

Abstract
Background
Distal arthrogryposis (DA) is a group of congenital autosomal‐dominant disorders secondary to defects in joint and muscle function, characterized by multiple joint contractures of the hands and feet. DA can be divided into 10 types according to clinical features. DA has been confirmed to be caused by mutations in genes encoding components of the contractile apparatus of skeletal muscle fibers, such as troponin I2 (TNNI2).
Methods
In this study, we report a three‐generation DA family belonging to the DA2B type. The clinical characteristics of affected members are genetically stable and consistent, with severe deformities in hands and feet, and two affected adults had short stature. None exhibited facial abnormalities. Blood from three affected and three healthy members were collected for whole‐exome sequencing and Sanger sequencing.
Results
A missense variant in TNNI2 (NM_003282.4: c.525G>T: p.K175N) was successfully identified, which resulted in the substitution of amino acid at position 175 of TNNI2 from lysine to asparagines.
Conclusion
The variant c.525G>T in TNNI2 explains the cause of DA in the family. This variant was identified in Chinese people for the first time, and the same variant had been reported in another study but no description of clinical symptoms. Our study comprehensively characterized the c.525G>T variant in TNNI2.
Keywords: distal arthrogryposis, TNNI2, variant, whole‐exome sequencing
A three‐generation Chinese family with Distal arthrogryposis type 2B show short stature and severe deformity of the hands and feet, but without the typical facial abnormality phenotype of DA2B. We identified a missense variant in TNNI2 c.525G>T(p.K175N) in this family.
1. INTRODUCTION
Distal arthrogryposis (DA) is a group of clinically and genetically heterogeneous disorders characterized by multiple joint contracture deformities in the distal extremities. Patients with DA typically present with two or more of the following deformities: camptodactyly, vertical talus, ulnar deviation, clubfoot, overlapping fingers or toes, congenital flexion of the wrist, elbow, and knee. In addition, some patients may be accompanied by abnormalities in the face, height, spine, hip, and skin texture, such as the triangular face, small mouth, arched palate, scoliosis, and short stature (Bamshad et al., 1996, 2009).
DA can be classified into 10 different types (DA1, DA2A, DA2B, DA3, DA4, DA5, DA7, DA8, DA9, and DA10) based on clinical characteristics, with some overlapping features among them (Bamshad et al., 1996, 2009). DA1 (OMIM 108120) and DA2B (OMIM 601680) are the two most common types of DA. The hallmarks of DA1 include limb deformities, such as camptodactyly and clubfoot, with no other abnormalities. DA2B is also known as Sheldon‐Hall syndrome (SHS), which has more severe clinical characteristics than DA1, with deeper involvement of extremities, as well as facial deformities such as the triangular face, small mouth, prominent nasolabial folds, downslanting palpebral fissures, and prominent chin, and some patients are accompanied by scoliosis (Krakowiak et al., 1998). Some researchers have suggested that DA1 and DA2B may be considered phenotypic extremes of the same disorder because they share similar phenotypes, and the same mutation caused DA1 in some families, but DA2B in others (Beck et al., 2013).
Mutations in TNNI2 can cause DA1 or DA2B (Jiang et al., 2006; Shrimpton & Hoo, 2006; Wang et al., 2016). Located at 11p15.5 in the human genome, TNNI2 encodes the fast‐twitch skeletal muscle isoform of troponin I (TnI), which is specifically expressed in fast‐twitch skeletal muscle fibers (Barton et al., 1997). As an inhibitory subunit, TnI forms a ternary troponin complex (Tn) with two other subunits, a calcium‐binding subunit: troponin C (TnC) and a tropomyosin (TM)‐binding subunit: troponin T (TnT). Tn is required for the regulation of calcium‐dependent fast‐twitch muscle skeletal fibers contraction and relaxation (Ochala, 2008), and abnormalities in any of its subunits may disturb muscle function.
In our study, we identified a three‐generation Chinese family with DA2B. The proband was a 2‐month‐old female infant whose mother and grandfather were symptomatic. To screen the pathogenic variants, we performed whole‐exome sequencing and Sanger sequencing and found a missense variant in the TNNI2 gene (c.525G>T: p.K175N). To our knowledge, this is the first report confirming that this variant occurred in Chinese people with a detailed clinical description.
2. MATERIALS AND METHODS
2.1. Genomic DNA preparation and whole‐exome sequencing
A total of six peripheral blood samples from patients and their relatives were collected for genomic DNA extraction, including three affected individuals (I‐1, II‐4, and III‐4) and three unaffected individuals (II‐1, II‐3, and III‐1). Peripheral blood was collected with an EDTA anticoagulation vacuum blood collection tube, and the peripheral blood was stored in a refrigerator at −80°C. According to the protocol, genomic DNA was extracted from peripheral blood samples using a DNA extraction kit (GBCBIO, D1105).
For whole‐exome sequencing, genomic DNA samples were sonicated into DNA fragments of 150–300 bp. After purification of the DNA fragments, adaptor ligation and end repair were performed on the exonic DNA fragments. The DNA library was constructed according to standard Illumina protocols. After the library was qualified, it was sequenced, and the raw data was collected using Illumina Base Calling software. Variants with a frequency >1% were excluded, nonexonic and spliced region variants were excluded, synonymous variants were excluded, and the remaining variants were subjected to pathogenicity prediction using SIFT, Polyphen2, and Mutation Taster software.
2.2. Variant verification
A variant in TNNI2 was confirmed by Sanger sequencing. Genomic DNA sequences were amplified according to the Takara rTaq protocol. PCR products were sequenced by Shanghai Sangon Biotechnology (Shanghai, China). Primers (Forward: GGAGGACATGAACCAGAAGCTATT; Reverse: TCAAATCCTTTATTGACGGTGGTG). TNNI2 reference sequence: NM_003282.4.
2.3. Construction of protein structure
The amino acid sequence of Homo sapiens TNNI2 protein was obtained from UniProtKB Database (TNNI2_HUMANP48788). AlphaFold Protein Structure Database website (https://alphafold.ebi.ac.uk/) was used to predict the 3D structure of protein and generate a PDB file. Pymol, Swiss PDB‐viewer software, and Missense3d website (http://missense3d.bc.ic.ac.uk/~missense3d/) were used to evaluate the structural changes of the mutated amino acid.
3. RESULTS
3.1. Clinical report
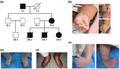
In a three‐generation Chinese family, five members suffered from DA (Figure 1a). The proband was a 2‐month‐old female Chinese infant (III‐4). She was born with obvious symptoms on the distal limb including overlapping fingers (Figure 1b), congenital vertical talus on the right foot, and clubfoot on the left foot (Figure 1e). Similar clinical features were also found in her mother (II‐4) and grandfather (I‐1). Mother and grandfather appeared short stature. Their hands showed severe ulnar deviation and camptodactyly. Their feet showed congenital vertical talus characterized by nonweight‐bearing heel, elongated and convex medial longitudinal column, and elevated and clawed lateral toes (Figure 1c,d). The mother had two boys (III‐2 and III‐3) previously who were also found with phenotypes of DA. None of the affected individuals exhibited any additional facial abnormalities. We concluded that this family was affected by DA2B.
FIGURE 1.

(a) Pedigree tree of a three‐generation Chinese family with DA2B. The III‐4 is the proband, a 2‐month‐old female infant. (b) The right hand of proband shows overlapping fingers. (c,d) The feet of proband's mother (II‐4) and grandfather (I‐1) shows severe ulnar deviation, camptodactyly, vertical talus and clawed lateral toes. (e) The left foot of proband showing clubfoot
3.2. Variant analysis
To identify the genetic factors responsible for DA in this family, we collected genomic DNA samples from six family members and performed whole‐exome sequencing. A total of 340,062 genetic variants were initially detected. After filtering out noncompliant variants according to the pipeline (Figure 2a), 49 variants were retained. We then examined whether the screened variants matched the phenotypes of DA. A missense heterozygous variant (c.525G>T; p.K175N) in TNNI2 was the only eligible variant. In addition, according to the criteria of the American College of Medical Genetics and Genomic (ACMG), this TNNI2 variant was classified as a “Likely Pathogenic”. Finally, this variant was verified by Sanger sequencing in three affected individuals (Figure 2b).
FIGURE 2.
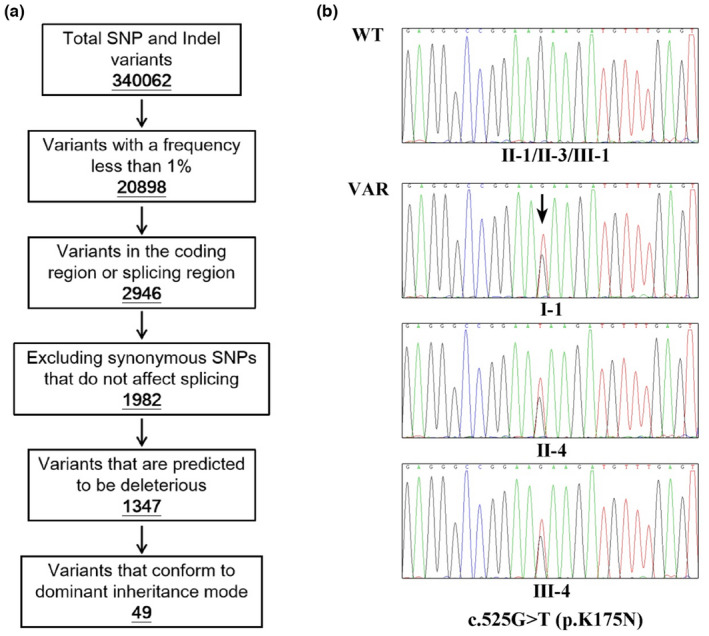
(a) Schematic representation of the filtering process of WES data. (b) Validation of genomic DNAs in the family. A heterozygous variant of TNNI2 (c.525G>T) was identified in the proband (III‐4), proband's mother (II‐4), and grandfather (I‐1) (Black arrows indicate variant sites)
The c.525G>T variant in TNNI2 results in the substitution of amino acid at position 175 of TNNI2 from lysine to asparagines, the basic amino acid (lysine) changing to the acidic amino acid (asparagines). To examine the influence of p.K175N on TNNI2 protein, we generated a PDB file of TNNI2 protein (Figure 3a) and analyzed the protein structure using Pymol, Swiss PDB‐viewer software, and the Missense3d website. The results showed that the lysine at position 175 would form hydrogen bonds with the adjacent amino acids at positions 171, 172, 178, and 179 (Figure 3b). And the mutated asparagines at position 175 would not change the connection and distance of these hydrogen bonds (Figure 3c). However, the p.K175N substitution would disrupt the salt bridge detected at the wild‐type amino acid between NZ atom of lysine at position 175 and OE1 atom of Glutamate at position 179 (distance: 4.330 Å) (data not shown).
FIGURE 3.
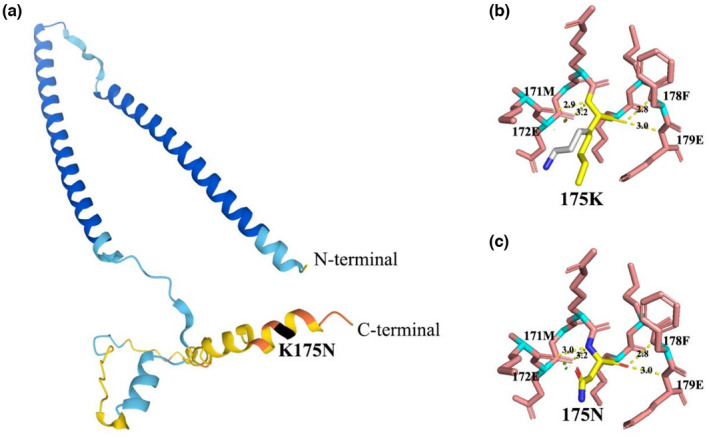
(a) 3D structure of TNNI2 protein. The black region is the position of the mutated amino acid. (b,c) Lysine and mutated asparagines at position 175 would form hydrogen bonds with the adjacent amino acids at positions 171, 172, 178, and 179 (hydrogen bonds are shown by a yellow dashed line and numbers indicate hydrogen bond distances)
4. DISCUSSION
DA is an autosomal‐dominant disorder with genetic and phenotypic heterogeneity that primarily affects the distal extremities. DA1 and DA2B are two DA types with similar clinical phenotypes. DA1 is characterized by mild deformities of the hands or feet. DA2B also is featured with limb deformity features, but the involvement is much deeper. The major subtyping features of DA2B are facial abnormalities, oropharyngeal abnormalities, spinal deformities, and short stature, with facial abnormalities being the most common. In this study, we recruited a three‐generation DA family. All affected individuals exhibited severe deformities of the hands and feet, but none exhibited facial abnormalities. At first, we thought they could be identified as DA1. But considering that both the mother and the grandfather were accompanied by short stature and severe limb deformities, they were eventually diagnosed as DA2B.
Through whole‐exome sequencing and screening, a heterozygous missense variant (c.525G>T: p.K175N) in TNNI2, a causative gene of DA, was identified to cosegregated with DA phenotypes in the family. The variant was classified as “Likely pathogenic” according to ACMG criteria (Richards et al., 2015). The pathogenic evidence included one strong pathogenic evidence (PS1), one moderate pathogenic evidence (PM2), and three supporting pathogenic evidence (PP1, PP3, and PP5).
Multiple pathogenic variants in the TNNI2 had been widely reported. And the same variant was identified in 2013 in a Western patient who was also diagnosed with DA2B, but no case report and detailed clinical description were available (Beck et al., 2013). Thus, we believed c.525G>T variant in TNNI2 is responsible for DA in the Chinese family. To our knowledge, this study for the first time comprehensively described the clinical features caused by this variant which was identified for the first time in Chinese people.
TNNI2 encodes the fast skeletal muscle isoform of TnI containing 182 amino acids. TnI, a subunit of the heteromeric protein complex Tn, plays an inhibition role in controlling calcium‐dependent skeletal muscle contraction. In the presence of low Ca2+ concentration, TnI binds to actin, which inhibits the interaction between actin and myosin, and actomyosin ATPase activity, consequently muscle keeping relaxation state. When the concentration of Ca2+ increases and exceeds the threshold, TnC binds to Ca2+ and induces a conformational change of TnI, causing TnI to dissociate from actin, then myofilaments sliding relatively, thereby muscle contraction.
In previous reports of DA caused by TNNI2 variants, all pathogenic variants were located at the C‐terminus of TNNI2 (Table S1). The C‐terminus of TNNI2 contains the binding domain of TnI to actin and TnC, which are required for the inhibitory function of TnI (Ramos, 1999) and are highly conserved in the amino acid sequences among the three TnI isoforms (Zhang et al., 2011). In this study, c.525G>Tvariant in TNNI2 resulted in the replacement of amino acid at position 175 of TnI from lysine to asparagines, which also located in the actin‐ and TnC‐binding domain. Through structural bioinformatics analysis, we found that the amino acid substitution would not destroy the hydrogen bonds with adjacent amino acids and its own protein structure, but disrupt the salt bridge between an atom of amino acids at positions 175 and 179. We suggested that the variant may alter the surface properties of the protein and impair its molecular function, such as affecting the molecular interaction of TnI with actin and TnC, affecting the sensitivity of Tn to calcium ions (Robinson et al., 2007), consequently affecting the muscle contractile function. The specific mechanism is expected to be further explored.
AUTHOR CONTRIBUTIONS
Mingwei Zhu, Yue Li, Tianying Nong, Jingchun Li, and Hui Lv conceived and designed the experiments. Jingchun Li and Hongwen Xu performed clinical assessment and data collection of patients. Tianying Nong, Yiqiang Li, Xia Li, and Zhaohui Li performed experiments and analyzed data. Yue Li drafted the manuscript. Tianying Nong, Mingwei Zhu, and Jingchun Li prepared the manuscript. All authors read and approved the final manuscript.
CONFLICT OF INTEREST
The authors declare that they have no conflict of interest.
ETHICS STATEMENT
All studies were approved by the Human Ethics Committee of Guangzhou Women and Children's Medical Center and Guangzhou Medical University. Written informed consent for peripheral blood of all participants was obtained from each individual or their legal guardians. All methods were carried out in accordance with the approved guidelines and regulations.
Supporting information
TABLE S1
ACKNOWLEDGMENTS
We thank the patients and their family members for participation in this study. We would like to thank Qian Gong from Guangzhou Women and Children's Medical Center for collecting the blood samples. This work was supported by the National Natural Science Foundation of China (grant no. 81972038) and an investigator‐initiated starter project grant from Guangzhou Women and Children's Medical Center to M.Z.
Li, Y. , Nong, T. , Li, Y. , Li, X. , Li, Z. , Lv, H. , Xu, H. , Li, J. , & Zhu, M. (2022). A TNNI2 variant c.525G>T causes distal arthrogryposis in a Chinese family. Molecular Genetics & Genomic Medicine, 10, e2042. 10.1002/mgg3.2042
Yue Li and Tianying Nong contributed equally to this work.
Contributor Information
Jingchun Li, Email: haiqingkun@126.com.
Mingwei Zhu, Email: mweizh@gzhmu.edu.cn.
DATA AVAILABILITY STATEMENT
The data that support the findings of this study are availablefrom the corresponding author upon reasonable request.
REFERENCES
- Bamshad, M. , Jorde, L. B. , & Carey, J. C. (1996). A revised and extended classification of the distal arthrogryposes. American Journal of Medical Genetics, 65(4), 277–281. [DOI] [PubMed] [Google Scholar]
- Bamshad, M. , Van Heest, A. E. , & Pleasure, D. (2009). Arthrogryposis: A review and update. The Journal of Bone and Joint Surgery. American Volume, 91(Suppl. 4), 40–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barton, P. J. , Townsend, P. J. , Brand, N. J. , & Yacoub, M. H. (1997). Localization of the fast skeletal muscle troponin I gene (TNNI2) to 11p15.5: Genes for troponin I and T are organized in pairs. Annals of Human Genetics, 61(Pt 6), 519–523. [DOI] [PubMed] [Google Scholar]
- Beck, A. E. , McMillin, M. J. , Gildersleeve, H. I. , Kezele, P. R. , Shively, K. M. , Carey, J. C. , Regnier, M. , & Bamshad, M. J. (2013). Spectrum of mutations that cause distal arthrogryposis types 1 and 2B. American Journal of Medical Genetics: Part A, 161A(3), 550–555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang, M. , Zhao, X. , Han, W. , Bian, C. , Li, X. , Wang, G. , Ao, Y. , Li, Y. , Yi, D. , Zhe, Y. , Lo, W. H. , Zhang, X. , & Li, J. (2006). A novel deletion in TNNI2 causes distal arthrogryposis in a large Chinese family with marked variability of expression. Human Genetics, 120(2), 238–242. [DOI] [PubMed] [Google Scholar]
- Krakowiak, P. A. , Bohnsack, J. F. , Carey, J. C. , & Bamshad, M. (1998). Clinical analysis of a variant of Freeman‐Sheldon syndrome (DA2B). American Journal of Medical Genetics, 76(1), 93–98. [DOI] [PubMed] [Google Scholar]
- Ochala, J. (2008). Thin filament proteins mutations associated with skeletal myopathies: Defective regulation of muscle contraction. Journal of Molecular Medicine (Berl), 86(11), 1197–1204. [DOI] [PubMed] [Google Scholar]
- Ramos, C. H. (1999). Mapping subdomains in the C‐terminal region of troponin I involved in its binding to troponin C and to thin filament. The Journal of Biological Chemistry, 274(26), 18189–18195. [DOI] [PubMed] [Google Scholar]
- Richards, S. , Aziz, N. , Bale, S. , Bick, D. , Das, S. , Gastier‐Foster, J. , Grody, W. W. , Hegde, M. , Lyon, E. , Spector, E. , Voelkerding, K. , Rehm, H. L. , & Laboratory Quality Assurance Committee, A. C. M. G. (2015). Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genetics in Medicine, 17(5), 405–424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson, P. , Lipscomb, S. , Preston, L. C. , Altin, E. , Watkins, H. , Ashley, C. C. , & Redwood, C. S. (2007). Mutations in fast skeletal troponin I, troponin T, and beta‐tropomyosin that cause distal arthrogryposis all increase contractile function. The FASEB Journal, 21(3), 896–905. [DOI] [PubMed] [Google Scholar]
- Shrimpton, A. E. , & Hoo, J. J. (2006). A TNNI2 mutation in a family with distal arthrogryposis type 2B. European Journal of Medical Genetics, 49(2), 201–206. [DOI] [PubMed] [Google Scholar]
- Wang, B. , Zheng, Z. , Wang, Z. , Zhang, X. , Yang, H. , Cai, H. , & Fu, Q. (2016). A novel missense mutation of TNNI2 in a Chinese family cause distal arthrogryposis type 1. American Journal of Medical Genetics: Part A, 170A(1), 135–141. [DOI] [PubMed] [Google Scholar]
- Zhang, Z. , Akhter, S. , Mottl, S. , & Jin, J. P. (2011). Calcium‐regulated conformational change in the C‐terminal end segment of troponin I and its binding to tropomyosin. The FEBS Journal, 278(18), 3348–3359. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
TABLE S1
Data Availability Statement
The data that support the findings of this study are availablefrom the corresponding author upon reasonable request.