

Abstract
Background and Objectives
Observational studies suggest low levels of 25-hydroxyvitamin D (25[OH]D) may be associated with increased disease activity in people with multiple sclerosis (PwMS). Large-scale genome-wide association studies (GWAS) suggest 25(OH)D levels are partly genetically determined. The resultant polygenic scores (PGSs) could serve as a proxy for 25(OH)D levels, minimizing potential confounding and reverse causation in analyses with outcomes. Herein, we assess the association of genetically determined 25(OH)D and disease outcomes in MS.
Methods
We generated 25(OH)D PGS for 1,924 PwMS with available genotyping data pooled from 3 studies: the CombiRx trial (n = 575), Johns Hopkins MS Center (n = 1,152), and Immune-Mediated Inflammatory Diseases study (n = 197). 25(OH)D-PGS were derived using summary statistics (p < 5 × 10−8) from a large GWAS including 485,762 individuals with circulating 25(OH)D levels measured. We included clinical and imaging outcomes: Expanded disability status scale (EDSS), timed 25-foot walk (T25FW), nine-hole peg test (9HPT), radiologic activity, and optical coherence tomography-derived ganglion cell inner plexiform layer (GCIPL) thickness. A subset (n = 935) had measured circulating 25(OH)D levels. We fitted multivariable models based on the outcome of interest and pooled results across studies using random effects meta-analysis. Sensitivity analyses included a modified p value threshold for inclusion in the PGS (5 × 10−5) and applying Mendelian randomization (MR) rather than using PGS.
Results
Initial analyses demonstrated a positive association between generated 25(OH)D-PGS and circulating 25(OH)D levels (per 1SD increase in 25[OH]D PGS: 3.08%, 95% CI: 1.77%, 4.42%; p = 4.33e-06; R2 = 2.24%). In analyses with outcomes, we did not observe an association between 25(OH)D-PGS and relapse rate (per 1SD increase in 25[OH]D-PGS: 0.98; 95% CI: 0.87–1.10), EDSS worsening (per 1SD: 1.05; 95% CI: 0.87–1.28), change in T25FW (per 1SD: 0.07%; 95% CI: −0.34 to 0.49), or change in 9HPT (per 1SD: 0.09%; 95% CI: −0.15 to 0.33). 25(OH)D-PGS was not associated with new lesion accrual, lesion volume or other imaging-based outcomes (whole brain, gray, white matter volume loss or GCIPL thinning). The results were similarly null in analyses using other p value thresholds or those applying MR.
Discussion
Genetically determined lower 25(OH)D levels were not associated with worse disease outcomes in PwMS and raises questions about the plausibility of a treatment effect of vitamin D in established MS.
Multiple sclerosis (MS) is an inflammatory and neurodegenerative disorder of the CNS.1,2 Disease course in people with MS typically follows a pattern of intermittent inflammatory disease activity that evolves into a state in which progressive neurologic deficits accumulate.3 Notably, disease course in people with MS is highly variable, and contributors to this variability remain poorly understood.4
Observations of higher MS risk in individuals with low vitamin D intake or low circulating 25-hydroxyvitamin D (25[OH]D) levels, and the results implicating a potential worse MS prognosis in individuals with low 25(OH)D levels suggest that vitamin D may contribute to MS disease processes.5-8 Small experimental vitamin D supplementation studies have also reported a beneficial role of supplementation on immunologic, radiologic, and clinical outcomes. However, the results from larger scale randomized clinical trials to date have been less clear.9-18
Genome-wide association studies (GWAS) have identified multiple loci associated with circulating 25[OH]D levels and suggest that a significant proportion of variation in 25(OH)D levels can be explained by common genetic variation.19,20 Resultant measures of polygenic inheritance of 25(OH)D can then be summarized using a polygenic score (PGS), which represent the sum of the number of alleles contributing to 25(OH)D levels possessed by a given individual weighted by their effect size on 25(OH)D levels.21 Such scores then can not only be used to predict a given person's 25(OH)D levels but also serve as a proxy for measured 25(OH)D levels, potentially minimizing confounding (e.g., geographical location, dietary intake, vitamin D supplementation) and reverse causation in the study of vitamin D levels and disease outcomes.22,23 PGS may also provide some insight into the expected level (rather than observed as with measured 25[OH]D levels) of vitamin D. Notably, studies using a PGS approach have consistently identified an association of vitamin D with MS susceptibility.24-26 In contrast, studies relating vitamin D-associated PGS to disease prognosis in MS are scarce.27 Although a possible association between genetically determined 25(OH)D levels and relapse risk was noted in a Belgian cohort of people with MS (PwMS), this study involved a single center and did not consider other clinical outcomes or objective imaging outcomes such as the rate of new lesion development, which has a strong body of support from observational studies.28 Thus, to address this gap, we explored whether genetically determined 25(OH)D levels are associated with disease outcomes in a large, multinational study of PwMS considering multiple radiologic and clinical outcomes. We hypothesized that genetically predicted higher 25(OH)D levels would be associated with better MS outcomes over time.
Methods
Standard Protocol Approvals, Registrations, and Patient Consents
The studies used in this manuscript were approved by all clinic or cohort-specific local Institutional Review Boards. All study participants provided written informed consent.
Study Population and Inclusion Criteria
In this multicenter study, 3 well-characterized cohorts of MS participants were included.
The CombiRx trial was a 3-arm double-blind placebo-controlled phase III clinical trial conducted in the United State with a 2:1:1 randomization scheme of combination therapy (interferon beta-1a [INFB] plus glatiramer acetate [GA]) vs single therapy (INFB or GA). The trial design and primary findings have been described in detail elsewhere.29 Briefly, eligible participants were treatment-naïve PwMS (McDonald 2005 revised criteria)30 with ≥2 clinical relapses or ≥1 clinically identified relapse with subsequent radiologic activity within 2 years before study onset. Participants were followed for 3 years as a part of the trial and had an option to enter the extension study in which follow-up continued for up to 7 years.
The Johns Hopkins (JHU) cohort included participants with a confirmed MS diagnosis who receive clinical care at the JHU MS center in Baltimore, Maryland. Participants are followed approximately biannually and complete functional assessments for research in a clinical care setting. Information on current 25(OH)D levels was available for a subset of the cohort using values obtained clinically.
The Immune-Mediated Inflammatory Diseases (IMIDs) Winnipeg cohort,31 included participants with MS, rheumatoid arthritis, inflammatory bowel disease, or a lifetime diagnosis of depression or anxiety disorder without an IMID residing in Winnipeg, Manitoba, Canada who were aged 18 years or older, able to provide consent, and had an adequate knowledge of English to complete questionnaires and interviews. Participants were enrolled from November 2014 through July 2016 and followed prospectively approximately annually over a 3-year period.
From these 3 cohorts, we included in our study participants with available genotyping who were of European ancestry.
Genotyping Information and Calculation of 25(OH)D PGS
For each cohort, blood samples from eligible participants underwent DNA extraction and subsequent genotyping using genome-wide Illumina genotyping chips: (CombiRx: HumanOmni1-Quad chip; IMID: Global Screening Array v2; JHU: Multi-Ethnic Global Array [MEGA]). After genotyping, we implemented an established quality control procedure for GWAS by excluding individuals with low genotyping success rates, related individuals, and those with excess heterozygosity. We also excluded individuals of non-European ancestry as identified using principal components (PCs) analysis (>2SD from PC1, PC2) mapping study populations on to the 1,000 Genomes population. We excluded variants with minor allele frequencies >0.05, missing genotype rates >0.05, and those with evidence of deviation from Hardy Weinberg equilibrium (p < 0.001). For each cohort, genotype imputation was applied using the Haplotype Reference Consortium as deployed on the Michigan Imputation Server and filtered for allele frequency (0.05–0.95) and imputation quality scores (imputation quality scores >0.70).32
To derive 25(OH) PGS, we used summary statistics from a large-scale meta-analysis combining a study linking genetic variants to 25(OH)D levels conducted in the UK Biobank and SUNLIGHT Consortium in 485,762 individuals (for body mass index [BMI]-adjusted analyses) and in the UK Biobank in 416,560 individuals (for BMI unadjusted analyses).20,33 Summary statistics from these studies were used to create the 25(OH)D PGSs for this study using Polygenic Risk Score software for Biobank-Scale Data (PGSice-2) with a p-value threshold of 5e-8, and linkage disequilibrium (LD) clumping (r2 < 0.10 in 250-kb window) and proxy R2 threshold of 0.80. Our primary analyses used the 25(OH)D PGS adjusted for BMI (as this was the original 25(OH)D GWAS with the larger sample size); sensitivity analyses considered the unadjusted 25(OH)D PGS. Sensitivity analyses also considered PGS using a p value threshold of 5e-05; some previous research suggests that this threshold may provide increased power when considering PGS and outcomes.34,35
Clinical Assessment, Data Collection, and Outcomes
Sociodemographic and clinical characteristics, disease-modifying therapies (DMTs), and concomitant medications were available for all participants through a combination of designated study visits, questionnaires, or the electronic medical record (EMR) depending on the cohort. A summary of available outcomes and associated sample size for each outcome considered across cohorts is provided in eTable 1 (http://links.lww.com/NXI/A766).
CombiRx
Participants underwent clinical assessments including Expanded Disability Status Scale (EDSS) and MSultiple Sclerosis Functional Composite (MSFC) examinations at baseline, and every 3 months until month 36, then every 6 months thereafter.29,36 MRIs were obtained at baseline, months 6, 12, 24, and 36, then annually thereafter. For relapses, we considered both protocol-defined relapses (PDE) and nonprotocol-defined relapse (NPDE). PDEs were defined as the development of new symptoms or worsening of old symptoms, lasting ≥24 hours in the absence of fever, preceded by 30 days of stability, and associated with changes in the EDSS score confirmed by the examining physician within 7 days of onset. NPDE were defined as relapses, which met these criteria but were not confirmed within 7 days of onset. For MRIs, brain volume was estimated using a standardized protocol that included 7 scan series; all MRIs obtained in CombiRx used Siemens, General Electric or Phillips scanners at 1.5 or 3T. Semiautomated processing was applied to estimate enhancing lesion volume, the strictly T2 hyperintense and T1 hypointense lesion volumes, total normal appearing white matter, gray matter and CSF (CSF) volumes, and normalized by the sum of all segmented values. We also considered a combined unique lesion activity, defined as the sum of the number of new enhancing lesions, new nonenhancing T2 lesions, and enlarged nonenhancing T2 lesions observed after enrollment.
JHU
Serum 25(OH)D levels were available for a subset of the cohort and were collected as a part of routine clinical monitoring. Disability status was determined by Neurostatus-certified raters for EDSS scores and was collected as a part of an on-going longitudinal observational study. Other clinical characteristics including the use of DMTs were also available. Similarly, information regarding walking speed, manual dexterity, and cognitive status was derived using the timed 25-foot walk, nine-hole peg, and the paced auditory serial addition (PASAT-3) tests of the MSFC, also collected as a part of on-going study visits. For neuroimaging, participant MRIs are acquired as a part of clinical care and are generally obtained for monitoring purposes. Each is obtained on any Siemens 3T Scanner after a standardized protocol. Two 3-D sagittal, whole-brain sequences are used: 1) fluid-attenuated inversion recovery (FLAIR; acquired resolution: 1 × 1 × 1mm; echo time [TE]: 392 ms; repetition time: 5,000 ms; inversion time [TI]: 1,800 ms) and 2) magnetization-prepared rapid gradient-echo (MPRAGE; acquired resolution: 1 × 1 × 1mm; TE: 2.96 ms; TR: 2,300 ms; TI: 900ms). Images are reviewed by neuroradiology to ensure correct sequence use, complete brain coverage, and adequate image quality. Quantitative MRI measures are derived from brain MRIs as part of a collaboration with the MS Partners Advancing Technology and Health Solutions (MS PATHS) network.37 Volumetric measures include brain parenchymal fraction (BPF), gray matter fraction (GMF), white matter fraction (WMF), and T2 lesion volume. These values are automatically calculated from MS PATHS MRIs via a software prototype developed jointly by Biogen and Siemens (MS PATHS Image Evaluation or MSPie).37 BPF is a normalized measure of whole brain volume that is calculated via a combined approach to segmentation of the brain parenchyma and total intracranial volume in the 3D FLAIR and 3D T1 images. The algorithm removes nonbrain tissue to create a total intracranial volume mask and then performs tissue classification incorporating partial volume effects using a Bayesian “mixel” model.38 In analyses of a scan-rescan substudy of MS-PATHS including 3 MS-PATHS sites at which 30 PwMS underwent 4 MRIs on 2 different Siemens 3T MRI scanners within 1 week, BPF estimated by MSPie exhibited excellent reproducibility, with a mean coefficient of variation of 0.18%. New T2 lesions are segmented automatically based on 3D FLAIR and 3D T1 images using a Bayesian partial volume estimation algorithm.
In addition, longitudinal retinal imaging data were available as a part of a long-term observational study. Optical coherence tomography (OCT) scans were acquired annually or biannually using CIRRUS HD-OCT device (model 5000; Carl Zeiss Meditec, Dublin, CA). Macular scans were obtained with the Macular Cube 512 x 128 protocol by experienced technicians, in accordance with the quality control criteria for (O) obvious problems, (S) poor signal strength, (C) centration of scan, (A) algorithm failure, (R) retinal pathology other than MS related, (I) illumination and (B) beam placement (OSCAR-IB), as described in detail elsewhere.39,40 Ganglion cell and inner plexus layer (GCIPL) thickness was quantified using a validated in-house segmentation algorithm.41 We focused on GCIPL thinning in this study, as changes in these layers are a highly sensitive marker reflecting global CNS neurodegeneration and disease progression in MS that may capture subtler changes than clinical outcomes.42-44 All scans underwent rigorous quality assurance evaluation by experienced raters to confirm the accuracy of segmentation boundaries and identify the presence of incidental pathologies impactful on retinal measures. Only scans with adequate quality were considered. Individuals with other known neurologic or ophthalmologic disorders, glaucoma, or refractive errors exceeding ±6 diopters were excluded. OCT methods and the results presented in this study were in accordance with the Advised Protocol for OCT Study Terminology and Elements criteria.45
IMID
Clinical and MS characteristics including the use of DMTs were collected at baseline and approximately annually thereafter. Likewise, functional assessments including the EDSS and MSFC components for timed 25-foot walk and 9-hole peg test scores were available approximately annually for each participant; the PASAT-3 was not obtained as a part of the IMID study protocol.
Statistical Analysis
Descriptive analyses characterized baseline demographic, clinical, and radiologic characteristics for each cohort. We first assessed the association between 25(OH)D PGS and circulating 25(OH)D levels in the JHU cohort to ensure that 25(OH)D PGS were a reasonable surrogate for 25(OH)D levels. For this analysis, we residual-adjusted 25(OH)D levels for BMI and the use of vitamin D supplements using generalized estimating equations to incorporate multiple circulating levels per person. Furthermore, because supplementation use was obtained from the EMR and could potentially be misclassified, we also excluded individuals with circulating levels exceeding 70 ng/mL. This threshold may constitute the physiologic maximum blood level of 25(OH)D, and some studies suggest people with MS may have a suboptimal response to supplementation (e.g., a person with MS may have a lower circulating level, despite use of supplements).46 Sensitivity analyses also considered 100 ng/mL as the physiologic threshold.47
Linear mixed effects models assessed the association between 25(OH)D PGS with the following continuous outcomes (1) clinical: EDSS, timed 25-foot walk, nine-hole peg, and PASAT-3 and (2) radiologic outcomes: T2 lesion volume, BPF, GMF, WMF, and GCIPL thickness. Models included subject-specific random intercepts and slopes (and eye-specific random slope for GCIPL analyses) to account for repeated measures. For analyses of 25(OH)D PGS and new lesions, we applied negative binomial regression models, and for relapses, we used an Andersen-Gill model for recurrent events. In secondary EDSS-based analyses, we defined disability progression as (1) EDSS increase by 1.5 points if baseline EDSS was zero, (2) increase in EDSS by 1 point if baseline EDSS was between 1 and 5.5, and (3) 0.5 point increase in EDSS if baseline EDSS was above 5.5, as previously described.48 We then assessed time to disability progression using a Cox proportional hazards model. Model fit and assumptions were verified using tests appropriate for the model in question.
We conducted all analyses in each individual cohort and pooled studies using random effects meta-analysis and calculated I2 statistics and associated tests of heterogeneity. For each study and outcome, we considered 25(OH)D PGS as a linear variable and categorically, using cohort-specific quartiles. We tested for trend by modeling the quartile variable as a linear covariate.49 We fit 3 series of models: model A, adjusted for age (continuous), sex, MS DMT (categorical), and genetic ancestry (as first 5 PC; continuous); model B, adjusted for model A covariates and the number of relapses in the previous 3 years and disease duration (both continuous); and a supplementary model C, adjusted for model B covariates and the use of vitamin D supplements and use of a medication associated with changes to vitamin D status (both binary covariates). These medications included corticosteroids, anticonvulsants, antidepressants, neuropathic pain medication, statins, and weight loss medication (e.g., orlistat).
We performed several sensitivity analyses assessing the consistency of our findings. First, we repeated all analyses using 25(OH)D PGS without adjusting for BMI. Second, we relaxed the p-value threshold to compute the 25(OH)D PGS to 5e-5 and repeated all analyses. Other analyses for relapse in the CombiRx data set were stratified by type of relapse (protocol defined exacerbation [PDE] and nonprotocol defined exacerbation [NPDE]). In addition, because some analyses are pooling results with only 2 studies (see eTable 1, http://links.lww.com/NXI/A766), we also performed sensitivity analyses applying Bayesian random effects meta-analysis with half-normal priors with a specified SD of 0.5; a previous analysis suggested these modeling parameters in the context of pooling 2 studies.50 We also performed sensitivity analyses to confirm consistent results were observed when applying Mendelian randomization (MR); this approach was previously applied in a study linking 25(OH)D and relapse hazard.28 To select instrumental variables (IVs) for 25(OH)D, we used the set of summary statistics for 25(OH)D used to create 25(OH)D PGS for our primary analyses and we aligned alleles to ensure directionality of effect estimates was maintained.20 We clumped 25(OH)D variants using a p-value threshold of 5e-8, distance of 10,000 kilobases, and LD of 0.001. For IVs not genotyped explicitly in the outcome cohort, we selected a proxy variant in LD (with R2 ≥ 0.9) with the variant of interest. Proxy variants were also included for palindromic IVs. For primary MR analyses, we applied multiplicative random effects inverse variance (IVW) to estimate the effects of 25(OH)D IVs on relapse risk and rate of new lesion development. We selected these outcomes as a relatively large body of observational evidence implicates a potential link between higher 25(OH)D and a lower rate of new lesion development and a previous study linked genetic liability to 25(OH)D and relapse risk. Secondary analyses calculated radial IVW with modified second order weights, MR-Egger, and weighted median-based effect estimates.51-53 Cochran Q test and I2 statistic were calculated to assess heterogeneity. We removed potential outlying variants identified using the modified weights from the radial IVW estimate. Pleiotropy was also assessed by testing whether the intercept obtained from the MR Egger analyses differed from 0. MR analyses were implemented using the TwoSampleMR package (v.0.5.6).53-55 Last, sensitivity analyses considered measured 25(OH)D levels available from the JHU cohort and their association with the rate of new lesion development (e.g., to confirm that the results do not merely reflect that the PGS explains an insufficient amount of variation in 25[OH]D levels). For this analysis, we regressed new lesions on the average 25(OH)D levels in the 5 years before the first MRI using similarly adjusted negative binomial regression models.
All statistical analyses were conducted using R programming (R version 4.1.0). Statistical significance was defined as p < 0.05.
Data Availability
Components of the IMID data set may be made accessible to qualified investigators with the appropriate ethical approvals and data use agreements upon request.
Results
Study Population and Baseline Characteristics
Table 1 describes demographic and clinical characteristics of included cohorts. Overall, 1924 participants were included in this study (from JHU: n = 1,152; from CombiRx: n = 575; from IMID: n = 197). The average age of the participants at first visit varied across cohorts (JHU: 44.1 years, SD [SD]:12.2; CombiRx: 40.9 [12.5], IMID: 50.4 [12.7]). The cohorts also varied about disease duration, with JHU and IMID participants having longer disease duration. Across all cohorts, participants were largely female (1,425; 74.1%) and had relapsing remitting MS (n = 1,670; 87%). Cohort-specific subpopulations contributing to individual outcomes of interest are included in eTable 1 (http://links.lww.com/NXI/A766).
Table 1.
Demographic Characteristics of Included Study Participants

25(OH)D PGS Scores and Circulating 25(OH)D Levels
Initial analyses assessed the association between continuous 25(OH)D PGS with circulating 25(OH)D levels in 935 participants in whom clinical values were available (n = 935). The subgroup of participants with measured 25(OH)D levels was generally similar to the overall JHU cohort regarding age, sex, BMI, and MS characteristics (eTable 2, http://links.lww.com/NXI/A766). 25(OH)D PGS was strongly associated with circulating 25(OH)D levels; each 1 SD increase in 25(OH)D PGS was associated with a 3.08% increase in circulating 25(OH)D level (95% CI: 1.77%, 4.42%; p = 4.33e-06; R2 = 2.24%) (Figure 1).
Figure 1. Decile of 25(OH)D PGS and Measured 25(OH)D Levels in 935 People With MS From the JHU Cohort.
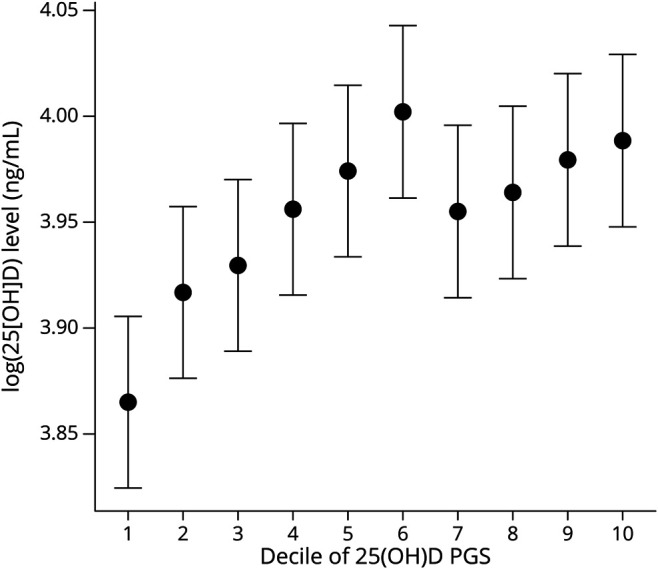
25(OH)D PGS are derived from summary statistics in which the primary model was adjusted for BMI. The association between circulating 25(OH)D levels and 25(OH)D PGS are adjusted for also BMI among people with MS. We excluded individuals with measured levels exceeding 70 ng/mL, as previous studies suggest this threshold may be the physiologic limit of 25(OH)D derived from nonsupplemental sources. Similar results were obtained when applying a threshold of 100 ng/mL (not shown). 25[OH]D = 25-hydroxyvitamin D; PGS = polygenic score.
25(OH)D PGS Scores and Clinical Outcomes
The pooled results for analyses examining the association between quartiles of 25(OH)D PGS and clinical outcomes are presented in Table 2. We did not observe an association between 25(OH)D PGS and any clinical outcomes (EDSS progression, relapse, MSFC), and the results were consistent for all models considered (eTable 3, http://links.lww.com/NXI/A766). In models considering continuous 25(OH)D PGS, we similarly did not observe any association between 25(OH)D PGS and outcomes in any cohort or overall (Figure 2). Although we did note some potential heterogeneity for analyses of EDSS progression, formal tests were not significant and could be related to relatively few progression events in JHU and IMID cohorts.
Table 2.
Pooled Results for 25(OH)D PGSa and Clinical Outcomes

Figure 2. Results for Continuous 25(OH)D PGS1 Clinical Outcomes for Individual Studies and the Pooled Estimate2 Across Studies.

125(OH)D PGS are derived from summary statistics in which the primary model was adjusted for BMI. 2Effect estimates displayed are for a 1 SD increase in 25(OH)D PGS. They are adjusted for age, 5 ancestry PCs, MS DMT, disease duration, and the number of relapses in previous 3 years. The pooled effect estimate is results from a random effects meta-analysis. (A) The results for rate of EDSS progression (heterogeneity I2 = 0.00%; p het=0.45). (B) The results for annualized percent change T25FW (I2 = 0.0%; p het=0.91). (C) The results for annualized percent change 9HPT (I2 = 0.0%; p het=0.67). 25[OH]D = 25-hydroxyvitamin D; 9HPT = nine-hole peg test; EDSS = expanded disability status scale; PGS = polygenic score; T25FW = timed 25-foot walk; DMT = disease-modifying therapies; JHU = Johns Hopkins; IMID = immune-mediated inflammatory diseases.
25(OH)D PGS Scores and Imaging Outcomes
Table 3 presents the pooled analysis assessing the association between imaging outcomes and quartiles of 25(OH)D PGS. Similar to clinical outcomes, we did not detect an association between 25(OH)D PGS and any MRI outcome. For example, individuals in the top quartile of 25(OH)D PGS did not have a slower rate of new lesion development relative to individuals in the bottom quartile in either multivariable model considered (model B: pooled rate ratio (RR): 0.98; 95% CI: 0.63, 1.51; model B: pooled RR: 0.99; 95% CI: 0.63, 1.58). The results were similar when we evaluated continuous measures of 25(OH)D PGS (Figure 3). Last, when inner retinal layer atrophy was evaluated as a surrogate of disease progression individuals with available OCT data, rates of retinal atrophy did not substantially differ across 25(OH)D PGS quartiles during pooled cohort analysis (Table 4).
Table 3.
Pooled Results for 25(OH)D PGSa and MRI Outcomes

Figure 3. Results for Continuous 25(OH)D PGS1 and MRI Outcomes for Individual Studies and the Pooled Estimate2 Across Studies.

125(OH)D PGS are derived from summary statistics in which the primary model was adjusted for BMI. 2Effect estimates displayed are for a 1 SD increase in 25(OH)D PGS. 2Effect estimates displayed are for a 1 SD increase in 25(OH)D PGS. They are adjusted for age, 5 ancestry PCs, MS DMT, disease duration, and the number of relapses in previous 3 years. The pooled effect estimate is results from a random effects meta-analysis. (A) The results for annualized percent change in BPF (Heterogeneity I2 = 66.0%; p het=0.09). (B) The results for annualized percent change in lesion volume (I2 = 0.0%; p het=0.78). (C) Relative rate for new lesions (I2 = 38.8%; p het=0.20). 25[OH]D = 25-hydroxyvitamin D; BPF = brain parenchymal fraction; DMT = disease-modifying therapies; JHU = Johns Hopkins; PGS = polygenic score.
Table 4.
Results for 25(OH)D PGS and Rate of Change in GCIPL (n = 1,105)

Sensitivity Analyses
The results were also similar when we used a 25(OH)D PGS that was not adjusted for BMI (eFigures 1 and 2, http://links.lww.com/NXI/A766) and when we considered 25(OH)D PGS derived using a p value threshold of 5e-05 to compute the PGS (instead of 5e-08; eFigures 3 and 4); no associations were observed between 25(OH)D PGS and clinical or imaging outcomes. The results were similar when we modeled EDSS progression using survival analysis (pooled n = 1,066; 287 progression events) or in stratified analyses by relapse type. We also observed no association between 25(OH)D and relapse risk or new lesion formation when applying MR (eTable 4; eFigures 5). The results were also similar when we applied Bayesian meta-analysis methods to pool results.
As a last step, and in accordance with current standards in the field, we assessed the association between serum 25(OH)D levels and radiologic disease activity as an extra layer of validation of the presented findings. Although there was a substantial correlation between circulating 25(OH)D levels and 25(OH)D PGS, we did not find an association between circulating 25(OH)D levels and rate of new lesions (n = 208 with MRI and 25[OH]D levels; per 1SD increase in circulating 25(OH)D: 1.04; 95% CI: 0.76, 1.44).
Discussion
In this large binational study, we observed a strong association between 25(OH)D PGS and circulating 25(OH)D levels, but 25(OH)D PGS did not relate to the clinical outcomes in MS including relapses, disability progression, or imaging outcomes including measures of change in brain or T2 lesion volume and development of new lesions. These results were consistent in analyses considering continuous 25(OH)D PGS, as well as in analyses using quartiles, and analyses where we derived Mendelian randomization estimators.
Having low circulating 25(OH)D levels is an established risk factor for developing MS, where strong observational evidence links low vitamin D intake or low 25(OH)D levels and subsequent risk of MS.6,7,56 Previous Mendelian randomization (MR) studies have also found a strong association between genetically determined 25(OH)D levels and risk of MS.24-26 For example, a two-sample MR study incorporating summary information from large-scale genome-wide association studies (GWAS) for MS and 25(OH)D levels implicated low 25(OH)D levels as a causal risk factor for MS. These results are similar to a follow-up analysis in which summary statistics from GWAS for 25(OH)D were derived from larger populations and the most up to date GWAS of MS.57
Several observational studies and small clinical trials have reported a link between low 25(OH)D levels and adverse disease outcomes in MS, as well as suggested a potential favorable effect of vitamin D supplementation on MS disease activity.9,11,13,14 Small experimental vitamin D supplementation studies have reported a beneficial role of supplementation on immunologic, radiologic, and clinical outcomes. However, the results from larger scale randomized clinical trials to date have been less clear, with some positive findings in subgroups or for secondary outcomes but not for the primary outcomes of relapse rate or no evidence of disease activity.9-12,14 Notably, each trial included varying supplementation dosages, different disease-modifying therapies as adjunctive therapies, and diverse primary outcomes, all of which could have contributed to differences in the downstream results and conclusions. As a result, equipoise persists as to whether vitamin D supplementation confers any benefit about MS outcomes including relapse risk and new lesion formation. Our results add to this body of evidence suggesting a lack of benefit of modulation of 25(OH)D levels for improving MS outcomes.
Limited studies have examined disease heterogeneity in MS using PGS. In a study by Vandebergh et al.,58 the “response to vitamin D” pathway was identified by Gene Ontology as affecting susceptibility to relapses. In a more recent study, 25(OH)D-related genetic variants were identified by GWAS (using similar studies as in the current report) and 2 sample Mendelian Randomization (MR) was applied to link 25(OH)D with relapse risk in a cohort of 506 PwMS. The authors reported a protective effect of 25(OH)D PGS regarding relapse hazard.28 In our study, an association between 25(OH)D PGS and clinical relapses was not observed, although we included a larger sample size and more diverse set of MS outcomes. This remained true when numerous other clinical and radiologic outcomes were considered as potentially more sensitive proxies of disease activity. Reasons for this discrepancy could be related to the different regions (Europe vs the US and Canada) for which the study population was derived. Although we restricted to individuals with European ancestry, its possible different distributions of underlying contributors to vitamin D status (e.g., obesity) could contribute. We also did not have relapse (the primary outcome in Vandebergh et al.58) in all cohorts, although relapse in our study was derived using a rigorously collected clinical trial cohort. Notably, we also did not apply an identical set of methods in that we considered 25(OH)D PGS instead of MR in our primary analysis; the PGS approach is expected to be more powerful (although has a higher probability of false positives). Here, we applied both techniques and observed a lack of association using both PGS and MR and included other notable outcomes such as the rate of new lesion development which have been noted in previous large observational studies. Brain and retinal imaging data were also included as surrogates for disability because they have been shown to be highly linked with clinical disease progression and long-term MS prognosis.59 We did not find an association between 25(OH)D PGS and any radiographic measures, including change in GCIPL. GCIPL thinning is a highly sensitive marker reflecting global CNS neurodegeneration and disease progression in MS, with the potential to more readily capture subtle changes than clinical outcomes.42,60 Thus, our findings were consistent across a spectrum of clinical and radiographic outcomes.
Our study has some important strengths. The data were obtained from geographically diverse populations with a broad range of clinical presentations and disease durations, enhancing our capacity to quantify the impact of 25(OH)D PGS and replicate our finding in 3 clinically distinct populations. The large total number of participants, long follow-up period, and detailed characterization of each group allowed us to assess the effect of vitamin D on disease heterogeneity after accounting for numerous known clinical and comorbid confounding factors. Nonetheless, the study does have some notable limitations. Namely, the availability of clinical and imaging data were variable across cohorts and outcomes, which could contribute to excess heterogeneity in pooled estimates, although it is worth noting that the I2 were generally low. Data on supplementation and use of medications for some of the cohorts were derived from the EMR and could be vulnerable to misclassification or inaccuracy. We also lacked circulating 25(OH)D levels across cohorts to verify 25(OH)D PGS were consistently associated with measured levels in all cohorts. Notably, the R2 from the model linking circulating 25(OH)D was lower than previous reports in other populations, which could have also contributed to the null results. It is possible underlying differences between the populations (e.g., about environmental or lifestyle factors) in the previous study and in our study may play a role. Genotyped participants were of predominantly European genetic ancestry; therefore, the observed results may not be applicable to more racially and ethnically diverse cohorts. We applied this restriction as similarly sized large multiethnic GWAS for 25(OH)D levels were not available. Horizontal pleiotropy, that is, that the PGS of interest may affect traits that influence the outcome other than the exposure (25[OH]D) of interest is also a potential concern. Although the results were consistent when we applied MR and in sensitivity tests assessing this potential bias, the potential for pleiotropy cannot be eliminated entirely. Furthermore, by design, the 25(OH)D PGS is a time-fixed exposure when 25(OH)D levels vary over the life-course. It is possible that there may be a specific time window in which modulation of 25(OH)D levels would be impactful; this limitation also applies to analyses using MR. Last, low 25(OH)D levels are associated with a higher risk of developing MS; thus, among patients with MS, the distribution of 25(OH)D or 25(OH)PRGS may be truncated, possibly limiting the potential to observe associations with outcomes.
Although 25(OH)D PGS were highly associated with circulating 25(OH)D levels, 25(OH)D was not associated with measures of clinical or radiographic disease progression in people with MS. This finding is largely in-line with larger randomized clinical trials on vitamin D supplementation and raises questions about the plausibility of a treatment effect of vitamin D in established MS. Future research is warranted to understand whether similar findings exist in more racially and ethnically representative populations.
Acknowledgment
This study was supported in part by the NIH (1K01MH121582-01 to K.C. Fitzgerald) and the National Multiple Sclerosis Society (TA-1805-31136 to K.C. Fitzgerald). The IMID study was funded by the Canadian Institutes of Health Research (THC-135234), Crohn's and Colitis Canada, Research Manitoba, and the Waugh Family Chair in Multiple Sclerosis (to R.A. Marrie), with genotyping supported by the CMSC, the University of Manitoba, and by the Department of Defense Congressionally Directed Medical Research Program, through the Multiple Sclerosis Research Program under Award No. W81XWH2010566 (PI: Kowalec). C.N. Bernstein is supported in part by the Bingham Chair in Gastroenterology. K. Kowalec is supported by the NIMH (MH123724) and the University of Manitoba. The authors acknowledge Shared Health and Health Sciences Centre Winnipeg. The authors acknowledge the use of the Genome QC/Genome QC Innovation Centre/Genome Canada for genotyping facilities for the IMID study.
Glossary
- 25[OH]D
25-hydroxyvitamin D
- BMI
body mass index
- BPF
brain parenchymal fraction
- DMT
disease modifying therapies
- EDSS
Expanded disability status scale
- EMR
electronic medical record
- FLAIR
fluid-attenuated inversion recovery
- GA
glatiramer acetate
- GCIPL
ganglion cell inner plexiform layer
- GMF
gray matter fraction
- GWAS
genome-wide association studies
- IMID
immune-mediated inflammatory diseases
- INFB
interferon beta-1a
- JHU
Johns Hopkins
- LD
linkage disequilibrium
- MR
Mendelian randomization
- MSFC
MS Functional Composite
- OCT
Optical coherence tomography
- PC
principal components
- PGS
polygenic score
- TE
echo time
- TI
inversion time
- WMF
white matter fraction
Appendix. Authors
Study Funding
1K01MH121582-01; TA-1805-31,136; THC-135234; W81XWH2010566; MH123724.
Disclosure
Dr. Vasileiou reports no disclosures; Ms. Hu reports no disclosures; Dr. Bernstein reports no disclosures; Dr. Wolinsky received compensation for consulting, scientific advisory boards, or other activities with Avotres, Brainstorm Cell Therapeutics, Cleveland Clinic Foundation, EMD Serono, Inmagene, Novartis/Sandoz, Roche/Genentech, Sanofi Genzyme, University of Alabama. Royalties are received for out licensed monoclonal antibodies through UTHealth to Millipore (Chemicon International) Corporation; Dr. Lublin reports receiving reseach funding from Novartis, Actelion, Biogen, Sanofi, Brainstorm Cell Therapeutics, and has served as a consultant or on an advisory board or DSMB for Biogen, EMD Serono, Novartis, Teva, Actelion/Janssen, Sanofi/Genzyme, Acorda, Roche/Genentech, MedImmune/Viela Bio/Horizon Therapeutics, Receptos/Celgene/BMS, TG Therapeutics, Medday, Atara Biotherapeutics, Mapi Pharma, Apitope, Orion Biotechnology, Brainstorm Cell Therapeutics, Jazz Pharmaceuticals, GW Pharma, Mylan, Immunic, Population Council, Avotres, Neurogene, Banner Life Sciences, Labcorp, Entelexo Biotherapeutics, Neuralight, SetPoint Medical. He also has stock options Avotres and Neuralight, and has received non-promotional speaking fees from Sanofi; Dr. Cutter reports serving on data and safety monitoring boards for AstraZeneca, Avexis, Biolinerx, Brainstorm Cell Therapeutics, Bristol-Myers Squibb–Celgene, CSL Behring, Galmed, GreenValley Pharma, Mapi, Merck, Merck–Pfizer, Opko Biologics, OncoImmune, Neurim, Novartis, Orphazyme, Sanofi, Reata, Teva Pharmaceuticals, VielaBio, Vivus, the National Heart, Lung, and Blood Institute (Protocol Review Committee), the Eunice Kennedy Shriver National Institute of Child Health and Human Development (Obstetric Pharmacology Research Unit oversight committee); consulting or advisory boards for Biodelivery Sciences International, Biogen, Click Therapeutics, Genzyme, Genentech, GW Pharmaceuticals, Immunic, Klein-Buendel, MedDay, MedImmune, Neurogenesis, Novartis, Osmotica, Perception Neurosciences, Recursion–Cerexis, Rekover, Roche, and TG Therapeutics; is employed by the University of Alabama at Birmingham, Birmingham, AL, USA; and is president of Pythagoras, a private consulting company located in Birmingham, AL, USA; Dr. Sotirchos has served on scientific advisory boards for Horizon Therapeutics, Viela Bio, Alexion and Genentech, and has received speaker fees from Alexion, Viela Bio and Biogen; Dr. Salter reports no disclosures; Dr. Kowalec reports no disclosures; Dr. Saidha has received consulting fees from Medical Logix for the development of CME programs in neurology, and has served on scientific advisory boards for Biogen, Genentech Corporation, TG therapeutics & Bristol Myers Squibb, and he has received consulting fees from Carl Zeiss Meditec and Novartis, and he is the PI of investigator-initiated studies funded by Genentech Corporation and Biogen, and he has previously received support from the Race to Erase MS foundation, and has received equity compensation for consulting from JuneBrain LLC, a retinal imaging device developer, and he was also the site investigator of a trial sponsored by MedDay Pharmaceuticals; Dr. Mowry reports research support from Biogen, Teva and Genentech, and royalties for editorial duties from UpToDate; Dr. Calabresi has received consulting fees from Disarm, Nervgen, and Biogen, and is PI on grants to JHU from Genentech, Principia, Biogen and Annexon; Dr. Marrie reports no disclosures; Dr. Fitzgerald reports no disclosures.
References
- 1.Reich DS, Lucchinetti CF, Calabresi PA. Multiple sclerosis. N Engl J Med. 2018;378(2):169-180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ludwin SK. The pathogenesis of multiple sclerosis: relating human pathology to experimental studies. J Neuropathol Exp Neurol. 2006;65(4):305-318. [DOI] [PubMed] [Google Scholar]
- 3.Lublin FD, Reingold SC, Cohen JA, et al. . Defining the clinical course of multiple sclerosis: the 2013 revisions. Neurology. 2014;83(3):278-286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Pinto MF, Oliveira H, Batista S, et al. . Prediction of disease progression and outcomes in multiple sclerosis with machine learning. Sci Rep. 2020;10(1):21038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Vickaryous N, Jitlal M, Jacobs BM, et al. . Remote testing of vitamin D levels across the UK MS population—a case control study. Plos One. 2020;15(12):e0241459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Munger KL, Levin LI, Hollis BW, Howard NS, Ascherio A. Serum 25-hydroxyvitamin D levels and risk of multiple sclerosis. JAMA. 2006;296(23):2832-2838. [DOI] [PubMed] [Google Scholar]
- 7.Munger KL, Zhang SM, O'Reilly E, et al. . Vitamin D intake and incidence of multiple sclerosis. Neurology. 2004;62(1):60-65. [DOI] [PubMed] [Google Scholar]
- 8.Snellman G, Melhus H, Gedeborg R, et al. . Seasonal genetic influence on serum 25-hydroxyvitamin D levels: a twin study. Plos One. 2009;4(11):e7747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sotirchos ES, Bhargava P, Eckstein C, et al. . Safety and immunologic effects of high- vs low-dose cholecalciferol in multiple sclerosis. Neurology. 2016;86(4):382-390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Simpson S, Taylor B, Blizzard L, et al. . Higher 25-hydroxyvitamin D is associated with lower relapse risk in multiple sclerosis. Ann Neurol. 2010;68(2):193-203. [DOI] [PubMed] [Google Scholar]
- 11.Mowry EM, Krupp LB, Milazzo M, et al. . Vitamin D status is associated with relapse rate in pediatric-onset multiple sclerosis. Ann Neurol. 2010;67(5):618-624. [DOI] [PubMed] [Google Scholar]
- 12.Mowry EM, Pelletier D, Gao Z, Howell MD, Zamvil SS, Waubant E. Vitamin D in clinically isolated syndrome: evidence for possible neuroprotection. Eur J Neurol. 2016;23(2):327-332. [DOI] [PubMed] [Google Scholar]
- 13.Mowry EM, Waubant E, McCulloch CE, et al. . Vitamin D status predicts new brain magnetic resonance imaging activity in multiple sclerosis. Ann Neurol. 2012;72(2):234-240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Fitzgerald KC, Munger KL, Köchert K, et al. . Association of vitamin D levels with multiple sclerosis activity and progression in patients receiving interferon beta-1b. JAMA Neurol. 2015;72(12):1458-1465. [DOI] [PubMed] [Google Scholar]
- 15.Camu W, Lehert P, Pierrot-Deseilligny C, et al. . Cholecalciferol in relapsing-remitting MS: a randomized clinical trial (CHOLINE). Neurol Neuroimmunol Neuroinflamm. 2019;6(5):e597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hupperts R, Smolders J, Vieth R, et al. . Randomized trial of daily high-dose vitamin D3 in patients with RRMS receiving subcutaneous interferon β-1a. Neurology. 2019;93(20):e1906-e1916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kampman MT, Steffensen LH, Mellgren SI, Jørgensen L. Effect of vitamin D3 supplementation on relapses, disease progression, and measures of function in persons with multiple sclerosis: exploratory outcomes from a double-blind randomised controlled trial. Mult Scler J. 2012;18(8):1144-1151. [DOI] [PubMed] [Google Scholar]
- 18.Stein MS, Liu Y, Gray OM, et al. . A randomized trial of high-dose vitamin D2 in relapsing-remitting multiple sclerosis. Neurology. 2011;77(17):1611-1618. [DOI] [PubMed] [Google Scholar]
- 19.Stridh P, Kockum I, Huang J. Seasonal variability of serum 25-hydroxyvitamin D on multiple sclerosis onset. Sci Rep. 2021;11(1):20989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Revez JA, Lin T, Qiao Z, et al. . Genome-wide association study identifies 143 loci associated with 25 hydroxyvitamin D concentration. Nat Commun. 2020;11(1):1647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Choi SW, Mak TSH, O'Reilly PF. Tutorial: a guide to performing polygenic risk score analyses. Nat Protoc. 2020;15(9):2759-2772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hunter D, De Lange M, Snieder H, et al. . Genetic contribution to bone metabolism, calcium excretion, and vitamin D and parathyroid hormone regulation. J Bone Mineral Res. 2001;16(2):371-378. [DOI] [PubMed] [Google Scholar]
- 23.Mimpen M, Rolf L, Poelmans G, et al. . Vitamin D related genetic polymorphisms affect serological response to high-dose vitamin D supplementation in multiple sclerosis. Plos One. 2021;16(12):e0261097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Mokry LE, Ross S, Ahmad OS, et al. . Vitamin D and risk of multiple sclerosis: a mendelian randomization study. Plos Med. 2015;12(8):e1001866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gianfrancesco MA, Stridh P, Rhead B, et al. . Evidence for a causal relationship between low vitamin D, high BMI, and pediatric-onset MS. Neurology. 2017;88(17):1623-1629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Rhead B, Bäärnhielm M, Gianfrancesco M, et al. . Mendelian randomization shows a causal effect of low vitamin D on multiple sclerosis risk. Neurol Genet. 2016;2(5):e97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lin R, Taylor BV, Simpson S, et al. . Association between multiple sclerosis risk-associated SNPs and relapse and disability—a prospective cohort study. Mult Scler J. 2014;20(3):313-321. [DOI] [PubMed] [Google Scholar]
- 28.Vandebergh M, Dubois B, Goris A. Effects of vitamin D and body mass index on disease risk and relapse hazard in multiple sclerosis: a mendelian randomization study. Neurol Neuroimmunol Neuroinflamm. 2022;9(3):e1165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lublin FD, Cofield SS, Cutter GR, et al. . Randomized study combining interferon and glatiramer acetate in multiple sclerosis. Ann Neurol. 2013;73(3):327-340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Polman CH, Reingold SC, Edan G, et al. . Diagnostic criteria for multiple sclerosis: 2005 revisions to the “McDonald Criteria”. Ann Neurol. 2005;58(6):840-846. [DOI] [PubMed] [Google Scholar]
- 31.JMIR research protocols—effects of psychiatric comorbidity in immune-mediated inflammatory disease: protocol for a prospective study. Accessed April 22, 2022. researchprotocols.org/2018/1/e15/. [DOI] [PMC free article] [PubMed]
- 32.Das S, Forer L, Schönherr S, et al. . Next-generation genotype imputation service and methods. Nat Genet. 2016;48(10):1284-1287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Jiang X, O'Reilly PF, Aschard H, et al. . Genome-wide association study in 79, 366 European-ancestry individuals informs the genetic architecture of 25-hydroxyvitamin D levels. Nat Commun. 2018;9(1):260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Fritsche LG, Gruber SB, Wu Z, et al. . Association of polygenic risk scores for multiple cancers in a phenome-wide study: results from the Michigan genomics initiative. Am J Hum Genet. 2018;102(6):1048-1061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Richardson TG, Harrison S, Hemani G, Davey Smith G. An atlas of polygenic risk score associations to highlight putative causal relationships across the human phenome. eLife. 2019;8:e43657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lindsey J, Scott T, Lynch S, et al. . The CombiRx trial of combined therapy with interferon and glatiramer acetate in relapsing remitting MS: design and baseline characteristics. Mult Scler Relat Disord. 2012;1(2):81-86. [DOI] [PubMed] [Google Scholar]
- 37.Mowry EM, Bermel RA, Williams JR, et al. . Harnessing real-world data to inform decision-making: multiple sclerosis Partners Advancing Technology and Health Solutions (MS PATHS). Front Neurol. 2020;11:632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Roche A, Forbes F; Alzheimer's Disease Neuroimaging Initiative. Partial volume estimation in brain MRI revisited. Med Image Comput Assist Interv. 2014;17(pt 1):771-778. [DOI] [PubMed] [Google Scholar]
- 39.Tewarie P, Balk L, Costello F, et al. . The OSCAR-IB consensus criteria for retinal OCT quality assessment. Plos One. 2012;7(4):e34823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Syc SB, Saidha S, Newsome SD, et al. . Optical coherence tomography segmentation reveals ganglion cell layer pathology after optic neuritis. Brain. 2012;135(2):521-533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Lang A, Carass A, Al-Louzi O, et al. . Combined registration and motion correction of longitudinal retinal OCT data. Proc SPIE. 2016;9784:97840X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Saidha S, Al-Louzi O, Ratchford JN, et al. . Optical coherence tomography reflects brain atrophy in multiple sclerosis: a four-year study. Ann Neurol. 2015;78(5):801-813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Saidha S, Sotirchos ES, Oh J, et al. . Relationships between retinal axonal and neuronal measures and global central nervous system pathology in multiple sclerosis. JAMA Neurol. 2013;70(1):34-43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Martinez-Lapiscina EH, Arnow S, Wilson JA, et al. . Retinal thickness measured with optical coherence tomography and risk of disability worsening in multiple sclerosis: a cohort study. Lancet Neurol. 2016;15(6):574-584. [DOI] [PubMed] [Google Scholar]
- 45.Cruz-Herranz A, Balk LJ, Oberwahrenbrock T, et al. . The APOSTEL recommendations for reporting quantitative optical coherence tomography studies. Neurology. 2016;86(24):2303-2309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Bhargava P, Steele SU, Waubant E, et al. . Multiple sclerosis patients have a diminished serologic response to vitamin D supplementation compared to healthy controls. Mult Scler J. 2016;22(6):753-760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Hollis BW. Circulating 25-hydroxyvitamin D levels indicative of vitamin D sufficiency: implications for establishing a new effective dietary intake recommendation for vitamin D. J Nutr. 2005;135(2):317-322. [DOI] [PubMed] [Google Scholar]
- 48.Kalincik T, Cutter G, Spelman T, et al. . Defining reliable disability outcomes in multiple sclerosis. Brain. 2015;138(11):3287-3298. [DOI] [PubMed] [Google Scholar]
- 49.Fitzgerald KC, O'Reilly ÉJ, Falcone GJ, et al. . Dietary ω-3 polyunsaturated fatty acid intake and risk for amyotrophic lateral sclerosis. JAMA Neurol. 2014;71(9):1102-1110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Friede T, Röver C, Wandel S, Neuenschwander B. Meta-analysis of two studies in the presence of heterogeneity with applications in rare diseases. Biometrical J. 2017;59(4):658-671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Bowden J, Davey Smith G, Haycock PC, Burgess S. Consistent estimation in mendelian randomization with some invalid instruments using a weighted median estimator. Genet Epidemiol. 2016;40(4):304-314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Bowden J, Del Greco M F, Minelli C, et al. . Improving the accuracy of two-sample summary-data Mendelian randomization: moving beyond the NOME assumption. Int J Epidemiol. 2019;48(3):728-742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Bowden J, Davey Smith G, Burgess S. Mendelian randomization with invalid instruments: effect estimation and bias detection through Egger regression. Int J Epidemiol. 2015;44(2):512-525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Burgess S, Thompson SG. Interpreting findings from Mendelian randomization using the MR-Egger method. Eur J Epidemiol. 2017;32(5):377-389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Hemani G, Zheng J, Elsworth B, et al. . The MR-Base platform supports systematic causal inference across the human phenome. eLife. 2018;7:e34408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Cortese M, Riise T, Bjørnevik K, et al. . Timing of use of cod liver oil, a vitamin D source, and multiple sclerosis risk: the EnvIMS study. Mult Scler J. 2015;21(14):1856-1864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.International Multiple Sclerosis Genetics Consortium. Multiple sclerosis genomic map implicates peripheral immune cells and microglia in susceptibility. Science. 2019;365(6460):eaav7188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Vandebergh M, Andlauer TFM, Zhou Y, et al. . Genetic variation in WNT9B increases relapse hazard in multiple sclerosis. Ann Neurol. 2021;89(5):884-894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Fisniku LK, Chard DT, Jackson JS, et al. . Gray matter atrophy is related to long-term disability in multiple sclerosis. Ann Neurol. 2008;64(3):247-254. [DOI] [PubMed] [Google Scholar]
- 60.Rothman A, Murphy OC, Fitzgerald KC, et al. . Retinal measurements predict 10-year disability in multiple sclerosis. Ann Clin Transl Neurol. 2019;6(2):222-232. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Components of the IMID data set may be made accessible to qualified investigators with the appropriate ethical approvals and data use agreements upon request.