

Abstract
Annexins (Anxs) are a family of highly homologous proteins that bind and aggregate lipid vesicles in the presence of calcium. All members of the family contain a variable N-terminus determining specific functions, followed by a conserved core region responsible for the general calcium-dependent lipid-binding property. The core structure consists of four homologous domains (DI–DIV), each consisting of a right-handed super-helix of five α-helices. We present data from a combination of site-directed mutagenesis, NMR, and circular dichroism showing that the G25–D34 region of the N-terminus as well as the contacts between residues D38A, R63A, and Q67A of AnxA2-DI are crucial for the autonomous folding and stability of DI of AnxA2. However, we also show that the folding of the full-length protein is very robust in that mutations and truncations that disrupted the folding of AnxA2-DI did not abolish the folding of full-length AnxA2, only lowering its thermal stability. This robustness of the folding of full-length AnxA2 is likely to be mediated by the existence of at least one transient nonnative intermediate as suggested by our kinetic data using stopped-flow fluorescence experiments. We also show that hydrophobic amino acids in AnxA2-DI involved in interfacial contacts with AnxA2-DIV are important for the cooperative folding and stability of the full-length protein. Mutating all of the V57E-V98R-G101Y residues in AnxA2-DI did not affect the folding of the domain, only its stability, but prevented the cooperative folding of the full-length protein. Our collective results favor a highly cooperative and robust folding process mediated by alternative intermediate steps. Since AnxA2 is a multifunctional protein involved in several steps of the progression of cell transformation, these data on structure and folding pathways are therefore crucial to designing anticancer drugs targeting AnxA2.
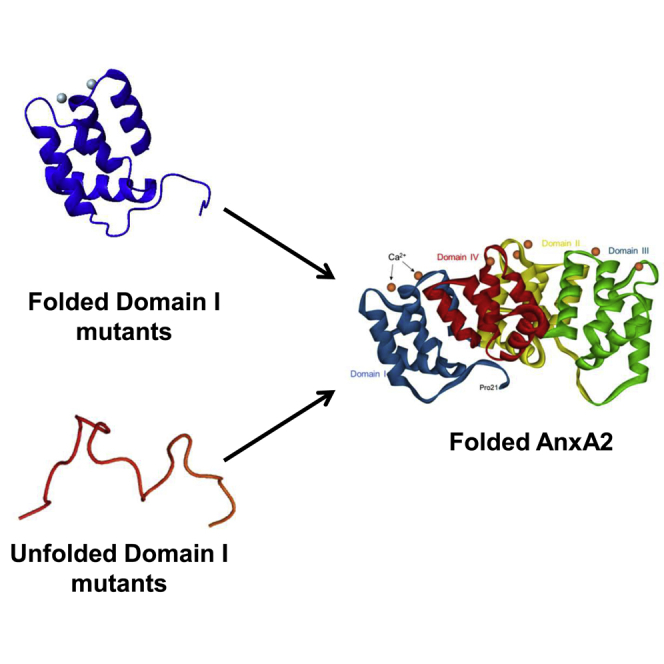
Graphical abstract
Significance
Annexin A2 (AnxA2) is associated with tumorigenesis, neo-angiogenesis, metastasis, and chemoresistance. Since it is a multifunctional protein, function-targeted peptides/domains of AnxA2 corresponding to specific ligand-interaction sites can and have been used as biological competitors of endogenous AnxA2. To develop such competitors further, it is important to understand the folding process of AnxA2 and its isolated domains to generate more soluble forms of the protein. More generally, but as importantly, AnxA2, like other members of the annexin family, represents an interesting model to study the determinants governing cooperative folding of a purely α-helical protein. Thus, the identification of an obligatory transient intermediate in the folding pathway adds greatly to our understanding of the folding of AnxA2 and possibly other annexins.
Introduction
Annexins are defined as a family of α-helical proteins that interact with vesicles containing negatively charged phospholipids in a Ca2+-dependent manner (1). Annexin A2 (AnxA2) is a multifunctional protein implicated in numerous cellular processes such as signal transduction, membrane trafficking, membrane/cytoskeleton interactions, and mRNA transport and translation (1,2,3,4,5,6). Its involvement in multiple different functions is regulated by posttranslational modifications (7). AnxA2 is associated with tumorigenesis, neo-angiogenesis, metastasis, and chemoresistance, processes all related to a poor prognosis (8). Inhibitors of angiogenesis have been associated with increased metastasis (9,10), so AnxA2 appears to be an ideal target for anticancer drugs, since the protein is involved in both processes (11). Initial attempts to employ isolated DI or DIV or peptides derived from these domains in in vitro assays resembling VEGF-induced angiogenesis were promising (12,13). In addition, a hexapeptide harboring the tissue plasminogen activator-binding site in the AnxA2 N-terminus competed successfully with endogenous AnxA2 for tissue plasminogen activator, thereby lowering the generation of plasmin, a protease important in tumor progression (14).
All annexins contain a conserved core region of about 300–310 residues harboring the Ca2+- and membrane-binding sites. The general structure of the annexin core consists of four domains (repeats) of about 75 residues each (denoted DI to DIV) that form a planar-cyclic arrangement (15,16). Each domain contains five α-helices (denoted A to E) arranged in a right-handed super-helix, in which helices A, B, D, and E form a bundle closed on one side by helix C lying perpendicular to the other helices. Within the bundle, helix pairs A-B and D-E are each arranged in an antiparallel mode. The Ca2+-binding sites are located in interhelical loops situated on the convex membrane-binding surface of the annexin core.
The N-terminal “tail” preceding the core region has a highly variable sequence and confers specific properties to each particular annexin (1). In the obtained wild-type AnxA2 structures (1,13,17,18,19), the first 20 residues could never be observed. The maximal observable portion of the tail spanned residues P21 to F33 (the next residue, D34, is the capping residue for helix A of AnxA2-DI). The extreme N-terminus of the visible part of the tail (residues 21–26) interacts with DIV and runs in the direction of DI. This conserved region in most annexins including AnxA2 was found to function as a velcro-like bridging segment in AnxA11 stabilizing the core structure (20). V27 (counting M as residue 1) and S26 both insert in between DI and DIV of AnxA2 (19).
Despite their overall structural similarity, each of the four domains of the annexin core displays some specific structural features. Accordingly, sequence divergence between the four domains within any given annexin is higher than that observed for a given domain between different annexin cores (21). In particular, the interfacial contacts between the domains are not evenly distributed. The domains DI and DII are engaged in tight hydrophobic contacts with DIV and DIII, respectively, resulting in two modules, DI–DIV and DII–DIII, which interact with each other via weaker, largely electrostatic interactions (Figs. 1 and S1) (16). There are also variations in the ability of the annexin domains to acquire a native tertiary structure when expressed separately from the rest of the protein. It has been suggested that the folding process of the domains of full-length AnxA1 follows an ordered and sequential series of events (22,23). In the proposed model, DI folds first, whereas DII and DIII remain partly unfolded until DIV docks on DI, thereby initiating the complete folding of the protein (22,23,24). In this model, the establishment of the hydrophobic interface between already folded DI and DIV is a prerequisite for the completion of the folding of DII and DIII. In AnxA2, DIV could be solubilized only at the expense of mutating several hydrophobic amino acids involved in interfacial contacts with DI (25). However, the resulting construct was only partly folded, indicating the importance of the hydrophobic contacts between DI and DIV in stabilizing the folding of DIV.
Figure 1.

(A) Ribbon plot of domain I (DI) of AnxA2 (from Pro21) showing the four conserved residues that appear to constitute a structural tetrad at the N-terminus of DI. The four conserved residues are F33, D38, R63, and Q67, counting the first M as residue 1. (B) Ribbon plot of DI of AnxA2 (from P21) indicating the side chains of the three mutated hydrophobic residues (V57, V98, and G101) at the interface of DI with α-helices in DII and DIV of AnxA2. The figures were prepared using Biovia Studio Visualizer (http://accelrys.com/products/collaborative-science/biovia-discovery-studio/visualization.html) and based on the PDB structures 1XJL and 4X9P. (C) Schematic representation of wild-type and different mutated/truncated forms of AnxA2-DI and full-length AnxA2. To investigate the role of a folded AnxA2-DI in the folding of the full-length AnxA2, single mutations (F33A, D38A, R63A, and Q67A) were introduced in the context of [25–104] and [35–104] (not F33A) AnxA2-DI as well as in [1–339] full-length AnxA2 and [35–339] Δ1-34-truncated AnxA2 (not F33A). Furthermore, a multiple mutation (F33A-D38A-R63A-Q67A) was introduced in [1–339] full-length AnxA2. Subsequently, to investigate the role of interfacial hydrophobic amino acid residues in AnxA2-DI for the folding of full-length AnxA2, single mutations (V57E, V98R, and G101Y) and a multiple mutation (V57E-V98R-G101Y) were introduced in the context of [25–104] AnxA2-DI and [1–339] full-length AnxA2 as indicated in the figure. The numbers represent the number of the particular amino acid in the bovine and human AnxA2 sequence, and the four domains are shown as black boxes, denoted DI–DIV. The color code for the respective mutated amino acids as indicated in (C) is also used in (A) and (B).
In the present work, we decided to investigate whether, as proposed for AnxA1 (22,23), AnxA2-DI forms an autonomous folding unit that subsequently induces the cooperative folding of the full-length AnxA2. To this aim, we produced wild-type and various mutated constructs of the AnxA2-DI as well as of full-length AnxA2. The solubility, the temperature-dependent stability, and the conformational status of the different constructs were studied by circular dichroism (CD) and proton-nitrogen heteronuclear single quantum correlation nuclear magnetic resonance (1H-15N HSQC NMR) spectroscopies. Furthermore, we provide a thorough characterization of the folding and unfolding kinetics of full-length AnxA2 in comparison to the mutant proteins. We observed that several mutations/truncations that disrupted the autonomous folding of AnxA2-DI did not disrupt the capacity of the full-length AnxA2 to fold. Conversely, some mutations that preserved the autonomous folding of AnxA2-DI strongly disrupted the thermostability of full-length AnxA2. However, these mutations at the AnxA2 DI–DIV interface render the full-length protein less stable, which does not mean that the folding pathway is affected. Therefore, we propose a new model of annexin folding, in which the autonomous folding of AnxA2-DI is not a precondition for the folding of the full-length AnxA2 protein. Of additional interest, we provide evidence for the presence of a previously undetected obligatory transient intermediate in the folding of AnxA2. Analysis of the effect of the mutations on the stability of the intermediate versus that of the native state suggests this intermediate to be misfolded, with residues of AnxA2-DI involved in nonnative contacts. In fact, we observed that most mutations have a more pronounced effect on the stability of the intermediate compared with that of the native state. Thus, the results presented suggest the folding of AnxA2 to be a cooperative process, rather than a stepwise reaction, undergoing transient misfolding events, possibly involving AnxA2-DI. This does not preclude the possibility that AnxA2-DI must be folded during the folding events of full-length AnxA2.
Materials and methods
Construction of wild-type and mutants of full-length AnxA2, truncated Δ1-34AnxA2, and AnxA2-DI
The bovine anxA2 cDNA (RefSeq: NM_174716.1) for the coding region in the pGEM-3Zf(+) vector (Promega Corporation, Madison, USA) was used as the template for the construction of wild-type (wt) and mutant full-length AnxA2, truncated Δ1-34AnxA2, and AnxA2-DI (for details, see Fig. 2). This was done by BbsI seamless cloning (26) using TA cloning of the cDNA fragments into the TOPO vector (Thermo Fisher Scientific, Waltham, USA) as an intermediate step or by QuickChange Site-Directed Mutagenesis (Agilent Technologies, Santa Clara, USA). The full-length and the Δ1-34 truncated AnxA2 forms were expressed with a 6His tag from the pETM10 vector, whereas the wild-type and mutant forms of AnxA2-DI were expressed from the pETM41 vector fused to 6HisMBP, and subsequently separated by Tobacco Etch Virus (TEV) cleavage of a TEV-protease sensitive linker region (ENLYFQG). Both vectors were kindly provided by Gunter Stier (University of Heidelberg, Germany). Oligonucleotide primers, 38–59 oligomers, were obtained from Sigma-Aldrich (Saint-Louis, USA). The final constructs were verified by DNA sequencing at the Haukeland University Hospital.
Figure 2.

Characterization of the solubility of wild-type and mutated (A) [1–339] (wild-type, F33A, D38A, R63A, Q67A, and F33A-D38A-R63A-Q67A), (B) [35–339] (wild-type, D38A, R63A, Q67A), and (C) [1–339] (wild-type, V57E, V98R, G101Y, V57-V98R-G101Y) AnxA2 forms as determined by 10% SDS-PAGE analysis. The 6His-tagged AnxA2 forms were expressed for 3 h at 37°C after induction with 1 mM IPTG. Subsequently, aliquots were retrieved and sonicated in breakage buffer (total, T). After centrifugation at 16,000 g for 15 min, aggregated proteins were recovered as the pellet (P) fraction and the soluble proteins as the supernatant (S) fraction. The protein bands were visualized by Coomassie Brilliant Blue staining. AnxA2 is indicated by an arrowhead to the right.
Protein expression and purification
The expression and purification of the AnxA2 wild-type and mutants of full-length, Δ1-34 truncated and DI forms were done as described previously (25), with some modifications. The sonicated homogenate was centrifuged in 2-mL tubes at 16,000 gav for 30 min at 4°C, and the supernatant was filtrated through a sterile 0.2-μ-pore-size filter (Merck Millipore, Burlington, USA) before loading onto a column (Bio-Rad Laboratories, Hercules, USA) with equilibrated His-Select Nickel Affinity Gel (Qiagen, Hilden, Germany). The resin and the supernatant were mixed and incubated for 15 min, until the resin settled in the column and the supernatant was eluted by the force of gravity. The flow-through was again mixed with the resin before elution of protein. Subsequently, the 6HisMBP- AnxA2-DI fusion proteins were cut by TEV and purified again as described (25,27). The TEV cleavage site results in four additional residues (GAMG) at the N-terminus of the peptide constructs.
Proteins used in NMR experiments were expressed in M9 15N minimal medium (4.3 mM Na2HPO4 x 2H2O, 22 mM KH2PO4, 8.6 mM NaCl, 9.2 mM 15NH4Cl, 30 μM FeCl3 x 6H2O, 134 μM EDTA, 6.2 μM ZnCl2, 1 μM CuCl2, 0.4 μM CoCl2 x 6H2O, 1.6 μM H3BO3, 68.6 μM MnCl2 x 2H2O, 0.4% glucose, 1 mM MgSO4, 0.3 mM CaCl2, 1 μg/mL biotin, 1 μg/mL thiamine, and 35 μg/mL kanamycin). All reagents were from Sigma-Aldrich (Saint-Louis, USA). The labeled proteins were finally eluted with 20 mM Tris (pH 7).
Solubility assays
Cultures (25 mL) were expressed at 37°C for 3 h. 1 mL of the expressed cultures was pelleted and resuspended in 500 μL breakage buffer (50 mM Na2HPO4 (pH 8.0), 0.5 M NaCl, 10 mM imidazole, 5% (w/v) glycerol, 0.5 μg/mL DNase I, 0.25 μg/mL RNase A, protease inhibitor cocktail (1 tablet/50 mL) (Complete, EDTA-free, Roche Diagnostics, Basel, Switzerland) and 1 mM DTT). All reagents were from Sigma-Aldrich (Saint-Louis, USA) if not otherwise stated. The bacteria were disrupted by sonication cycles at 4°C (4 x 20 pulses) with a thin probe. Sonicated cells (T) (100 μL) were centrifuged (16,000x g for 15 min) to obtain a supernatant (S) and a pellet (P) fraction. Proteins in equal volumes of total extract (T), supernatant fraction (S), and the pellet (P) (resuspended in 100 μL breakage buffer) were separated by 10% SDS-PAGE.
CD measurements
Thermal transition curves (25°C–75°C, 30°C–80°C, or 25°C–95°C temperature ranges) were determined by monitoring the decrease in ellipticity at 222 nm at a scanning rate of 100°C/h using a 1-mm pathlength cell in a Jasco J-810 spectropolarimeter equipped with a Peltier temperature control unit. The proteins were diluted to 15–40 μM in 20 mM Tris (pH 7.0). Since we purified several constructs at the same time to standardize conditions as much as possible, it was important to determine an apparent Tm as soon as possible after purification. Therefore, the scanning rate was 100°C/h for all constructs, so the profiles and apparent Tms were comparable, providing data sufficiently informative for drawing qualitative conclusions about the folding status and the thermostability of the constructs. All samples were freshly prepared, and the CD measurements were performed with single runs on the same day to obtain the relative differences between the apparent Tm values.
NMR spectroscopy
The NMR spectra were acquired using sample concentrations of 150–700 μM at 25°C and 700 MHz (1H frequency) on a Bruker DRX700 spectrometer equipped with a z-gradient triple resonance cryoprobe, in 20 mM Tris (pH 7.0). 1H-15N HSQC were recorded with spectral widths set to avoid folding of 15N resonances. In all spectra, the water signal was suppressed using a WATERGATE sequence (28). Typically, 256 t1 increments were acquired and quadrature was achieved (29). Spectra were processed and analyzed with TopSpin Bruker Software Package Version 2.1 (RRID: SCR_014227).
Stopped-flow measurements and analysis of kinetic data
The kinetics of unfolding and folding of AnxA2 were detected by monitoring the fluorescence emission after excitation at 280 nm, using a cutoff filter at 320 nm on a stopped-flow instrument (Applied Photophysics, Leatherhead, UK). For each experiment, an average calculated from three to five independent data acquisitions was fitted to the following equation:
| (1) |
which allows to calculate the observed rate constant (kobs) at different denaturant concentrations for each variant analyzed (Eq. 1). The dependence of the kobs as a function of denaturant concentration was fitted using an equation implying a three-state scenario, with the presence of an intermediate accumulating in the unfolding reaction:
| (2) |
where kf0 and ku0 represent the folding and unfolding microscopic rate constant in the absence of denaturant, and KN–I is the equilibrium rate constant between the native and intermediate state (Eq. 2) (30).
The difference in free energy between the denatured and native state for each mutant analyzed was calculated as
| (3) |
| (4) |
and
| (5) |
| (6) |
The beta Tanford (βT) value for the intermediate state was calculated as follows:
| (7) |
| (8) |
Accession numbers
Bovine AnxA2 was used in these experiments; UniProt: P04272, GenBank: BT021010.1, MMDB: 126161, and PDB 1: 4X9P.
Results
Design of mutated AnxA2-DI, Δ1-34AnxA2, and full-length AnxA2 forms
In a former study, we showed that calculation of the cumulated surface hydrophobicity of each isolated AnxA2 domain indicated that AnxA2-DI is the most hydrophilic domain (25). In the AnxA2 crystal structure (PDBs: 1XJL (18) and 4X9P (13)), we observed that four highly conserved amino acids (F33, D38, R63, and Q67; counting the first M as residue 1) appear to form a “structural tetrad,” which stands on the periphery of AnxA2-DI, yet establishes a network of specific mutual contacts (Fig. 1 A). The side chain of F33 is shielded from the solvent by the side chains of D38 and R63, which extend toward each other due to a favorable charge interaction. In addition, the aromatic ring of F33 establishes an antiparallel interaction with the side chain of Q67. The residue Q67, in turn, establishes, via its side chain, specific contacts that connect the N-terminal tail to AnxA2-DI. The side chain carboxamide oxygen of Q67 makes a bidentate H-bond, one to the main-chain amino group of residue Y30 that belongs to the N-terminal tail, and the other one to the main-chain amino group of S64, a residue situated at the connection between helix B and helix C of AnxA2-DI. Based on these observations, we hypothesized that the network of contacts established by the F33/D38/R63/Q67 tetrad could be important for stabilizing the folding of AnxA2-DI and thus of full-length AnxA2. The four positions were therefore mutated into alanine residues, either individually (mutations F33A, D38A, R63A, Q67A) or simultaneously (mutation F33A-D38A-R63A-Q67A) (Fig. 1 C). These mutations were introduced in the context of a construct of DI, corresponding to residues [25–104] of AnxA2 encompassing the five helices of DI preceded by 10 residues of the N-terminal tail (Fig. 1 C), exactly like in the previously published AnxA1-DI construct (22,23). The same mutations were also performed in the context of full-length AnxA2 (construct [1–339]).
The C-terminal part of the N-terminal tail that is visible in the crystal structures (residues 25–34) is involved in several contacts with DI. Thus, a deletion of the N-terminal tail (residues 1–34) would be expected to disrupt the capacity of DI to fold cooperatively. Such a deletion was therefore performed both in the context of DI (construct [35–104]) and in the context of full-length protein (construct [35–339]). We also generated constructs that combined the N-terminal deletion G25-D34 with the single-point mutations D38A, R63A, and Q67A, both in the context of isolated DI and of the full-length protein.
AnxA2-DI is engaged in strong contacts with other domains within the annexin core. To identify the amino acid residues that are involved in the interaction of AnxA2-DI with the rest of the protein, we examined the crystal structures of AnxA2 (PDBs: 1XJL and 4X9P) and found that its helices B and E are involved in the interfacial contact with the rest of AnxA2 (Fig. 1 B). Key residues at the interface are V57, H94, L95, V98, G101, L102, and K104. However, in the isolated AnxA2-DI, L95 and L102 appear to be too buried to be mutated, since it is likely that mutation of these residues will affect the folding of AnxA2-DI. H94 and K104 are not conserved in AnxA2 (Fig. 1 B in Aukrust et al. (25)). By contrast, V57 (at the interface between DI and DIV), V98 (interface between DI and DIV), and G101 (interface between DI and DII and DIV) are quite conserved and exposed in the isolated AnxA2-DI. We therefore mutated these three positions to polar residues (V57E and V98R) and bulky, yet polar residue (G101Y) in the context of [25–104] AnxA2-DI and full-length AnxA2 (Fig. 1 C).
Cloning, expression, and purification of the wild-type and mutated AnxA2-DI, Δ1-34-AnxA2, and full-length AnxA2 forms
Constructs of AnxA2-DI were fused via a TEV-protease-sensitive linker to the C-terminus of the highly soluble bacterial maltose binding protein. Constructs of full-length AnxA2 were produced with an N-terminal 6His tag.
The His-tagged constructs of point-mutated and N-terminally deleted full-length AnxA2 all showed decreased solubility upon bacterial overexpression at 37°C, as compared with the wild-type full-length AnxA2 (Fig. 2). Although about 60%–70% of the wild-type construct [1–339] was retrieved in the soluble fraction of the sonicated lysate, no more than 10% of the other constructs was retrieved in the soluble fraction, except for construct AnxA2 [1–339] G101Y, which was almost as soluble as the wild-type construct. These differences in solubility were no more apparent when the expression was performed at 15°C (data not shown). In this condition, the amount of overexpressed protein retrieved in the soluble fraction was comparable for all mutants and for the wild-type AnxA2. The proteins were therefore all expressed at 15°C for further purification.
Lowering the expression temperature from 37°C to 15°C is expected to decrease 1) the rate of ribosomal peptide synthesis, 2) the concentration of newly expressed AnxA2 polypeptide chains exploring their folding at any given time point, and 3) the diffusion rate of these polypeptide chains undergoing folding. The decreased efficiency of folding of AnxA2 mutants at 37°C as compared with 15°C suggests that the corresponding mutations (or deletion) resulted in misfolded intermediates with high propensity to self-associate. Being more concentrated and prone to faster diffusion at 37°C, these misfolded intermediates form a higher proportion of insoluble aggregates at 37°C, whereas at 15°C, they have an increased chance to explore the folding and reach the correctly folded final state.
Role of the N-terminal tail and of the structural tetrad F33-D38-R63-Q67 in the folding and stability of AnxA2-DI
To explore the role of the “structural tetrad” formed by F33, D38, R63, and Q67, the thermal stability and the folding status of the purified protein constructs were analyzed by thermal denaturation CD measurements and 1H-15N HSQC NMR.
Thermal denaturation was assessed by following the change in ellipticity at 222 nm. The 222-nm signal is mainly contributed by α-helical secondary structure, yet other structures including random coil can also contribute some signal. Moreover, the presence of α-helical secondary structure does not warrant that the domain has acquired a folded tertiary structure. For some constructs, we could only obtain low-concentrated samples, so that the spectral quality was sometimes suboptimal (see Fig. S2), and therefore subsequent determination of the apparent transition temperature (Tm) was error-prone (see Table 1). However, even in such cases, the profiles were sufficiently informative for drawing qualitative conclusions about the folding status and the thermostability of the domain.
Table 1.
Estimated Tms of [25–104], [35–104], [1–339], and [35–339] recombinant wild-type or mutated AnxA2 forms
| AnxA2-DI |
Full-length/truncated AnxA2 |
||
|---|---|---|---|
| Construct | Estimated Tm | Construct | Estimated Tm |
| [25–104] wild-type | 65°C | [1–339] wild-type | 58°C |
| [25–104] F33A | 63°C | [1–339] F33A | 56°C |
| [25–104] D38A | Not folded | [1–339] D38A | 51°C |
| [25–104] R63A | 55°C | [1–339] R63A | 54°C |
| [25–104] Q67A | Not Folded | [1–339] Q67A | 53°C |
| [1–399] F33A-D38D-R63A-Q67A | 49°C | ||
| [35–104] wild-type | Not folded | [35–339] wild-type | 47°C |
| [35–104] D38A | Not folded | [35–339] D38A | 41°C |
| [35–104] R63A | Not folded | [35–339] R63A | 47°C |
| [35–104] Q67A | Not folded | [35–339] Q67A | 45°C |
| [25–104] V57E | 58°C | [1–339] V57E | 52°C |
| [25–104] V98R | 64°C | [1–339] V98R | 41°C |
| [25–104] G101Y | 56°C | [1–339] G101Y | 50°C |
| [25–104] V57E-V98R-G101Y | 62°C | [1–339] V57E-V98R-G101Y | Not folded |
The folded tertiary structure can be tracked by observing a wide distribution of the amide 1H frequencies in the 1H-15N HSQC NMR (Fig. 3), as well as by observing a cooperative transition in the temperature-induced decrease of the absolute value of the negative ellipticity signal in the CD instrument (Fig. S2).
Figure 3.

Impact of mutating amino acid residues (F33, D38A, R63A, Q67A) or deleting the N-terminus on the folding of AnxA2-DI. 1H-15N HSQC NMR spectra of [25–104] AnxA2; folded wild-type (600 μM), F33A (550 μM) and R63A (150 μM) (upper row), and misfolded D38A (275 μM), Q67A (275 μM) as well as misfolded [35–104] AnxA2; wild-type (50 μM), D38A (700 μM), R63A (700 μM), and Q67A (700 μM), were recorded at pH 7, 25°C, and 700 MHz (1H frequency). To see this figure in color, go online.
According to these approaches, the isolated wild-type AnxA2-DI (construct [25–104] wild-type) was fully folded at 25°C (Fig. 3, upper left panel), in agreement with published data on a corresponding construct of AnxA1-DI (22,23). The thermal transition of wild-type AnxA2-DI followed a broad temperature range with an apparent transition temperature (Tm) of ∼65°C (Table 1) as determined from the first derivative of the transition curve (Fig. S2). Among the mutants of isolated AnxA2-DI constructs, [25–104] F33A and [25–104] R63A were also folded (Fig. 3, upper central and right panels), albeit with a lower Tm (Table 1; Fig. S2), indicating a loss of thermodynamic stability, particularly in the case of the R63A mutant. By contrast, the other mutants of isolated AnxA2-DI, [25–104] D38A and [25–104] Q67A, were not folded (Fig. 3, middle-left and central panels; Fig. S2). This was corroborated by data from ellipticity scans at 25°C, which showed a high contribution of random coil and thus low helical structure (results not shown). Furthermore, all the wild-type and mutated [35–104] AnxA2-DI constructs, corresponding to the strict five-helix AnxA2-DI without any N-terminal tail, were also not folded at 25°C (Fig. 3, middle-right panel and all lower panels). Altogether, these results point to the importance of the G25-D34 region of the N-terminal tail, as well as of the structural tetrad F33-D38-R63-Q67 in the autonomous folding of the isolated AnxA2-DI.
Role of the N-terminal tail and of the structural tetrad F33-D38-R63-Q67 in the folding and stability of full-length AnxA2
Next, we introduced the same mutations, namely F33A, D38A, R63A, Q67A, and Δ1-34, separately or combined, in the context of full-length AnxA2. The thermal unfolding of wild-type full-length AnxA2 (Fig. S2) showed a sharp transition (Tm = 55.4°C) over a range of less than 10°C, characteristic of a strong cooperative transition from a specific native tertiary structure to an unfolded state. Interestingly, all our constructs of AnxA2 bearing the above-mentioned mutations were still able to fold according to their 1H-15N HSQC NMR spectra acquired at 25°C (Fig. 4). They also retained the highly cooperative thermal unfolding behavior of wild-type AnxA2 (Fig. S2), albeit with variable decrease in thermodynamic stability, illustrated by lower melting temperatures (Tm) (Table 1). Therefore, although mutations Δ1-34, D38A, and Q67A abolished the folding capacity of isolated AnxA2-DI, they did not prevent full-length AnxA2 to acquire a folded conformation, demonstrating that the folding of the core four-domain structure can occur in the presence of N-terminal deletions or mutations that prevent the folding of the isolated DI domain. It remains possible that DI–DIV docking and subsequent formation of a rigidified DI–DIV module would be the first step of AnxA2 folding, as proposed earlier in the sequential folding model (22,24).
Figure 4.

Impact of mutating amino acid residues (D38A, Q67A), causing collapse of the folding of AnxA2-DI, and of deleting the N-terminus on the folding of full-length AnxA2. 1H-15N HSQC NMR spectra of [1–339] AnxA2; wild-type D38A and Q67A as well as [35–339] AnxA2, all at 200 μM were recorded at pH 7, 30°C, and 700 MHz (1H frequency). To see this figure in color, go online.
Nonetheless, although the full-length F33A and R63A mutants only have slightly lowered Tms, the full-length D38A and in particular the multiple full-length AnxA2 mutant (F33A-D38A-R63A-Q67A) show decreased thermodynamic stability (Table 1; Fig. S2). This is in agreement with the decreased solubility, upon bacterial overexpression at 37°C, of the mutated full-length AnxA2 constructs compared with the wild-type full-length AnxA2 (Fig. 2). This suggests that the full-length AnxA2 mutants, which harbor folding-defective DI, display slower kinetics of folding than the wild type.
The importance of hydrophobic interfacial amino acids of DI in the folding of full-length AnxA2
Subsequently, we focused on several amino acids located in AnxA2-DI, which were targeted to disrupt the folding of full-length AnxA2, but not of the isolated AnxA2-DI. Thus, a new series of mutations, this time targeting hydrophobic amino acids involved in interfacial contacts with other domains of AnxA2, was performed: three single-point mutants V57E, V98R, and G101Y and the triple mutant V57E-V98R-G101Y in the full-length protein. Neither the single-point mutations nor the combined triple mutation altered the capacity of the isolated AnxA2-DI domain to acquire its folded tertiary structure (Fig. 5). However, all mutations affected the thermodynamic stability of the isolated domain to various degrees (Table 1; Fig. S2). When the same interfacial mutations were introduced into full-length AnxA2, all mutants could be purified in a soluble form and retained, to some extent, a cooperative thermal unfolding process (Fig. S2). However, the unfolding profile of the multiple mutant (V57E-V98R-G101Y) of full-length AnxA2 was seriously altered (Fig. S2), suggesting a less cooperative process than that observed for the wild-type protein. Furthermore, the Tms of the three single mutants of full-length AnxA2 decreased significantly. The multiple mutant (V57E-V98R-G101Y) of full-length AnxA2 was also found to precipitate when prepared for 1H-15N HSQC NMR spectroscopy measurements, so that no spectrum could be obtained. Altogether, these data point to the contribution of the interfacial residues V57, V98, and G101 of AnxA2-DI, mainly interacting with interfacial residues in AnxA2-DIV, to the stability of the full-length AnxA2 protein.
Figure 5.

Mutating amino acid residues (V57E, V98R, G101Y, V57E-V98R-G101Y) in AnxA2-DI involved in interfacial contact with other domains does not unfold this domain. 1H-15N HSQC NMR spectra of [25–104] AnxA2; wild-type (600 μM), V57E (250 μM), V98R (250 μM), G101Y (250 μM), and V57E-V98R-G101Y (150 μM) were recorded at pH 7, 25°C, and 700 MHz (1H frequency). To see this figure in color, go online.
The folding and unfolding kinetics of AnxA2
Prompted by the detailed characterization of the unfolding stability of AnxA2 and its site-directed mutants reported above, we resorted to analyze quantitatively their folding and unfolding kinetics by stopped-flow fluorescence. In particular, we carried out 11-fold dilution experiments in which 1) the native protein diluted in buffer (25 mM Tris-HCl, 150 mM NaCl, 2 mM EDTA, 2 mM Tris(2-carboxyethyl)phosphine (TCEP), pH 8.0) was rapidly mixed with guanidinium-HCl (GdnHCl) at different concentrations (unfolding), and 2) the protein in denaturing conditions (buffer with added GdnHCl) was mixed with refolding buffer to reach different final concentrations of GdnHCl. Samples were excited at 280 nm, and the fluorescence emission was collected using a 320-nm cutoff filter. Typical folding and unfolding time courses are reported in Fig. 6. Interestingly, although in both cases the apparent decay appears consistent with a single exponential function, both folding and unfolding correspond to a decrease of fluorescence versus time, suggesting the rapid accumulation of a burst phase intermediate in the dead-time of the stopped-flow in either folding or unfolding (31,32).
Figure 6.

Typical unfolding and refolding time courses of AnxA2. (A) AnxA2 diluted in buffer (25 mM Tris-HCl, 150 mM NaCl, 2 mM EDTA, 2 mM Tris(2-carboxyethyl)phosphine (TCEP), pH 8.0) was rapidly mixed with GdnHCl to reach 5.4 M final concentration (unfolding); (B) the protein in denaturing conditions (buffer with added GdnHCl) was mixed with refolding buffer to reach 0.5 M final concentration of GdnHCl.
A semilogarithmic plot of the observed (un)folding rate constants (chevron plot) for wild-type AnxA2 is shown in Fig. 7. A visual inspection of the observed dependence might appear consistent with a V-shape chevron plot, a typical signature of two-state folding (33). Fitting the observed rate constant to a simple two-state equation, however, indicates a folding m value of 2.2 ± 0.1 kcal mol−1 M−1 and an unfolding m value of 0.9 ± 0.05 kcal mol−1 M−1, with a total m value of 3.1 ± 0.1 kcal mol−1 M−1. The m value, defined as δΔG/δ[denaturant], represents a quantitative measure of the cooperativity of the (un)folding reaction, and it reflects the change in accessible surface area upon denaturation (34). The m value calculated from the two-state analysis of wild-type AnxA2 appears to be much lower than that expected from a protein of 339 amino acids, which should be around 6 kcal mol−1 M−1 (34). This result suggests the presence of additional undetected states along the reaction pathway that influence the calculation of the structural transition occurring between native and denatured state. Therefore, the unexpected lower m value suggests the presence of (at least) one intermediate.
Figure 7.

A semilogarithmic plot of the observed (un)folding rate constants (chevron plot) for wild-type AnxA2. The chevron plot of the observed rate of folding and unfolding is shown as a function of final denaturant concentration (GdnHCl). The total chevron plot is fit to a two-state equation.
Keeping in line with the stability experiments reported above, we subjected the mutants reported in Table 2 to kinetic experiments. The chevron plot of all the mutants in comparison to that of wild-type AnxA2 is reported in Fig. 8. A pronounced curvature of the unfolding limb of the chevron plot is clearly observed in all cases, strongly indicating the presence of an intermediate. Therefore, the stopped-flow experiments clearly point to the accumulation of a reaction intermediate in the folding of AnxA2, for the wild-type protein as well as all for its mutants. Quantitative analysis of the chevron plots presented in Fig. 8, by fitting kinetic data with a three-state model (Eq. 2), allows to calculate the mf, mu, and mI-N parameters, which represent the dependence of folding and unfolding microscopic rate constants and equilibrium rate constant between the intermediate and the native state, from denaturant concentration. The mI-N value calculated for the intermediate is 2.4 ± 0.3 kcal mol−1 M−1, with a total mD-N value of 5.9 ± 0.2 kcal mol−1 M−1. As mentioned above, these kinetic parameters correlated to the change in accessible surface area upon unfolding and allow to pinpoint the position of the intermediate state along the reaction coordinate. The calculation of the so called Tanford β value (βT) for the intermediate state, as formalized by Eq. 8, reports a value of βT = 0.59 ± 0.06. This value, typically lying between 0 (for the denatured state) and 1 (for native state), suggests the intermediate to be populated as a relatively late event in the reaction, implying a possible involvement of more than one of the four domains of AnxA2. Importantly, the observed robustness of kinetic m values and consequently of βT values clearly points to the evidence that, although destabilized, all variants explored appear to conserve a similar folding mechanism.
Table 2.
Kinetic and thermodynamic parameters for [1–339] and [35–339] wild-type AnxA2 and the indicated mutant AnxA2 variants
| kD-I (s−1) | kN-D (10−5 s−1) | KI-N (104) | ΔΔGD-N (kcal mol−1) | ΔΔGD-I (kcal mol−1) | |
|---|---|---|---|---|---|
| [1–339] wild-type | 6.5 ± 1.0 | 1.3 ± 0.7 | 0.2 ± 0.1 | ||
| [1–339] F33A-D38A-R63A-Q67A | 1.3 ± 0.2 | 0.8 ± 3.7 | 5.5 ± 0.5 | 0.7 ± 0.2 | 2.5 ± 0.3 |
| [1–339] D38A | 1.1 ± 0.2 | 1.2 ± 0.6 | 2.3 ± 0.2 | 1.0 ± 0.2 | 2.4 ± 0.3 |
| [1–339] V57E | 2.8 ± 0.5 | 2.1 ± 1.0 | 6.3 ± 0.6 | 0.8 ± 0.2 | 2.7 ± 0.3 |
| [1–339] R63A | 3.7 ± 0.6 | 1.3 ± 0.6 | 1.9 ± 0.2 | 0.3 ± 0.2 | 1.6 ± 0.2 |
| [1–339] Q67A | 2.2 ± 0.4 | 4.8 ± 3.2 | 0.2 ± 0.1 | 1.4 ± 0.2 | 1.4 ± 0.2 |
| [1–339] V98R | 2.9 ± 0.5 | 60 ± 20 | 0.6 ± 0.1 | 2.7 ± 0.3 | 3.3 ± 0.3 |
| [1–339] G101Y | 3.0 ± 0.5 | 1.7 ± 0.8 | 1.7 ± 0.2 | 0.6 ± 0.1 | 1.8 ± 0.2 |
| [35–339] wild-type | 1.4 ± 0.2 | 2.4 ± 1.0 | 0.9 ± 0.1 | 1.2 ± 0.2 | 2.0 ± 0.2 |
| [35–339] R63A | 1.4 ± 0.2 | 2.9 ± 1.3 | 4.2 ± 0.4 | 1.4 ± 0.2 | 3.0 ± 0.3 |
| [35–339] Q67A | 1.3 ± 0.2 | 3.2 ± 1.4 | 1.8 ± 0.2 | 1.5 ± 0.2 | 2.7 ± 0.3 |
Figure 8.

The chevron plots of wild-type AnxA2 and the indicated AnxA2 variants. Lines represent the best fit to an equation that describes a three-state folding mechanism, implying the presence of an intermediate along the reaction pathway. Chevron plots were globally fitted by sharing kinetic m values for all datasets (see materials and methods and results sections for details).
A quantitative analysis of the chevron plots reported in Fig. 8 allows calculation of the native and intermediate stability for each of the measured mutants (Table 2). Of particular interest, it may be noted that the mutations result in a clear destabilization of the intermediate state versus the native state. Importantly, in all cases, with the sole exception of the Q67A mutant, the analysis of thermodynamic parameters reports that such destabilization appears to be more evident in the intermediate state compared with the native state. This peculiar effect, whereby a mutation has a more pronounced effect on a transient intermediate state rather than on the native state, represents a characteristic signature of transient misfolding, suggesting that AnxA2-DI is misfolded in the intermediate state.
Discussion
AnxA2 is a genuinely multifunctional protein (1,2,3,4,5), involved in a variety of cellular processes ranging from membrane-cytoskeleton interactions and actin dynamics to mRNA biogenesis. The latter process involves its binding to specific mRNAs and participation in the regulation of their transport (as exemplified by c-myc mRNA (35)) and/or translation (as exemplified by p53 (36) and c-myc (6) mRNAs). Moreover, AnxA2 appears to have several roles in the nucleus (37). Furthermore, it functions as an extracellular protein (38,39) and constitutes a marker of exosomes derived from the endocytic pathway (www.exocarta.org/). Thus, it comes as no surprise that AnxA2 is closely involved in tumor progression by participating, for example, in metalloproteinase activation (40), cell migration (41), cell proliferation (42), and epithelial-mesenchymal transition (43). In several tumor types, it even serves as a specific marker for the more advanced/progressive states of the cancer (40,44). It has been proposed that AnxA2 serves as an ideal target for anticancer drugs (45). However, to target the full-length protein by knockout or knockdown may not be an adequate strategy due to its multifunctionality and/or compensatory effects (46). Thus, a better approach may be to use peptides representing specific regions of AnxA2 to compete with specific cellular ligands that bind to this particular region. The proper design of such peptides highly depends on our understanding of the structure of the full-length protein and of its individual domains. This also implies understanding the effects of perturbation of the AnxA2 structure on its folding and the underlying kinetics.
When considering the folding and stability of globular proteins or domains, one should distinguish two very distinct properties. A first property is the ability to adopt a cooperatively folded state, encoded by the protein sequence. The cooperatively folded state displays various characteristics, including a wide distribution of 1H frequencies and a cooperative denaturation profile. The second property is the thermostability of the folded state, characterized—at least in part—by the temperature of the denaturation mid-point. Some mutations may alter the thermostability of the domain while preserving the ability to adopt a folded state. Other mutations, directed at particular “hot spots,” may completely disrupt the ability to adopt a folded state (47). In the present work, the two types of effects have been observed, both for isolated AnxA2-DI and for full-length AnxA2.
A sequential folding model had been previously proposed for the annexin core structure (22,23). In this model, DI folds first, whereas DII and DIII remain partly unfolded until DIV folds by docking on DI, which in turns induces the complete folding of DII and DIII. This model was primarily based on the observation that, within AnxA1, only the isolated domain DI was capable of folding independently (22,23). While working on AnxA2, we also observed that AnxA2-DI was also able to fold independently (12,25). This observation, together with the high conservation of annexin core sequences across the annexin family members, suggested that the folding process of the annexin core structure should be conserved throughout the entire family. We therefore decided to investigate if the proposed annexin core sequential folding model also applies to AnxA2.
As a first step, we identified mutations that are likely to disrupt the autonomous folding of isolated AnxA2-DI. One possible approach would have been to mutate one or several residues within the central hydrophobic core of the domain, which hold together the five conserved helices. However, mutations within the hydrophobic core sometimes just decrease thermostability, without really disrupting the folding process. By carefully inspecting the structure, we realized that the network of contacts established by the F33/D38/R63/Q67 tetrad constitute a “structural knot,” which might represent a critical bottleneck in the folding process of AnxA2-DI. This hypothesis was indeed confirmed, as we found that the single-point mutations D38A and Q67A, directed at the tetrad, were both sufficient to completely disrupt the folding ability of DI. In addition, we found that deletion of the N-terminal region of DI until residue 34 also completely disrupted the folding ability of this domain. This can be explained by the fact that this deletion eliminated both the side chain of F33 and the main chain of Y30, which both provide contacts to the above-mentioned “structural knot.”
As a second step, we showed that re-introduction of the mutated DI bearing any of the disrupting mutations (single-point, deletion, or combined) within the full-length annexin did not prevent the folding of the latter. This unambiguously demonstrated that, unexpectedly and in contradiction with the previously proposed folding model (22,23), the ability of DI to fold independently is not a prerequisite for the folding of the full-length AnxA2.
As a third step, we performed other mutations of DI, which were not intended to disrupt its folding, but rather aimed at disrupting the interface of DI with DIV. According to the stepwise folding model previously proposed for AnxA1, the protein can achieve its complete folding only when DIV is allowed to dock onto DI. We generated three mutations of DI, which replaced interface residues of DI with bulkier charged or polar residues, expected to perturb the interface. None of these mutations, either single-point or combined in a multiple mutant, disrupted the independent folding of the isolated DI. Neither did they prevent full-length AnxA2 to acquire a cooperatively folded structure. Nevertheless, some of these mutations strongly impacted the thermostability of full-length AnxA2, pointing to a role of interfacial residues in holding together the four-domain cooperative arrangement. Going more in depth, we observed an inverse correlation between the impact of G101Y and V98R mutations on the thermostability of the individual DI and their impact on the thermostability of the entire protein. G101Y strongly destabilized the domain yet destabilized only slightly the full-length protein. In full contrast, V98R hardly altered the stability of the isolated domain, yet it strongly destabilized the full-length AnxA2 and suggests that V98 holds a key position in the interfacial contacts between the domains (DI with DIV). Hence, we found no correlation between the folding behaviors of DI and full-length AnxA2, neither regarding their ability to fold nor regarding their thermostabilities. The ensemble of these data is in favor of a highly cooperative and robust folding process for AnxA2, which may be able to go through various alternative intermediate steps. What about other vertebrate annexins? AnxA7 and AnxA11 both harbor a very long N-terminus (∼20 kDa) and are difficult to obtain in a soluble state ((20) unpublished observations). Likewise, AnxA9 is notoriously prone to aggregation. Thus, it is expected that annexins that can easily be obtained in a soluble form also present a robust folding process stabilized by interfacial contacts like that seen in AnxA2. However, other annexins, less stabilized by interfacial contacts, may follow a different folding path.
The analysis of the kinetic data reported in this work appears to reconcile the stepwise mechanism, invoked for AnxA2, with a more cooperative view of folding. In fact, the accumulation of a misfolded intermediate involving more than one domain (with a βT of 0.59), implies that, although DI may represent a folding nucleus of AnxA2, it is prone to engage some nonnative interactions with the other domains before folding, as suggested by the analysis of thermodynamic parameters. In fact, although it is not surprising to observe that mutations disrupting the structure of DI in isolation correspond to a change in stability in the case of the full-length protein, a comparison of ΔΔG values obtained from the analysis of kinetic data highlights a more pronounced destabilizing effect upon mutation on the intermediate state compared with the native state. This finding points to the evidence of the presence of a misfolded intermediate on the folding pathway of AnxA2. Future mutational analysis combined with kinetic (un)folding experiments may provide additional information useful to characterize these transient misfolding events in greater detail. Importantly, mutations affecting the structure of DI in isolation appear to have no effect on kinetic m values calculated from the analysis of chevron plots. This aspect further corroborates the scenario of a robust cooperative folding reaction and suggests the folding pathway of the protein to be not affected by mutations.
In conclusion, we show that the folding process of full-length AnxA2 is highly cooperative and robust and does not rely on an initially folded DI. By contrast, hydrophobic interfacial amino acids of DI are important in the folding of full-length AnxA2. Furthermore, our kinetic data indicate that AnxA2 folds through a transient nonnative intermediate, which is a novel finding. This novel insight into the folding process of AnxA2, and possibly also other annexins, provides the basis to dissect the cooperative folding process of proteins, in particular with repeating subunits.
Author contributions
H.H.: methodology, validation, data curation, formal analysis, investigation, visualization, writing – part of original draft, writing – review & editing. J.R.: methodology, validation, data curation, formal analysis, investigation, writing – review & editing. Y.N.: methodology, validation, formal analysis, investigation, writing – review & editing. C.K.: methodology, validation, formal analysis, writing – review & editing. A.T.: methodology, validation, formal analysis, writing – review & editing. S.G.: methodology, validation, data curation, formal analysis, investigation, resources, supervision, writing – original draft, writing – review & editing. G.T.: conceptualization, funding acquisition, investigation, visualization, project administration, resources, supervision, writing – original draft, writing – review & editing. A.V.: conceptualization, funding acquisition, investigation, project administration, resources, supervision, writing – original draft, writing – review & editing.
Acknowledgments
The study was supported by grants from the Research Council of Norway (Aurora program; no. 199747) and Helse Vest (no. 911499). We are grateful to Hanne Ravneberg (now Equinor) for excellent technical assistance and to Gunter Stier (Heidelberg University, Germany) for the pETM10 and pETM41 vectors.
Declaration of interests
The authors declare no competing interests.
Editor: Doug Barrick.
Footnotes
Gilles Travé and Anni Vedeler contributed equally to this work.
Supporting material can be found online at https://doi.org/10.1016/j.bpj.2022.10.043.
Contributor Information
Gilles Travé, Email: gilles.trave@unistra.fr.
Anni Vedeler, Email: anni.vedeler@biomed.uib.no.
Supporting material
material
References
- 1.Gerke V., Moss S.E. Annexins: from structure to function. Physiol. Rev. 2002;82:331–371. doi: 10.1152/physrev.00030.2001. [DOI] [PubMed] [Google Scholar]
- 2.Gerke V., Creutz C.E., Moss S.E. Annexins: linking Ca2+ signalling to membrane dynamics. Nat. Rev. Mol. Cell Biol. 2005;6:449–461. doi: 10.1038/nrm1661. [DOI] [PubMed] [Google Scholar]
- 3.Rescher U., Gerke V. Annexins--unique membrane binding proteins with diverse functions. J. Cell Sci. 2004;117:2631–2639. doi: 10.1242/jcs.01245. [DOI] [PubMed] [Google Scholar]
- 4.Bharadwaj A., Bydoun M., et al. Waisman D. Annexin A2 heterotetramer: structure and function. Int. J. Mol. Sci. 2013;14:6259–6305. doi: 10.3390/ijms14036259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Vedeler A., Hollås H., et al. Raddum A.M. Multiple roles of annexin A2 in post-transcriptional regulation of gene expression. Curr. Protein Pept. Sci. 2012;13:401–412. doi: 10.2174/138920312801619402. [DOI] [PubMed] [Google Scholar]
- 6.Strand E., Hollås H., et al. Vedeler A. Annexin A2 binds the internal ribosomal entry site of c-myc mRNA and regulates its translation. RNA Biol. 2021;18:337–354. doi: 10.1080/15476286.2021.1947648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Grindheim A.K., Saraste J., Vedeler A. Protein phosphorylation and its role in the regulation of Annexin A2 function. Biochim. Biophys. Acta. Gen. Subj. 2017;1861:2515–2529. doi: 10.1016/j.bbagen.2017.08.024. [DOI] [PubMed] [Google Scholar]
- 8.Sharma M.C. Annexin A2 (ANX A2): an emerging biomarker and potential therapeutic target for aggressive cancers. Int. J. Cancer. 2019;144:2074–2081. doi: 10.1002/ijc.31817. [DOI] [PubMed] [Google Scholar]
- 9.Loges S., Mazzone M., et al. Carmeliet P. Silencing or fueling metastasis with VEGF inhibitors: antiangiogenesis revisited. Cancer Cell. 2009;15:167–170. doi: 10.1016/j.ccr.2009.02.007. [DOI] [PubMed] [Google Scholar]
- 10.Thomas S., Harding M.A., et al. Theodorescu D. CD24 is an effector of HIF-1-driven primary tumor growth and metastasis. Cancer Res. 2012;72:5600–5612. doi: 10.1158/0008-5472.CAN-11-3666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zhang H.J., Yao D.F., et al. Lu S.L. Annexin A2 silencing inhibits invasion, migration, and tumorigenic potential of hepatoma cells. World J. Gastroenterol. 2013;19:3792–3801. doi: 10.3748/wjg.v19.i24.3792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Raddum A.M., Evensen L., et al. Vedeler A. Domains I and IV of annexin A2 affect the formation and integrity of in vitro capillary-like networks. PLoS One. 2013;8 doi: 10.1371/journal.pone.0060281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Raddum A.M., Hollås H., et al. Vedeler A. The native structure of annexin A2 peptides in hydrophilic environment determines their anti-angiogenic effects. Biochem. Pharmacol. 2015;95:1–15. doi: 10.1016/j.bcp.2015.02.013. [DOI] [PubMed] [Google Scholar]
- 14.Valapala M., Thamake S.I., Vishwanatha J.K. A competitive hexapeptide inhibitor of annexin A2 prevents hypoxia-induced angiogenic events. J. Cell Sci. 2011;124:1453–1464. doi: 10.1242/jcs.079236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Huber R., Schneider M., et al. Paques E.P. The calcium binding sites in human annexin V by crystal structure analysis at 2.0 A resolution. Implications for membrane binding and calcium channel activity. FEBS Lett. 1990;275:15–21. doi: 10.1016/0014-5793(90)81428-q. [DOI] [PubMed] [Google Scholar]
- 16.Liemann S., Huber R. Three-dimensional structure of annexins. Cell. Mol. Life Sci. 1997;53:516–521. doi: 10.1007/s000180050065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Tran J.T., Rosengarth A., Luecke H. Cloning, purification and crystallization of full-length human annexin 2. Acta Crystallogr. D Biol. Crystallogr. 2002;58:1854–1857. doi: 10.1107/s0907444902013288. [DOI] [PubMed] [Google Scholar]
- 18.Rosengarth A., Luecke H. Annexin A2. Does it induce membrane aggregation by a new multimeric state of the protein? Annexins. 2004;1:129–136. [Google Scholar]
- 19.Ecsedi P., Kiss B., et al. Nyitray L. Regulation of the equilibrium between closed and open conformations of annexin A2 by N-terminal phosphorylation and S100A4-binding. Structure. 2017;25:1195–1207.e5. doi: 10.1016/j.str.2017.06.001. [DOI] [PubMed] [Google Scholar]
- 20.Lillebostad P.A.G., Raasakka A., et al. Kursula P. Structure of the ALS mutation target annexin A11 reveals a stabilising N-terminal segment. Biomolecules. 2020;10:E660. doi: 10.3390/biom10040660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Morgan R.O., Martin-Almedina S., et al. Fernandez M.P. Evolutionary perspective on annexin calcium-binding domains. Biochim. Biophys. Acta. 2004;1742:133–140. doi: 10.1016/j.bbamcr.2004.09.010. [DOI] [PubMed] [Google Scholar]
- 22.Cordier-Ochsenbein F., Guerois R., et al. Sanson A. Exploring the folding pathways of annexin I, a multidomain protein. I. non-native structures stabilize the partially folded state of the isolated domain 2 of annexin I. J. Mol. Biol. 1998;279:1163–1175. doi: 10.1006/jmbi.1998.1829. [DOI] [PubMed] [Google Scholar]
- 23.Cordier-Ochsenbein F., Guerois R., et al. Sanson A. Exploring the folding pathways of annexin I, a multidomain protein. II. Hierarchy in domain folding propensities may govern the folding process. J. Mol. Biol. 1998;279:1177–1185. doi: 10.1006/jmbi.1998.1828. [DOI] [PubMed] [Google Scholar]
- 24.Cordier-Ochsenbein F., Guerois R., et al. Sanson A. Folding properties of an annexin I domain: a 1H-15N NMR and CD study. Biochemistry. 1996;35:10347–10357. doi: 10.1021/bi960747v. [DOI] [PubMed] [Google Scholar]
- 25.Aukrust I., Evensen L., et al. Vedeler A. Engineering, biophysical characterisation and binding properties of a soluble mutant form of annexin A2 domain IV that adopts a partially folded conformation. J. Mol. Biol. 2006;363:469–481. doi: 10.1016/j.jmb.2006.08.042. [DOI] [PubMed] [Google Scholar]
- 26.Zanier K., Nominé Y., et al. Travé G. Formation of well-defined soluble aggregates upon fusion to MBP is a generic property of E6 proteins from various human papillomavirus species. Protein Expr. Purif. 2007;51:59–70. doi: 10.1016/j.pep.2006.07.029. [DOI] [PubMed] [Google Scholar]
- 27.Aukrust I., Hollås H., et al. Vedeler A. The mRNA-binding site of annexin A2 resides in helices C-D of its domain IV. J. Mol. Biol. 2007;368:1367–1378. doi: 10.1016/j.jmb.2007.02.094. [DOI] [PubMed] [Google Scholar]
- 28.Piotto M., Saudek V., Sklenár V. Gradient-tailored excitation for single-quantum NMR spectroscopy of aqueous solutions. J. Biomol. NMR. 1992;2:661–665. doi: 10.1007/BF02192855. [DOI] [PubMed] [Google Scholar]
- 29.States D.J., Haberkorn R.A., Ruben D.J. A two-dimensional nuclear overhauser experiment with pure absorption phase in 4 quadrants. J. Magn. Reson. 1982;48:286–292. [Google Scholar]
- 30.Parker M.J., Spencer J., Clarke A.R. An integrated kinetic analysis of intermediates and transition states in protein folding reactions. J. Mol. Biol. 1995;253:771–786. doi: 10.1006/jmbi.1995.0590. [DOI] [PubMed] [Google Scholar]
- 31.Gianni S., Ivarsson Y., et al. Travaglini-Allocatelli C. Identification and characterization of protein folding intermediates. Biophys. Chem. 2007;128:105–113. doi: 10.1016/j.bpc.2007.04.008. [DOI] [PubMed] [Google Scholar]
- 32.Khorasanizadeh S., Peters I.D., Roder H. Evidence for a three-state model of protein folding from kinetic analysis of ubiquitin variants with altered core residues. Nat. Struct. Biol. 1996;3:193–205. doi: 10.1038/nsb0296-193. [DOI] [PubMed] [Google Scholar]
- 33.Jackson S.E., Fersht A.R. Folding of chymotrypsin inhibitor 2. 1. Evidence for a two-state transition. Biochemistry. 1991;30:10428–10435. doi: 10.1021/bi00107a010. [DOI] [PubMed] [Google Scholar]
- 34.Myers J.K., Pace C.N., Scholtz J.M. Denaturant m values and heat capacity changes: relation to changes in accessible surface areas of protein unfolding. Protein Sci. 1995;4:2138–2148. doi: 10.1002/pro.5560041020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Mickleburgh I., Burtle B., et al. Hesketh J. Annexin A2 binds to the localization signal in the 3' untranslated region of c-myc mRNA. FEBS J. 2005;272:413–421. doi: 10.1111/j.1742-4658.2004.04481.x. [DOI] [PubMed] [Google Scholar]
- 36.Sharathchandra A., Lal R., et al. Das S. Annexin A2 and PSF proteins interact with p53 IRES and regulate translation of p53 mRNA. RNA Biol. 2012;9:1429–1439. doi: 10.4161/rna.22707. [DOI] [PubMed] [Google Scholar]
- 37.Grindheim A.K., Hollås H., et al. Vedeler A. Reactive oxygen species exert opposite effects on Tyr23 phosphorylation of the nuclear and cortical pools of annexin A2. J. Cell Sci. 2016;129:314–328. doi: 10.1242/jcs.173195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kim J., Hajjar K.A. Annexin II: a plasminogen-plasminogen activator co-receptor. Front. Biosci. 2002;7:d341–d348. doi: 10.2741/kim. [DOI] [PubMed] [Google Scholar]
- 39.Mai J., Waisman D.M., Sloane B.F. Cell surface complex of cathepsin B/annexin II tetramer in malignant progression. Biochim. Biophys. Acta. 2000;1477:215–230. doi: 10.1016/s0167-4838(99)00274-5. [DOI] [PubMed] [Google Scholar]
- 40.Wang C.Y., Lin C.F. Annexin A2: its molecular regulation and cellular expression in cancer development. Dis. Markers. 2014;2014 doi: 10.1155/2014/308976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Hayes M.J., Shao D., et al. Moss S.E. Regulation of actin dynamics by annexin 2. EMBO J. 2006;25:1816–1826. doi: 10.1038/sj.emboj.7601078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Xu X.H., Pan W., et al. Song Y.Q. Association of annexin A2 with cancer development (Review) Oncol. Rep. 2015;33:2121–2128. doi: 10.3892/or.2015.3837. [DOI] [PubMed] [Google Scholar]
- 43.Rocha M.R., Barcellos-de-Souza P., et al. Morgado-Diaz J.A. Annexin A2 overexpression associates with colorectal cancer invasiveness and TGF-ss induced epithelial mesenchymal transition via Src/ANXA2/STAT3. Sci. Rep. 2018;8 doi: 10.1038/s41598-018-29703-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Christensen M.V., Høgdall C.K., et al. Høgdall E.V.S. Annexin A2 and cancer: a systematic review. Int. J. Oncol. 2018;52:5–18. doi: 10.3892/ijo.2017.4197. [DOI] [PubMed] [Google Scholar]
- 45.Li Z., Yu L., et al. Zhao L. Advances in cancer treatment: a new therapeutic target, Annexin A2. J. Cancer. 2021;12:3587–3596. doi: 10.7150/jca.55173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Aareskjold E., Grindheim A.K., et al. Vedeler A. Two tales of Annexin A2 knock-down: one of compensatory effects by antisense RNA and another of a highly active hairpin ribozyme. Biochem. Pharmacol. 2019;166:253–263. doi: 10.1016/j.bcp.2019.05.028. [DOI] [PubMed] [Google Scholar]
- 47.Reddy B.V., Datta S., Tiwari S. Use of propensities of amino acids to the local structural environments to understand effect of substitution mutations on protein stability. Protein Eng. 1998;11:1137–1145. doi: 10.1093/protein/11.12.1137. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
material