

Abstract
Introduction:
The effect of random error on performance of blood-based biomarkers for Alzheimer’s disease must be determined before clinical implementation.
Methods:
We measured test-retest variability of plasma Aβ42/Aβ40, NfL, GFAP, and P-tau217 and simulated effects of this variability on biomarker performance when predicting either CSF Aβ-status or conversion to AD dementia in 399 non-demented participants with cognitive symptoms.
Results:
Clinical performance was highest when combining all biomarkers. Among single-biomarkers, P-tau217 performed best. Test-retest variability ranged from 4.1% (Aβ42/Aβ40) to 25% (GFAP). This variability reduced the performance of the biomarkers (~ΔAUC −1% to −4%) with least effects on models with P-tau217. The percent of individuals with unstable predicted outcomes was lowest for the multi-biomarker combination (14%).
Discussion:
Clinical prediction models combining plasma biomarkers – particularly P-tau217 – exhibit high performance and are less effected by random error. Individuals with unstable predicted outcomes (“gray zone”) should be recommended for further tests.
Introduction
The field of Alzheimer’s disease (AD) has been transformed in recent years by development of several clinically relevant blood-based markers (BBMs), including plasma amyloid-β (Aβ) and phosphorylated tau (p-tau), along with neurofilament light (NfL, a marker of neurodegeneration) and glial fibrillary acidic protein (GFAP, a marker of astrocytic activation)1. In large, independent cohort studies, these biomarkers have consistently been shown to provide useful prognostic information with respect to longitudinal cognitive decline and risk for AD dementia2–6. Recent work has even demonstrated the superiority of plasma biomarkers (combined with other accessible measures) compared to clinicians’ predictions of AD-related outcome in a population with subjective cognitive decline (SCD) and mild cognitive impairment (MCI)7.
BBMs also show high diagnostic performance, particularly in differentiating individuals based on abnormal amyloid or tau status as measured by cerebrospinal fluid (CSF) or positron emission tomography (PET)8–10. These promising results have led to expectations that BBMs may eventually serve as a complement or even replacement for more invasive and expensive modalities, like CSF- and PET-based methods, in scenarios where high throughput or low cost is a priority11.
However, one obstacle to the implementation of BBMs at the patient level is the large overlap in biomarker levels observed between normal and disease groups. As an example, the plasma Aβ42/40 ratio is only 10% lower in amyloid-PET positive individuals, while the same ratio is 43% lower when measured in CSF12. Due to this overlap, random error in biomarker measurements may cause individuals who are close to diagnostic cutoffs to be classified as having normal levels of AD biomarkers at one timepoint but abnormal levels at another, hampering the detection of meaningful biological changes. Such random error is caused by a combination of 1) intra-individual variability in the biomarker levels in the blood over time (“biological variation”), 2) uncontrolled factors associated with sample collection/handling (“pre-analytical variation”), and 3) intra- and inter-assay variability (“analytical variation”)13. The extent to which the overlap between diagnostic groups interacts with observed levels of random error is still an open question since test-retest variability for core BBMs has not been empirically measured due to their novelty.
In the present study we aimed to gather information on this topic by collecting and analyzing test-retest plasma and CSF samples from 38 study participants, at different occasions close in time, to derive the total random error estimates for plasma Aβ42/Aβ40, P-tau217, NfL, and GFAP. We then simulated the impact of these random error estimates in a larger group of non-demented patients with cognitive symptoms from the Swedish BioFINDER study (n=399) when predicting AD-related outcomes (Figure 1). We hypothesized that predictions from a model combining multiple BBMs together would be perturbed less by simulated random error when compared to using only individual BBMs.
Figure 1. Study flowchart.
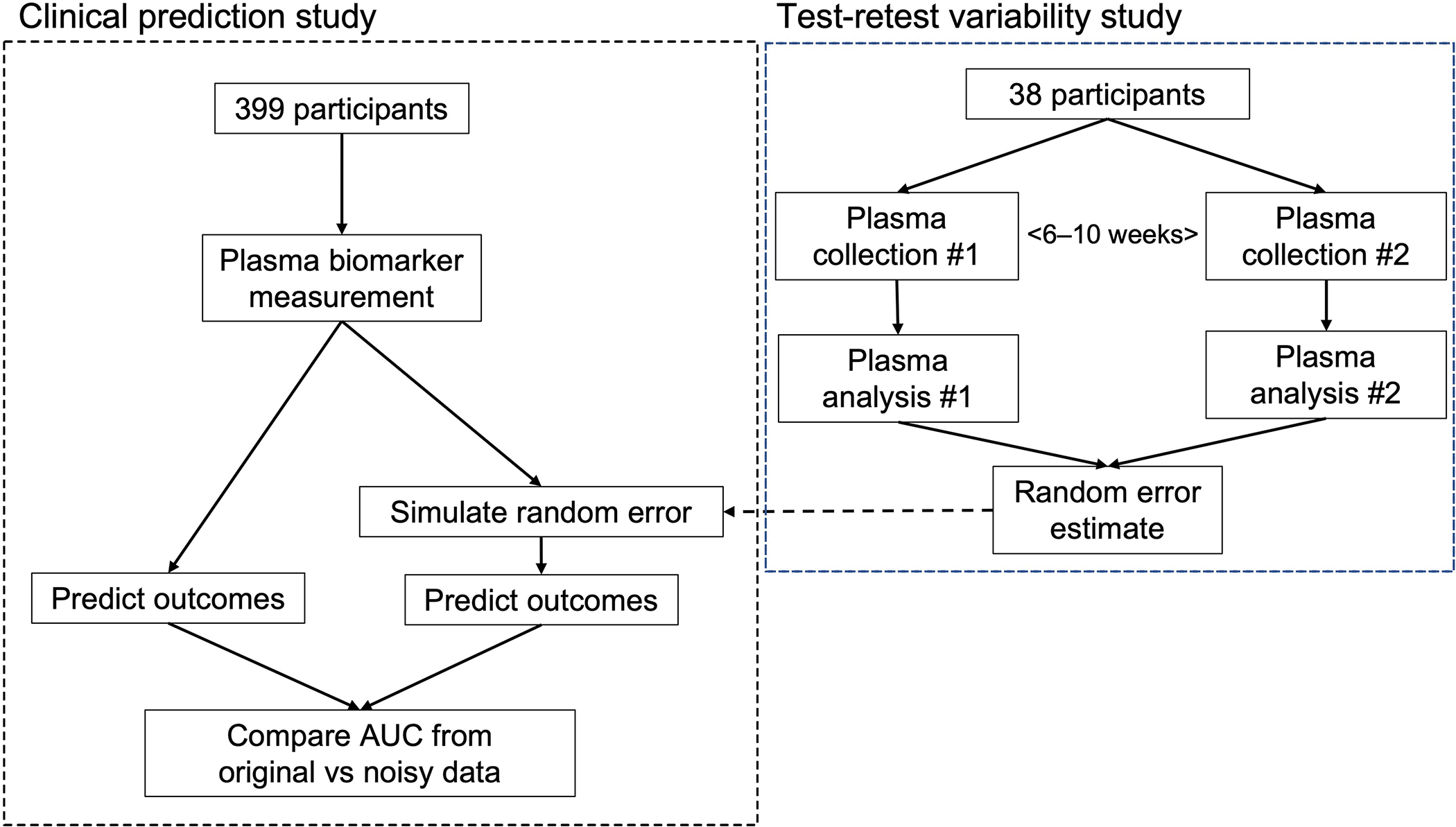
This figure gives an overview of the workflow involved in the present study. Briefly, random error estimates for each plasma biomarker from the test-retest variability studies were used to simulate the effect on prediction of abnormal CSF amyloid status using plasma biomarker values collected from the BioFINDER study.
Methods
Study design and participants
An overview of the study design in presented in Figure 1.
Participants (n=399) from the Swedish BioFINDER-1 study (http://biofinder.se; NCT01208675) consisted of consecutively included non-demented patients with mild cognitive symptoms referred to the participating memory clinics as previously described7. The inclusion criteria were (i) referred to the memory clinic due to cognitive symptoms experienced by the patient and/or informant; (ii) age between 60 and 80 years; (iii) MMSE score of 24–30 points at the baseline visit; (iv) do not fulfill the criteria for any dementia; and (v) speaks and understands Swedish to the extent that an interpreter was not necessary for the patient to fully understand the study information and neuropsychological tests. The exclusion criteria included (i) significant unstable systemic illness or organ failure, such as terminal cancer, that makes it difficult to participate in the study; (ii) current significant alcohol or substance misuse; and (iii) refusing lumbar puncture or neuropsychological assessment.
Participants in the test-retest study (n=38) were selected from the clinical practice of the Memory Clinic at Skåne University Hospital such that the percentage of participants who were amyloid positive was approximately equal (actual = 47.4%). For each participant, CSF and plasma samples were collected at a first visit and at a second visit which occurred 6 – 10 weeks later (average 7.4 ± 1.05 weeks). The collection procedure, amount of fluid collected, and pre-analytical handling protocol was identical across visits.
All patients gave their written informed consent to participate in the BioFINDER study. Separate written informed consent was given to participate in the test-retest study. The study was approved by the regional ethics committee in Lund, Sweden.
Biomarker measurements
As described previously, CSF was collected according to routine clinical procedures following the Alzheimer’s Association Flow Chart for lumbar puncture, centrifuged, frozen at −80C on dry ice, and shipped for analysis16. Plasma was collected in EDTA-plasma tubes and centrifuged (2,000g, +4 C) for 10 minutes. Following centrifugation, plasma from all tubes were transferred into one 50 ml polypropylene tube and mixed, after which 1ml was aliquoted into 1.5ml polypropylene tubes and stored at −80C within 30 – 60 minutes of collection. All plasma samples underwent one freeze-thaw cycle when 200μl were further aliquoted into 0.5ml LoBind tubes and the 200μl aliquots were stored at −80C as described previously14. Prototype immunoassays on a cobas e 601 and e 411 analyzer (Roche Diagnostics International Ltd, Rotkreuz, Switzerland) were used at the Clinical Neurochemistry laboratory in Gothenburg to analyze Aβ42, Aβ40, NfL and GFAP 17–19. Plasma and CSF P-tau217 were measured using an assay developed by Eli Lilly and analyzed at Lund University as previously described14.
Outcomes
The primary outcome of the clinical prediction modelling was normal versus abnormal levels of amyloid pathology as determined by CSF Aβ42/Aβ40 levels measured using enzyme-linked immunosorbent assay (ELISA) kits (Euroimmun). The cutoff for a positive (“abnormal”) CSF Aβ42/Aβ40 status (“CSF Aβ+”) versus a negative (“normal”) CSF Aβ42/Aβ40 status (“CSF Aβ−“) was 0.091 pg/mL as determined previously using gaussian mixture modelling20. This CSF measure has been validated extensively against both Aβ PET21,22 and neuropathology. The secondary outcome was conversion to AD dementia within four years of the baseline visit, based on the DSM-5 criteria for major neurocognitive disorder due to probable AD along with confirmation of abnormal amyloid accumulation according to the 2011 NIA-AA criteria for AD dementia23. Follow-up diagnosis was based on the treating physician’s assessments and reviewed by a consensus group of memory clinical physicians and a neuropsychologist.
Statistical Analysis
Random error estimates for each biomarker were derived in the test-retest study by calculating the relative percent change (100 * [x − y] / y) of biomarker values between the first and second sample for each participant (which were both collected and analyzed separate in time). The biomarker test-retest variability was then calculated as the standard deviation of this distribution of percent change values.
In the larger group of study participants, ROC analysis was used to calculate the overall classification performance (area under the curve, AUC) of each individual plasma biomarker to identify CSF Aβ status (with conversion to AD dementia as secondary outcome). Youden’s index was used to identify the optimal cutoff independently for each individual plasma biomarker that best distinguished Aβ− and Aβ+ participants. Percent agreement (i.e., accuracy) and AUC was then calculated for each biomarker at its respective cutoff. Additionally, a logistic regression model was fit that included all plasma biomarkers and optimal cutoffs were derived from individual-level predicted risk values.
Next, we randomly varied plasma biomarker values for each participant based on a random sample from a normal distribution with mean equal to zero and standard deviation equal to the variability estimate for each biomarker, obtained from the test-retest study. The model performance of these “noisy” (estimated) biomarker values was evaluated and compared to the performance of original biomarker values. The primary metric was change in AUC value between noisy and original biomarker models. We also reported the percentage of study participants whose predicted outcome changed when biomarkers were randomly varied. This simulation was run over 1000 bootstrap trials to obtain confidence intervals.
We performed the same analysis using the same biomarkers measured in CSF based on their corresponding estimates of test-retest variability. A sensitivity analysis was also performed for the primary outcome of CSF Aβ status whereby test-retest variability estimates were a priori specified as 5%, 10%, 20%, and 30% for all biomarkers and the effect on model performance was investigated. All statistical analysis was performed using the R programming language (v5.0.0) with an alpha level of 0.05.
Results
Characterizing study participants
A total of 399 participants were included in the clinical prediction analysis. The average age was 70.8 ± 5.5 years, and the average educational attainment was 11.7 ± 3.6 years, with 46.9% of participants being female (Table 1). A total of 196 (49.1%) participants were CSF Aβ+ and 96 (24.1%) participants developed AD dementia within four years of baseline.
Table 1:
Cohort characteristics
| Overall | CSF Aβ− | CSF Aβ+ | p | ||
|---|---|---|---|---|---|
| n | 399 | 203 | 196 | ||
| AGE (mean (SD)) | 70.78 (5.50) | 69.63 (5.59) | 71.96 (5.17) | <0.001 | |
| EDUCATION (mean (SD)) | 11.69 (3.62) | 11.90 (3.71) | 11.48 (3.52) | 0.250 | |
| GENDER (%) | 0 | 212 (53.1) | 108 (53.2) | 104 (53.1) | 1.000 |
| 1 | 187 (46.9) | 95 (46.8) | 92 (46.9) | ||
| Diagnosis (%) | SCD | 175 (43.9) | 111 (54.7) | 64 (32.7) | <0.001 |
| MCI | 224 (56.1) | 92 (45.3) | 132 (67.3) | ||
| Four-year AD dementia (%) | No | 195 (48.9) | 124 (61.1) | 71 (36.2) | <0.001 |
| Yes | 96 (24.1) | 4 (2.0) | 92 (46.9) | ||
| Not eligible | 108 (27.1) | 75 (36.9) | 33 (16.8) | ||
| CSF Aβ42/Aβ40 (mean (SD)) | 69.31 (30.55) | 95.62 (15.72) | 41.28 (11.75) | <0.001 | |
| Plasma Aβ42/Aβ40 (mean (SD)) | −0.11 (0.02) | −0.12 (0.02) | −0.11 (0.01) | <0.001 | |
| Plasma P-tau217 (mean (SD)) | 0.24 (0.22) | 0.14 (0.13) | 0.35 (0.25) | <0.001 | |
| Plasma NfL (mean (SD)) | 2.99 (2.28) | 2.82 (2.35) | 3.17 (2.20) | 0.125 | |
| Plasma GFAP (mean (SD)) | 0.11 (0.07) | 0.09 (0.07) | 0.13 (0.07) | <0.001 |
This table displays characteristics for participants in the clinical modelling analysis. All continuous values are reported as mean and standard deviation, while all categorical variables are reported as total counts and percentage in the entire study population. All variables are described in the entire study population and separately in Aβ− and Aβ+ individuals (as defined using CSF Aβ42/Aβ40). Individuals who were “not eligible” in the four-year AD dementia analysis were individuals who did not convert to AD dementia but did not have at least four years’ follow-up time. P-values represent the result of statistical tests (t-test for continuous, chi-square for categorical) when comparing variable values between Aβ− and Aβ+ participants. Abbreviations: CSF Aβ− = normal CSF Aβ42/Aβ40 levels; CSF Aβ+ = abnormal CSF Aβ42/Aβ40 levels; SCD = Subjective Cognitive Decline; MCI = Mild Cognitive Impairment; SD = standard deviation; n = number of participants
Estimating test-retest biomarker variability
The observed test-retest variability was 4.1% for plasma Aβ42/Aβ40, 20.0% for plasma P-tau217, 23.7% for plasma NfL, and 25.0% for plasma GFAP. Individual-level relative change values across test-retest measurements for each plasma biomarker are displayed visually in Figure 2, which can be compared to the test-retest variability for CSF markers in Supplementary Figure 1.
Figure 2. Test-retest variability of plasma biomarkers.
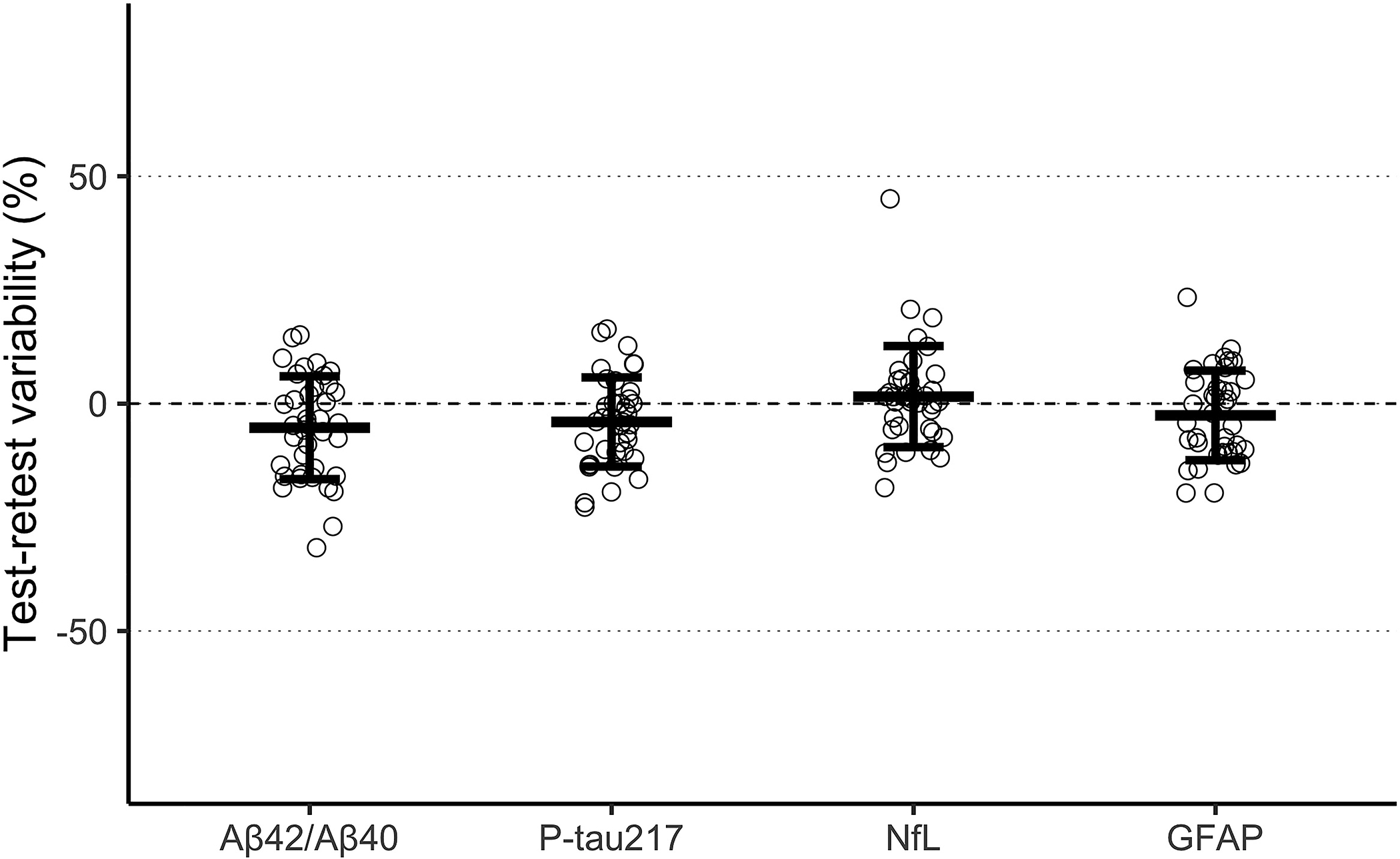
This figure shows the observed test-retest variability at the individual level for each plasma biomarker along with the mean and 95% confidence interval. Test-retest variability for each biomarker was derived by first calculating the relative percent change (100 * [x − y] / y) of biomarker values for each participant across the two samples and then using the standard deviation of this distribution as the overall estimate of random error.
Modelling Alzheimer-related outcomes
When using baseline samples of participants from the BioFINDER study (n=399), the highest performing individual biomarker model in terms of separating Aβ− from Aβ+ participants was plasma P-tau217 (AUC = 0.82, 95% CI [0.80, 0.85]), followed by plasma Aβ42/Aβ40 (AUC = 0.79, CI [0.76, 0.82]), plasma GFAP (AUC = 0.72, CI [0.70, 0.74]), and finally plasma NfL (AUC = 0.60, CI [0.57, 0.64]). All individual biomarker models were outperformed by the multi-biomarker model (AUC = 0.86, CI [0.85, 0.88]; P < 0.05 for all comparisons). The performance of the biomarkers was qualitatively similar with conversion to AD dementia at 4 years as outcome. The ROC curves from these results are displayed graphically in Figure 3A. Moreover, the performance of CSF biomarkers was generally somewhat higher for predicting conversion to AD dementia, except for CSF GFAP (Supplementary Figure 2A).
Figure 3. Modelling performance of plasma biomarkers and the simulated effect of test-retest variability.
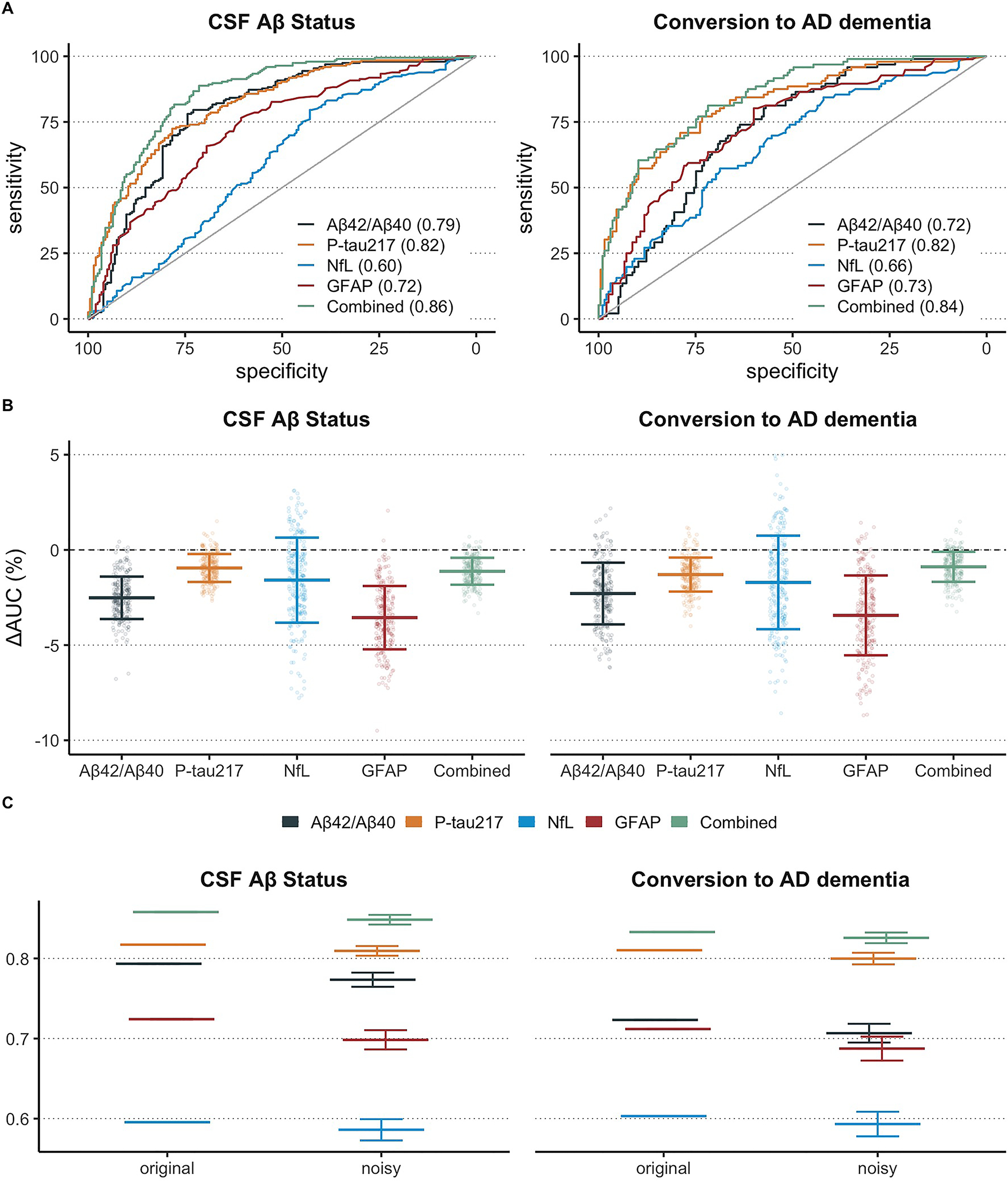
This figure shows the ability of plasma biomarkers (individually and combined) to predict abnormal amyloid pathology in CSF and conversion to AD dementia within four years from baseline (panel A). This figure also shows the effect on AUC values when test-retest variability for each biomarker was simulated over 1000 trials (panel B). The change in AUC represents the mean difference between the model performance with original (i.e., true) biomarker values versus the model performance with random error added to each biomarker. Finally, this figure shows AUC results with original, unperturbed plasma biomarker data and AUC results after simulation (panel C).
Simulating effects of variability on model performance
We next simulated the effect on model performance in the same participants from the BioFINDER study when random adding noise to each biomarker based on the corresponding test-retest variability for each biomarker measured in the first analysis (n=38). Here, we found that plasma P-tau217 was affected least by simulation of test-retest variability (ΔAUC = −0.98%), followed by the combined biomarker model (ΔAUC = −1.2%), plasma NfL (ΔAUC = −1.7%), plasma Aβ42/Aβ40 (ΔAUC = −2.5%), and plasma GFAP (ΔAUC = −3.7%). The results were similar with conversion to AD dementia as outcome: ΔAUC = −0.88% for combined model, ΔAUC = −1.29% for plasma P-tau217, ΔAUC = −2.29% for plasma Aβ42/Aβ40, ΔAUC = −1.64% for plasma NfL, ΔAUC = −3.43% for plasma GFAP. The difference in AUC values from each of the 1000 simulation trials is displayed graphically in Figure 3B and the AUC values for both original and noise-simulated models are presented in Figure 3C. A sensitivity analysis using Cox regression instead of logistic regression for the longitudinal conversion to AD outcome is also presented in Supplementary Figure 5.
The decrease in AUC values when simulating test-retest variability in the same manner on CSF biomarkers was generally much lower than seen in plasma and is displayed graphically in Supplementary Figure 2B–C. Additionally, a sensitivity analysis in which all possible models with plasma P-tau217 were investigated and compared against a model with all biomarkers besides plasma P-tau217 (see Supplementary Figure 4). Here, we found that models with plasma P-tau217 always contained similar levels of performance-related robustness to test-retest variability as the model with plasma P-tau217 by itself. Moreover, the model with all plasma biomarkers besides plasma P-tau217 had a worse robustness to test-retest variability than any model with plasma P-tau217.
While our primary analysis focused on empirical estimates of biomarker test-retest variability, we also performed a sensitivity analysis in which we investigated a scenario where each plasma biomarker had the same test-retest variability which was defined in advance. Test-retest variability levels were varied from 5%, 10%, 20%, and 30%. Here, we found that plasma P-tau217, plasma NfL, and plasma GFAP had similar decreases in AUC value while plasma Aβ42/Aβ40 had a significantly larger decrease than all other biomarkers. The combined model also had a similar decrease in AUC value as individual biomarkers despite also including plasma Aβ42/Aβ40. These results are visualized in Supplementary Figure 6.
Estimating individual-level uncertainty of predictive models
Finally, we calculated the percentage of participants with uncertain predicted outcomes as estimated when simulating test-retest variability. These individuals are those whose biomarker values place them in the “gray zone” where test-retest variability means they have a >5% chance of having a different predicted outcome if they were to have two plasma samples collected and analyzed close in time with some weeks apart. First, we developed a 95% confidence interval for thresholds of each biomarker model (Figure 4). Next, we derived the percentage of participants who fell within this uncertain interval for each biomarker model (individual and combined). With Aβ-status as outcome, we found that the individual biomarkers with lowest prediction uncertainty were plasma P-tau217 (uncertain = 20.3%) and plasma Aβ42/Aβ40 (uncertain = 20.3%; note: equal to plasma P-tau217), followed by plasma NfL (uncertain = 30.1%) and plasma GFAP (uncertain = 30.6%). The combined plasma biomarker model had a lower percentage of uncertain participant predictions than all individual biomarkers (uncertain = 14.3%). These results were similar with conversion to AD as outcome, except that plasma P-tau217 (uncertain = 19.5%) had lower prediction uncertainty than plasma Aβ42/Aβ40 (uncertain = 22.3%). These results are displayed graphically in Figure 5. The individual-level uncertainty for CSF biomarkers was generally much lower than compared to plasma biomarkers and is displayed in Supplementary Figure 7.
Figure 4. Distribution of plasma biomarker values across diagnostic/prognostic groups and optimal cutoff variability.
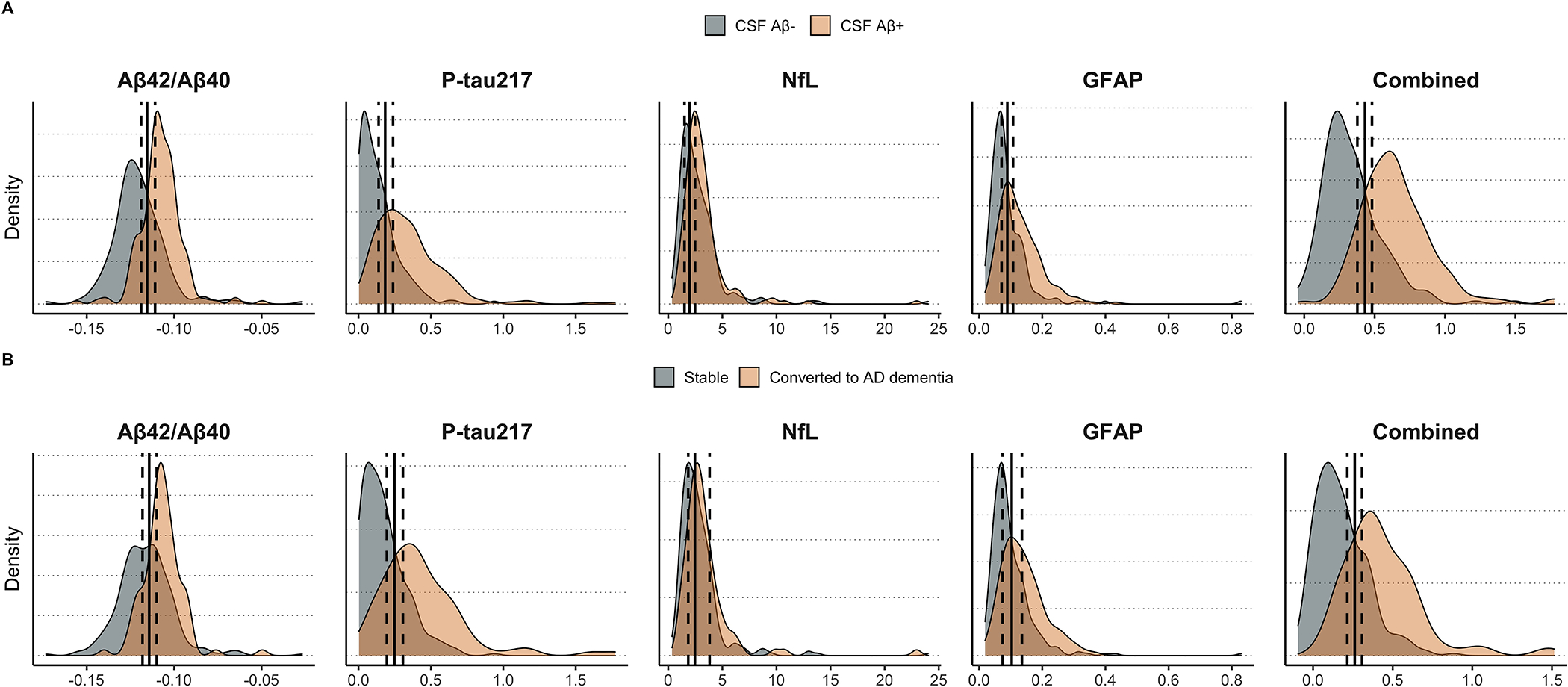
This figure demonstrates the distribution of plasma biomarker values (or risk predictions from logistic regression for the combined model) across CSF amyloid-negative and CSF amyloid-positive groups (panel A) or across participants who remained stable versus those who developed AD dementia within four years from baseline (panel B). The optimal cutoff derived from Youden’s index is also plotted for each model, along with the 95% confidence interval of the cutoff as derived from 1000 trials of simulating random error for each biomarker according to empirical estimates of test-retest variability.
Figure 5. Individual-level uncertainty in predicted outcomes due to test-retest variability of plasma biomarkers.
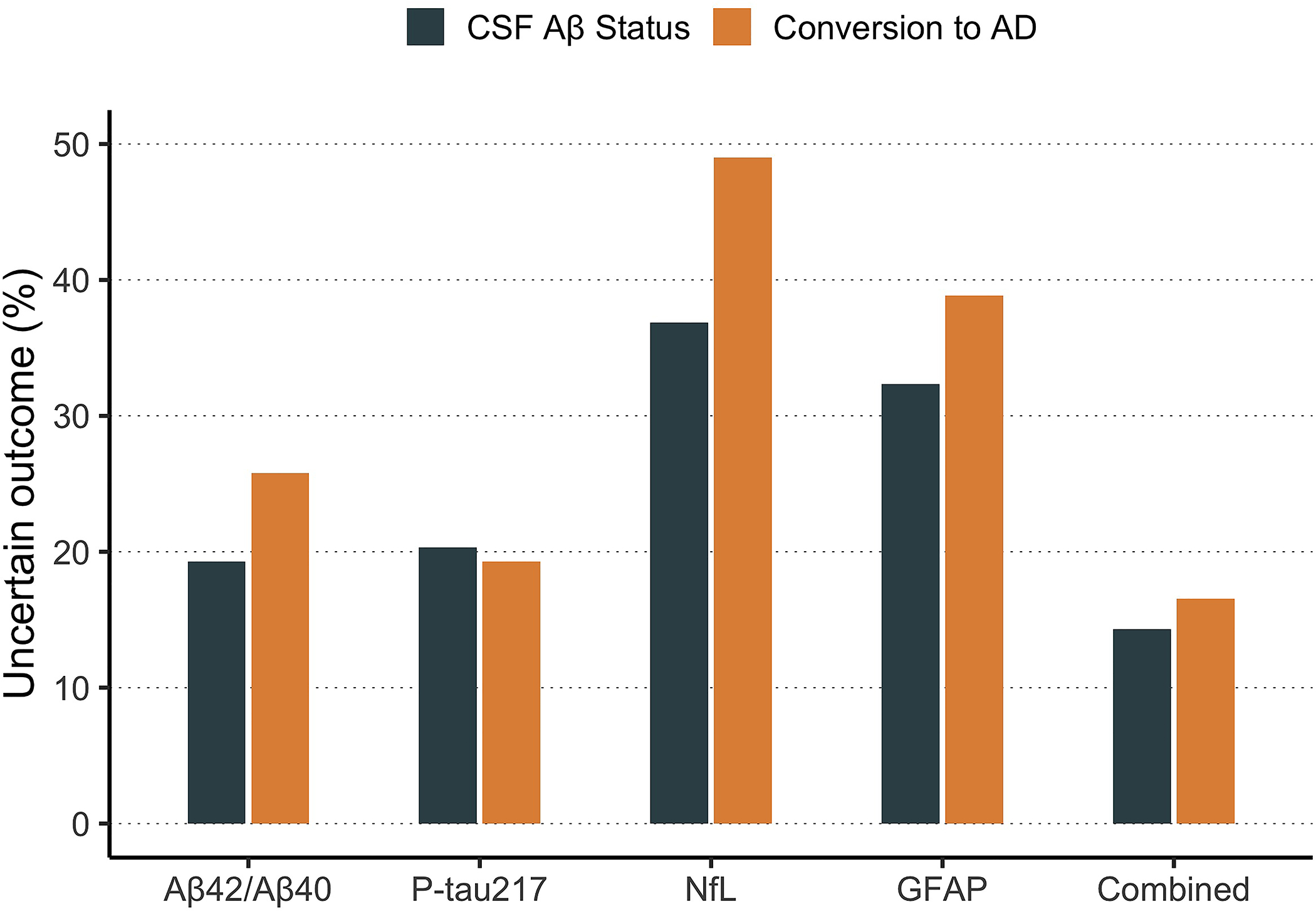
This figure shows the percentage of participants whose predicted outcome (CSF amyloid status or conversion to AD dementia) would (theoretically) have a greater than 5% chance of varying back-and-forth across the cutoff threshold due to random error of each plasma biomarker.
Discussion
The results of the present study showed that plasma biomarkers of AD exhibit varying levels of test-retest variability, with plasma Aβ42/Aβ40 having the lowest levels of variability. However, the effect of this variability on clinical performance depended greatly on how well-separated the biomarker distributions were between individuals with and without the outcome of interest (here, abnormal cerebral amyloid accumulation or development of AD dementia). This was evidenced by the finding that plasma P-tau217 was least influenced by simulating the additional of test-retest variability to real clinical data. Moreover, our results suggest that the effects of test-retest variability on clinical performance can be largely neutralized by combining plasma biomarkers into a multi-variable panel.
This study contributes to a better understanding of how random error affects the uncertainty of predicting AD-related outcomes from core plasma biomarkers measured in non-demented patients with cognitive symptoms. The specific test-retest variability estimates for each plasma biomarker provided here can also be used by other researchers to perform similar analyses to understand how random error affects clinical prediction models. Understanding effects of random error is of outmost importance for implementing BBMs for prospective use in clinical practice and trials
We directly quantified the estimated percentage of cases where predicted outcome would have a >5% chance to be non-concordant between two visits in a short time span. This type of analysis could lead to the development of a “gray zone” model used in clinical practice whereby individuals whose plasma biomarkers provide an uncertain diagnostic or prognostic prediction can be referred for other tests such as through PET to determine AD biomarker status. Note that because we did not include demographic variables (e.g., age and APOE4 genotype) in our models and these variables have no test-retest variables, the gray zone measured here likely provides a maximum bound on the real gray zone. Including stable variables are likely to shrink the gray zone, but such investigations are outside the scope of the present analysis where we aimed to isolate effects on a specific set of biomarkers.
The relationship between random error and overlap between normal and abnormal groups is not specific to AD BBMs but is a general problem throughout the field of clinical chemistry. It has therefore been suggested to define gray zones for diagnostic biomarkers, where biomarker results should be interpreted with caution and need to be confirmed with orthogonal methods24–27.
Note that we chose CSF amyloid status as the primary outcome because this represents in our view the most likely outcome of interest to be used when implementing plasma biomarkers for two major reasons: 1) evidence of cerebral Aβ pathology is very often required as an inclusion criteria in clinical trials, and plasma biomarker are likely to be used as pre-screeners identifying individuals likely to exhibit an abnormal CSF Aβ42/Aβ40 (or Aβ-PET) status, and 2) detection of cerebral Aβ in clinical practice will likely be important in the future considering the possible clinical implementation of anti-Aβ therapies. Plasma biomarkers represent an inexpensive, first-line risk screening tool for determining whether individuals have abnormal amyloid accumulation, and this may only be the first step in a long workflow towards diagnosis or inclusion in clinical trials.
Besides using empirical measurements of test-retest variability, we also simulated effects on performance in a scenario where all plasma biomarkers had the same level of test-retest variability. We found that plasma P-tau217, plasma NfL, and plasma GFAP were all about equally influenced by the same levels of test-retest variability and plasma P-tau217 may have performed better in the primary analysis because it has lower empirical test-retest variability. This result suggests that plasma assays should optimize for test-retest variability in addition to model performance. Importantly, this analysis provided even more evidence that the performance of a combined biomarker model is not greatly degraded even when included one biomarker which was essentially just noise (e.g., plasma Aβ42/Aβ40 at 20% and 30% variability). Thus, a combined biomarker model will continue to perform even if one biomarker becomes completely unusable due to random error. Still, diagnostic model combining multiple plasma biomarkers may be more complicated to implement than a model with only one biomarker. A multi-biomarker model would require careful work to standardize and control all factors that may contribute to random noise across several biomarkers. We also found that CSF biomarkers generally had less test-retest variability at the individual level and that performance of CSF biomarkers decreased less when random error was simulated.
In this study, we primarily considered only one type of error in the present analysis – random error estimated by collecting and analyzing samples from the same individuals at different occasions but within a short time span. Another source of noise which can greatly affect biomarker values is systematic error caused by assay-related changes in the analytical performance of the methods such as when changing lots of key materials (such as antibodies or calibrators)28,29. However, systematic error is much more difficult to quantify empirically given its unpredictable nature. Systematic error can also greatly degrade performance of diagnostic models when the assay is characterized by low dynamic range or when there is high overlap between positive and negative groups.
The major strength of the study is the availability of real test-retest variability measurements on which to base our investigation into how clinical prediction models are influenced by such noise. This means that our assumptions are based in real experience and are more likely to be applicable. The duration between sample visits was short on an AD timescale and there was no significant shift in biomarker levels across sample visits, indicating that disease progression was unlikely to affect test-retest estimates. However, a major limitation of the study is the fact that the participants used to derive the test-retest variability estimates was not the same population used when evaluating clinical prediction models. Although there were no significant differences in the participant characteristics between groups, it is not entirely possible to rule out that test-retest variability estimates in the clinical population may have been different.
In all, our results provide a first step towards a better understanding of the effects of plasma biomarker assay variability in a clinical prediction context. This is an important area of research given the potential use of plasma biomarkers at the earliest stage of AD detection. The potential impact of these results on clinical practice is two-fold. For one, these findings suggest that implementing a multi-biomarker panel for use in prediction of AD-related outcomes could potentially lead to fewer misclassifications. Whether the improved performance outweighs the increased cost of a multi-biomarker panel requires further investigation. Secondly, these findings may impact clinical practice by better establishing “gray zones” for each biomarker where patients may be recommended for additional testing if their biomarker levels fall into these intervals. Taken together, this work represents a step towards improving performance of AD plasma biomarkers in clinical practice.
In the future, we plan to apply this type of test-retest analysis to other plasma biomarker assays (e.g., more accurate IP-MS assays20), biomarker modalities (e.g., PET), clinical scenarios (e.g., shorter or longer time horizons for AD-related outcomes), and statistical models (e.g., Cox regression, mixed effects models).
Supplementary Material
Research in Context.
Systematic review:
The authors reviewed the literature using traditional sources and found that there is a lack of studies investigating how individuals along the Alzheimer’s disease (AD) spectrum will shift from having “normal” values for core plasma biomarkers to having “abnormal” values (or the other way around) if blood is collected, processed, and analyzed at different occasions.
Interpretation:
The predictive performance of plasma biomarkers is largely unaffected by test-retest variability when biomarkers are combined (compared to used individually) or when plasma P-tau217 is included in the panel.
Future directions:
This work will spur more interest in the concept of “gray zones” – i.e., ranges of biomarker values for which prediction of relevant outcomes is uncertain, thereby requiring further testing by more invasive biomarkers such as CSF or PET.
Acknowledgments
The study was supported by the Swedish Research Council (2016-00906), the Knut and Alice Wallenberg foundation (2017-0383), the Marianne and Marcus Wallenberg foundation (2015.0125), the Strategic Research Area MultiPark (Multidisciplinary Research in Parkinson’s disease) at Lund University, the Swedish Alzheimer Foundation (AF-939932), the Swedish Brain Foundation (FO2021-0293), The Parkinson foundation of Sweden (1280/20), the Konung Gustaf V:s och Drottning Victorias Frimurarestiftelse, the Skåne University Hospital Foundation (2020-O000028), Regionalt Forskningsstöd (2020-0314) and the Swedish federal government under the ALF agreement (2018-Projekt0279). HZ is a Wallenberg Scholar supported by grants from the Swedish Research Council (#2018-02532), the European Research Council (#681712), Swedish State Support for Clinical Research (#ALFGBG-720931), the Alzheimer Drug Discovery Foundation (ADDF), USA (#201809-2016862), the AD Strategic Fund and the Alzheimer’s Association (#ADSF-21-831376-C, #ADSF-21-831381-C and #ADSF-21-831377-C), the Olav Thon Foundation, the Erling-Persson Family Foundation, Stiftelsen för Gamla Tjänarinnor, Hjärnfonden, Sweden (#FO2019-0228), the European Union’s Horizon 2020 research and innovation programme under the Marie Skłodowska-Curie grant agreement No 860197 (MIRIADE), European Union Joint Program for Neurodegenerative Disorders (JPND2021-00694), and the UK Dementia Research Institute at UCL. KB is supported by the Swedish Research Council (#2017-00915), the Alzheimer Drug Discovery Foundation (ADDF), USA (#RDAPB-201809-2016615), the Swedish Alzheimer Foundation (#AF-742881), Hjärnfonden, Sweden (#FO2017-0243), the Swedish state under the agreement between the Swedish government and the County Councils, the ALF-agreement (#ALFGBG-715986), the European Union Joint Program for Neurodegenerative Disorders (JPND2019-466-236), the National Institute of Health (NIH), USA, (grant #1R01AG068398-01), and the Alzheimer’s Association 2021 Zenith Award (ZEN-21-848495).
Footnotes
DISCLOSURES
NCC, SJ, ES, NMC have no disclosures. OH has acquired research support (for the institution) from AVID Radiopharmaceuticals, Biogen, Eli Lilly, Eisai, GE Healthcare, Pfizer, and Roche. In the past 2 years, he has received consultancy/speaker fees from Roche, Genentech, Siemens, Biogen, Alzpath, and Cerveau. TB is a full time employee of F. Hoffman La-Roche. AJ and GK are full time employees of Roche Diagnostics GmbH. IS is a fulltime employee and shareholder of Roche Diagnostics International. HZ has served at scientific advisory boards and/or as a consultant for Abbvie, Alector, Annexon, Artery Therapeutics, AZTherapies, CogRx, Denali, Eisai, Nervgen, Pinteon Therapeutics, Red Abbey Labs, Passage Bio, Roche, Samumed, Siemens Healthineers, Triplet Therapeutics, and Wave, has given lectures in symposia sponsored by Cellectricon, Fujirebio, Alzecure, Biogen, and Roche, and is a co-founder of Brain Biomarker Solutions in Gothenburg AB (BBS), which is a part of the GU Ventures Incubator Program. KB has served as a consultant, at advisory boards, or at data monitoring committees for Abcam, Axon, BioArctic, Biogen, JOMDD/Shimadzu. Julius Clinical, Lilly, MagQu, Novartis, Pharmatrophix, Prothena, Roche Diagnostics, and Siemens Healthineers, and is a co-founder of Brain Biomarker Solutions in Gothenburg AB (BBS), which is a part of the GU Ventures Incubator Program. SP has served on scientific advisory boards and/or given lectures in symposia sponsored by F. Hoffmann-La Roche, Biogen, and Geras Solutions.
References
- 1.Hansson O Biomarkers for neurodegenerative diseases. Nat Med. 2021;27(6):954–963. doi: 10.1038/s41591-021-01382-x [DOI] [PubMed] [Google Scholar]
- 2.Cullen NC, Leuzy A, Palmqvist S, et al. Individualized prognosis of cognitive decline and dementia in mild cognitive impairment based on plasma biomarker combinations. Nat Aging. Published online 2020:1–10. doi: 10.1038/s43587-020-00003-5 [DOI] [PubMed] [Google Scholar]
- 3.Cullen NC, Leuzy A, Janelidze S, et al. Plasma biomarkers of Alzheimer’s disease improve prediction of cognitive decline in cognitively unimpaired elderly populations. Nat Commun. 2021;12(1):3555. doi: 10.1038/s41467-021-23746-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Janelidze S, Mattsson N, Palmqvist S, et al. Plasma P-tau181 in Alzheimer’s disease: relationship to other biomarkers, differential diagnosis, neuropathology and longitudinal progression to Alzheimer’s dementia. Nat Med. 2020;26(3):379–386. doi: 10.1038/s41591-020-0755-1 [DOI] [PubMed] [Google Scholar]
- 5.Cicognola C, Janelidze S, Hertze J, et al. Plasma glial fibrillary acidic protein detects Alzheimer pathology and predicts future conversion to Alzheimer dementia in patients with mild cognitive impairment. Alzheimer’s Res Ther. 2021;13(1):68. doi: 10.1186/s13195-021-00804-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Verberk IMW, Laarhuis MB, van den Bosch KA, et al. Serum markers glial fibrillary acidic protein and neurofilament light for prognosis and monitoring in cognitively normal older people: a prospective memory clinic-based cohort study. Lancet Heal Longev. 2021;2(2):e87–e95. doi: 10.1016/s2666-7568(20)30061-1 [DOI] [PubMed] [Google Scholar]
- 7.Palmqvist S, Tideman P, Cullen N, et al. Prediction of future Alzheimer’s disease dementia using plasma phospho-tau combined with other accessible measures. Nat Med. 2021;27(6):1034–1042. doi: 10.1038/s41591-021-01348-z [DOI] [PubMed] [Google Scholar]
- 8.Mofrad RB, Scheltens P, Kim S, et al. Plasma amyloid-β oligomerization assay as a pre-screening test for amyloid status. Alzheimer’s Res Ther. 2021;13(1):133. doi: 10.1186/s13195-021-00873-w [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Verberk IMW, Thijssen E, Koelewijn J, et al. Combination of plasma amyloid beta(1–42/1–40) and glial fibrillary acidic protein strongly associates with cerebral amyloid pathology. Alzheimer’s Res Ther. 2020;12(1):118. doi: 10.1186/s13195-020-00682-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Grothe MJ, Moscoso A, Ashton NJ, et al. Associations of Fully Automated CSF and Novel Plasma Biomarkers With Alzheimer Disease Neuropathology at Autopsy. Neurology. 2021;97(12):e1229–e1242. doi: 10.1212/wnl.0000000000012513 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zetterberg H, Blennow K. Blood Biomarkers: Democratizing Alzheimer’s Diagnostics. Neuron. 2020;106(6):881–883. doi: 10.1016/j.neuron.2020.06.004 [DOI] [PubMed] [Google Scholar]
- 12.Schindler SE, Bollinger JG, Ovod V, et al. High-precision plasma β-amyloid 42/40 predicts current and future brain amyloidosis. Neurology. Published online 2019:10.1212/WNL.0000000000008081. doi: 10.1212/wnl.0000000000008081 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Aylward LL, Hays SM, Smolders R, et al. Sources of Variability in Biomarker Concentrations. J Toxicol Environ Heal Part B. 2014;17(1):45–61. doi: 10.1080/10937404.2013.864250 [DOI] [PubMed] [Google Scholar]
- 14.Palmqvist S, Janelidze S, Quiroz YT, et al. Discriminative Accuracy of Plasma Phospho-tau217 for Alzheimer Disease vs Other Neurodegenerative Disorders. Jama. 2020;324(8):772–781. doi: 10.1001/jama.2020.12134 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Blennow K, Hampel H, Weiner M, Zetterberg H. Cerebrospinal fluid and plasma biomarkers in Alzheimer disease. Nat Rev Neurol. 2010;6(3):131–144. doi: 10.1038/nrneurol.2010.4 [DOI] [PubMed] [Google Scholar]
- 16.Palmqvist S, Zetterberg H, Blennow K, et al. Accuracy of Brain Amyloid Detection in Clinical Practice Using Cerebrospinal Fluid β-Amyloid 42: A Cross-Validation Study Against Amyloid Positron Emission Tomography. Jama Neurol. 2014;71(10):1282–1289. doi: 10.1001/jamaneurol.2014.1358 [DOI] [PubMed] [Google Scholar]
- 17.Palmqvist S, Janelidze S, Stomrud E, et al. Performance of Fully Automated Plasma Assays as Screening Tests for Alzheimer Disease–Related β-Amyloid Status. Jama Neurol. 2019;76(9):1060–1069. doi: 10.1001/jamaneurol.2019.1632 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Pereira JB, Janelidze S, Smith R, et al. Plasma GFAP is an early marker of amyloid-β but not tau pathology in Alzheimer’s disease. Brain. Published online 2021:awab223–. doi: 10.1093/brain/awab223 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hulle CV, Jonaitis EM, Betthauser TJ, et al. An examination of a novel multipanel of CSF biomarkers in the Alzheimer’s disease clinical and pathological continuum. Alzheimer’s Dementia. 2021;17(3):431–445. doi: 10.1002/alz.12204 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Janelidze S, Teunissen CE, Zetterberg H, et al. Head-to-Head Comparison of 8 Plasma Amyloid-β 42/40 Assays in Alzheimer Disease. Jama Neurol. 2021;78(11). doi: 10.1001/jamaneurol.2021.3180 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Janelidze S, Stomrud E, Palmqvist S, et al. Plasma β-amyloid in Alzheimer’s disease and vascular disease. Sci Rep-uk. 2016;6(1):26801. doi: 10.1038/srep26801 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Janelidze S, Pannee J, Mikulskis A, et al. Concordance Between Different Amyloid Immunoassays and Visual Amyloid Positron Emission Tomographic Assessment. Jama Neurol. 2017;74(12):1492. doi: 10.1001/jamaneurol.2017.2814 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.McKhann GM, Knopman DS, Chertkow H, et al. The diagnosis of dementia due to Alzheimer’s disease: Recommendations from the National Institute on Aging-Alzheimer’s Association workgroups on diagnostic guidelines for Alzheimer’s disease. Alzheimer’s Dementia. 2011;7(3):263–269. doi: 10.1016/j.jalz.2011.03.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Landsheer JA. Interval of Uncertainty: An Alternative Approach for the Determination of Decision Thresholds, with an Illustrative Application for the Prediction of Prostate Cancer. Plos One. 2016;11(11):e0166007. doi: 10.1371/journal.pone.0166007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Landsheer JA. The Clinical Relevance of Methods for Handling Inconclusive Medical Test Results: Quantification of Uncertainty in Medical Decision-Making and Screening. Diagnostics. 2018;8(2):32. doi: 10.3390/diagnostics8020032 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Coste J, Jourdain P, Pouchot J. A Gray Zone Assigned to Inconclusive Results of Quantitative Diagnostic Tests: Application to the Use of Brain Natriuretic Peptide for Diagnosis of Heart Failure in Acute Dyspneic Patients. Clin Chem. 2006;52(12):2229–2235. doi: 10.1373/clinchem.2006.072280 [DOI] [PubMed] [Google Scholar]
- 27.Lazzati JM, Zaidman V, Maceiras M, Belgorosky A, Chaler E. The use of a “gray zone” considering measurement uncertainty in pharmacological tests. The serum growth hormone stimulation test as an example. Clin Chem Laboratory Medicine Cclm. 2016;54(11):e349–e351. doi: 10.1515/cclm-2015-0954 [DOI] [PubMed] [Google Scholar]
- 28.Mattsson N, Andreasson U, Persson S, et al. CSF biomarker variability in the Alzheimer’s Association quality control program. Alzheimer’s Dementia. 2013;9(3):251–261. doi: 10.1016/j.jalz.2013.01.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Bastard NL, Deyn PPD, Engelborghs S. Importance and Impact of Preanalytical Variables on Alzheimer Disease Biomarker Concentrations in Cerebrospinal Fluid. Clin Chem. 2015;61(5):734–743. doi: 10.1373/clinchem.2014.236679 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.