

Abstract
The pharmacometric analysis of the double‐blind, randomized, phase II study (NCT02975349) investigating the safety and efficacy of evobrutinib, explored exposure–response relationships and suitable dosing regimens of evobrutinib for relapsing multiple sclerosis. Population pharmacokinetic (PK)/pharmacodynamic modeling was applied to data collected in fasted patients treated with placebo or evobrutinib (25 mg once‐daily [q.d.], 75 mg q.d., or 75 mg twice‐daily [b.i.d.]) for 24 weeks, followed by a 24‐week blinded extension (placebo patients switched to 25 mg q.d.). Model‐based exposures for PK and Bruton's tyrosine kinase occupancy (BTKO) were used for exposure–response analyses (maximum 207 patients). PK, BTKO profiles, and annualized relapse rate (ARR) after 48 weeks of treatment under alternative dosing regimens were simulated. Exposure–response modeling identified a relationship between evobrutinib exposure and clinical response for total number of T1 Gd+ and new/enlarging T2 lesions at weeks 12–24, and ARR at week 48. Area under the concentration–time curve over 24 h at steady‐state (AUC0–24,SS) of 468 and ≥400 ng/ml h was associated with T1 Gd+/T2 lesion reduction and ARR improvement, respectively. These exposures were associated with steady‐state (SS) predose BTKO ≥95%. Based on PK and BTKO profile simulations, evobrutinib 75 mg b.i.d. while fasted is predicted to maintain SS predose BTKO >95% in 92% of patients. Evobrutinib 45 mg b.i.d. with food is predicted to achieve similar exposure as 75 mg b.i.d. while fasted (predose BTKO >95% in 93% of patients). Evobrutinib 45 mg b.i.d. with food is predicted to have comparable exposure and BTKO to 75 mg b.i.d. without food (phase II) and will be pharmacologically effective and appropriate for clinical use in phase III multiple sclerosis studies.
Study Highlights.
WHAT IS THE CURRENT KNOWLEDGE ON THE TOPIC?
Evobrutinib is a Bruton's tyrosine kinase (BTK) inhibitor, with a mechanism of action that uniquely impacts a range of key cell types involved in the pathogenesis of multiple sclerosis (MS), such as B cells and myeloid cells, including macrophages and microglia.
WHAT QUESTION DID THIS STUDY ADDRESS?
The main objective was to identify the exposure–response relationship and to explore potential alternative dosing regimens via pharmacokinetics (PK) and BTK occupancy (BTKO) simulations, and eventually identifying a suitable dosing regimen for further clinical development.
WHAT DOES THIS STUDY ADD TO OUR KNOWLEDGE?
Based on PK and BTKO profile simulations performed in this study, evobrutinib dose of 75 mg twice daily (b.i.d.) while fasted is predicted to maintain predose BTKO of >95% in 92% of patients at steady‐state. Importantly, evobrutinib dose of 45 mg b.i.d. with food is predicted to achieve similar efficacy (predose BTKO of >95% in 93% of patients) as 75 mg b.i.d. while fasted. No once daily dose fasted or taken with food is predicted to achieve the desired predose BTKO level at steady‐state.
HOW MIGHT THIS CHANGE CLINICAL PHARMACOLOGY OR TRANSLATIONAL SCIENCE?
Our findings indicate that evobrutinib dose of 45 mg b.i.d. with food will be pharmacologically effective and is appropriate for clinical use in phase III studies in patients with relapsing MS (RMS). Based on projected target engagement and efficacy and the fact that taking evobrutinib with food increases the relative bioavailability by about 50%, a dose of 45 mg b.i.d. taken with food is currently being administered in the evolutionRMS 1 (NCT04338022) and 2 (NCT04338061) phase III RMS studies.
INTRODUCTION
Bruton's tyrosine kinase (BTK), a member of the Tec family of non‐receptor tyrosine kinases, is expressed in B cells, macrophages, and microglia (although not in T cells) and is involved in signal propagation and modulation of signal responsiveness in these cells. 1 , 2 , 3 , 4 , 5 Overexpression of BTK has been shown to be associated with certain autoimmune diseases due to an increase in autoreactive B cells and autoantibodies. 2 Thus, BTK represents a potential therapeutic target in autoimmune diseases, such as multiple sclerosis (MS).
Evobrutinib is a highly selective, central nervous system (CNS)‐penetrant, orally administered, covalent BTK inhibitor. 6 , 7 Evobrutinib targets B cells and myeloid cells, including macrophages and microglia that are involved in the pathogenesis of MS; and has shown therapeutic potential in relapsing MS (RMS). 7 The high selectivity of evobrutinib for BTK indicates that it may have a low potential for off‐target‐related adverse effects. 6 , 8 The covalent binding of evobrutinib to BTK has resulted in continued target inhibition even after evobrutinib has been cleared from the circulation; this inhibition declined gradually over 1 week following drug withdrawal due to the continuous synthesis of new endogenous BTK protein. 7 , 9
The relationship of BTK occupancy (BTKO) and systemic drug exposure can help in the selection of doses required to achieve maximal occupancy of BTK, inferring maximal inhibition of B cell receptor signaling. 10 In a first‐in‐human phase I study of evobrutinib in healthy participants, BTKO was found to be dose‐dependent, with maximum occupancy of >90% within 4 h of single doses ≥200 mg while fasted. After 14 days administration of evobrutinib 25 mg once daily (q.d.) while fasted, very high BTKO was achieved (88% median at trough), in contrast to serum evobrutinib concentrations that were all below the limit of quantification (BLQ) at trough, demonstrating slow turnover of BTK protein in vivo. 9 In the phase II study (NCT02975349) that investigated the clinical efficacy of evobrutinib in patients with RMS, 11 the observed BTKO in a subset of the study population (74 patients in all treatment arms) was very high (>95% at trough with evobrutinib 75 mg twice daily [b.i.d.]; Table 1) and in line with population model predictions. 12 In addition, a reduction in the total cumulative number of T1 gadolinium‐enhancing (Gd+) lesions was observed with evobrutinib 75 mg q.d. while fasted compared with placebo over 24 weeks; the magnitude of this effect was maintained through week 48. 11 , 13 Moreover, the efficacy of evobrutinib 75 mg b.i.d. in the 48‐week double‐blind period on annualized relapse rate (ARR 0.11) was shown to be maintained over a 108‐week period (ARR 0.12), including the 48‐week double‐blind period plus the 60‐week open‐label extension. 11 , 12 Evobrutinib 25 mg q.d. dose did not show clinical efficacy on any end points compared with placebo. 11 , 12 An exposure–response analysis may provide a better understanding of the effect of evobrutinib dose on clinical efficacy and BTKO.
TABLE 1.
Percentage of patients with predose occupancy at steady state above 70%, 80%, 90%, and 95% thresholds – observed data
| BTKO threshold | Food state | Dosing regimen (%) | ||
|---|---|---|---|---|
| 25 mg q.d. | 75 mg q.d. | 75 mg b.i.d. | ||
| 0.70 | Fasted | 91 | 100 | 100 |
| 0.80 | 80 | 98 | 100 | |
| 0.90 | 51 | 87 | 100 | |
| 0.95 | 23 | 48 | 98 | |
Abbreviations: b.i.d., twice daily; BTKO, Bruton's tyrosine kinase occupancy; q.d., once daily.
The main objective of this pharmacometrics analysis was to evaluate the exposure–response relationship for the data from the phase II study and eventually inform dose selection for phase III studies in patients with RMS. Intermediate objectives were to describe the pharmacokinetic (PK) profile of evobrutinib in patients with RMS, describe the PK/pharmacodynamic (PD) relationship for BTKO, and explore potential alternative dosing regimens for various end points via simulations.
METHODS
Ethical considerations
An independent ethics committee or institutional review board at each study site approved the protocols of the studies included in this analysis. The studies were conducted in accordance with the Declaration of Helsinki and International Conference on Harmonization Good Clinical Practice Guidelines. Written informed consent was obtained from each participant before any study‐related activities were performed.
Study population
The population PK/PD modeling was performed using data from two phase I studies in healthy participants and one phase II study in patients with RMS, to obtain full PK and BTKO profiles. In studies MS200527_0019 and MS200527_0017, the population PK/PD modeling was performed on the safety population. Details of these two studies have been published previously 14 and are described in brief in article doi: 10.1111/cts.13417. In study MS200527_0086, any patient with at least one dose of evobrutinib and at least one quantifiable postdose plasma concentration of evobrutinib was evaluable for inclusion in the analysis.
Study MS200527_0086 (NCT02975349)
This was a phase II, randomized, placebo‐controlled study, comprised of a 48‐week double‐blind period followed by an open‐label extension in patients with RMS. 11 , 12 Patients were randomized 1:1:1:1:1 to receive either evobrutinib 25 mg q.d., 75 mg q.d., 75 mg b.i.d., placebo (switched to evobrutinib 25 mg q.d. in a blinded manner after week 24), or open‐label dimethyl fumarate (at a dose of 120 mg b.i.d. for the first week and 240 mg b.i.d. thereafter). Evobrutinib, per protocol, was administered while fasting (>1 h pre‐meal or >2 h post‐meal). Serial blood samples for PK and PD assessments were collected at scheduled timepoints throughout the study. PK sampling occurred in patients who received evobrutinib or placebo. PK samples were collected on day 1 and at weeks 4, 12, and 24. On day 1, samples were collected predose (within 30 min prior to dosing) and at 0.25, 0.5, 1, 1.5, 2 h, and 4 h postdose. On weeks 4, 12, and 24, samples were collected predose (within 30 min prior to dosing) and between 2 and 8 h postdose. The vast majority (all but five) of available evobrutinib concentrations for modeling were associated with fasted state. PD samples (predose and 2 h postdose) for BTKO (in peripheral blood mononuclear cells) were collected on day 1 and at weeks 4, 12, and 24. Magnetic resonance imaging (MRI) scans were performed at screening, at 4‐week intervals from weeks 12 to 24, and at the end of treatment visit at week 48. MS relapse was assessed at 4‐week intervals from weeks 4 to 24, at week 36, at the end of treatment visit at week 48, and at end of trial visit at week 52. Relapse was also assessed at any unscheduled visit for neurological worsening and relapse assessment.
Bioanalysis
Measurement of evobrutinib concentration in plasma and BTKO was performed by high performance liquid chromatography (HPLC) coupled to tandem mass spectrometry (MS/MS). Plasma samples were extracted using a liquid/liquid extraction with ethyl acetate and subsequently analyzed. The lower limit of quantification for evobrutinib concentrations was 0.600 ng/ml.
Sample preparation involved addition of internal standard (evobrutinib‐d3) to plasma samples. Ammonium bicarbonate (1 M) was added to samples which were then extracted with ethyl acetate. Aliquots of the ethyl acetate layer were recovered, evaporated under nitrogen, and then the samples were reconstituted in acetonitrile:HPLC water (30:70). Aliquots were injected onto a C18, 50 × 2.1 mm, 1.6 μM HPLC column. HPLC was conducted with a flow rate of 800 μl/min and gradient conditions using HPLC‐water/1 M ammonium formate (1000/10; v/v) and acetonitrile. Evobrutinib and internal standard were detected by triple quadrupole MS detection, positive‐ion electrospray, and multiple reaction monitoring (MS/MS) scan type. Calibration curve (range: 0.6–600 ng/ml) was determined by linear regression with 1/x 2 weighting.
Population PK and BTKO model development
Data from patients in the phase II RMS study (48‐week data base lock) were used to update the evobrutinib population PK and BTKO models first built on healthy participant data. 14 A detailed description of these models can be found in article doi: 10.1111/cts.13417.
The PK model structure used in this study was a two‐compartment model with sequential zero and first‐order absorption and first‐order elimination (Figure S1), identical to the previous model in healthy participants. 14 The BTKO model structure was an irreversible binding population model (Figure S2) that included interindividual variability on k out (first order elimination rate of BTK protein) and k irrev (the second order irreversible binding rate constant) with no covariate; this was identical to the previous preclinical model 6 and the model in healthy participants. 14
The NONMEM program (version 7.3.0) was used for PK and BTKO model fitting. The M3 method was used in the PK modeling, as slightly more than 20% of the observed PK data were BLQ. 15 SAS (version 9.04, proc nlmixed) was used for the exposure–response models.
Exposure–response analysis
The relationship between evobrutinib exposure and key MS end points (i.e., total cumulative number of T1 Gd+ lesions and new/enlarging T2 lesions at weeks 12–24 and ARR at week 48) was investigated to determine the distribution best describing the data and to characterize the exposure–response using standard statistical criteria. The relevant modeling investigation was performed on the modified intention‐to‐treat (mITT) population (randomized patients who had received at least one dose of evobrutinib, analyzed as randomized and who had at least one baseline and one post‐baseline MRI assessment). The clinical end points were investigated with cross‐sectional negative‐binomial regression distribution.
Key exposure and occupancy metrics were derived. Based on standard statistical criteria, area under the concentration–time curve (AUC) was optimal among the various exposure metrics investigated in the exposure–response models. The selection was based on Akaike's information criterion (AIC), and AUC was the metric that provided the optimal AIC across different exposure metrics and functional forms of the exposure–response relationship investigated, perhaps not surprisingly given that the 75 mg b.i.d. regimen was proved to be the best in the phase II study and that AUC is the metric that differentiates this regimen from the 75 mg q.d. regimen, due to the lack of accumulation.
The mathematical formulations of the log‐linear models investigating the total cumulative number of T1 Gd+ lesions (Equation 1), total cumulative number of new/enlarging T2 lesions (Equation 2), and ARR (Equation 3) were:
| (1) |
where AUC0–24,SS is the area under the concentration–time curve over 24 h at steady‐state, b01 and b02 intercepts corresponding to 0 T1 Gd+ lesions and >0 T1 Gd+ lesions at baseline, respectively; b1 is the slope of the exposure–response relationship; and λ is the mean total number of T1 Gd+ lesions of the negative‐binomial distribution.
| (2) |
where AUC0–24,SS is the area under the concentration–time curve over 24 h at steady state; b0 and b1 are intercept and slope, respectively, of the exposure–response relationship; and λ is the mean total number of new/enlarging T2 lesions of the negative‐binomial distribution.
| (3) |
where the log time in trial (in years) for a patient was used as an offset in the model; ARR is the annualized relapse rate; AUC0–24,SS is the area under the concentration–time curve over 24 h at steady state; exp(B0) and exp(B1) are the parameters denoting the levels of ARR for low and high values of the AUC (estimates of 0.52 and 0.11 respectively); STEP_AUC is the value of AUC associated with 50% of the maximum ARR decrease; and λ is the mean ARR of the negative‐binomial distribution. The model for ARR introduces nonlinearity in the exposure–response relationship.
Other functional exposure–response relationships were also tested for Equation 3, including linear and maximum effect (Emax; with or without sigmoidicity parameter) functions. The selected function was based on its optimal value of AIC and the fact that the 95% confidence intervals of corresponding model parameters did not include zero.
The parameter estimates for these models are shown in Tables S1–S3, respectively. In parallel, to define the effect of food and different types of meals on evobrutinib exposure, information from the phase I program and a dedicated food effect clinical pharmacology study was used.
Simulation of alternative regimens
The components from the population models developed on MS200527‐0086 study data (fasted patients) were used to simulate PK and BTKO profiles at steady‐state under alternative dosing regimens (10–200 mg q.d. and b.i.d. with food or while fasted). The simulations were performed to determine dose levels/regimens to be used in future studies or to support dose levels/regimens being used in ongoing evobrutinib studies. Simulated profiles were obtained for the tablet formulation with food or under fasted states in patients with RMS.
The PK and BTKO profiles of 1000 patients per dosing regimen were simulated, including the percentage of patients above BTKO thresholds of interest (70%, 80%, 90%, and 95%).
RESULTS
Data availability for population PK model
The available data sets from studies MS200527_0019, MS200527_0017, and MS200527_0086 are outlined in Table S4. BLQ observations (total of 800 out of 3850, i.e., 21%) were used in the modeling data set.
Population PK model
Population PK model parameter estimates at week 48 are shown in Table 2. The model included between‐participant variability (BPV) on apparent clearance (CL/F), apparent central and peripheral volumes of distribution (V2/F and V3/F, respectively), apparent intercompartmental clearance (Q/F), lag time (ALAG1), and duration of zero‐order input (D1; Figure S1). The model also incorporated correlation between CL/F and ALAG1, as well as Q/F and V3/F, and different random effect for D1 on the same patient for different food states.
TABLE 2.
Parameter estimates of the final PK model for evobrutinib at week 48
| Parameter | Estimate | RSE % | 95% CI |
|---|---|---|---|
| CL/F (L/h) | 269 | 4.05 | 248–290 |
| V2/F (L) | 50.8 | 9.37 | 41.5–60.1 |
| Q/F (L/h) | 33.2 | 11.7 | 25.6–40.8 |
| V3/F (L) | 385 | 18.7 | 244–526 |
| K a (/h) | 0.793 | 2.57 | 0.753–0.833 |
| ALAG (h) | 0.197 | 7.11 | 0.170–0.224 |
| D1 (h) | 0.241 | 18.6 | 0.153–0.329 |
| F1 in fed state a | 1.49 | 3.81 | 1.38–1.60 |
| D1 in fed state a | 5.41 | 15.2 | 3.80–7.02 |
| CL/F patients | 0.594 | 6.57 | 0.518–0.670 |
| Parameter | Estimate (RSE %) | Etabar (SE) | CV % | Shr % |
|---|---|---|---|---|
| ω2 CL/F | 0.257 (18.1) | 0.00000538 (0.0314) | 54.1 | 4.67 |
| Cov CL/F + ALAG | 0.0572 (120) | |||
| ω2 ALAG | 0.317 (83.3) | 0.00121 (0.0323) | 61.1 | 17.3 |
| ω2Q/F | 1.25 (18.2) | −0.00215 (0.0602) | 158 | 17.8 |
| Cov Q/F + V3/F | 1.09 (17.3) | |||
| ω2 V3/F | 1.82 (15.2) | −0.000715 (0.0722) | 227 | 33.4 |
| ω2 V2/F | 0.566 (32.9) | 0.00199 (0.0288) | 87.52 | 68.9 |
| ω2 D1 fasted | 0.784 (38.3) | 0.0181 (0.0384) | 109 | 4.67 |
| ω2 D1 fed | 0.784 (38.3) | 0.00911 (0.0179) | 109 | 11.9 |
| Residual variability | Estimate | RSE (%) | 95% CI | CV% |
|---|---|---|---|---|
| Additive error (ng/ml) | 0.272 | 33.1 | 0.0958–0.448 | – |
| Proportional error | 0.509 | 2.87 | 0.480–0.538 | 50.9 |
Note: Covariates: F1 = 1 for fasted; D1(h) = 0.241 for fasted; D1(h) = 0.241*5.41 for fed; CL(L/h) = 269 for healthy participants; CL(L/h) = 269*0.594 for MS patients.
Abbreviations: ALAG1, lag time; CI, confidence interval; CL/F, apparent clearance; Cov, covariance; CV%, coefficient of variation (%); D1, duration of zero order input; Etabar, arithmetic mean of the η estimates; F1, bioavailability; K a, absorption rate constant; MS, multiple sclerosis; PK, pharmacokinetic; %RSE, percent relative standard error of the estimate (standard error/parameter estimate × 100); Q/F, apparent intercompartmental clearance; SE, standard error; Shr, shrinkage; V2/F, apparent volume of distribution of the central compartment; V3/F, apparent volume of distribution of the peripheral compartment.
Fractional change estimated. Residual error terms were estimated as thetas (point estimate is √ω2). CV% = 100* √ω2 for log‐normally distributed variability terms. If ω2 > 0.15, CV% = 100 *√eω2 − 1.
Covariate effects included in the model were, food state effect on bioavailability and duration of zero‐order input, and study population (healthy participants vs. patients with RMS) on apparent clearance. Evobrutinib bioavailability was increased by 49% and duration of the zero‐order input was increased by 441% with food compared with when fasted. Apparent clearance in patients with RMS was reduced by 40.6% compared with healthy participants (Table 2). A higher CL/F in healthy participants compared with patients with rheumatoid arthritis (RA) has also been reported for another BTK inhibitor, 16 but the exact mechanism has not been elucidated. However, pro‐inflammatory cytokines observed in MS and RA 17 , 18 are known to alter CYP3A4/5 expression 19 and have been reported to be associated with altered apparent clearance for some drugs. 18 , 20
All model parameters were accurately estimated, where the highest relative standard error (RSE) for fixed effects parameters was 18.7%, whereas for many of the parameters the RSE was <10% (Table 2).
Together, the final two‐compartment model with sequential zero and first‐order absorption and first‐order elimination adequately described the PK profiles of evobrutinib.
Data availability for BTKO model
The available BTKO data sets from studies MS200527_0017 and MS200527_0086 are outlined in Table S5. The total available data set model consisted of 909 observations.
BTKO model parameter estimates
The irreversible binding model adequately described the BTKO profiles, with model parameter estimates shown in Table 3.
TABLE 3.
Parameter estimates of the final BTKO model for evobrutinib at week 48
| Parameter | Estimate | RSE (%) | 95% CI |
|---|---|---|---|
| K out (1/h) | 0.00346 | 5.98 | 0.00305–0.00387 |
| K irrev (ml/ng/h) | 0.0146 | 6.27 | 0.0128–0.0164 |
| Inter‐individual variability | Estimate (RSE%) | Etabar (SE) | CV% | Shr% |
|---|---|---|---|---|
| ω2 K out | 0.350 (30.3) | 0.00774 (0.0442) | 64.7 | 20.5 |
| ω2 K irrev | 0.209 (29.7) | 0.000315 (0.0329) | 48.2 | 23.5 |
| Residual variability | Estimate | RSE % | 95% CI | CV% |
|---|---|---|---|---|
| Additive error | 0.00180 | 9.89 | 0.00145–0.00215 | – |
| Proportional error | 0.617 | 9.69 | 0.500–0.734 | 61.7 |
Note: Residual error terms were estimated as thetas (point estimate is √ω2). As ω2 > 0.15, CV% = 100 *√eω2 − 1.
Abbreviations: BTKO, Bruton's tyrosine kinase occupancy; CI, confidence interval; CV%, coefficient of variation (%); Etabar, arithmetic mean of the η estimates; K out, first‐order elimination rates of BTK protein; K irrev, second‐order irreversible binding rate constant; %RSE, percent relative standard error of the estimate (standard error/parameter estimate × 100); Shr, shrinkage; ω2 K out and ω2 K irrev, variance of random effect of K out and K irrev, respectively.
The parameter estimates were of good accuracy: all fixed effect parameter estimates RSE were <7%, and the proportional residual error was estimated to be 61.7% (Table 3). The updated BTKO model demonstrated that the exposure to BTKO relationship was identical between healthy participants and patients with RMS (i.e., the same evobrutinib exposure resulted in the same occupancy in the two groups).
Exposure–response relationship with clinical outcomes
Exposure–response models showed a significant relationship between evobrutinib exposure (AUC0–24,SS) and ARR at week 48 (Figure 1). A step function exposure–response curve was identified for the ARR data, with the lower step (associated with maximal efficacy, i.e., ARR improvement) reached at an evobrutinib exposure of 400 ng/ml h (Figure 1). When evobrutinib exposure was categorized by quintiles, the lowest two quintiles of the distribution of exposure were associated with a mean ARR of 0.63 and 0.42, whereas the highest three (starting at an AUC of 356 ng/ml h) were associated with a mean ARR of 0.14 or less (Table S6). AUC0–24,SS > 400 ng/ml h was exceeded by all patients in the 75 mg b.i.d. treatment arm in the phase II study. Overall, maximal efficacy is achieved when evobrutinib exposure exceeds 400 ng/ml h.
FIGURE 1.
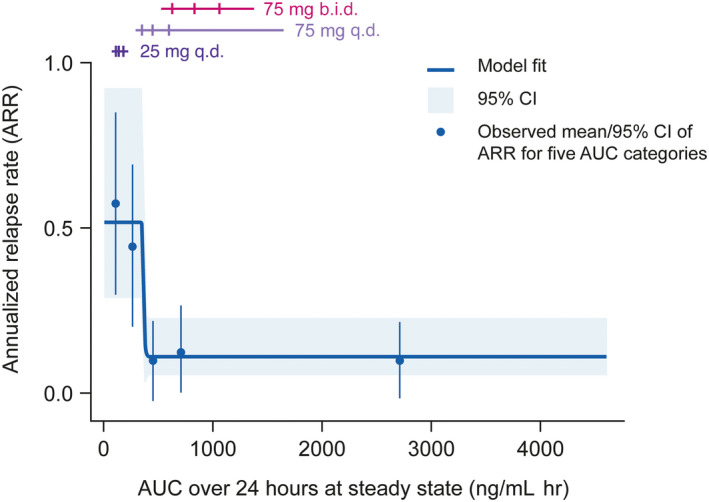
Relationship between ARR and evobrutinib exposure (mITT population). Horizontal lines correspond to the middle 80% of the distribution of AUC by dose/regimen, the three ticks are the first, second (median), and third quartiles. Observed means (blue points) are plotted at the mid‐point of the corresponding AUC exposure group. Data shown correspond to n = 154. ARR, annualized relapse rate; AUC, area under the curve; b.i.d., twice daily; CI, confidence interval; mITT, modified intent‐to‐treat; q.d., once daily.
Exposure–response models showed a significant relationship between evobrutinib exposure (AUC0–24,SS) and total cumulative number of T1 Gd+ and total cumulative number of T2 lesions at weeks 12–24 (Figure 2). The exposure–response relationships with T1 Gd+/T2 lesions were linear, with an AUC0–24,SS of 468 ng/ml h, which was associated with a substantially lower number of T1 Gd+ and new/enlarging T2 lesions. When evobrutinib exposure was categorized by quartiles within subgroups defined by the presence or absence of T1 Gd+ lesions at baseline, in patients with T1 Gd+ lesions at baseline, an AUC of 468 ng/ml h was associated with a reduction in T1 Gd+ lesion count (Table S7). Similarly, when evobrutinib exposure was categorized by quartiles within subgroups defined by baseline T2 lesion volume (≤13 cc or >13 cc), in patients with a baseline T2 lesion volume >13 cc, an AUC of 468 ng/ml h was associated with a reduction in new/enlarging T2 lesion count (Table S8). Overall, maximal response in T1 Gd+ and new/enlarging T2 lesion count was achieved with an exposure (AUC) >400 ng/ml h.
FIGURE 2.
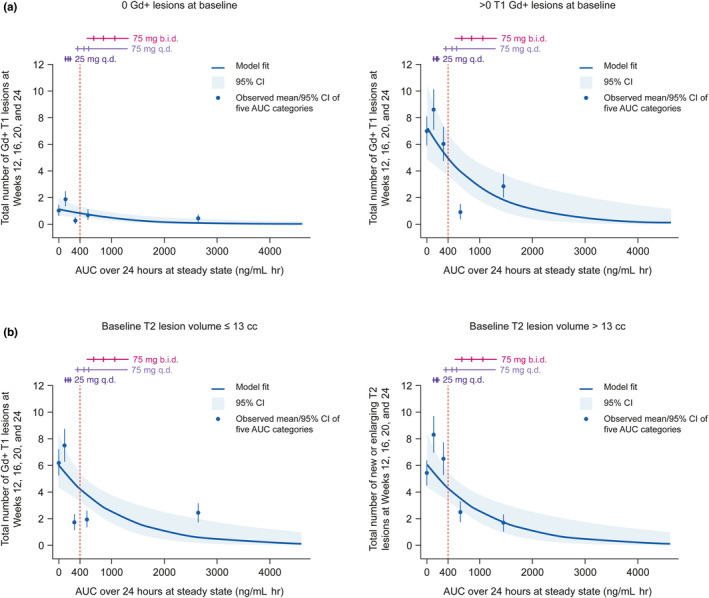
Relationship between (a) total T1 Gd+ and (b) total new/enlarging T2 lesion count and evobrutinib exposure (mITT population). Horizontal lines correspond to the middle 80% of the distribution of AUC by dose/regimen (not split by baseline T1 Gd+ lesion covariate), the three ticks are the first, second (median), and third quartiles. The vertical, dashed lines correspond to an evobrutinib exposure of 400 ng/mL h. Observed means (blue points) are plotted at the mid‐point of the corresponding AUC exposure group. In the T1 Gd+ lesion analysis, data shown correspond to n = 123 with 0 T1 Gd+ lesions at baseline and n = 84 with >0 T1 Gd+ lesions at baseline. In the new/enlarging T2 lesion analysis, data shown correspond to n = 108 with ≤13 cc T2 lesion volume at baseline and n = 99 with >13 cc T2 lesion volume at baseline. AUC, area under the curve; b.i.d., twice daily CI, confidence interval; Gd+, gadolinium‐enhancing; mITT, modified intent‐to‐treat; q.d., once daily.
Simulation of alternative dosing regimens
In the phase II RMS study, BTKO increased in a dose‐dependent manner based on predose observations at weeks 4, 12, and 24 (Table 1). 12 The highest predose BTKO was observed with the 75 mg b.i.d. dose. The largest and most sustained change in ARR was achieved when BTKO was >95%, observed in nearly all patients receiving 75 mg b.i.d. Based on these observed data, 95% BTKO is necessary to reach maximum efficacy.
The simulated percentage of participants with predose BTKO above four thresholds (70%, 80%, 90%, and 95%) over 24 h at steady‐state are presented for the different dosing regimens and food states in Table 4.
TABLE 4.
Simulated percentage of participants with predose BTKO above the four thresholds (70%, 80%, 90%, and 95%) over 24 h at steady‐state for different dose/regimen and food state combinations
| BTKO threshold | Food state | Simulated % of participants | |||||||
|---|---|---|---|---|---|---|---|---|---|
| 10 mg | 25 mg | 35 mg | 45 mg | 50 mg | 75 mg | 100 mg | 200 mg | ||
| q.d. dosing regimen | |||||||||
| 0.70 | Fasted | 93 | 99 | 100 | 100 | 100 | 100 | 100 | 100 |
| 0.80 | 79 | 95 | 97 | 98 | 98 | 99 | 99 | 100 | |
| 0.90 | 43 | 70 | 77 | 81 | 83 | 88 | 90 | 94 | |
| 0.95 | 12 | 31 | 39 | 46 | 48 | 55 | 60 | 72 | |
| 0.70 | Fed | 97 | 100 | 100 | 100 | 100 | 100 | 100 | 100 |
| 0.80 | 89 | 97 | 98 | 99 | 99 | 100 | 100 | 100 | |
| 0.90 | 57 | 79 | 85 | 87 | 88 | 92 | 93 | 96 | |
| 0.95 | 20 | 43 | 51 | 55 | 57 | 64 | 70 | 79 | |
| b.i.d. dosing regimen | |||||||||
| 0.70 | Fasted | 99 | 100 | 100 | 100 | 100 | 100 | 100 | 100 |
| 0.80 | 96 | 100 | 100 | 100 | 100 | 100 | 100 | 100 | |
| 0.90 | 78 | 94 | 97 | 98 | 99 | 99 | 100 | 100 | |
| 0.95 | 45 | 72 | 81 | 85 | 87 | 92 | 94 | 96 | |
| 0.70 | Fed | 100 | 100 | 100 | 100 | 100 | 100 | 100 | 100 |
| 0.80 | 98 | 100 | 100 | 100 | 100 | 100 | 100 | 100 | |
| 0.90 | 88 | 97 | 99 | 99 | 99 | 100 | 100 | 100 | |
| 0.95 | 60 | 84 | 89 | 93 | 93 | 95 | 96 | 98 | |
Abbreviations: b.i.d., twice daily; BTKO, Bruton's tyrosine kinase occupancy; q.d., once daily.
The simulations showed that an evobrutinib dose of 45 mg b.i.d. with food in patients with RMS is expected to result in AUC0–24,SS and steady‐state predose BTKO similar to that observed for the 75 mg b.i.d. fasted arm of the phase II study (Table 4 and Figure 3). The 45 mg b.i.d. dose is projected to achieve an AUC0–24,SS > 400 ng/ml h and steady‐state predose BTKO of >95% in more than 90% of patients (Figure 3). The expected ARR is anticipated to be similar to that observed for the 75 mg b.i.d. fasted arm of the phase II study.
FIGURE 3.
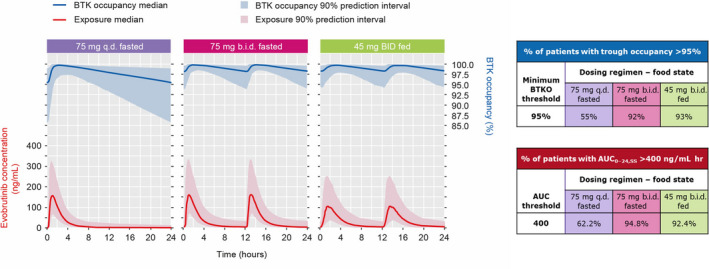
Simulation of evobrutinib concentration and BTK occupancy at steady state. AUC0–24,SS, area under the concentration–time curve over 24 h at steady‐state; b.i.d., twice daily; BTK, Bruton's tyrosine kinase; q.d., once daily.
DISCUSSION
Exposure–response models showed a significant relationship between evobrutinib exposure (AUC0–24,SS) and clinical response. Increasing evobrutinib (75 mg q.d. and b.i.d.) exposure resulted in an increase in clinical response (total cumulative number of T1 Gd+ and new/enlarging T2 lesions at weeks 12–24, and ARR at week 48). A steady‐state AUC over 24 h of 468 and ≥400 ng/ml h was associated with reductions in the total cumulative number of T1 Gd+/T2 lesions and maximal efficacy, respectively. Importantly, these exposures were found to be associated with steady‐state predose BTKO of ≥95%.
Simulations performed in this study allowed the estimation of evobrutinib dose required to achieve BTKO of any level considered necessary to attain efficacy, or to better define the relevant dose–response (BTKO) curves. The simulated percentage of patients with predose BTKO has been found to be increased in a dose‐dependent manner across the BTKO thresholds. Importantly, the modeling predicted that >90% of patients administered 75 mg b.i.d. while fasted would achieve exposure and BTKO thresholds necessary for maximal efficacy.
The simulations performed based on data from phase I studies showed that a dose of 45 mg b.i.d., when given with food, is predicted to provide a (geometric) mean evobrutinib daily exposure (AUC0–24,SS) of 840 ng/ml h in patients with RMS, with only 7.6% of the participants expected to have a daily exposure of <400 ng/ml h. Dosing with food has been shown to increase evobrutinib bioavailability by about 50% (data not shown for high‐fat or moderate‐fat meal; internal report). 11 Consequently, a dose of 45 mg b.i.d. when given with food is predicted to achieve similar exposure as 75 mg b.i.d. while fasted (835 ng/ml hl), which provided a significant reduction in ARR. Mean peak evobrutinib exposure (Cmax,SS) for 45 mg b.i.d. with food is expected to be 24% less (128 ng/ml) than the observed (geometric) mean peak exposure (168 ng/ml) for 75 mg b.i.d. under fasted state. High BTKO (≥95% occupancy in 93% of the patients) is expected at the end of a dosing interval with 45 mg b.i.d. administration with food, and similar efficacy to that of 75 mg b.i.d. (BTKO of at least 95% in 92% of the patients) is expected. Therefore, based on projected target engagement and efficacy and that taking evobrutinib with food increases the relative bioavailability by about 50%, a dose of 45 mg b.i.d. taken with food was proposed for use in the phase III RMS program.
Our simulation data show that an evobrutinib dose of 45 mg b.i.d. with food in patients with RMS is expected to result in a similar steady‐state AUC0–24,SS over 24 h and steady‐state predose BTKO as the 75 mg b.i.d. fasted dose of the phase II trial. As shown in Figure 3, the first table illustrates the percentage of the simulated population that will be above the threshold of 95% BTKO for each dosing regimen. The second table illustrates the percentage of the simulated AUC that will be above the threshold of 400 ng/ml h for each dosing regimen. With the 75 mg q.d. fasted dosing regimen, insufficient numbers of patients were projected to achieve the required exposure and BTKO thresholds, whereas with both the 75 mg b.i.d. fasted and 45 mg b.i.d. with food dosing regimens more than 90% of the patients will reach or exceed the required thresholds. As can be seen, the relevant percentages are very similar for these two b.i.d. dosing regimens.
LIMITATIONS
The exposure–response models were based on the phase II study schedule (for MRI) and few relapse events occurred during the double‐blind period of the study. Additional relapse data from the open‐label extension may allow further description of exposure–response longitudinally. In addition, the simulations performed in this study did not include residual error and parameter uncertainty was not used. Therefore, it is possible that the simulation results presented here might underestimate uncertainty.
CONCLUSIONS
Exposure–response modeling demonstrated a significant relationship between evobrutinib exposure and clinical efficacy. High steady‐state predose BTKO (≥95%) was associated with maximal ARR improvement. The modeling predicted that >90% of patients administered with evobrutinib 75 mg b.i.d. without food will achieve exposure and BTKO thresholds necessary for maximal efficacy. Evobrutinib 45 mg b.i.d. administered with food is predicted to be comparable to evobrutinib 75 mg b.i.d. without food, with similar exposure and BTKO levels and, therefore, is currently being administered in two phase III RMS trials (evolutionRMS 1 and 2).
AUTHOR CONTRIBUTIONS
O.P., D.M., P.G., F.D., and M.D. wrote the manuscript. O.P., D.M., P.G., F.D., and M.D. designed the research. O.P., D.M., P.G., F.D., and M.D. performed the research. O.P. analyzed the data.
FUNDING INFORMATION
This study was funded by the healthcare business of Merck KGaA, Darmstadt, Germany (CrossRef Funder ID: 10.13039/100009945).
CONFLICT OF INTEREST
O.P. and P.G. are employees of Merck Institute for Pharmacometrics, Lausanne, Switzerland, an affiliate of Merck KGaA, Darmstadt, Germany. F.D. and M.D. are employees of EMD Serono. D.M. has received compensation for his institution for serving as a consultant for EMD Serono.
Supporting information
Appendix S1
ACKNOWLEDGMENTS
The authors would like to thank Richard Mills and Colm Farrell of ICON plc for the update of the PK and BTKO models built on healthy participants with the data from the phase II study (NCT02975349), and Eleanor Harrison‐Moench from Merck KGaA, Darmstadt, Germany, for overseeing and coordinating the data set preparation tasks for the modeling work presented in this manuscript. Medical writing and editorial support were provided by Ankit Turakhiya and Leanne Cummings (Bioscript Group Ltd., Macclesfield, United Kingdom), supported by the healthcare business of Merck KGaA, Darmstadt, Germany (CrossRef Funder ID: 10.13039/100009945).
Papasouliotis O, Mitchell D, Girard P, Dangond F, Dyroff M. Determination of a clinically effective evobrutinib dose: Exposure–response analyses of a phase II relapsing multiple sclerosis study. Clin Transl Sci. 2022;15:2888‐2898. doi: 10.1111/cts.13407
[Corrections added on 5 October 2022, after first online publication: Affiliation 1 has been modified and few minor edits have been made to this version that do not affect the meaning of the content.]
REFERENCES
- 1. Bender AT, Gardberg A, Pereira A, et al. Ability of Bruton's tyrosine kinase inhibitors to sequester Y551 and prevent phosphorylation determines potency for inhibition of fc receptor but not B‐cell receptor signaling. Mol Pharmacol. 2017;91:208‐219. [DOI] [PubMed] [Google Scholar]
- 2. Crofford LJ, Nyhoff LE, Sheehan JH, Kendall PL. The role of Bruton's tyrosine kinase in autoimmunity and implications for therapy. Expert Rev Clin Immunol. 2016;12:763‐773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Lopez‐Herrera G, Vargas‐Hernandez A, Gonzalez‐Serrano ME, et al. Bruton's tyrosine kinase‐an integral protein of B cell development that also has an essential role in the innate immune system. J Leukoc Biol. 2014;95:243‐250. [DOI] [PubMed] [Google Scholar]
- 4. Smith CI, Islam TC, Mattsson PT, et al. The tec family of cytoplasmic tyrosine kinases: mammalian Btk, Bmx, Itk, Tec, Txk and homologs in other species. Bioessays. 2001;23:436‐446. [DOI] [PubMed] [Google Scholar]
- 5. Keaney J, Gasser J, Gillet G, Scholz D, Kadiu I. Inhibition of Bruton's tyrosine kinase modulates microglial phagocytosis: therapeutic implications for Alzheimer's disease. J Neuroimmune Pharmacol. 2019;14:448‐461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Haselmayer P, Camps M, Liu‐Bujalski L, et al. Efficacy and pharmacodynamic modeling of the BTK inhibitor Evobrutinib in autoimmune disease models. J Immunol. 2019;202:2888‐2906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Boschert U, Crandall T, Pereira A, et al. T cell mediated experimental CNS autoimmunity induced by PLP in SJL mice is modulated by Evobrutinib (M2951) a novel Bruton's tyrosine kinase inhibitor. Mult Scler. 2017;23:327. [Google Scholar]
- 8. Caldwell RD, Qiu H, Askew BC, et al. Discovery of Evobrutinib: an Oral, potent, and highly selective, covalent Bruton's tyrosine kinase (BTK) inhibitor for the treatment of immunological diseases. J Med Chem. 2019;62:7643‐7655. [DOI] [PubMed] [Google Scholar]
- 9. Becker A, Martin EC, Mitchell DY, et al. Safety, tolerability, pharmacokinetics, target occupancy, and concentration‐QT analysis of the novel BTK inhibitor Evobrutinib in healthy volunteers. Clin Transl Sci. 2020;13:325‐336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Marostica E, Sukbuntherng J, Loury D, et al. Population pharmacokinetic model of ibrutinib, a Bruton tyrosine kinase inhibitor, in patients with B cell malignancies. Cancer Chemother Pharmacol. 2015;75:111‐121. [DOI] [PubMed] [Google Scholar]
- 11. Montalban X, Arnold DL, Weber MS, et al. Placebo‐controlled trial of an Oral BTK inhibitor in multiple sclerosis. N Engl J Med. 2019;380:2406‐2417. [DOI] [PubMed] [Google Scholar]
- 12. Montalban X, Arnold DL, Weber MS, et al. Clinical relapse rates in relapsing MS patients treated with the BTK inhibitor evobrutinib: results of an open‐label extension to a phase II study. Mult Scler J. 2020;26:118‐659. [Google Scholar]
- 13. Montalban X, Arnold DL, Weber MS, et al. Efficacy and safety of the Bruton's tyrosine kinase inhibitor Evobrutinib (M2951) in patients with relapsing multiple sclerosis over 48 weeks: a randomized, placebo‐controlled, phase 2 study (S56.004). Neurology. 2019;92(Suppl. 15):S56.004. [Google Scholar]
- 14. Papasouliotis O, Mitchell DY, Dyroff M, Girard P. Population pharmacokinetic model of Evobrutinib, a Bruton's tyrosine kinase inhibitor – an analysis of two phase I clinical trials in healthy subjects. Clin Pharmacol Ther. 2020;107:S5‐S121.32060909 [Google Scholar]
- 15. Beal SL. Ways to fit a PK model with some data below the quantification limit. J Pharmacokinet Pharmacodyn. 2001;28:481‐504. [DOI] [PubMed] [Google Scholar]
- 16. Chan P, Yu J, Chinn L, et al. Population pharmacokinetics, efficacy exposure–response analysis, and model‐based meta‐analysis of Fenebrutinib in subjects with rheumatoid arthritis. Pharm Res. 2020;37:25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Martins TB, Rose JW, Jaskowski TD, et al. Analysis of proinflammatory and anti‐inflammatory cytokine serum concentrations in patients with multiple sclerosis by using a multiplexed immunoassay. Am J Clin Pathol. 2011;136:696‐704. [DOI] [PubMed] [Google Scholar]
- 18. Coutant DE, Hall SD. Disease‐drug interactions in inflammatory states via effects on CYP‐mediated drug clearance. J Clin Pharmacol. 2018;58:849‐863. [DOI] [PubMed] [Google Scholar]
- 19. Mimura H, Kobayashi K, Xu L, et al. Effects of cytokines on CYP3A4 expression and reversal of the effects by anti‐cytokine agents in the three‐dimensionally cultured human hepatoma cell line FLC‐4. Drug Metab Pharmacokinet. 2015;30:105‐110. [DOI] [PubMed] [Google Scholar]
- 20. Morgan ET. Impact of infectious and inflammatory disease on cytochrome P450‐mediated drug metabolism and pharmacokinetics. Clin Pharmacol Ther. 2009;85:434‐438. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix S1
Data Availability Statement
The available data sets from studies MS200527_0019, MS200527_0017, and MS200527_0086 are outlined in Table S4. BLQ observations (total of 800 out of 3850, i.e., 21%) were used in the modeling data set.
The available BTKO data sets from studies MS200527_0017 and MS200527_0086 are outlined in Table S5. The total available data set model consisted of 909 observations.