

Summary
An Xq22.2 region upstream of PLP1 has been proposed to underly a neurological disease trait when deleted in 46,XX females. Deletion mapping revealed that heterozygous deletions encompassing the smallest region of overlap (SRO) spanning six Xq22.2 genes (BEX3, RAB40A, TCEAL4, TCEAL3, TCEAL1, and MORF4L2) associate with an early-onset neurological disease trait (EONDT) consisting of hypotonia, intellectual disability, neurobehavioral abnormalities, and dysmorphic facial features. None of the genes within the SRO have been associated with monogenic disease in OMIM. Through local and international collaborations facilitated by GeneMatcher and Matchmaker Exchange, we have identified and herein report seven de novo variants involving TCEAL1 in seven unrelated families: three hemizygous truncating alleles; one hemizygous missense allele; one heterozygous TCEAL1 full gene deletion; one heterozygous contiguous deletion of TCEAL1, TCEAL3, and TCEAL4; and one heterozygous frameshift variant allele. Variants were identified through exome or genome sequencing with trio analysis or through chromosomal microarray. Comparison with previously reported Xq22 deletions encompassing TCEAL1 identified a more-defined syndrome consisting of hypotonia, abnormal gait, developmental delay/intellectual disability especially affecting expressive language, autistic-like behavior, and mildly dysmorphic facial features. Additional features include strabismus, refractive errors, variable nystagmus, gastroesophageal reflux, constipation, dysmotility, recurrent infections, seizures, and structural brain anomalies. An additional maternally inherited hemizygous missense allele of uncertain significance was identified in a male with hypertonia and spasticity without syndromic features. These data provide evidence that TCEAL1 loss of function causes a neurological rare disease trait involving significant neurological impairment with features overlapping the EONDT phenotype in females with the Xq22 deletion.
Keywords: TCEAL1, neurodevelopmental disorder, NDD, early-onset neurological disease trait, EONDT, X-linked disease trait, developmental delay/intellectual disability, DD/ID, hypotonia, Xq22.2 deletion
Graphical abstract
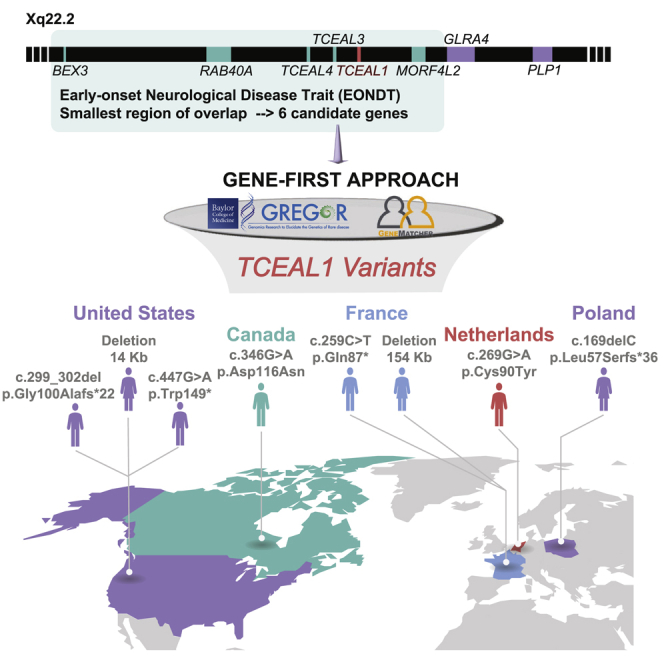
Applying a gene-first approach and worldwide gene-matching, we identify eight individuals with variants in TCEAL1, a candidate gene for the early-onset neurological disease trait (EONDT) in females with Xq22.2 deletion. The neurodevelopmental disorder observed overlaps that described in Xq22.2 deletion females, implicating TCEAL1 as the driver gene.
Main text
The Xq22.2 sub-band is ∼1.2 Mb in size and contains 19 annotated genes of which one, proteolipid protein 1 (PLP1 [MIM: 300401]), is associated with two allelic Mendelian neurological disease traits, Pelizaeus-Merzbacher disease (PMD [MIM: 312080]) and spastic paraplegia type 2 (SPG2 [MIM: 312920]).1 No other monogenic associations with Mendelian diseases have been reported in OMIM for genes mapped to the Xq22.2 interval. However, we and others have described the association of heterozygous intergenic Xq22.2 deletions that encompass the smallest region of overlap (SRO) containing six contiguous genes (BEX3, RAB40A, TCEAL4, TCEAL3, TCEAL1, and MORF4L2) with the emerging early-onset neurological disease trait (EONDT) in 46,XX females, consisting of hypotonia at birth, severe intellectual disability, neurobehavioral abnormalities, and mildly dysmorphic facial features.2,3 Two of the six genes, TCEAL1 and MORF4L2, have been specifically prioritized in previous studies as potential candidates because of their inclusion in a smaller Xq22.2 deletion that was reported in a female with an EONDT-like phenotype.4 To date, no individuals with damaging variants impacting MORF4L2 alone have been reported.
TCEAL1 (transcription elongation factor A-like 1 [MIM: 300237]) is a single coding-exon gene that encodes a 21-kDa nuclear phosphoprotein, referred to as TCEAL1 (or p21/SIIR). The encoded protein is related to the S-II class of transcription elongation factors as it consists of three main functional domains along its length of 157 amino acids (aa), an arginine/serine (RS) domain, a zinc-finger-like (ZnF-L) domain, and a helix-turn-helix (HTH) domain, and has a predicted RNA polymerase II binding site.5,6,7 Previous knockout functional studies of the different domains of TCEAL1 in the context of the Rous sarcoma virus were conducted in transfected COS-1 cells where it was shown that loss of function (LoF) of the C-terminal domain of TCEAL1, RS, and the middle domain, ZnF-L, lead to down-regulation of the promoter activity of the virus; whereas LoF of the N-terminal domain, HTH, had minimal effects on the promoter activity of the virus.6 These data suggest that TCEAL1, particularly the RS and ZnF-L domains, may play a role in transcriptional regulation.6
A total of four males and three females were identified with de novo disruption of TCEAL1 (Table 1 and supplemental note, individuals 1–7) along with an inherited variant of uncertain significance in an additional male (individual 8). Written informed consent for research studies and/or reporting of clinical features was obtained for all individuals, including photo publication if applicable; all study procedures were approved by a local research review board and adhered to the Declaration of Helsinki. The seven individuals with de novo variants presented with neurological anomalies, which overlap those observed in EONDT females with contiguous gene deletions that include TCEAL1 and PLP1. In particular, both males and females with de novo TCEAL1 variants, as well as EONDT females, presented with DD/ID, behavioral abnormalities including autism or autistic-like features, abnormal muscle tone, and gait disturbance (Table 1).2,3,4 Strabismus, a thin corpus callosum, and delayed or hypomyelination were less common in the present cohort compared to EONDT females who have deletions that include both TCEAL1 and PLP1.2,3,4
Table 1.
Clinical phenotypes of individuals with TCEAL1 variants
| Sex |
Individual 1 |
Individual 2 |
Individual 3 |
Individual 4 |
Individual 5 |
Individual 6 |
Individual 7 |
Individual 8 |
|---|---|---|---|---|---|---|---|---|
| male | male | male | male | female | female | female | male | |
| Ancestry | White (US) | White (European) | White (European) | White (European) | White/Chinese | Algerian | White (European) | White (European) |
| Consanguinity | − | − | − | two AOH regions totaling 27 Mb by SNP array | − | − | − | − |
| Age | 5 y/o | 10 y/o | 17 y/o | 7 y/o | 17 y/o | 9 y/o | 6 y/o | 12 y/o |
| Variant type | nonsense | frameshift | nonsense | missense | CNV | CNV | frameshift | missense |
| Variant | c.447G>A (p.Trp149∗) | c.299_302del (p.Gly100Alafs∗22) | c.259C>T (p.Gln87∗) | c.269G>A (p.Cys90Tyr) | ∼14 kb DEL of TCEAL1 | ∼154 kb DEL of TCEAL1, TCEAL3, TCEAL4 | c.169delC (p.Leu57Serfs∗36) | c.346G>A (p.Asp116Asn) |
| In silico prediction | damaging | damaging | damaging | mixed | damaging | damaging | damaging | mixed |
| Zygosity | hemi | hemi | hemi | hemi | het | het | het | hemi |
| Coordinates (hg19) | ChrX: 102,885,291 | ChrX: 102,885,138 | ChrX: 102,885,103a | ChrX: 102,885,113 | ChrX: 102,879,326–102,893,312 | ChrX: 102,774,750–102,929,222 | ChrX: 102,885,012 | ChrX: 102,885,190 |
| Inheritance | de novo | de novo | de novo | de novo | de novo | de novo | de novo | maternal |
| Birth history | ||||||||
| Prenatal complications | maternal HELLP | maternal PUPPP | none | none | − | − | − | − |
| Gestational age, delivery | 37 weeks, C-section | 41 weeks, C-section | term | 40 weeks, NSVD | 37 weeks, NSVD | 40 weeks | 38 weeks, C-section | term |
| Postnatal complications | − | +, hyperbilirubinemia | − | difficulty feeding (short frenulum) | +, hyperbilirubinemia | − | − | − |
| Neurological anomalies | ||||||||
| DD/ID (HP: 0012758/HP: 0001249) | ++, moderate | +++, severe | +++, severe | ++, moderate to severe | +, mild to moderate | +++, severe | ++, mild | − |
| Neurodevelopmental regression (HP: 0002376) | − | +, lost single words | − | − | + | − | − | − |
| Seizures (HP: 0001250) | − | + (developed in early childhood) | − | + (one absence with apnea) | − | − | + | − |
| Verbal skills | single words only, communication device and signs | currently non-verbal | non-verbal | single words only, max 30 words | single words, short sentences | single words only, ∼10 words | single words, short sentences | full sentences and normal vocabulary for age |
| Abnormal muscle tone (HP: 0003808) | hypotonia (HP: 0001319) | hypotonia (HP: 0001319) | hypotonia (HP: 0001319) | hypotonia (HP: 0001319) | N/A | hypotonia (HP: 0001319) | hypotonia (HP: 0001319), lower leg spasticity | hypertonia (HP: 0001276), spasticity (HP: 0001257), ankle clonus |
| Gait disturbance (HP: 0001288) | + | +, non-ambulatory | +, ataxic with support | + | + | − | + | +, toe walking |
| Behavioral abnormalities (HP: 0000708) | + | + | + | + | + | + | + | − |
| Autism/autistic-like behavior (HP: 0000717) | + | + | + | − | + | − | − | − |
| Abnormal myelination (HP: 0012447) | − | − | N/A | + | + | − | + | − |
| Structural brain anomalies | possible mild foreshortening of corpus callosum | − | N/A | delayed myelinization of terminal zones of lateral ventricles, slight diminishing of white matter parietooccipital | abnormal myelination for age (HP: 0012447) | − | − | − |
| Ocular anomalies | ||||||||
| Astigmatism (HP: 0000483) | + | + | + | − | + | + | + | − |
| Nystagmus (HP: 0000639) | + | − | − | − | − | − | − | − |
| Strabismus (HP: 0000486) | + | + | + | − | − | + | − | − |
| Myopia/hyperopia (HP: 0000545/HP: 0000540) | +, mild myopia | +, hyperopia | − | − | − | − | − | − |
| Iris coloboma (HP: 0000612) | − | − | − | + | − | − | − | − |
| Dysmorphic features | ||||||||
| Broad forehead (HP: 0000337) | + | + | + | + | − | + | − | − |
| Other dysmorphic features | + | + | deep-set eyes, very bright blue eyes | − | + | + | + | − |
| Other | ||||||||
| Gastrointestinal abnormality (HP: 0011024) | − | +, GERD, constipation, G-tube | +, chewing difficulty, constipation | +, constipation | N/A | +, regurgitation | − | − |
| Abnormality of the immune system (HP: 0002715) | +, recurrent ear infections | +, recurrent chest infections (aspiration) | +, recurrent ear infections | +, recurrent respiratory and ear infections | +, oral allergy syndrome, recurrent infection | − | − | − |
| Other findings | − | − | growth retardation (onset 5 y/o), no puberty onset, hyperlaxity | not toilet trained at age 7, hypermobile fingers | hypertriglyceridemia, microcytic anemia | − | premature puberty | urinary incontinence |
Abbreviations (alphabetical order) and symbols: AOH, absence of heterozygosity; CNV, copy-number variant; DD, developmental delay; DEL, deletion; HELLP, hemolysis, elevated liver enzymes, and low platelets; ID, intellectual disability; N/A, not available; NSVD, normal spontaneous vaginal delivery; PUPPP, pruritic urticarial papules and plaques of pregnancy; SNV, single-nucleotide variant; y/o, years old. GenBank: NM_001006639.2 is used for variant nomenclature; in silico: SIFT/PolyPhen (damaging, tolerated, or mixed predictions).
Converted from hg38 coordinates (chrX: 103,630,175).
In males, DD/ID ranged from moderate to severe with particular weakness in expressive language; the female individuals in this study showed similar but perhaps somewhat milder impairment. Individuals were identified independently through trio-exome or genome sequencing or review of the Baylor Hopkins Center for Mendelian Genomics (BHCMG) research exome sequencing (rES) database8 (∼15,000 exomes), and the Baylor Genetics (BG) diagnostic laboratory (clinical ES, cES, ∼15,000 exomes) and CMA databases (>75,000 personal genomes) and connected through GeneMatcher9,10 and the Matchmaker Exchange.11,12 Careful review identified an overlapping phenotype of developmental delay/intellectual disability (DD/ID) including affected expressive language (7/7, 100%), neurobehavioral abnormalities (7/7, 100%) including autism or autistic-like behavior (4/7, 57.1%), and dysmorphic craniofacial features (7/7, 100%) that include a broad forehead, deep-set eyes, telecanthus, a prominent bow-shaped upper lip, slightly low-set ears, mild coarsening of facies, and brachycephaly (Figure 1). Individuals also demonstrated hypotonia (6/6, 100%), motor stereotypies (5/5, 100%), and abnormal gait or non-ambulatory status (6/7, 85.7%). Abnormal myelination or structural brain anomalies were observed in 3 of 6 individuals (50.0%, Figures 2A and 2B). Three individuals (3/7, 42.9%) reported seizures (Figure 2C). Additional affected organ systems and features (Table S1) present in more than half of individuals included ocular anomalies (astigmatism, nystagmus, strabismus, myopia or hyperopia, iris coloboma; 6/7, 85.7%), gastrointestinal abnormalities (gastroesophageal reflux disease [GERD], constipation, or regurgitation; 4/7, 57.1%), and recurrent infections (ear, respiratory; 5/7, 71.4%).
Figure 1.

Clinical photographs
(A and B) Individual 1 at 5 years old showing mildly dysmorphic facial features including broad forehead, deep-set eyes, telecanthus, prominent bow-shaped upper lip, mildly coarse facial features, and mildly low-set ears.
(C and D) Individual 2 at 3 years old with mildly dysmorphic features including long palpebral fissures, deep-set eyes, prominent bow-shaped upper lip, and brachycephaly.
(E–G) Individual 3 at 7 (E) and 17 (F and G) years old with similarly mild dysmorphic features including broad forehead, deep-set eyes, and bow-shaped upper lip.
(H) Individual 4 demonstrating a broad forehead, deep-set eyes, and bow-shaped upper lip.
(I and J) Individual 6 showing frontal bossing, bilateral epicanthus, hypertelorism, deep-set eyes, horizontal eyebrows, and fleshy earlobes.
(K and L) Individual 7 demonstrating a broad forehead, telecanthus, low-set ears, and widely spaced teeth.
Figure 2.

Brain MRIs and EEG
(A) Individual 1 brain MRI (i) T1 sequence axial view showing mildly reduced corpus callosum (CC) length (arrows) measuring 50.7 mm (between −1.0 and −2.0 SD). The CC thickness appears appropriate for age. (ii) T2 sequence midsagittal view showing normal myelination of the internal capsule (arrow).
(B) Individual 5 brain MRI (i) rapid sequence axial image showing lack of myelination of the internal capsule (arrows) and a bilateral T2 hyperintensity at the posterior limb of the internal capsule suggestive of gliosis (arrowheads). (ii) Rapid sequence axial image showing lack of myelination of the internal capsule (arrows). (iii) Rapid sequence sagittal image showing borderline to low-normal corpus callosum thickness (arrows).
(C) Individual 7 electroencephalogram (EEG) performed at 4 years of age, showing focal paroxysmal activity in the left frontocentrotemporal area with sharp waves and slow waves. y/o, years old.
In contrast, individual 8 (Table 1, Table S1, supplemental note), a male with a maternally inherited missense variant (c.346G>A [p.Asp116Asn] [GenBank: NM_001006639.2]), demonstrated a distinct phenotype characterized by spasticity with hypertonia, hyperreflexia, bilateral ankle clonus, and a toe-walking gait in the absence of any developmental delay, intellectual disability, or dysmorphic craniofacial features. Notably, his mother, who is heterozygous for the same TCEAL1 missense variant, was unaffected—a finding that is distinct from the observation of heterozygous LoF variants leading to disease trait expression observed in female individuals 5, 6, and 7 in the present cohort. The observation of a distinct neurological phenotype in individual 8, in combination with the unaffected status of this individual’s mother who is heterozygous for the variant, raises the possibility that an alternative molecular mechanism (i.e., LoF hypomorphic or null allele; or antimorphic in carrier females versus gain of function [GoF hypermorphic] or novel function [neomorphic allele]) may be responsible for his unique phenotype compared to individuals 1–7. It is also possible that this variant is a rare benign allele with a CADD score = 24 or the personal genome of individual 8 has another unrecognized variant mapping at this or another locus that contributes to the phenotype. Detailed clinical descriptions of all eight individuals are provided as supplemental text.
TCEAL1 variants were identified by exome sequencing (ES), genome sequencing (GS), array comparative genomic hybridization (aCGH), and/or single-nucleotide polymorphism (SNP)-array (Table S2, see supplemental information for detailed methods). Six de novo putative LoF variants (four truncating and two deletions) were identified in TCEAL1 (GenBank: NM_001006639.2); the gene contains three exons, of which only the third exon is coding (Figure 3). The truncating variants were two hemizygous nonsense variants (c.447G>A [p.Trp149∗] [GenBank: NM_001006639.2] and c.259C>T [p.Gln87∗] [GenBank: NM_001006639.2]) and two frameshift variants (hemizygous c.299_302del [p.Gly100Alafs∗22] [GenBank: NM_001006639.2] and heterozygous c.169delC [p.Leu57Serfs∗36] [GenBank: NM_001006639.2]) in individuals 1, 3, 2, and 7, respectively. These truncating variants occurred at the C-terminal end of the RS domain, within the ZnF-L and HTH domains, or just outside the HTH domain (Figure 3I). These variants may lead to complete LoF of TCEAL1 in males (individuals 1–3). However, because TCEAL1 is a single-coding-exon gene, it may escape nonsense-mediated decay (NMD).13 It is possible that these truncating variants with PTCs (premature termination codons) might lead to proteins with partial function or even a GoF involving loss of the HTH domain of TCEAL1. Little is known about the function of this domain in TCEAL1, although HTH domains generally act as DNA-binding sites that regulate transcription.14
Figure 3.

Description of TCEAL1 variants and segregation within families
(A) Individual 1 with a de novo hemizygous nonsense variant that maps to the HTH domain.
(B) Individual 2 with a de novo hemizygous frameshift variant that maps to the HTH domain.
(C) Individual 3 with a de novo hemizygous nonsense variant that maps to the ZnF-L domain.
(D) Individual 4 with a de novo hemizygous missense variant that maps to the ZnF-L domain.
(E) HD-aCGH on individual 5 and parents revealing an ∼14 kb de novo heterozygous deletion that encompasses the entirety of TCEAL1.
(F) Individual 6 with an ∼154 kb de novo heterozygous deletion that encompasses TCEAL1, TCEAL3, and TCEAL4.
(G) Individual 7 with a de novo heterozygous frameshift variant that maps to the RS domain.
(H) Individual 8 with a maternally inherited hemizygous missense variant that maps to the HTH domain.
(I) Three domains are indicated: the arginine/serine (RS) rich domain (red), the zinc finger-like (ZnF-L) domain (green), and the helix-turn-helix (HTH) domain (blue). The RNA polymerase II binding site is indicated by vertical stripes within the HTH domain. Described missense, nonsense, and frameshift variants are mapped to the linear protein structure of TCEAL1. Q15170 is the UniProt accession ID used.
(J) Location of deletions identified in individuals 5 and 6 within the Xq22.2 region.
Abbreviations: tR, total read depth; vR, variant read depth. GenBank: NM_001006639.2 is used for variant nomenclature.
While the numbers of individuals are too small to draw definitive conclusions, there did appear to be a correlation between the location of the truncating intragenic variant and the severity of the neurological phenotype in males, i.e., a “polarity effect” (see Figure 1 of Inoue et al.15) as has been observed at the SOX10 locus with peripheral demyelinating neuropathy, central dysmyelination, Waardenburg syndrome, and Hirschsprung disease (PCWH [MIM: 609136]). Individuals 2 and 3 with truncating variants within the ZnF-L or early in the HTH domain both had severe/profound cognitive impairment, no independent ambulation or meaningful words, and gastrointestinal abnormalities, while individual 1, with truncation just after the HTH domain, had moderate delay, abnormal but independent gait, single words, and normal gastrointestinal functioning. This observation is consistent with the contention that the mRNA will most likely escape nonsense-mediated decay (because the variants occur in the third and final exon, which is the only coding exon), so the longer proteins would be expected to be more functional. It also suggests that the C terminus of the protein is important in the function of TCEAL1, despite the lack of identified domains within this region. Further identified variants will be needed to determine whether this deduction of association remains true and suggests a focus for possible future functional experiments on the HTH domain of TCEAL1.
In contrast to the male subjects, two of the three females reported herein had de novo heterozygous TCEAL1 deletions and one had a frameshift variant allele located N-terminal to the termination codons identified in male individuals. Overall, these three females had a somewhat milder clinical presentation than the male individuals despite their heterozygous LoF variants, supporting a role for TCEAL1 gene dosage16,17 in the observed variable severity of disease.
Individual 5 was found to have a heterozygous 14 kb TCEAL1 deletion. Clinically performed X chromosome inactivation (XCI) studies were indeterminate as the studied markers were not informative. No CNVs with boundaries comparable to the deletion identified in individual 5 were found in public structural variant databases, including gnomAD, DECIPHER, and the Database of Genomic Variants (DGV). While partial gene deletions have not been reported to date, this may be due to methodological limitations of CNV detection in the studied cohorts. Given its small single-coding-exon size (coding exon is ∼900 bp), partial (i.e., intragenic) CNVs in TCEAL1 are likely to be missed. The average probe coverage of clinical-grade CMA does not meet the validation threshold within the coding interval of TCEAL1 (∼900 bp); therefore, it is possible that affected individuals with intragenic TCEAL1 deletions are missed due to technical limitations, a challenge often faced in studies of short single-coding-exon genes.18 Given the frequency of gene deletion and inherent genomic instability of this region, future screening of TCEAL1 in affected individuals that are optimized for both DNA sequencing and copy-number variant assessment are warranted.
Mapping of the 14 kb deletion in individual 5 via high density aCGH (HD-aCGH, Figure 3E) and subsequent junction-PCR and Sanger sequencing (Figure 4) identified the precise (nucleotide-level) coordinates as chrX: 102,879,326–102,893,312 (GRCh37/hg19) and a 77 bp homology defined by the presence of 77 bp of sequence shared by the distal and proximal breakpoints. The deletion was de novo, confirmed via HD-aCGH and trio junction PCR of the parent-proband trio (Figures 3E and 4). Repeat sequences, specifically highly similar intrachromosomal repeats (HSIRs), have been implicated in predisposing a 90 kb hotspot on Xq22.2 to genomic instability and the formation of potentially pathogenic deletions.2 HSIRs are defined as intrachromosomal repeat sequences that are typically >700 bp in length with 95%–100% identity between pairs.2 An HSIR that is ∼140 kb in length, RepX-i1010, is of particular note as ∼50% of proximal breakpoints of pathogenic Xq22.2 deletions are clustered within it.2 Notably, TCEAL1 is entirely embedded within RepX-i1010.
Figure 4.

Demonstration of de novo status and breakpoint sequence of TCEAL1 deletion and local genomic architecture at the TCEAL1 locus in individual 5
(A) Junction PCR confirms the de novo nature of the TCEAL1 deletion in individual 5.
(B) Nucleotide resolution of the deletion breakpoint-junction demonstrates a 77 bp homology.
(C) The human genome reference sequence (GRCh37/hg19) surrounding the proximal and distal breakpoint sites is provided (top and bottom), mapped to the sequence of the deletion breakpoint-junction (middle). Chromosome coordinates are provided for the start and end points of the displayed portions of the proximal and distal genome reference sequence, as well as for the nucleotide positions at the start and end of the 77 bp homology. Breakpoints map to SelfChain repeat pairs that are in direct orientation (indicated by ">>>" highlighted in red). Furthermore, the entire deletion and both SelfChain repeats are embedded within the larger repeat, RepX-i1010 (indicated by ">>>" highlighted in blue).
(D) The positions of RepX-i1010 and both SelfChain repeats within Xq22.2 are demonstrated. Black dotted lines illustrate the positions of the breakpoints in individual 5.
We therefore sought to examine the breakpoints of the TCEAL1 deletion in individual 5 for the possibility of overlapping repeats. The breakpoints of the deletion were aligned to the haploid reference genome and examined for overlapping repeats in four different repeat datasets, (1) the (HSIR) dataset,2 (2) the Segmental Dup dataset,19,20 (3) the Repbase (Repeat Masker) dataset,21 and (4) the SelfChain dataset.22,23 Intriguingly, we found both breakpoints overlap a pair of SelfChain repeats (101408 and 101409), i.e., the junction forms a fusion SelfChain, and the deletion along with the SelfChain pair are entirely embedded within RepX-i1010. The fusion SelfChain resulting from the deletion suggests that homology of the two SelfChains may have stimulated genomic instability and deletion formation.24 These findings emphasize the unique enrichment of RepX-i1010 for smaller repeat constituents and the role for repeats generally in predisposing Xq22.2 to the formation of pathogenic structural variants, specifically genomic deletions.
Individual 6 was found to have a heterozygous 154 kb deletion of TCEAL1, TCEAL3, and TCEAL4. No comparable deletions were identified in public structural variant databases, including gnomAD, DECIPHER, and the DGV. Mapping of the 154 kb deletion in individual 6, initially identified by SNP-array and genomic sequencing, confirmed the de novo status of the deletion and confirmed deletion coordinates of chrX: 102,774,750–102,929,222 marked by a 1 bp microhomology (GRCh37/hg19, Figure 5). XCI studies performed in a clinical diagnostic laboratory on individual 6 further indicated a skewed XCI of 85%/15% at the AR locus (data not shown), although the identity of the preferentially inactivated allele (reference allele or variant allele) was not determined through this testing.
Figure 5.

Demonstration of de novo deletion CNV and breakpoint sequencing in individual 6
(A) Junction PCR confirms the de novo nature of the deletion CNV in individual 6.
(B) Nucleotide resolution of the deletion breakpoint-junction demonstrates a 1 bp microhomology.
(C) The human genome reference sequence (GRCh37/hg19) surrounding the proximal and distal breakpoint sites is provided (top and bottom), mapped to the sequence of the deletion breakpoint-junction (middle). Chromosome coordinates are provided for the start and end points of the displayed portions of the proximal and distal genome reference sequence, as well as for the nucleotide position of the 1 bp microhomology.
Two additional hemizygous missense variants were identified including one de novo (individual 4, c.269G>A [p.Cys90Tyr] [GenBank: NM_001006639.2]) and one maternally inherited (individual 8, c.346G>A [p.Asp116Asn] [GenBank: NM_001006639.2]). Notably, the maternally inherited missense variant was identified in individual 8, who also demonstrated a phenotype distinctive from the remainder of the cohort. This variant was found to have a CADD phred score of 24.1 and was present in one heterozygous individual (one female out of 183,451 individuals) in gnomAD (MAF = 5.451 × 10−6), suggesting the potential that this variant could be damaging in hemizygous individuals.
In mice, the knockout of Tceal1 has been studied in female and male mice where affected offspring appeared to exhibit a neurological phenotype, involving behavioral abnormalities (in homozygous females), abnormal response to stimulus (in hemizygous males), and ophthalmologic anomalies (in homozygous females) (data sourced from the International Mouse Phenotyping Consortium; IMPC25,26). Overall, the reported phenotypes (neurological anomalies, autistic-like behavioral abnormalities, strabismus, and refractive vision abnormalities) in the present cohort show overlap with those reported in homozygous female and hemizygous male mice with Tceal1 knockout.26 Although phenotype data for heterozygous Tceal1 knockout mice have not been reported, the neurological phenotypes described in hemizygous males and homozygous females support a potential role of TCEAL1 in the human nervous system.
TCEAL1 was previously implicated as a possible contributor to the contiguous gene deletion syndromes caused by Xq22.2 intergenic microdeletions, but without clear evidence of its monogenic involvement.2,3,4 Indeed, in DECIPHER, there are a total of 70 individuals with deletions or duplications encompassing the TCEAL1 locus. Despite this large number of individuals with CNVs in this region, each of these CNVs either extends proximally beyond the TCEAL genes in this region to include RAB40A and/or distally to include PLP1, an established disease gene. Thus, these data alone are not informative to conclude that TCEAL1 is solely responsible for the phenotypes reported in these individuals in DECIPHER.
The present study identifies the role of TCEAL1 in Mendelian disease through identification of de novo TCEAL1 variants in seven unrelated individuals with neurological disease as well as one variant of uncertain significance (VUS) in a male individual harboring a maternally inherited variant allele. These data provide evidence that TCEAL1 loss of function causes a rare disease trait involving significant neurological impairment with features overlapping the EONDT phenotype in females with the Xq22 deletion. Notably, the TCEAL gene family shares a common ancestral sequence with the BEX gene family, and their protein structures share a common C-terminal domain that can support homodimerization and heterodimerization of TCEAL and BEX proteins.27,28 This shared C-terminal domain suggests the possibility that other members of the TCEAL and BEX gene families may be associated with rare human disease traits in the future. TCEAL1 is one of nine members of the TCEAL protein family (TCEAL1 through TCEAL9), and the genes for these proteins all map to the Xq22.1-Xq22.2 region. Despite the encoding genes' proximity, TCEAL1 has limited overall protein sequence identity with a majority of the TCEAL/BEX proteins, whereas TCEAL8 and TCEAL9 share the highest amino acid similarity (52.83%, 53.33% respectively; Table S3). Compared to other TCEAL protein family members encoded within the SRO, TCEAL1 shares 31.25% homology with TCEAL4 and no significant similarity with TCEAL3. This limited sequence similarity supports the possibility that TCEAL1 may not share redundant function with other TCEAL protein family members, supporting a role for haploinsufficiency and LoF in human neurological disease traits.
Importantly, TCEAL1 is expected to be subject to the effect of XCI in females by virtue of its X-linked location. The process of XCI can greatly influence ChrX gene expression and X-linked disease penetrance in 46,XX females because of the random chance of silencing or expressing heterozygous mutant alleles.29,30 Thus, the expression of pathogenic heterozygous TCEAL1 variants in females is influenced not only by gene dosage but also expression of that gene dosage: the direction and influence of XCI (i.e., whether the mutated or the wild-type X chromosome is inactivated), specifically in disease-relevant organs and tissues. It is possible that unaffected females heterozygous for putatively damaging variants in TCEAL1 may benefit from skewed XCI favoring expression of the wild-type allele. In contrast, affected females may demonstrate skewed XCI favoring expression of the damaging TCEAL1 allele. XCI studies performed in a clinical diagnostic laboratory on individual 6 further indicated a skewed XCI of 85%/15% at the AR locus (data not shown), although the identity of the preferentially inactivated allele (reference allele or variant allele) was not determined through this testing. The detection of XCI skewing (85%/15%) at the AR locus in female individual 6 supports the possibility that both TCEAL1 gene dosage and expression of that gene dosage, modulated by XCI, may influence the severity and expression of disease associated with TCEAL1.
The X chromosome, like its heterologous/heterogametic ChrY sex chromosome, is enriched with repeat sequences, specifically long interspersed nuclear elements (LINEs) that facilitate spreading of XCI to all parts of the chromosome.31,32 However, the enrichment for LINEs is a double-edged sword, as these repetitive sequences also render an increased susceptibility to the formation of copy-number variants (CNVs).33,34,35,36 We and others have previously pointed to the prominence of repeat sequences in predisposing Xq22 to the formation of pathogenic CNVs,2,37,38,39 of which perhaps the most significant to this study is the ∼140 kb repeat, RepX-i1010, that was implicated in a 90 kb genomic instability hotspot within Xq22.2 where nearly half of all breakpoints of pathogenic Xq22 deletions cluster.2 Here, we identified an additional individual with an Xq22.2 deletion with breakpoints that also map to RepX-i1010.
These data provide compelling evidence that loss of gene function, specifically of TCEAL1, contributes to the EONDT-like neurological disease traits in females with Xq22 deletions, potentially acting as a “driver gene” in the microdeletion syndrome. Comparison of clinical features of affected individuals suggests a more defined X-linked dominant rare disease trait consisting of neurological impairment, ocular and gastrointestinal anomalies, and mildly dysmorphic facial features and warrants further consideration of this gene in the molecular diagnosis of similarly affected individuals. Furthermore, the TCEAL1 locus appears to be highly susceptible to the formation of genomic deletions by virtue of the overlapping and surrounding genomic architecture, specifically the RepX-i1010 HSIR, and thus may be a de novo structural variant mutagenesis hotspot.
Acknowledgments
We thank all individuals, their families, and the referring physicians who submitted samples for testing. No additional compensation was received for these contributions. The authors thank the contributors to MyGene2, GeneMatcher, and other Matchmaker Exchange databases and the Genome Aggregation Database (gnomAD) and the groups that provided exome and genome variant data to these resources. A full list of contributing groups to gnomAD can be found at https://gnomad.broadinstitute.org/about. This work was supported in part by the US National Human Genome Research Institute (NHGRI)/National Heart, Lung, and Blood Institute (NHLBI) grant UM1 HG006542 to the Baylor-Hopkins Center for Mendelian Genomics (BHCMG); US National Institute of Neurological Disorders and Stroke (NINDS) grants R01 NS058529 and R35 NS105078 and National Institute of General Medical Sciences (NIGMS) grant R01 GM106373 to J.R.L.; NHGRI U01 HG011758 to the Baylor College of Medicine Genomics Research Elucidate Genetics of Rare disease (BCM-GREGoR) consortium and grant K08 HG008986 to J.E.P.; by NIGMS T32 GM007526-42 to D.M.; and by the National Eye Institute grants R01EY025718 and EY015518 to E.V.S. and 1UL1RR031973 from the Clinical and Translational Science Award (CTSA) program. D.P. is supported by International Rett Syndrome Foundation (IRSF grant #3701-1). This study makes use of data generated by the DECIPHER40 community and the Deciphering Developmental Disorders (DDD) study. Please refer to the supplemental acknowledgments for full acknowledgment and details.
Declaration of interests
J.R.L. serves on the Scientific Advisory Board of Baylor Genetics (BG); J.A.R. and W.B. report affiliation with BG. Baylor College of Medicine (BCM) and Miraca Holdings have formed a joint venture with shared ownership and governance of Baylor Genetics (BG), which performs clinical microarray analysis (CMA) and clinical exome sequencing (cES) and molecular diagnostic whole-genome sequencing (WGS). J.R.L. has stock ownership in 23andMe, is a paid consultant for the Regeneron Genetics Center, and is a co-inventor on multiple United States and European patents related to molecular diagnostics for inherited neuropathies, eye diseases, genomic disorders, and bacterial genomic fingerprinting. H.H., D.P., Y.L., J.M.F., D.M., J.A.R., Z.H.C.A., W.B., R.A.G., C.M.B.C., J.E.P., and J.R.L. report affiliation with the Department of Molecular and Human Genetics at Baylor College of Medicine. The Department of Molecular and Human Genetics at Baylor College of Medicine derives revenue from molecular genetic and personal genome (CMA, cES, WGS) genomic testing offered in BG. Z.G.O. serves on the scientific advisory boards and receives consultancy fees from Bial Biotech Inc. and Handl Therapeutics. He has received consultancy fees from Neuron23, Ono Therapeutics, and UCB.
Published: November 10, 2022
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.ajhg.2022.10.007.
Contributor Information
Elena V. Semina, Email: esemina@mcw.edu.
James R. Lupski, Email: jlupski@bcm.edu.
Web resources
CADD – Combined Annotation Dependent Depletion, https://cadd.gs.washington.edu/snv
DECIPHER, https://decipher.sanger.ac.uk
DGV – Database of Genomic Variants, http://dgv.tcag.ca/
GeneMatcher, https://genematcher.org/
Matchmaker Exchange, https://matchmakerexchange.org/
MyGene2, https://mygene2.org
Online Mendelian Inheritance in Man, https://www.omim.org/
UCSC Genome Browser, https://genome.ucsc.edu/
Supplemental information
Data and code availability
All TCEAL1 variants reported herein have been deposited to ClinVar, accession IDs ClinVar: SCV002558856; ClinVar: SCV002558857; ClinVar: SCV002558858; ClinVar: SCV002558859; ClinVar: SCV002558860; ClinVar: SCV002558861; ClinVar: SCV002558862; ClinVar: SCV002558863. For subjects who have provided written informed consent for sharing of their genomic data in controlled access databases, these data will be deposited to AnVIL and/or dbGaP under accession dbGAP: phs000711.v5.p1.
References
- 1.Lupski J.R. Biology in balance: human diploid genome integrity, gene dosage, and genomic medicine. Trends Genet. 2022;38:554–571. doi: 10.1016/j.tig.2022.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hijazi H., Coelho F.S., Gonzaga-Jauregui C., Bernardini L., Mar S.S., Manning M.A., Hanson-Kahn A., Naidu S., Srivastava S., Lee J.A., et al. Xq22 deletions and correlation with distinct neurological disease traits in females: further evidence for a contiguous gene syndrome. Hum. Mutat. 2020;41:150–168. doi: 10.1002/humu.23902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Yamamoto T., Wilsdon A., Joss S., Isidor B., Erlandsson A., Suri M., Sangu N., Shimada S., Shimojima K., Le Caignec C., et al. An emerging phenotype of Xq22 microdeletions in females with severe intellectual disability, hypotonia and behavioral abnormalities. J. Hum. Genet. 2014;59:300–306. doi: 10.1038/jhg.2014.21. [DOI] [PubMed] [Google Scholar]
- 4.Labonne J.D.J., Graves T.D., Shen Y., Jones J.R., Kong I.K., Layman L.C., Kim H.G. A microdeletion at Xq22.2 implicates a glycine receptor GLRA4 involved in intellectual disability, behavioral problems and craniofacial anomalies. BMC Neurol. 2016;16:132. doi: 10.1186/s12883-016-0642-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Pillutla R.C., Shimamoto A., Furuichi Y., Shatkin A.J. Genomic structure and chromosomal localization of TCEAL1, a human gene encoding the nuclear phosphoprotein p21. Genomics. 1999;56:217–220. doi: 10.1006/geno.1998.5705. [DOI] [PubMed] [Google Scholar]
- 6.Yeh C.H., Shatkin A.J. Down-regulation of Rous sarcoma virus long terminal repeat promoter activity by a HeLa cell basic protein. Proc. Natl. Acad. Sci. USA. 1994;91:11002–11006. doi: 10.1073/pnas.91.23.11002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Yeh C.H., Shatkin A.J. A HeLa-cell-encoded p21 is homologous to transcription elongation factor SII. Gene. 1994;143:285–287. doi: 10.1016/0378-1119(94)90112-0. [DOI] [PubMed] [Google Scholar]
- 8.Posey J.E., O'Donnell-Luria A.H., Chong J.X., Harel T., Jhangiani S.N., Coban Akdemir Z.H., Buyske S., Pehlivan D., Carvalho C.M.B., Baxter S., et al. Insights into genetics, human biology and disease gleaned from family based genomic studies. Genet. Med. 2019;21:798–812. doi: 10.1038/s41436-018-0408-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sobreira N., Schiettecatte F., Valle D., Hamosh A. GeneMatcher: a matching tool for connecting investigators with an interest in the same gene. Hum. Mutat. 2015;36:928–930. doi: 10.1002/humu.22844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wohler E., Martin R., Griffith S., Rodrigues E.D.S., Antonescu C., Posey J.E., Coban-Akdemir Z., Jhangiani S.N., Doheny K.F., Lupski J.R., et al. PhenoDB, GeneMatcher and VariantMatcher, tools for analysis and sharing of sequence data. Orphanet J. Rare Dis. 2021;16:365. doi: 10.1186/s13023-021-01916-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sobreira N.L.M., Arachchi H., Buske O.J., Chong J.X., Hutton B., Foreman J., Schiettecatte F., Groza T., Jacobsen J.O., Haendel M.A., et al. Matchmaker Exchange. Curr. Protoc. Hum. Genet. 2017;95 doi: 10.1002/cphg.50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Philippakis A.A., Azzariti D.R., Beltran S., Brookes A.J., Brownstein C.A., Brudno M., Brunner H.G., Buske O.J., Carey K., Doll C., et al. The matchmaker exchange: a platform for rare disease gene discovery. Hum. Mutat. 2015;36:915–921. doi: 10.1002/humu.22858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Cusack B.P., Arndt P.F., Duret L., Roest Crollius H. Preventing dangerous nonsense: selection for robustness to transcriptional error in human genes. PLoS Genet. 2011;7:e1002276. doi: 10.1371/journal.pgen.1002276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Aravind L., Anantharaman V., Balaji S., Babu M.M., Iyer L.M. The many faces of the helix-turn-helix domain: transcription regulation and beyond. FEMS Microbiol. Rev. 2005;29:231–262. doi: 10.1016/j.femsre.2004.12.008. [DOI] [PubMed] [Google Scholar]
- 15.Inoue K., Khajavi M., Ohyama T., Hirabayashi S.i., Wilson J., Reggin J.D., Mancias P., Butler I.J., Wilkinson M.F., Wegner M., Lupski J.R. Molecular mechanism for distinct neurological phenotypes conveyed by allelic truncating mutations. Nat. Genet. 2004;36:361–369. doi: 10.1038/ng1322. [DOI] [PubMed] [Google Scholar]
- 16.Lupski J.R. Structural variation mutagenesis of the human genome: Impact on disease and evolution. Environ. Mol. Mutagen. 2015;56:419–436. doi: 10.1002/em.21943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ricard G., Molina J., Chrast J., Gu W., Gheldof N., Pradervand S., Schütz F., Young J.I., Lupski J.R., Reymond A., Walz K. Phenotypic consequences of copy number variation: insights from Smith-Magenis and Potocki-Lupski syndrome mouse models. PLoS Biol. 2010;8:e1000543. doi: 10.1371/journal.pbio.1000543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Boone P.M., Bacino C.A., Shaw C.A., Eng P.A., Hixson P.M., Pursley A.N., Kang S.H.L., Yang Y., Wiszniewska J., Nowakowska B.A., et al. Detection of clinically relevant exonic copy-number changes by array CGH. Hum. Mutat. 2010;31:1326–1342. doi: 10.1002/humu.21360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bailey J.A., Gu Z., Clark R.A., Reinert K., Samonte R.V., Schwartz S., Adams M.D., Myers E.W., Li P.W., Eichler E.E. Recent segmental duplications in the human genome. Science. 2002;297:1003–1007. doi: 10.1126/science.1072047. [DOI] [PubMed] [Google Scholar]
- 20.Bailey J.A., Yavor A.M., Viggiano L., Misceo D., Horvath J.E., Archidiacono N., Schwartz S., Rocchi M., Eichler E.E. Human-specific duplication and mosaic transcripts: the recent paralogous structure of chromosome 22. Am. J. Hum. Genet. 2002;70:83–100. doi: 10.1086/338458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Jurka J. Repbase update: a database and an electronic journal of repetitive elements. Trends Genet. 2000;16:418–420. doi: 10.1016/s0168-9525(00)02093-x. [DOI] [PubMed] [Google Scholar]
- 22.Chiaromonte F., Yap V.B., Miller W. Scoring pairwise genomic sequence alignments. Pac. Symp. Biocomput. 2002:115–126. doi: 10.1142/9789812799623_0012. [DOI] [PubMed] [Google Scholar]
- 23.Schwartz S., Kent W.J., Smit A., Zhang Z., Baertsch R., Hardison R.C., Haussler D., Miller W. Human-mouse alignments with BLASTZ. Genome Res. 2003;13:103–107. doi: 10.1101/gr.809403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zhou W., Zhang F., Chen X., Shen Y., Lupski J.R., Jin L. Increased genome instability in human DNA segments with self-chains: homology-induced structural variations via replicative mechanisms. Hum. Mol. Genet. 2013;22:2642–2651. doi: 10.1093/hmg/ddt113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.IMPC . 2020. International mouse phenotyping consortium, Tceal1 knockout mice.https://www.mousephenotype.org/data/genes/MGI:2385317 [Google Scholar]
- 26.Dickinson M.E., Flenniken A.M., Ji X., Teboul L., Wong M.D., White J.K., Meehan T.F., Weninger W.J., Westerberg H., Adissu H., et al. High-throughput discovery of novel developmental phenotypes. Nature. 2016;537:508–514. doi: 10.1038/nature19356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Manford A.G., Mena E.L., Shih K.Y., Gee C.L., McMinimy R., Martínez-González B., Sherriff R., Lew B., Zoltek M., Rodríguez-Pérez F., et al. Structural basis and regulation of the reductive stress response. Cell. 2021;184:5375–5390.e16. doi: 10.1016/j.cell.2021.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Navas-Pérez E., Vicente-García C., Mirra S., Burguera D., Fernàndez-Castillo N., Ferrán J.L., López-Mayorga M., Alaiz-Noya M., Suárez-Pereira I., Antón-Galindo E., et al. Characterization of an eutherian gene cluster generated after transposon domestication identifies Bex3 as relevant for advanced neurological functions. Genome Biol. 2020;21:267. doi: 10.1186/s13059-020-02172-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Carrel L., Willard H.F. X-inactivation profile reveals extensive variability in X-linked gene expression in females. Nature. 2005;434:400–404. doi: 10.1038/nature03479. [DOI] [PubMed] [Google Scholar]
- 30.Lupski J.R., Garcia C.A., Zoghbi H.Y., Hoffman E.P., Fenwick R.G. Discordance of muscular dystrophy in monozygotic female twins: evidence supporting asymmetric splitting of the inner cell mass in a manifesting carrier of Duchenne dystrophy. Am. J. Med. Genet. 1991;40:354–364. doi: 10.1002/ajmg.1320400323. [DOI] [PubMed] [Google Scholar]
- 31.Chow J.C., Ciaudo C., Fazzari M.J., Mise N., Servant N., Glass J.L., Attreed M., Avner P., Wutz A., Barillot E., et al. LINE-1 activity in facultative heterochromatin formation during X chromosome inactivation. Cell. 2010;141:956–969. doi: 10.1016/j.cell.2010.04.042. [DOI] [PubMed] [Google Scholar]
- 32.Lyon M.F. The Lyon and the LINE hypothesis. Semin. Cell Dev. Biol. 2003;14:313–318. doi: 10.1016/j.semcdb.2003.09.015. [DOI] [PubMed] [Google Scholar]
- 33.Stankiewicz P., Lupski J.R. The genomic basis of disease, mechanisms and assays for genomic disorders. Genome Dyn. 2006;1:1–16. doi: 10.1159/000092496. [DOI] [PubMed] [Google Scholar]
- 34.Startek M., Szafranski P., Gambin T., Campbell I.M., Hixson P., Shaw C.A., Stankiewicz P., Gambin A. Genome-wide analyses of LINE-LINE-mediated nonallelic homologous recombination. Nucleic Acids Res. 2015;43:2188–2198. doi: 10.1093/nar/gku1394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Szafranski P., Kośmider E., Liu Q., Karolak J.A., Currie L., Parkash S., Kahler S.G., Roeder E., Littlejohn R.O., DeNapoli T.S., et al. LINE- and Alu-containing genomic instability hotspot at 16q24.1 associated with recurrent and nonrecurrent CNV deletions causative for ACDMPV. Hum. Mutat. 2018;39:1916–1925. doi: 10.1002/humu.23608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Stankiewicz P., Lupski J.R., editors. The Genomic Basis of Medicine. Oxford University Press; 2020. [DOI] [Google Scholar]
- 37.Beck C.R., Carvalho C.M.B., Banser L., Gambin T., Stubbolo D., Yuan B., Sperle K., McCahan S.M., Henneke M., Seeman P., et al. Complex genomic rearrangements at the PLP1 locus include triplication and quadruplication. PLoS Genet. 2015;11:e1005050. doi: 10.1371/journal.pgen.1005050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lee J.A., Inoue K., Cheung S.W., Shaw C.A., Stankiewicz P., Lupski J.R. Role of genomic architecture in PLP1 duplication causing Pelizaeus-Merzbacher disease. Hum. Mol. Genet. 2006;15:2250–2265. doi: 10.1093/hmg/ddl150. [DOI] [PubMed] [Google Scholar]
- 39.Bahrambeigi V., Song X., Sperle K., Beck C.R., Hijazi H., Grochowski C.M., Gu S., Seeman P., Woodward K.J., Carvalho C.M.B., et al. Distinct patterns of complex rearrangements and a mutational signature of microhomeology are frequently observed in PLP1 copy number gain structural variants. Genome Med. 2019;11:80. doi: 10.1186/s13073-019-0676-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Firth H.V., Richards S.M., Bevan A.P., Clayton S., Corpas M., Rajan D., Van Vooren S., Moreau Y., Pettett R.M., Carter N.P. DECIPHER: Database of Chromosomal Imbalance and Phenotype in Humans Using Ensembl Resources. Am. J. Hum. Genet. 2009;84:524–533. doi: 10.1016/j.ajhg.2009.03.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All TCEAL1 variants reported herein have been deposited to ClinVar, accession IDs ClinVar: SCV002558856; ClinVar: SCV002558857; ClinVar: SCV002558858; ClinVar: SCV002558859; ClinVar: SCV002558860; ClinVar: SCV002558861; ClinVar: SCV002558862; ClinVar: SCV002558863. For subjects who have provided written informed consent for sharing of their genomic data in controlled access databases, these data will be deposited to AnVIL and/or dbGaP under accession dbGAP: phs000711.v5.p1.