

Abstract
Little is known about factors which enable Salmonella serotypes to circulate within populations of livestock and domestic fowl. We have identified a DNA region which is present in Salmonella serotypes commonly isolated from livestock and domestic fowl (S. enterica subspecies I) but absent from reptile-associated Salmonella serotypes (S. bongori and S. enterica subspecies II to VII). This DNA region was cloned from Salmonella serotype Typhimurium and sequence analysis revealed the presence of a 6,105-bp open reading frame, designated shdA, whose product's deduced amino acid sequence displayed homology to that of AIDA-I from diarrheagenic Escherichia coli, MisL of serotype Typhimurium, and IcsA of Shigella flexneri. The shdA gene was located adjacent to xseA at 52 min, in a 30-kb DNA region which is not present in Escherichia coli K-12. A serotype Typhimurium shdA mutant was shed with the feces in reduced numbers and for a shorter period of time compared to its isogenic parent. A possible role for the shdA gene during the expansion in host range of S. enterica subspecies I to include warm-blooded vertebrates is discussed.
Salmonella serotypes are a frequent constituent of the intestinal flora of poikilothermic animals. The percentage of apparently healthy, cold-blooded vertebrates which harbor Salmonella serotypes ranges from 74 to 94% (20, 28, 32, 34, 59), and these bacteria could thus be considered part of the normal intestinal flora (23, 48). Salmonella serotypes are also commonly isolated from a fraction (usually <20%) of warm-blooded animal hosts (15, 31, 49, 54). Although chronic carriers, which appear healthy, are observed within the human population and among warm-blooded animals (22, 27, 35, 40), Salmonella serotypes are commonly associated with illness in these hosts (55). Consequently Salmonella serotypes are regarded as pathogens rather than part of the normal intestinal flora of homeothermic animals.
On the basis of multilocus enzyme electrophoresis and comparative sequence analysis of orthologous genes, two species, S. enterica and S. bongori, have been assigned to the genus Salmonella (18, 46). S. enterica is further subdivided into seven subspecies designated with roman numerals (18, 44). While S. bongori and S. enterica subspecies II, IIIa, IIIb, IV, VI, and VII are mainly associated with cold-blooded vertebrates, members of S. enterica subspecies I are frequently isolated from avian and mammalian hosts (44). For instance, of the 90,201 Salmonella isolates collected between 1977 and 1992 by the German National Reference Center for Enteric Pathogens from humans and warm-blooded animals, 89,798 isolates (99.55%) belonged to S. enterica subspecies I (1). Currently it is not clear which virulence mechanisms are responsible for the apparent adaptation of S. enterica subspecies I to circulation within populations of warm-blooded animals.
S. bongori or S. enterica subspecies II to VII are able to infect humans, colonize the intestine and cause disease (1). Human infections with serotypes of S. bongori and S. enterica subspecies II to VII are rare and are usually the result of contact with reptiles (21, 29, 42, 60). The symptoms of intestinal and extraintestinal infections caused by reptile-associated Salmonella serotypes in humans are, however, indistinguishable from those produced by nontyphoidal serotypes of S. enterica subspecies I (1). These data demonstrate that S. bongori or S. enterica subspecies II to VII are pathogenic for humans and suggest that the scarcity of clinical cases of illness is a result of the absence of these serotypes from animals from which we draw our food supply. This raises the question as to which genetic traits enable serotypes of S. enterica subspecies I to establish themselves in populations of livestock or domestic fowl. Theoretical models to describe the general principles concerning the ability of pathogens to invade, persist, and spread within a host population are well developed (3). In order to become established in populations of domesticated animals, a pathogen must generate on average more than one secondary case of infection from a primary case. The average number of animals in a susceptible host population which become infected from a single case can be defined as the basic case reproductive number, R0 (3). The basic case reproductive number of S. enterica subspecies I serotypes for higher vertebrates must therefore be greater than one, since these pathogens circulate in warm-blooded host populations. The absence of S. bongori and S. enterica subspecies II to VII serotypes from populations of livestock or domestic fowl, on the other hand, suggests that their basic case reproductive number for higher vertebrates is less than one, a property apparently independent of their ability to cause illness in these hosts (1). Thus, an expansion in host range may have involved the acquisition of one or more genetic determinants by a common ancestor of S. enterica subspecies I which increased the basic case reproductive number (but not necessarily the lethality) of this organism for warm-blooded vertebrates. To predict how acquisition of new genetic material by a common ancestor of the S. enterica subspecies I lineage may have contributed to its expansion in host range, it is helpful to apply theoretical models which combine epidemiology with population biology (3). In the case of direct transmission (by the fecal-oral route or any other route), the basic case reproductive number of a pathogen is directly proportional to the duration, D, for which an infected host can transmit the disease; the probability, β, at which the disease is transmitted from an infected animal to a susceptible host; and the density of susceptible hosts, X (2):
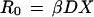 |
1 |
An infected host can transmit the disease until it either dies of natural causes (at the natural mortality rate, b), is killed by the pathogen (at the disease-induced mortality rate, α), or is able to clear the infection (at the clearance rate, ν) (3). Thus the average lifespan of an infectious host, D, can be described as follows:
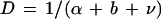 |
2 |
After combining equations 1 and 2 it becomes clear that a reduction of either the clearance rate, ν, or the disease induced mortality rate, α, will result in an increase in the basic case reproductive number, R0, of a pathogen:
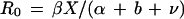 |
3 |
There is no evidence that members of S. enterica subspecies I are less virulent or cause lower mortality rates in warm-blooded hosts than serotypes of S. bongori or S. enterica II to VII. However, it is possible that a common ancestor of the S. enterica subspecies I lineage may have increased its basic case reproductive number for warm-blooded animals by reducing the rate at which the infection is cleared from the feces. The genetic determinants responsible for this phenotype are expected to be present in S. enterica subspecies I but absent from serotypes of S. bongori and S. enterica subspecies II to VII.
Here we describe the identification of a gene, termed shdA, which is specific to S. enterica subspecies I serotypes and study its role in fecal shedding during S. enterica serotype Typhimurium infection of mice. This analysis is relevant for human health, since little is known about virulence determinants required for circulation of enteric pathogens within animal reservoirs from which we draw our food supply.
MATERIALS AND METHODS
Bacterial strains and culture conditions.
Strains CL1509 (aroA::Tn10), IR715 (virulent nalidixic acid resistant derivative), AJB82 (aroA::Tn10 invA::TnphoA) and AJB75 (IR715 invA::TnphoA) are derivatives of serotype Typhimurium strain ATCC 14028 (10, 56, 57). The Salmonella reference B and SARC collections have been published recently (17, 18). Escherichia coli strains S17-1 λpir, and DH5α are described elsewhere (26, 53). Strains were cultured aerobically at 37°C in Luria-Bertani (LB) broth supplemented with the following antibiotics as appropriate at the indicated concentrations: carbenicillin, 100 mg/liter (LB+Cb); chloramphenicol, 30 mg/liter (LB+Cm); tetracycline, 20 mg/liter (LB+Tc); kanamycin 60 mg/liter (LB+Km); or nalidixic acid, 50 mg/liter (LB+Nal).
Southern hybridization.
Isolation of genomic DNA and Southern transfer of DNA onto a nylon membrane was performed as recently described (5). Hybridization was performed at 65°C in solutions without formamide. Two 15-min washes were performed under nonstringent conditions at room temperature in 2× SSC (1× SSC is 0.15 M NaCl plus 0.015 M sodium citrate)–0.1% sodium dodecyl sulfate. Labeling of DNA probes and detection of nucleotide sequences were performed using the labeling and detection kit (nonradioactive) from NEN.
Cloning of shdA.
A cosmid bank of serotype Typhimurium strain ATCC 14028 constructed in pLAFR2 and propagated in E. coli LE392 has been described previously (38). The bank was spread on LB+Tc plates, and 456 colonies were picked and grown individually overnight. Cosmid DNA was prepared from 19 pools, each containing 24 overnight cultures. Each pool was digested with EcoRI, separated on an agarose gel and hybridized with probe p5A8. The DNA of one pool hybridized with probe p5A8. The 24 strains representing this pool were then grown individually, and cosmid DNA was isolated, digested with EcoRI, and separated on an agarose gel. Southern hybridization was performed to identify a cosmid hybridizing with probe p5A8. Plasmid DNA for sequencing was isolated using ion-exchange columns from Qiagen. Sequencing was performed by the dideoxy chain termination method (50), using an AutoRead Sequencing Kit (Pharmacia) and an ALF automatic sequenator. The nucleotide sequences were analyzed using the MacVector 6.0.1 software package (Oxford Molecular Group).
Construction of mutants.
Bacteriophage P22 HT105/1 int− was used for generalized transduction of the aroA::Tn10 marker from serotype Typhimurium strain CL1509 into RAK1 or AJB75 (10). Transductants were routinely purified from contaminating phage by streaking the strain twice for single colonies on Evans blue uridine plates (16). Subsequently, strains were tested in a cross streak for P22 sensitivity. Transductants were tested for growth on M9 minimal medium agar plates and on minimal medium agar plates supplemented with aromatic amino acids (39). For construction of a shdA mutant, a 3-kb fragment of cosmid pRK824 was cloned into pBluescript KS(−) (52) to give rise to plasmid pRA38. A chloramphenicol acetyltransferase (cat) gene was ligated into the BamHI site of the shdA open reading frame cloned in pRA38 (see Fig. 3). The insert of the resulting plasmid (pRA55) was excised with EcoRI and SalI and cloned into the EcoRI- and SalI-restricted suicide vector pGP704 (33) to give rise to plasmid pRA56. Exconjugants of a mating between serotype Typhimurium strain IR715 and E. coli strain S17-1 λpir(pRA56) were selected on LB+Cm+Nal plates. An exconjugant which was resistant to chloramphenicol (shdA::cat allele) but sensitive to carbenicillin (through loss of pGP704) was identified by patching individual colonies on LB+Cb plates and was termed RAK1.
FIG. 3.

(Top) Pustell alignment of the ShdA amino acid sequence against itself (window size, 10; minimum identity, 60%). Lines parallel to the diagonal identified direct amino acid repeats. A predicted signal peptide and the C-terminal domain, which has homology to AIDA-I, MisL, and IcsA, are indicated in the ShdA primary structure shown as an arrow below the Pustell alignment. The positions of nine copies of a 63-amino-acid repeat (hatched bars numbered 1 to 9) and three copies of a 102-amino-acid repeat (closed bars numbered I to III) are indicated in the N-terminal domain of ShdA. The location of four direct repeats of a 12-amino-acid sequence in the C-terminal domain of ShdA are indicated (A-D). (Bottom) A CLUSTAL alignment of repeats I to III, 1 to 9, and A to D is shown. Identical residues (shaded boxes) and residues with similar biochemical properties (open boxes) are indicated.
Animal experiments.
Six- to eight-week-old female BALB/c (ByJ; Jackson Laboratory) mice were used throughout this study. Bacteria were routinely cultured as standing overnight cultures prior to infection. In all experiments the bacterial titer of the inoculum was determined by spreading serial 10-fold dilutions on agar plates containing appropriate antibiotics and determining the number of CFU.
The intestinal organ culture model has been described previously (7). The intestine was ligated at the distal end, filled with 1 ml of a bacterial suspension containing 5 × 107 to 8 × 107 CFU of a mixture of the two competing strains, and then ligated at the proximal end and incubated for 30 min at 37°C in 5% CO2. Nonadherent bacteria were removed by five washes in phosphate-buffered saline (PBS), and sections of intestinal wall were homogenized in 5 ml of PBS. Dilutions were spread on LB plates containing the appropriate antibiotics. Experiments were repeated with organs from three different animals.
For competitive infection experiments, groups of four mice were infected by oral gavage with an approximately 1:1 mixture of mutant and isogenic parent strains at a dose of approximately 109 CFU/mouse. Fecal pellets were collected daily and homogenized in 1 ml of PBS. The limit of detection was approximately 0.08 CFU/mg of feces. Dilutions of fecal pellets were plated on LB plates containing the appropriate antibiotics. Data were normalized by dividing the output ratio (CFU of mutant/CFU of wild type) by the input ratio (CFU of mutant/CFU of wild type). In case only one bacterial strain was recovered from fecal pellets, the limit of detection was determined for the missing strain and used to calculate of a minimum mutant/wild type ratio. All data were converted logarithmically prior to the calculation of averages and statistical analysis. Student's t test was used to determine whether the mutant/wild type ratio in specimens recovered from infected organs or fecal pellets was significantly different from the mutant/wild type ratio present in the inoculum.
For single infection experiments, groups of 12 BALB/c mice were inoculated with 109 CFU of either CL1509 (aroA::Tn10-tetr) or RAK7 (aroA::Tn10-tetr shdA::Cmr). The presence of inoculum strain in fecal pellets was determined on 29 days during the first 79 days postinoculation (days 1 to 16, 18, 21, 24, 27, 31, 34, 37, 44, 51, 58, 65, 72, and 79 postinoculation). Approximately 20 mg of fresh fecal pellets were resuspended in PBS (pH 7.4), and bacteria were enumerated on LB agar plates containing tetracycline (20 μg/ml) or tetracycline (20 μg/ml) and chloramphenicol (50 μg/ml). The presence or absence of the test strain in fecal pellets was scored for each mouse (limit of detection, 0.1 CFU/mg of feces). The proportion of mice in each inoculum group on each day tested that contained serotype Typhimurium in fecal pellets was subjected to statistical analysis using the Wilcoxon's signed-rank test (n = 29).
The 50% lethal morbidity dose (LD50) of serotype Typhimurium mutants was estimated by infecting groups of four mice intragastrically with serial 10-fold dilutions of bacterial cultures in a 0.2-ml volume. Lethal morbidity was recorded at 28 days postinfection, and the estimated LD50 was calculated by the method of Reed and Muench (45).
RESULTS
Identification of a DNA region restricted to S. enterica subspecies I.
We have recently screened a bank of 400 S. enterica serotype Gallinarum Mud-Cam transposon mutants for virulence in day-of-hatch White Leghorn chicks. Virulence data obtained for individual mutants during the initial screen were inconsistent with experiments performed using this animal model to confirm attenuation of individual mutants. We determined the inconsistency of data to be the result of the antibiotic history from battery-reared chicks. The fact that virulence defects could not be confirmed for mutants identified in this screen prompted us to discontinue the study. However, prior to this, the DNA flanking the Mud-Cam insertion was cloned from one mutant, labeled, and used as a probe to determine its phylogenetic distribution. Southern blot analysis was performed with genomic DNA prepared from SARC (18). This collection consists of 16 Salmonella serotypes representing S. bongori (formerly S. enterica subspecies V) and S. enterica subspecies I to VII. The 500-bp p5A8 DNA probe, derived from mutant G5A8, hybridized with genomic DNA of strains from subspecies I, but no hybridization signal was obtained with genomic DNA from isolates of S. bongori or S. enterica subspecies II to VII. Since the host range of S. enterica subspecies I differs from that of S. bongori or S. enterica subspecies II to VII we decided to further characterize this DNA region.
A cosmid (pRK824) containing the S. enterica subspecies I specific DNA region was cloned from an S. enterica serotype Typhimurium bank (38) by hybridization with probe p5A8. Restriction analysis of cosmid pRK824 indicated that it carried an insert of approximately 28 kb. To confirm that the cloned DNA region was restricted to S. enterica subspecies I, a 3-kb ClaI restriction fragment hybridizing with probe p5A8 was cloned to give rise to plasmid pRA38. The DNA probe derived from plasmid pRA38 hybridized with genomic DNA from S. enterica subspecies I but not genomic DNA from S. bongori or S. enterica subspecies II to VII (Fig. 1). To determine the distribution of the pRA38 DNA probe within S. enterica subspecies I, we used the SARB collection, which includes 72 strains representing 37 serotypes of S. enterica subspecies I (17). A DNA probe derived from plasmid pRA38 hybridized with 69 of the 72 strains of SARB collection. Southern blot analysis of 21 representative strains from SARB collection is shown in Fig. 1. No hybridization signal was obtained with genomic DNA from strains Dt1 (S. enterica serotype Decatur), Ts3 (S. enterica serotype Typhisuis) and En2 (S. enterica serotype Enteritidis). The multilocus enzyme electrophoresis profile of strain En2 is only distantly related to that of other S. enterica serotype Enteritidis clones (En1, En3, and En7) which are present in SARB and do hybridize with pRA38 (17).
FIG. 1.

Phylogenetic distribution of the shdA gene within the genus Salmonella. Southern blot analysis using representative serotypes of S. enterica (subspecies are indicated by roman numerals) and S. bongori is shown. Genomic DNA prepared from the serotypes indicated on the left (strain designations are indicated in parentheses) was hybridized with DNA probes pRA58 (left panel) and pRA38 (right panel). The location of these DNA probes (closed bars) relative to xseA and shdA (arrows) is indicated on the map shown at the top.
The shdA gene is located in the xseA-hisS intergenic region.
The cosmid (pRK824) contained at least one border of the subspecies I specific DNA region, since a DNA probe (pRA58) generated from a 1.6-kb ClaI restriction fragment hybridized with all serotypes of the SARC and SARB collections (Fig. 1). Nucleotide sequence analysis of pRA58 revealed that it contained the 3′ end of the serotype Typhimurium xseA gene, which encodes the large subunit of exonuclease VII. Since this gene is shared by both E. coli and serotype Typhimurium, it is likely to account for the hybridization signal observed with the S. bongori or S. enterica subspecies II to VII serotypes. Sequence homology between the E. coli and serotype Typhimurium sequence ended 21 bp prior to the 3′ end of xseA, which defined the left end of the S. enterica subspecies I-specific DNA region. In the sequence of the E. coli K-12 genome, the xseA gene is located between the guaAB and hisS loci at 54 min (14). However, genetic mapping data suggest that in serotype Typhimurium, guaAB and hisS are separated by a 30-kb DNA region which is absent from E. coli (Fig. 2) (47). Our data are consistent with the presence of the shdA gene on a genetic island present in the xseA-hisS intergenic region, which may be as large as 30 kb.
FIG. 2.

(Top) Comparison of the nucleotide sequences from E. coli (E.c.) and S. enterica serotype Typhimurium (S.t.) at the left boundary of the island. A putative termination loop located downstream of the shdA gene is indicated by arrows. (Bottom) Comparison of the genetic maps of E. coli and S. enterica serotype Typhimurium flanking the xseA gene. An approximately 30-kb DNA loop in the guaAB-hisS intergenic region, which is present in serotype Typhimurium but absent from E. coli, has been described by Riley and Krawiec (47) and is shown as an open bar.
The nucleotide sequence analysis was extended to include a total of 6,831 bp (GenBank accession no. AF091269). A single open reading frame of 6,105 bp transcribed in the opposite direction to xseA was identified and designated shdA. A putative termination loop was identified downstream of the translational stop codon of shdA (stem, bp 6482 to 6495; loop, bp 6496 to 6499; stem, bp 6500 to 6514). The C-terminal domain (477 amino acids) of the predicted ShdA protein exhibited homology to the C-terminal 440 amino acids of AIDA (34% identity) from diffuse adhering E. coli (11), the C-terminal 501 amino acids of MisL (36% identity) from serotype Typhimurium (13), and the C-terminal 353 amino acids of IcsA (VirG) (30% identity) from S. flexneri (12, 36). The C terminus also contained five copies of a 12-amino-acid repeat, which exhibited no homology to sequences in available databases (Fig. 3). Using the SignalP program (41a), a putative signal peptide was identified at the amino terminus of ShdA. Similar to AIDA, MisL, and IcsA, the signal peptide was atypical, with the predicted cleavage site (indicated by a slash) following the alanine residue at position 60 (LAMA/DNQV). The N-terminal domain of ShdA (amino acids 61 to 1558) did not exhibit homology to sequences in GenBank and contained nine copies of a 63-amino-acid repeat and three copies of a 102-amino-acid repeat (Fig. 3).
Effect of a mutation in shdA on the disease-induced mortality rate.
A serotype Typhimurium strain in which the shdA open reading frame was disrupted by insertion of a cat gene was constructed by allelic exchange and was designated RAK1. The insertion mutant was confirmed by Southern blot analysis of chromosomal DNA prepared from RAK1 and hybridization with the pRA38 DNA probe (data not shown). It is unlikely that insertional inactivation of shdA resulted in a polar effect on the expression of the adjacent xseA gene, since the shdA and xseA genes are transcribed in opposite orientations. The LD50 of strain RAK1 and that of its isogenic parent (IR715) were determined and found to be identical. These data show that mutational inactivation of shdA did not alter the disease-induced mortality rate during serotype Typhimurium infection of mice.
Effect of a mutation in shdA on fecal shedding.
We determined the contribution of shdA to bacterial shedding in the mouse typhoid model of serotype Typhimurium infection. Serotype Typhimurium causes lethal signs of disease in mice starting at day 5 postinfection. Thus, in order to study fecal shedding beyond day 5 postinfection, strain RAK1 (shdA) was attenuated for mouse virulence by introducing a mutation in aroA. Serotype Typhimurium aroA mutants are able to attach to and invade the intestinal mucosa and colonize deeper tissues but are unable to multiply rapidly at these sites. Since bacterial shedding results from bacterial colonization of an animal, we reasoned that inactivation of aroA was unlikely to mask the effect of other genes on shedding. To assess the effect of a mutation in shdA on bacterial shedding, a group of four mice was infected with equal numbers of CL1509 (aroA) and RAK7 (shdA aroA) bacteria, and the bacteria were recovered from fecal pellets on subsequent days. This analysis revealed that a mutation in shdA significantly decreased the number of serotype Typhimurium organisms shed in the feces (P < 0.01 at day 6 postinfection). The experiment was discontinued at day 6 postinfection, when RAK7 (shdA aroA) was not recovered from the fecal pellets of three mice, while strain CL1509 (aroA) was still shed with the feces of three animals.
The experiment was repeated with a group of six mice, and shedding was monitored until day 35 postinfection. All mice shed the inoculum on day 1 postinoculation, but on subsequent days shedding was intermittent and some animals cleared the inoculum. Again, CL1509 (aroA) was recovered in significantly higher numbers from fecal pellets than RAK7 (shdA aroA). More importantly, CL1509 was shed for a longer period than the shdA mutant (RAK7). Figure 4 shows the combined results of both shedding experiments. These data show that a mutation in shdA reduced the duration of bacterial shedding, which is indicative of an increased rate by which the host could clear serotype Typhimurium from intestinal contents shed with the feces. The shedding defect attributed to the shdA mutation was confirmed using single inoculation of groups of 12 mice with 109 CFU of either RAK7 (aroA shdA) or the CL1509 (aroA) parental strain. Shedding in fecal pellets was scored for the presence or absence of Salmonella in each inoculum group on 29 occasions over a 79-day period postinoculation. On 14 occasions during this period, a greater number of mice inoculated with the parental strain (CL1509) were shedding serotype Typhimurium than were those inoculated with the shdA mutant (RAK7). The opposite was true on only three occasions. Statistical analysis of these data using the Wilcoxon signed-rank test indicated that the shdA mutant was cleared significantly earlier than the parental strain (P < 0.01). Overall, these data suggest that a mutation in shdA increased the clearance rate of serotype Typhimurium from murine feces.
FIG. 4.

Recovery of bacteria from fecal pellets collected after inoculation of mice with an equal mixture of CL1509 (aroA) and RAK7 (shdA aroA). Data for days 1 to 6 were from 10 mice, and data for subsequent days were from 6 mice. (A) For each mouse, the output ratio (RAK7/CL1509) was determined daily. Data were converted logarithmically and are given as means ± standard errors (error bars). An asterisk below an error bar indicates that the RAK7/CL1509 output ratio was significantly different (P < 0.05) from that present in the inoculum. (B) Total numbers of CL1509 (open circles) and RAK7 (closed circles) recovered from fecal pellets of mice. The limit of detection (1.2 × 10−1 CFU/mg of feces) is indicated by a broken line. Each circle represents data for one strain from one animal. Animals for which no CFU of either CL1509 or RAK7 were detectable are indicated below the broken line along the x axis.
Effect of a mutation in invA on fecal shedding.
During competitive infection experiments, serotype Typhimurium strains carrying a mutation in invA are absent from feces more frequently than the wild type on days 3 and 5 postinoculation (10). However, the shedding defect previously reported for invA mutants was based on observation restricted to two occasions postinoculation. Observations from shedding experiments described above indicated that shedding is highly variable from day to day. In order to investigate whether a serotype Typhimurium strain containing a mutation in invA has a similar shedding defect to that observed for strains containing a mutation in shdA, eight mice were inoculated with an equal mixture of strain AJB82 (invA aroA) and its isogenic parental strain (CL1509). Fewer mice shed the invA mutant and at lower numbers at earlier time points. However, the opposite was observed at later time points (Fig. 5). Although a mutation in invA reduced bacterial shedding at early times postinfection, there was no evidence for an effect on bacterial clearance from the feces at the end of the experiment.
FIG. 5.

Recovery of bacteria from fecal pellets collected after inoculation of mice with an equal mixture of CL1509 (aroA) and AJB82 (invA aroA). (A) For each mouse, the output ratio (RAK82/CL1509) was determined daily. Data were converted logarithmically and are given as means ± standard errors (error bars). An asterisk below an error bar indicates that the RAK82/CL1509 output ratio was significantly different (P < 0.05) from that present in the inoculum. (B) Total numbers of CL1509 (open circles) and RAK82 (closed circles) recovered from fecal pellets of mice. The limit of detection (1.2 × 10−1 CFU/mg of feces) is indicated by a broken line. Each circle represents data for one strain from one animal. Animals for which no CFU of either CL1509 or RAK82 were detectable are indicated below the broken line along the x axis.
A mutation in shdA does not affect colonization of the villous intestine or Peyer's patches in an intestinal organ culture model.
It is known that invA is required for invasion of the mucosal epithelium, particularly at the Peyer's patches (25). To compare the contributions of shdA and invA to colonization of the small intestine, we used the intestinal organ culture model (7). Equal numbers of RAK1 (shdA) and its parent (IR715) were injected into loops formed from fresh mouse ileum, and following a 30-min incubation period, CFU of each strain were enumerated in the villous intestine and Peyer's patch regions. No significant difference in colonization of these tissues by the shdA strain and parental strain was observed. In a second experiment, an equal mixture of a serotype Typhimurium invA mutant (AJB75) and its isogenic parent (IR715) was inoculated into ligated intestinal loops. The invA mutant (AJB75) was recovered in significantly lower numbers from Peyer's patches than the parental strain, thus confirming the role of SPI1 in colonizing this organ.
DISCUSSION
A primary pathogen can be defined as an organism which is capable of entering a host, finding a unique niche in which to multiply, avoiding or subverting the host defenses, and being transmitted to a susceptible host (24). All members of the genus Salmonella fit this description, as they are pathogenic for humans (1). However, serotypes of S. enterica subspecies I differ from S. bongori and S. enterica subspecies II to VII serotypes with regard to animal reservoir. While human infections with S. bongori and S. enterica subspecies II to VII are rare and result from contact with reptiles (21, 29, 42, 60), serotypes of S. enterica subspecies I are frequently associated with disease, and most cases can be traced back to livestock or domestic fowl (4, 41). Thus, it could be speculated that serotypes of S. enterica subspecies I possess one or more genes which enable these pathogens to invade, persist, and spread within warm-blooded host populations, thereby resulting in their introduction into food items originating from domesticated animals. S. bongori and S. enterica subspecies II to VII, on the other hand, lack these genes and are unable to circulate in populations of livestock and domestic fowl.
We characterized shdA, a gene encoded on a genetic island which is present in serotypes of S. enterica subspecies I. Unlike previously identified virulence gene clusters, such as SPI1, SPI2, SPI3, agf, fim, lpf, and spv, the shdA gene was absent from lineages other than subspecies I (6, 13, 19, 30, 37). Although virulence determinants, which are restricted to subspecies I have been identified previously, these are present in only a small number of serotypes. For instance, the SARB collection which consists of 72 strains from S. enterica subspecies I, contains 3 isolates carrying the viaB region, 10 isolates possessing the sef operon, and 9 isolates hybridizing with the pef operon (6, 51). In contrast, shdA was present in 69 of the 72 strains of the SARB collection, suggesting that it was acquired early in the divergence of the S. enterica subspecies I lineage (Fig. 1). It has been postulated that the ability of S. enterica subspecies I serotypes to circulate in populations of warm-blooded animals is a new trait, since extant serotypes of all other phylogenetic lineages within the genus Salmonella are associated with cold-blooded vertebrates (8, 43). Our data suggest that this expansion in host range to include warm-blooded vertebrates was accompanied by acquisition of the shdA gene by a common ancestor of S. enterica subspecies I.
Our results show that mutational inactivation of shdA resulted in recovery of serotype Typhimurium at lower numbers and for a shorter period of time from murine fecal pellets than its isogenic parent (Fig. 4). It is unlikely that shdA is the only factor involved in prolonged fecal shedding of serotype Typhimurium from mice. Indeed, previous studies have shown that Typhimurium strains containing a mutation in invA are less likely to be recovered from fecal pellets of mice at days 3 and 5 postinfection than their isogenic parent (10). However, comparison of the shedding defects of strains AJB82 (invA) and RAK7 (shdA) demonstrated that inactivation of shdA reduced bacterial shedding at later time points (day 11 postinfection or subsequent days) to a greater extent than a mutation in invA (Fig. 4 and 5). Another group of virulence determinants previously implicated in bacterial shedding are fimbrial adhesins of serotype Typhimurium. A serotype Typhimurium agf pef fim lpf mutant is recovered in significantly lower numbers from fecal pellets at 5 days postinfection than the isogenic wild type during a competitive infection experiment (58). Thus, inactivation of genes required for attachment to or invasion of the intestinal mucosa may result in reduced bacterial shedding. While attachment of serotype Typhimurium to the murine small intestine can be detected using ligated ileal loops (7, 9), mutational inactivation of shdA did not reduce bacterial numbers recovered from this model (Fig. 6A). In contrast, a serotype Typhimurium invA mutant colonized Peyer's patches at reduced levels in the organ culture model, suggesting that bacterial invasion can be detected using this assay (Fig. 6B). These data suggest that unlike mutations in fimbrial biosynthesis or invasion genes, the shedding defect of a shdA mutant was not caused by decreased bacterial attachment to or invasion of the mucosa of the murine small intestine but may be due to a different mechanism.
FIG. 6.

Recovery of bacteria from the intestinal organ culture model. Intestinal loops were infected with an equal mixture of RAK1 (shdA) and IR715 (wild type) (A) or AJB75 (invA) and IR715 (wild type) (B). The output ratios were determined for Peyer's patches (PP) and villous intestine (VI). Data were converted logarithmically and are given as means ± standard errors (error bars).
A mutation in invA or the simultaneous inactivation of the agf, pef, fim, and lpf operons results in a 50- and 26-fold attenuation of serotype Typhimurium for mouse virulence, respectively (25, 58). The attenuating effect of these mutations suggests that the corresponding attachment or invasion genes increase the disease-induced mortality rate, α, which is expected to result in a reduction of the basic case reproductive number, R0 of serotype Typhimurium (equation 3). At the same time, however, invA, agf, pef, fim, and lpf may reduce the clearance rate, ν, which would be predicted to increase the basic case reproductive number. It is therefore difficult to predict whether the net result of expressing fimbriae or invasion genes is an increase or a decrease in the basic case reproductive number of serotype Typhimurium. In contrast, mutational inactivation of shdA did not reduce the disease-induced mortality rate, α, but decreased the duration of shedding (Fig. 4). This phenotype is consistent with a role of shdA in decreasing the clearance rate, ν, thereby resulting in an increase in the basic case reproductive number, R0, of serotype Typhimurium (equation 3). The phylogenetic distribution of shdA and its predicted effect on the basic case reproductive number are consistent with the idea that acquisition of this gene may have contributed to the expansion in host range of S. enterica subspecies I to include warm-blooded animals.
ACKNOWLEDGMENTS
We are grateful to Renée Tsolis for helpful suggestions on the manuscript and Kenneth Sanderson for providing strains from the SARB and SARC collections.
Work in A.B.'s laboratory is supported by Public Health Service grants AI40124 and AI44170 and grant 9802610 from the U.S. Department of Agriculture.
REFERENCES
- 1.Aleksic S, Heinzerling F, Bockemühl J. Human infection caused by salmonellae of subspecies II to VI in Germany, 1977-1992. Zentbl Bakteriol. 1996;283:391–398. doi: 10.1016/s0934-8840(96)80074-0. [DOI] [PubMed] [Google Scholar]
- 2.Anderson R M. Evolutionary pressures in the spread and persistence of infectious agents in vertebrate populations. Parasitology. 1995;111(Suppl.):S15–S31. doi: 10.1017/s003118200007579x. [DOI] [PubMed] [Google Scholar]
- 3.Anderson R M, May R M. Coevolution of host and parasites. Parasitology. 1982;85:411–426. doi: 10.1017/s0031182000055360. [DOI] [PubMed] [Google Scholar]
- 4.Aserkoff B, Schroeder S A, Brachman P S. Salmonellosis in the United States—a five-year review. Am J Epidemiol. 1970;92:13–24. doi: 10.1093/oxfordjournals.aje.a121175. [DOI] [PubMed] [Google Scholar]
- 5.Ausubel F M, Brent R, Kingston R E, Moore D D, Seidman J G, Smith J A, Struhl K, editors. Current protocols in molecular biology. J. New York, N.Y: Wiley & Sons; 1994. [Google Scholar]
- 6.Bäumler A J, Gilde A J, Tsolis R M, van der Velden A W M, Ahmer B M M, Heffron F. Contribution of horizontal gene transfer and deletion events to the development of distinctive patterns of fimbrial operons during evolution of Salmonella serotypes. J Bacteriol. 1997;179:317–322. doi: 10.1128/jb.179.2.317-322.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bäumler A J, Tsolis R M, Bowe F, Kusters J G, Hoffmann S, Heffron F. The pef fimbrial operon mediates adhesion to murine small intestine and is necessary for fluid accumulation in infant mice. Infect Immun. 1996;64:61–68. doi: 10.1128/iai.64.1.61-68.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bäumler A J, Tsolis R M, Ficht T A, Adams L G. Evolution of host adaptation in Salmonella enterica. Infect Immun. 1998;66:4579–4587. doi: 10.1128/iai.66.10.4579-4587.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bäumler A J, Tsolis R M, Heffron F. The lpf fimbrial operon mediates adhesion of Salmonella typhimurium to murine Peyer's patches. Proc Natl Acad Sci USA. 1996;93:279–283. doi: 10.1073/pnas.93.1.279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bäumler A J, Tsolis R M, Valentine P J, Ficht T A, Heffron F. Synergistic effect of mutations in invA and lpfC on the ability of Salmonella typhimurium to cause murine typhoid. Infect Immun. 1997;65:2254–2259. doi: 10.1128/iai.65.6.2254-2259.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Benz I, Schmidt M A. AIDA-I, the adhesin involved in diffuse adherence of the diarrhoeagenic Escherichia coli strain 2787 (O126:H27), is synthesized via a precursor molecule. Mol Microbiol. 1992;6:1539–1546. doi: 10.1111/j.1365-2958.1992.tb00875.x. [DOI] [PubMed] [Google Scholar]
- 12.Bernardini M L, Mounier J, d'Hauteville H, Coquis-Rondon M, Sansonetti P J. Identification of icsA, a plasmid locus of Shigella flexneri that governs bacterial intra- and intercellular spread through interaction with F-actin. Proc Natl Acad Sci USA. 1989;86:3867–3871. doi: 10.1073/pnas.86.10.3867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Blanc-Potard A B, Solomon F, Kayser J, Groisman E A. The SPI-3 pathogenicity island of Salmonella enterica. J Bacteriol. 1999;181:998–1004. doi: 10.1128/jb.181.3.998-1004.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Blattner F R, Plunkett G, Bloch C A, Perna N T, Burland V, Riley M, Colladovides J, Glasner J D, Rode C K, Mayhew G F, Gregor J, Davis N W, Kirkpatrick H A, Goeden M A, Rose D J, Mau B, Shao Y. The complete genome sequence of Escherichia coli K-12. Science. 1997;277:5331. doi: 10.1126/science.277.5331.1453. [DOI] [PubMed] [Google Scholar]
- 15.Blaxland J D, Sojka J W, Smither A M. Avian salmonellosis in England and Wales 1948-56 with comment on its prevention and control. Vet Rec. 1958;70:374–382. [Google Scholar]
- 16.Bochner B R. Curing bacterial cells of lysogenic viruses by using UCB indicator plates. BioTechniques. 1984;2:234–240. [Google Scholar]
- 17.Boyd E F, Wang F-S, Beltran P, Plock S A, Nelson K, Selander R K. Salmonella reference collection B (SARB): strains of 37 serovars of subspecies I. J Gen Microbiol. 1993;139:1125–1132. doi: 10.1099/00221287-139-6-1125. [DOI] [PubMed] [Google Scholar]
- 18.Boyd E F, Wang F-S, Whittam T S, Selander R K. Molecular genetic relationship of the Salmonellae. Appl Environ Microbiol. 1996;62:804–808. doi: 10.1128/aem.62.3.804-808.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Boyd F E, Hartl D L. Salmonella virulence plasmid: modular acquisition of the spv virulence region by an F-plasmid in Salmonella enterica subspecies I and insertion into the chromosome in subspecies II, IIIa, IV, and VII isolates. Genetics. 1998;149:1183–1190. doi: 10.1093/genetics/149.3.1183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Burnham B R, Atchley D H, DeFusco R P, Ferris K E, Zicarelli J C, Lee J H, Angulo F J. Prevalence of fecal shedding of salmonella organisms among captive green iguanas and potential public health implications. J Am Vet Med Assoc. 1998;213:48–50. [PubMed] [Google Scholar]
- 21.Chiodini R J, Sundberg J P. Salmonellosis in reptiles: a review. Am J Epidemiol. 1981;113:494–499. doi: 10.1093/oxfordjournals.aje.a113124. [DOI] [PubMed] [Google Scholar]
- 22.Devi S, Murray C J. Salmonella carriage rate amongst school children—a three year study. S E Asian J Trop Med Public Health. 1991;22:357–361. [PubMed] [Google Scholar]
- 23.Edwards P R, McWhorther A C, Fife M A. The Arizona group of enterobacteriaceae in animals and man. Bull WHO. 1956;14:511–528. [PMC free article] [PubMed] [Google Scholar]
- 24.Falkow S. What is a pathogen? ASM News. 1997;63:359–365. [Google Scholar]
- 25.Galán J E, Curtiss R., III Cloning and molecular characterization of genes whose products allow Salmonella typhimurium to penetrate tissue culture cells. Proc Natl Acad Sci USA. 1989;86:6383–6387. doi: 10.1073/pnas.86.16.6383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Grant S G N, Jessee J, Bloom F R, Hanahan D. Differential plasmid rescue from transgenic mouse DNAs into Escherichia coli methylation-restriction mutants. Proc Natl Acad Sci USA. 1990;87:4645–4649. doi: 10.1073/pnas.87.12.4645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Gronstol H, Osborne A D, Pethiyagoda S. Experimental Salmonella infection in calves. 1. The effect of stress factors on the carrier state. J Hyg London. 1974;72:155–162. doi: 10.1017/s0022172400023342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Habermalz D, Pietzsch O. Identification of Arizona bacteria. A contribution to the problem of salmonella infections among reptiles and amphibians in zoological gardens. Zentralbl Bakteriol Orig A. 1973;225:323–342. . (In German.) [PubMed] [Google Scholar]
- 29.Hall M L M, Rowe B. Salmonella arizonae in the United Kingdom from 1966 to 1990. Epidemiol Infect. 1991;108:59–65. doi: 10.1017/s0950268800049505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hensel M, Shea J E, Bäumler A J, Gleeson C, Blattner F, Holden D W. Analysis of the boundaries of Salmonella pathogenicity island 2 and the corresponding chromosomal region of Escherichia coli K-12. J Bacteriol. 1997;179:1105–1111. doi: 10.1128/jb.179.4.1105-1111.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hinz K H, Legutko P, Schroeter A, Lehmacher W, Hartung M. Prevalence of motile salmonellae in egg-laying hens at the end of the laying period. Zentbl Veterinarmed B. 1996;43:23–33. . (In German.) [PubMed] [Google Scholar]
- 32.Iveson J B, Mackay-Scollay E M, Bamford V. Salmonella and Arizona in reptiles and man in Western Australia. J Hyg (London) 1969;67:135–145. doi: 10.1017/s0022172400041516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kinder S A, Badger J L, Bryant G O, Pepe J C, Miller V L. Cloning of the YenI restriction endonuclease and methyltransferase from Yersinia enterocolitica serotype O:8 and construction of a transformable R−M+ mutant. Gene. 1993;136:271–275. doi: 10.1016/0378-1119(93)90478-l. [DOI] [PubMed] [Google Scholar]
- 34.Koopman J P, Janssen F G. The occurrence of salmonellas and lactose-negative Arizonas in reptiles in The Netherlands, and a comparison of three enrichment methods used in their isolation. J Hyg (London) 1973;71:363–371. doi: 10.1017/s0022172400022816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kotova A L, Kondratskaya S A, Yasutis I M. Salmonella carrier state and biological characteristics of the infectious agent. J Hyg Epidemiol Microbiol Immunol. 1988;32:71–78. [PubMed] [Google Scholar]
- 36.Lett M C, Sasakawa C, Okada N, Sakai T, Makino S, Yamada M, Komatsu K, Yoshikawa M. virG, a plasmid-coded virulence gene of Shigella flexneri: identification of the virG protein and determination of the complete coding sequence. J Bacteriol. 1989;171:353–359. doi: 10.1128/jb.171.1.353-359.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Li J, Ochman H, Groisman E A, Boyd E F, Solomon F, Nelson K, Selander R K. Relationship between evolutionary rate and cellular location among the Inv/Spa invasion proteins of Salmonella enterica. Proc Natl Acad Sci USA. 1995;92:7252–7256. doi: 10.1073/pnas.92.16.7252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Libby S J, Goebel W, Ludwig A, Buchmeier N, Bowe F, Fang F C, Guiney D G, Songer J G, Heffron F. A cytolysin encoded by Salmonella is required for survival within macrophages. Proc Natl Acad Sci USA. 1994;91:489–493. doi: 10.1073/pnas.91.2.489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Maniatis T, Sambrook J, Fritsch E F. Molecular cloning: a laboratory manual. 2nd ed. Cold Spring Harbor, N.Y: Cold Spring Harbor Laboratory Press; 1989. [Google Scholar]
- 40.McCain C S, Powell K C. Asymptomatic salmonellosis in healthy adult horses. J Vet Diagn Investig. 1990;2:236–237. doi: 10.1177/104063879000200318. [DOI] [PubMed] [Google Scholar]
- 41.McCoy J H. Trends in salmonella food poisoning in England and Wales 1941-72. J Hyg (London) 1975;74:271–282. doi: 10.1017/s0022172400024347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41a.Nielsen H, Engelbrecht J, Brunak S, von Heijne G. Identification of prokaryotic and eukaryotic signal peptides and prediction of cleavage sites. Protein Eng. 1997;10:1–6. doi: 10.1093/protein/10.1.1. [DOI] [PubMed] [Google Scholar]
- 42.Noskin G A, Clarke J T. Salmonella arizonae bacteremia as the presenting manifestation of human immunodeficiency virus infection following rattlesnake meat ingestion. Rev Infect Dis. 1990;12:514–517. doi: 10.1093/clinids/12.3.514. [DOI] [PubMed] [Google Scholar]
- 43.Ochman H, Wilson A C. Evolution in bacteria: evidence for a universal substitution rate in cellular genomes. J Mol Evol. 1987;26:74–86. doi: 10.1007/BF02111283. [DOI] [PubMed] [Google Scholar]
- 44.Popoff M Y, Le Minor L. Antigenic formulas of the Salmonella serovars. 5th ed. Paris, France: WHO Collaborating Center for Reference and Research on Salmonella, Institut Pasteur; 1992. [Google Scholar]
- 45.Reed L J, Muench H. A simple method for estimating fifty percent endpoints. Am J Hyg. 1938;27:493–497. [Google Scholar]
- 46.Reeves M W, Evins G M, Heiba A A, Plikaytis B D, Farmer J J., III Clonal nature of Salmonella typhi and its genetic relatedness to other salmonellae as shown by multilocus enzyme electrophoresis, and proposal of Salmonella bongori comb. nov. J Clin Microbiol. 1989;27:313–320. doi: 10.1128/jcm.27.2.313-320.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Riley M, Krawiec S. Genome organization. In: Neidhardt F C, editor. Escherichia coli and Salmonella typhimurium: cellular and molecular biology. Vol. 2. Washington, D.C.: American Society for Microbiology; 1987. pp. 967–981. [Google Scholar]
- 48.Roggendorf M, Muller H E. Enterobacteria from reptiles. Zentbl Bakteriol Parasitenkd Hyg Abt I Orig A. 1976;236:22–35. [PubMed] [Google Scholar]
- 49.Samuel J L, Eccles J A, Francis J. Salmonella in the intestinal tract and associated lymph nodes of sheep and cattle. J Hyg. 1981;87:225–232. doi: 10.1017/s0022172400069448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Sanger F, Nicklen S, Coulson A R. DNA sequencing with chain-terminating inhibitors. Proc Natl Acad Sci USA. 1977;74:5463–5467. doi: 10.1073/pnas.74.12.5463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Selander R K, Beltran P, Smith N H, Helmuth R, Rubin F A, Kopecko D J, Ferris K, Tall B D, Cravioto A, Musser J M. Evolutionary genetic relationships of clones of Salmonella serovars that cause human typhoid and other enteric fevers. Infect Immun. 1990;58:2262–2275. doi: 10.1128/iai.58.7.2262-2275.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Short J M, Fernandez J M, Sorge J A, Huse W D. λZAP: a bacteriophage expression vector with in vivo excision properties. Nucleic Acids Res. 1988;16:7583–7600. doi: 10.1093/nar/16.15.7583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Simon R, Priefer U, Puhler A. A broad host range mobilization system for in vivo genetic engineering: transposon mutagenesis in Gram-negative bacteria. Bio/Technology. 1983;1:784–791. [Google Scholar]
- 54.Smith B P, DaRoden L, Thurmond M C, Dilling G W, Konrad H, Pelton J A, Picanso J P. Prevalence of salmonellae in cattle and in the environment of California dairies. J Am Vet Med Assoc. 1994;205:467–471. [PubMed] [Google Scholar]
- 55.Sojka W J, Wray C. Incidence of Salmonella infection in animals in England and Wales, 1968-73. Vet Rec. 1975;96:280–284. doi: 10.1017/s0022172400055923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Stojiljkovic I, Bäumler A J, Heffron F. Ethanolamine utilization in Salmonella typhimurium: nucleotide sequence, protein expression and mutational analysis of the cchA cchB eutE eutJ eutG eutH gene cluster. J Bacteriol. 1995;177:1357–1366. doi: 10.1128/jb.177.5.1357-1366.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Tsolis R M, Bäumler A J, Stojiljkovic I, Heffron F. Fur regulon of Salmonella typhimurium: identification of new iron-regulated genes. J Bacteriol. 1995;177:4628–4637. doi: 10.1128/jb.177.16.4628-4637.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.van der Velden A W M, Baumler A J, Tsolis R M, Heffron F. Multiple fimbrial adhesins are required for full virulence of Salmonella typhimurium in mice. Infect Immun. 1998;66:2803–2808. doi: 10.1128/iai.66.6.2803-2808.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Wesselinoff W. Further studies of salmonella findings in reptiles. IV. Communication. Zentbl Bakteriol Orig A. 1977;239:483–487. . (In German.) [PubMed] [Google Scholar]
- 60.Woodward D L, Khakhria R, Johnson W M. Human salmonellosis associated with exotic pets. J Clin Microbiol. 1997;35:2786–2790. doi: 10.1128/jcm.35.11.2786-2790.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]