution (V3/F), absorption lag time (ALAG), and duration of zero‐order input (D1; Figure 1). The model also incorporated correlation between CL/F and ALAG, as well as Q/F and V3/F (i.e., block diagonal variance matrix for the random effects). It also included combined additive and proportional residual error, with the proportional residual error estimated to be 49%.
TABLE 2.
Parameter estimates of the final pharmacokinetic model for evobrutinib
| Parameter | Estimate | RSE % | 95% CI |
|---|---|---|---|
| CL/F (L/h) | 273 | 3.7 | 253.00–292.99 |
| V2/F (L) | 61.1 | 13.8 | 44.54–77.66 |
| Q/F (L/h) | 37 | 13.3 | 27.36–46.64 |
| V3/F (L) | 446 | 21.7 | 256.08–635.92 |
| K a (/h) | 0.784 | 2.5 | 0.75–0.82 |
| ALAG (h) | 0.235 | 4.4 | 0.22–0.26 |
| Additive error | 0.214 | 20.8 | 0.13–0.30 |
| Proportional error | 0.49 | 3.9 | 0.45–0.53 |
| Zero‐order absorption D 1 (h) | 0.181 | 20.8 | 0.11–0.26 |
| Food on F 1 | 1.49 | 3.8 | 1.38–1.60 |
| Food on D 1 | 4.27 | 20.4 | 2.57–5.97 |
| Parameter | Estimate (RSE %) | Etabar (SE) | CV % | Shr % |
|---|---|---|---|---|
| BPV CL/F | 0.0958 (23.0) | 0.002 (0.033) | 31.7 | 5.8 |
| Cov CL/F + ALAG | 0.0558 (45.3) | |||
| BPV ALAG | 0.156 (40.2) | 0.007 (0.041) | 41.1 | 8.4 |
| BPV Q/F | 1.17 (24.1) | −0.001 (0.113) | 149 | 8.6 |
| Cov Q/F + V3/F | 0.988 (22.9) | |||
| BPV V3/F | 1.86 (22.7) | 0.004 (0.128) | 233 | 17.3 |
| BPV V2/F | 0.627 (31.9) | 0.007 (0.061) | 93.3 | 32.7 |
| BPV D 1 Fasted | 1.39 (47.2) | 0.051 (0.113) | 174 | 16 |
| BPV D 1 Fed | 1.39 (47.2) | 0.096 (0.071) | 174 | 47.4 |
Abbreviations: ALAG, lag time before absorption starts; BPV, between‐participant variability; CL/F, apparent clearance; Cov, covariance; CV%, coefficient of variation (%); D 1, duration of zero‐order input; Etabar, arithmetic mean of the η estimates; F 1, oral bioavailability; k a , first‐order absorption rate constant; Q/F, apparent intercompartmental clearance; RSE, relative standard error; SE, standard error; Shr, shrinkage; V2, apparent central volume of distribution; V3, apparent peripheral volume of distribution; D 1 = 0.181 h under fasted condition; D 1 = 0.181 *(1 + 4.27) = 0.954 h under fed condition.
The effect of food conditions (i.e., those used in clinical studies: evobrutinib administered after an overnight fast or with food) on the PK of evobrutinib was assessed, based on the safety analysis set. According to the PK model parameter estimates, evobrutinib bioavailability depended on a prandial state, and was increased by 49% when given with food compared with when fasted (Table 2). The duration of the first‐order input also depended on prandial state and was increased by 427% with food compared with when fasted (Table 2).
Ethnicity (Japanese or non‐Japanese) was not a significant covariate for any of the population PK model parameters and was thus not included in the final model. In addition, none of the demographic covariates satisfied the criteria and were therefore not included in the final model. All model parameters were accurately estimated. The highest relative standard error (RSE) for fixed effects parameters was 22%, while for many of the parameters the RSE was <10% (Table 2). RSE for all variance/covariance parameters were higher, ranging from about 23% to 47%. Shrinkage was <33% for all random effects, except for D1 with food, which was 47%.
Concentration–time profiles observed and predicted by the model for evobrutinib after single doses with food and while fasted, and multiple doses while fasted, are presented in Figure 2. The final two‐compartment model with sequential zero‐first order absorption and first‐order elimination adequately described the PK profiles of evobrutinib tablet formulation (with food and while fasted; Figure 2).
FIGURE 2.
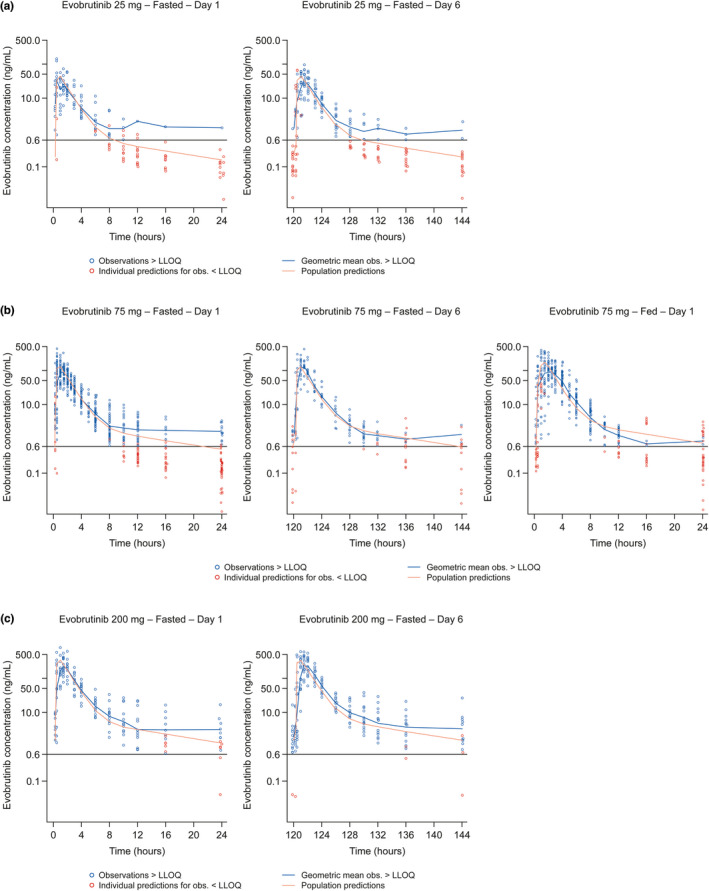
Model predictions for (a) evobrutinib 25 mg after Day 1 (single dose) and Day 6 (6× q.d. doses) while fasted; (b) evobrutinib 75 mg after Day 1 (single dose) and Day 6 (6× q.d. doses) while fasted and after Day 1 (single dose) with food; and (c) evobrutinib 200 mg after Day 1 (single dose) and Day 6 (6× q.d. doses) while fasted. LLOQ, lower limit of quantification; q.d., once‐daily.
Basic goodness‐of‐fit plots and plots of interindividual random error estimate (ETA) estimates versus covariates are presented in Figures S1 and S2, respectively. Prediction‐corrected visual predictive checks by food state and study day are depicted in Figure S3.
Data availability for BTKO model
The BTKO dataset consisted of 447 observations from 41 participants from study MS200527_0017 Part A, of which there were:
152 observations for 14 participants treated with 25 mg q.d. evobrutinib for 6 days
142 observations for 13 participants treated with 75 mg q.d. evobrutinib for 6 days
153 observations for 14 participants treated with 200 mg q.d. evobrutinib for 6 days.
Six records with dependent variable value (fraction unoccupied) ranging from 1.2 to 3.7 were omitted from the modeling set as implausible (corresponding to negative BTKO). Consequently, the total available dataset model consisted of 441 observations from 41 participants.
BTKO model parameter estimates
The irreversible binding model adequately described the BTKO profiles, with model parameter estimates shown in Table 3. The model included BPV on k out and k irrev. Ethnicity (Japanese versus non‐Japanese) was not a significant covariate for any of the two model parameters and was thus not included in the final model. In addition, none of the demographic covariates satisfied the criteria and were therefore not included in the final model. The irreversible binding model estimated the recovery half‐life to be 159 h.
TABLE 3.
Parameter estimates of the final Bruton's tyrosine kinase receptor occupancy model for evobrutinib
| Parameter | Estimate | RSE % | 95% CI |
|---|---|---|---|
| k out (L/h) | 0.00437 | 7.7 | 0.004–0.005 |
| k irrev (ml/ng/h) | 0.0135 | 8.1 | 0.011–0.016 |
| Additive error | 0.00317 | 16.7 | 0.002–0.004 |
| Proportional error | 0.43 | 8.4 | 0.359–0.501 |
| Parameter | Estimate (RSE %) | Etabar (SE) | CV % | Shr % |
|---|---|---|---|---|
| BPV k out | 0.217 (45.3%) | −0.002 (0.067) | 49.2 | 6.9 |
| BPV k irrev | 0.103 (40.5%) | −0.007 (0.043) | 32.9 | 12.9 |
Abbreviations: BPV, between‐participant variability; CI, confidence interval; CV%, coefficient of variation (%), Etabar, arithmetic mean of the η estimates; K irrev, second‐order irreversible binding rate constant; k out, first‐order elimination rate of Bruton's tyrosine kinase protein; RSE, relative standard error; SE, standard error; Shr, shrinkage.
The parameter estimates were of good accuracy. All fixed‐effect parameter estimates RSE were < 20% (Table 3). RSE for all variance parameters were rather higher, at up to 45%. Shrinkage was <15% for both random effects, and the proportional residual error was estimated to be 43%.
Basic goodness‐of‐fit plots for the BTKO model are presented in Figure S4. Visual predictive checks by dose and study day are depicted in Figure S5.
Simulation of alternative dosing regimens
The population PK and BTKO models were used as a basis to perform simulations of the profile of BTKO at steady‐state under alternative dosing regimens. The simulated percentage of participants with minimum BTKO above the four thresholds of interest (70%, 80%, 90%, and 95%) over 24 h at steady‐state are presented for the different dosing regimens in Table 4.
TABLE 4.
Simulated percentage of participants with minimum Bruton's tyrosine kinase receptor occupancy above the four thresholds of interest (70%, 80%, 90%, and 95%) over 24 h at steady‐state for different dose/regimen and food state combinations
| BTKO threshold | Prandial state | 25 mg | 50 mg | 75 mg | 100 mg | 200 mg |
|---|---|---|---|---|---|---|
| Simulated % of participants | ||||||
| q.d. dosing regimen | ||||||
| 0.70 | Fasted | 98 | 100 | 100 | 100 | 100 |
| 0.80 | 88 | 97 | 99 | 99 | 100 | |
| 0.90 | 36 | 59 | 71 | 77 | 87 | |
| 0.95 | 4 | 13 | 20 | 24 | 39 | |
| 0.70 | Fed | 99 | 100 | 100 | 100 | 100 |
| 0.80 | 95 | 99 | 99 | 99 | 100 | |
| 0.90 | 54 | 74 | 82 | 85 | 92 | |
| 0.95 | 10 | 22 | 29 | 34 | 51 | |
| b.i.d. dosing regimen | ||||||
| 0.70 | Fasted | 100 | 100 | 100 | 100 | 100 |
| 0.80 | 99 | 100 | 100 | 100 | 100 | |
| 0.90 | 88 | 97 | 99 | 100 | 100 | |
| 0.95 | 38 | 68 | 81 | 86 | 94 | |
| 0.70 | Fed | 100 | 100 | 100 | 100 | 100 |
| 0.80 | 100 | 100 | 100 | 100 | 100 | |
| 0.90 | 96 | 99 | 100 | 100 | 100 | |
| 0.95 | 59 | 85 | 90 | 93 | 97 | |
Abbreviations: b.i.d., twice‐daily; BTKO, Bruton's tyrosine kinase occupancy; q.d., once‐daily.
To maintain minimum occupancy over 24 h at steady‐state above different thresholds, evobrutinib 25 mg q.d. administered while fasted was predicted to maintain a minimum BTKO of 80% in 88% of participants, and a minimum BTKO of 90% in 36% of participants (Table 4, Figure 3). The corresponding percentages for 75 mg b.i.d. while fasted were 100% and 99% of participants and for the 50 mg b.i.d. while fasted regimen 100% and 97% of participants, respectively (Table 4, Figure 3). Assuming that BTKO of ≥70% at trough at steady‐state (independent of the proportion of patients achieving this BTKO level) is needed to achieve efficacy in the different indications considered, evobrutinib doses of 25 mg q.d., 50 mg b.i.d., or 75 mg b.i.d. while fasted seem plausible choices for further development.
FIGURE 3.
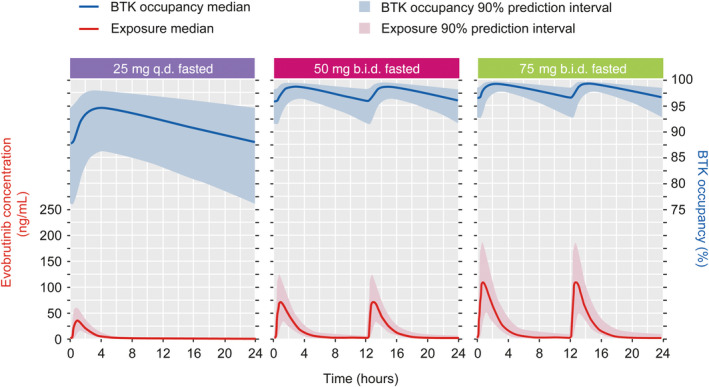
Simulation of evobrutinib concentration and BTK occupancy at steady‐state. Abbreviations: b.i.d., twice daily; BTK, Bruton's tyrosine kinase; q.d., once daily.
DISCUSSION
A two‐compartment model with sequential zero‐first order absorption and first‐order elimination was able to describe the observations obtained from participants treated under different conditions and at different dose levels with the tablet formulation. The model included BPV on CL/F, V2/F, Q/F, V3/F, ALAG, and D1, correlation between CL/F and ALAG, as well as Q/F and V3/F, and different random effect on D1 on the same participant for different food states.
According to the model parameter estimates, bioavailability increased by 49% after administration of a low‐fat meal compared with when fasted. The duration of the first‐order input also depended on prandial status and was increased by 427% (0.181 h, while fasted; 0.954 h, with food) after a low‐fat meal. None of the other covariates tested (age, sex, body size, ethnicity [Japanese vs. non‐Japanese]) were found to be a significant covariate for any of the model parameters, hence they were not included in the final population PK model.
Tolerability of evobrutinib was equivalent in fed and fasted states. Phase II studies were conducted under fasting conditions with information available at that time regarding PK variability and tolerability. While phase II studies were ongoing, clinical pharmacology studies were conducted to fully characterize PK under different fed/fasted conditions. The PK results and assessment of variability from the food effect studies, similar tolerability under fed and fasted conditions, and the feedback from patients which indicated they would prefer to take b.i.d. doses with meals, supported the decision to conduct phase III studies under fed conditions.
The BTKO profile of evobrutinib was well described by the irreversible binding population model. The model included BPV on k out and k irrev and no covariates. The population PK model was combined with the BTKO model, to simulate the PK and BTKO profiles under alternative dosing regimens. This model allowed the estimation of the evobrutinib dose required to achieve BTKO of any level considered necessary to attain efficacy, or to better define the relevant dose–response (BTKO) curves.
The simulated percentage of participants with minimum BTKO increased in a dose‐dependent manner across the BTKO thresholds. Assuming BTKO ≥70%, based on preclinical models, at trough at steady‐state is needed to achieve efficacy in the different indications considered, the results suggest that evobrutinib doses of 25 mg q.d., 50 mg b.i.d., or 75 mg b.i.d. while fasted are plausible choices for further development. These doses are projected to provide minimum BTKO of 70% at trough at steady‐state for 98%, 100%, and 100% of participants, respectively. Doses above 200 mg b.i.d. with food or under fasted conditions would be required if higher levels of occupancy (approaching 100%) were necessary for efficacy. Future studies should provide the data that will confirm this PK/PD relationship in different indications and investigate exposure and BTKO levels associated with efficacy.
Limitations
This study was conducted in healthy participants, therefore it is possible that exposure and BTKO might be different in a patient population. Steady‐state BTKO simulations performed in this study were based on data after a single dose and multiple doses for 5 days. As the PK/PD simulations did not include residual error and parameter uncertainty, the presented results represent the best‐case scenario for dose selection in terms of BTKO. Further dose optimization will be required for phase III dose selection and the relevant endpoints. In addition, the recommended doses for further development do not take into consideration the safety of evobrutinib, which may impact the choice of dose (or range of doses).
CONCLUSIONS
The proposed population PK model adequately described the plasma concentration–time profiles of evobrutinib. The irreversible binding population model described the BTKO profile of evobrutinib well. The simulated percentage of participants with minimum BTKO increased in a dose‐dependent manner across the BTKO thresholds, and evobrutinib doses of 25 mg q.d., 50 mg b.i.d., or 75 mg b.i.d. while fasted seem plausible choices for further development, assuming BTKO ≥70% at trough at steady‐state is needed to achieve efficacy in the different indications considered.
AUTHOR CONTRIBUTIONS
O.P., D.M., P.G., and M.D. wrote the manuscript. O.P., D.M., P.G., and M.D. designed the research. O.P., D.M., P.G., and M.D. performed the research. O.P. analyzed the data.
FUNDING INFORMATION
This study was funded by the healthcare business of Merck KGaA, Darmstadt, Germany (CrossRef Funder ID: 10.13039/100009945).
CONFLICT OF INTEREST
O.P. and P.G. are employees of Merck Institute for Pharmacometrics, Lausanne, Switzerland, an affiliate of Merck KGaA, Darmstadt, Germany. M.D. is an employee of EMD Serono. D.M. has received compensation for his institution for serving as a consultant for EMD Serono.
Supporting information
FIGURES S1–S5
ACKNOWLEDGMENTS
The authors would like to thank Tobias Feige from Merck KGaA, Darmstadt, Germany for overseeing and coordinating the dataset preparation tasks for the modeling of this article. Medical writing and editorial support was provided by Ankit Turakhiya and Leanne Cummings (Bioscript Group Ltd, Macclesfield, UK), supported by the healthcare business of Merck KGaA, Darmstadt, Germany (CrossRef Funder ID: 10.13039/100009945).
Papasouliotis O, Mitchell D, Girard P, Dyroff M. Population pharmacokinetic and pharmacodynamic modeling of evobrutinib in healthy adult participants. Clin Transl Sci. 2022;15:2899‐2908. doi: 10.1111/cts.13417
REFERENCES
- 1. Bradshaw JM. The Src, Syk, and Tec family kinases: distinct types of molecular switches. Cell Signal. 2010;22:1175‐1184. [DOI] [PubMed] [Google Scholar]
- 2. Schmidt U, Boucheron N, Unger B, Ellmeier W. The role of Tec family kinases in myeloid cells. Int Arch Allergy Immunol. 2004;134:65‐78. [DOI] [PubMed] [Google Scholar]
- 3. Estupiñán HY, Berglöf A, Zain R, Smith CIE. Comparative analysis of BTK inhibitors and mechanisms underlying adverse effects. Front Cell Dev Biol. 2021;9:630942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Dolgin E. BTK blockers make headway in multiple sclerosis. Nat Biotechnol. 2021;39:3‐5. [DOI] [PubMed] [Google Scholar]
- 5. Montalban X, Arnold DL, Weber MS, et al. Placebo‐controlled trial of an oral BTK inhibitor in multiple sclerosis. N Engl J Med. 2019;380:2406‐2417. [DOI] [PubMed] [Google Scholar]
- 6. Haselmayer P, Camps M, Liu‐Bujalski L, et al. Efficacy and pharmacodynamic modeling of the BTK inhibitor Evobrutinib in autoimmune disease models. J Immunol. 2019;202:2888‐2906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Boschert U, Crandall T, Pereira A, et al. T cell mediated experimental CNS autoimmunity induced by PLP in SJL mice is modulated by Evobrutinib (M2951) a novel Bruton's tyrosine kinase inhibitor. Mult Scler. 2017;23:327. [Google Scholar]
- 8. Marostica E, Sukbuntherng J, Loury D, et al. Population pharmacokinetic model of ibrutinib, a Bruton tyrosine kinase inhibitor, in patients with B cell malignancies. Cancer Chemother Pharmacol. 2015;75:111‐121. [DOI] [PubMed] [Google Scholar]
- 9. Becker A, Martin EC, Mitchell DY, et al. Safety, tolerability, pharmacokinetics, target occupancy, and concentration‐QT analysis of the novel BTK inhibitor Evobrutinib in healthy volunteers. Clin Transl Sci. 2020;13:325‐336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Beal SL. Ways to fit a PK model with some data below the quantification limit. J Pharmacokinet Pharmacodyn. 2001;28:481‐504. [DOI] [PubMed] [Google Scholar]
- 11. Jonsson EN, Karlsson MO. Automated covariate model building within NONMEM. Pharm Res. 1998;15:1463‐1468. [DOI] [PubMed] [Google Scholar]
- 12. Beal SLS, Lewis B, Boeckmann A, Bauer RJ. NONMEM users guides. NONMEM Project Group, University of California, San Francisco; 2013. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
FIGURES S1–S5
Data Availability Statement
The available datasets from studies MS200527_0019 and MS200527_0017 are outlined in Table 1. BLQ observations (total of 675 of 2326, i.e., 29%) were used in the modeling data set.
TABLE 1.
Available datasets from Study MS200527_0019 and Study MS200527_0017
| ID | Study MS200527_0019 | Study MS200527_0017 | Total | |||
|---|---|---|---|---|---|---|
| N | % | N | % | N | % | |
| 18 | 24 | 58 | 76 | 76 | 100 | |
| Observations | ||||||
| All | 576 | 100 | 1750 | 100 | 2326 | 100 |
| 25 mg q.d. | – | – | 434 | 25 | 434 | 19 |
| 75 mg q.d. | 576 | 100 | 882 | 50 | 1458 | 63 |
| 200 mg q.d. | – | – | 434 | 25 | 434 | 19 |
| BLQ observations | ||||||
| All | 166 | 29 | 509 | 29 | 675 | 29 |
| 25 mg q.d. | – | – | 195 | 11 | 195 | 8 |
| 75 mg q.d. | 166 | 29 | 265 | 15 | 431 | 19 |
| 200 mg q.d. | – | – | 49 | 3 | 49 | 2 |
| Predose | 36 | 6 | 74 | 4 | 110 | 5 |
| Postdose | 130 | 23 | 435 | 25 | 565 | 24 |
| Fed/fasted observations | ||||||
| Fed | 288 | 50 | 224 | 13 | 512 | 22 |
| Fasted | 288 | 50 | 1526 | 87 | 1814 | 78 |
Abbreviations: BLQ, below the limit of quantification; N, number of participants/observations; q.d., once‐daily.
The BTKO dataset consisted of 447 observations from 41 participants from study MS200527_0017 Part A, of which there were:
152 observations for 14 participants treated with 25 mg q.d. evobrutinib for 6 days
142 observations for 13 participants treated with 75 mg q.d. evobrutinib for 6 days
153 observations for 14 participants treated with 200 mg q.d. evobrutinib for 6 days.
Six records with dependent variable value (fraction unoccupied) ranging from 1.2 to 3.7 were omitted from the modeling set as implausible (corresponding to negative BTKO). Consequently, the total available dataset model consisted of 441 observations from 41 participants.