

Abstract
Transient receptor potential vanilloid 4 (TRPV4) ion channels on the endothelial cell membrane are widely regarded as a crucial Ca2+ influx pathway that promotes endothelium-dependent vasodilation. The downstream vasodilatory targets of endothelial TRPV4 channels vary among different vascular beds, potentially contributing to endothelial cell heterogeneity. Although numerous studies have examined the role of endothelial TRPV4 channels using specific pharmacological tools over the past decade, their physiological significance remains unclear, mainly due to a lack of endothelium-specific knockouts. Moreover, the loss of endothelium-dependent vasodilation is a significant contributor to vascular dysfunction in cardiovascular disease. The activity of endothelial TRPV4 channels is impaired in cardiovascular disease; therefore, strategies targeting the mechanisms that reduce endothelial TRPV4 channel activity may restore vascular function and provide therapeutic benefit. In this chapter, we discuss endothelial TRPV4 channel-dependent signaling mechanisms, the heterogeneity in endogenous activators and targets of endothelial TRPV4 channels, and the role of endothelial TRPV4 channels in the pathogenesis of cardiovascular diseases. We also discuss potentially interesting future research directions that may provide novel insights into the physiological and pathological roles of endothelial TRPV4 channels.
1. Introduction
Transient receptor potential (TRP) channels have emerged as important regulators of arterial diameter. Mammalian TRP channels are divided into six subfamilies based on their amino acid sequence homology—canonical (TRPC), vanilloid (TRPV), melastatin (TRPM), ankyrin (TRPA), mucolipin (TRPML), and polycystin (TRPP) (Earley & Brayden, 2015). Almost all the TRP channels are permeable to Ca2+ and influence Ca2+ signaling mechanisms in vascular endothelial cells (ECs) and smooth muscle cells (SMCs) (Earley & Brayden, 2015). Several TRP channels have been shown to regulate endothelial Ca2+, although direct recordings of Ca2+ influx through the channels have only been shown for TRPA1, TRPV3, and TRPV4 channels. Studies over the past decade firmly establish a crucial role for TRPV4 channels in controlling vascular reactivity (Ottolini, Hong, & Sonkusare, 2019). In particular, EC (Sonkusare et al., 2012, 2014) and SMC (Earley, Heppner, Nelson, & Brayden, 2005; Tajada et al., 2017) TRPV4 channels are widely regarded as promoters of vasodilation, a phenomenon that is directly associated with a lowering of vascular resistance. However, global TRPV4 channel knockout mice do not show a distinct vascular phenotype under resting conditions, possibly due to compensatory changes or the absence of the channel from multiple cell types (Hong, Cope, Marziano, & Sonkusare, 2016b; Zhang et al., 2009). In this regard, functional TRPV4 channels are also known to be expressed in astrocytes, neurons, ependymal cells of the choroid plexus, kidney epithelium, keratinocytes, adipocytes, inner ear, bladder urothelium, cardiac myocytes and fibroblast, skeletal myocytes, and immune cells (Jones et al., 2019; Shibasaki, Ikenaka, Tamalu, Tominaga, & Ishizaki, 2014; White et al., 2016). Thus, although numerous studies support a functional role for endothelial TRPV4 channels in causing vasodilation, a definitive answer to whether endothelial TRPV4 channel-mediated vasodilation is physiologically relevant awaits the development of endothelium-specific TRPV4 knockout animal models.
Numerous studies have provided crucial insights into the biophysical properties of TRPV4 channel. TRPV4 channel has been described to have a permeability ratio PCa/PNa of ~6, and single-channel conductance of ~90pS (Clapham, Montell, Schultz, & Julius, 2003). A single TRPV4 channel consists of four subunits, each with six transmembrane segments (S1–S6). Among the six transmembrane segments, S5, S6, and the interconnecting loop comprise the central cation-permeable pore, and S3 and S4 appear to form an agonist-binding pocket that influences channel gating (Nilius & Voets, 2013). The cytoplasmic amino- and carboxy-terminals of TRPV4 channels contain various functionally important domains, including an ankyrin repeat domain, a feature found in all channels of TRPC and TRPV subfamilies, a proline-rich region at the amino terminus, and a TRP box at the carboxy-terminal that is a binding site for calmodulin, actin, and tubulin. These functional domains enable protein–protein interactions that regulate channel assembly, channel activity and channel interaction with the cytoskeleton (Harteneck & Schultz, 2007; Nilius & Voets, 2013; White et al., 2016). Recently illustrated single-particle cryo-electron microscopy (cryo-EM) structure of Xenopus tropicalis TRPV4 channel under near-atomic resolution revealed unique interactions of transmembrane segments with the pore-forming domain. Moreover, the inner helices formed an intracellular gate in the ion-conduction pore but lacked an extracellular gate in the selectivity filter that was seen in other TRPV family members, possibly explaining the less selective nature of the channel compared to other TRPV channels. Anomalous X-ray diffraction analyses also identified a single ion-binding site in an unusually wide selectivity filter (Deng et al., 2018). Thus, unique structural features of TRPV4 channels may underlie their distinct gating mechanisms and selectivity features when compared to other ion channels of TRP family.
2. Methodologies for studying endothelial TRPV4 channel activity
Different methods have been used to study the activity of TRPV4 channels in native ECs. Direct studies of TRPV4 channel activity include the measurements of ionic currents through single TRPV4 channels or whole-cell TRPV4 channel in response to TRPV4 channel activators, and Ca2+ influx signals through individual TRPV4 channels on EC membranes. As an indirect way of estimating TRPV4 channel activity, numerous studies have also recorded whole-cell increases in EC Ca2+ in response to TRPV4 channel activation. The Ca2+ conductance of TRPV4 channels is approximately 100 times higher than that of l-type Ca2+ channel (Mercado et al., 2014). The large Ca2+ conductance of the channel makes it possible to record Ca2+ influx through individual TRPV4 channels using spinning disk confocal microscopy or total internal reflection fluorescence imaging (Sonkusare et al., 2012; Sullivan, Francis, Pitts, Taylor, & Earley, 2012). This technique has been termed “optical patch clamp” to indicate the optical measurements of single-channel activity (Demuro & Parker, 2005; Mercado et al., 2014; Sonkusare et al., 2014). The real-time recordings of Ca2+ influx, as opposed to whole-cell patch clamp, allow the use of intact tissue under physiological conditions, where cell–cell contacts and intercellular communications are maintained. Optical patch clamp also allows the determination of the spatial location of functional TRPV4 channels across the EC membrane. Ca2+ influx signals through endothelial TRPV4 channels were termed “TRPV4 sparklets” to imply their unique kinetic properties and single-channel behavior (Sonkusare et al., 2012). While TRPV4 channels are known to conduct cations other than Ca2+, TRPV4 channel-mediated functional effects have mostly been attributed to Ca2+ influx through the channel (Bagher et al., 2012; Marziano et al., 2017; Mendoza et al., 2010; Sonkusare et al., 2012; Wu et al., 2009; Yin et al., 2008). Depending on the methodology used, TRPV4 channels have also been shown to increase whole-cell Ca2+ levels, an effect that is absent in TRPV4−/− mice (White et al., 2016). Whole-cell Ca2+ is an integral of individual Ca2+ signals. Therefore, TRPV4 channel-induced increases in whole-cell Ca2+ may represent an additive or synergistic interaction among Ca2+ signals mediated by TRPV4 channels and other ion channels at the cell membrane or endoplasmic reticulum membrane. The measurements of TRPV4 sparklets were performed in the presence of a sarco-endoplasmic reticulum ATPase inhibitor or phospholipase C (PLC) inhibitor to eliminate the interference from intracellular Ca2+ release signals (Sonkusare et al., 2012). In ECs, activation of TRPV4 channels induces intermediate (IK) and small (SK) conductance Ca2+-sensitive K+ channel currents, which can interfere with the measurement of ionic currents through TRPV4 channels. As an alternative, TRPV4 channel currents can be recorded in the presence of non-selective TRPV4 channel antagonist ruthenium red (Sonkusare et al., 2012). Ruthenium red blocks TRPV4 channel at negative voltages, thereby inhibiting Ca2+ influx, and unblocks it at positive voltages so that outward K+ currents through the channel can be recorded at positive voltages without interference from Ca2+-activated IK/SK currents.
3. Signaling pathways for endothelial TRPV4 channel-mediated vasodilation
ECs are uniquely situated to sense the mechanical and humoral stimuli and convert them into vasodilation by increasing intracellular Ca2+. In resistance-sized arteries, ECs send out projections to SMCs, called myoendothelial projections (MEPs), which electrically connect the two cell types via myoendothelial gap junctions (MEGJs). An increase in EC Ca2+ activates one or more of the following targets: (1) endothelial nitric oxide synthase (eNOS) resulting in the formation of NO. NO diffuses to SMC layer, stimulates cyclic GMP (cGMP)-protein kinase G (PKG) signaling in SMC, which activates myosin light chain phosphatase (MLCP), resulting in a decrease in contractile force and causing vasodilation (Davis, Hill, & Kuo, 2008); (2) IK and SK channels, thereby causing EC membrane hyperpolarization that is transmitted to SMCs via MEGJs, leading to deactivation of SMC voltage-dependent Ca2+ channels (VDCCs) and vasodilation; and (3) release of endothelium-derived hyperpolarizing factors, including but not limited to epoxyeicosatrienoic acids (EETs) and hydrogen peroxide (H2O2) (Ellinsworth et al., 2016), which hyperpolarize SMCs and cause vasodilation. Pressure myography studies show that TRPV4-IK/SK channel linkage underlies endothelium-dependent vasodilation in systemic resistance arteries (Fig. 1) (Bagher et al., 2012; Sonkusare et al., 2012). Pressure myography studies in pulmonary resistance arteries show that TRPV4 channel-eNOS linkage is the major endothelial vasodilatory pathway in this vascular bed (Fig. 1) (Marziano et al., 2017). Thus, TRPV4 channels appear to couple with differential downstream targets in different vascular beds, although the precise mechanisms for specific channel-target coupling remain unknown. Localization of TRPV4 channels and IK/SK channels at MEPs appears to be responsible for the selective TRPV4-IK/SK channel coupling in systemic resistance arteries (Sonkusare et al., 2014). However, the mechanisms underlying (1) the lack of TRPV4-eNOS coupling in systemic arteries; (2) preferential TRPV4-eNOS coupling in pulmonary arteries, and (3) the lack of TRPV4-IK/SK channel coupling in pulmonary arteries remain elusive.
Fig. 1.
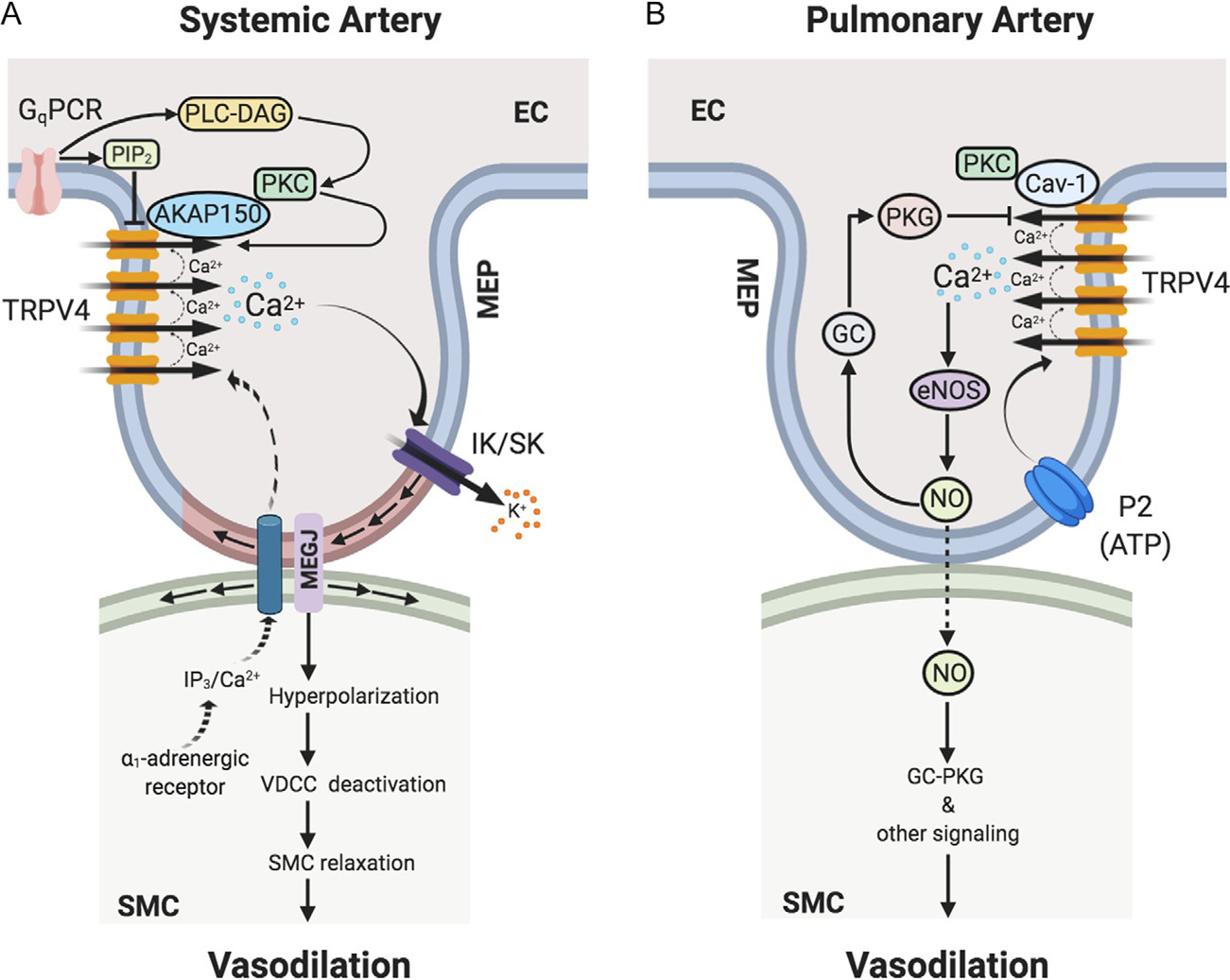
TRPV4 channel-dependent Ca2+ signaling at endothelial projections to smooth muscle cells or myoendothelial projections (MEPs) regulates vasodilation in (A) systemic and (B) pulmonary arteries. (A) In systemic artery, Gq protein-coupled receptor (GqPCR) stimulation activates phospholipase C (PLC)-diacylglycerol (DAG)-protein kinase C (PKC) signaling. PKC anchoring by AKAP150 enhances the activity and coupled openings of TRPV4 channels. In capillaries, GqPCR signaling can lower the levels of PIP2, thereby increasing TRPV4 channel activity. Ca2+ influx through TRPV4 channels selectively activates IK and SK channels at MEPs, thereby hyperpolarizing endothelial cell (EC) and smooth muscle cell (SMC) membranes. SMC membrane hyperpolarization leads to deactivation of voltage-dependent Ca2+ channels (VDCCs), thus relaxing SMCs and causing vasodilation. In response to SMC α1-adrenergic stimulation, IP3/Ca2+ from SMCs can cross MEGJs to enter EC layer to limit vasoconstriction via endothelial mechanisms. (B) In pulmonary artery, Ca2+ influx through TRPV4 channels selectively activates endothelial nitric oxide synthase (eNOS) to cause NO release and vasodilation. P2 purinergic receptor agonist ATP is an endogenous activator of the TRPV4-eNOS signaling. Endothelial caveolin-1 (Cav-1) enhances endothelial TRPV4 channel activity. Endothelium-derived NO subsequently causes the relaxation of SMCs leading to vasodilation via SMC guanylyl cyclase (GC)—protein kinase G (PKG)-dependent and -independent signaling mechanisms. NO also elicits activation of endothelial GC-PKG signaling, which lowers the coupled openings of TRPV4 channels, thereby limiting Ca2+ influx through TRPV4 channels. AKAP150, a-kinase anchoring protein 150; MEP, MEGJ, myoendothelial gap junction; IK/SK, intermediate/small-conductance Ca2+-activated K+ channels; SMC, smooth muscle cell; IP3, inositol trisphosphate.
4. Proteins regulators of endothelial TRPV4 channel activity
Several proteins are known to influence the activity of endothelial TRPV4 channels, including other ion channels, scaffolding proteins, and cytosolic signaling molecules. A recent study demonstrated the formation of a heteromeric TRPV4-TRPC1-TRPP2 (TRP polycystic 2) channel complex in primary rat mesenteric artery endothelial cells. Flow-induced shear stress is known to induce endothelium-dependent vasodilation that is associated with an increase in endothelial Ca2+ (Davies, 1995, 2009; Falcone, Kuo, & Meininger, 1993). Du et al. showed that TRPV4-TRPC1-TRPP2 complex is essential for flow-induced increase in intracellular Ca2+ (Du et al., 2014). TRPV4-TRPC1 channel complex was also shown to be responsible for increasing intracellular Ca2+ levels in human umbilical vein endothelial cells (Ma et al., 2010). Thus, the formation of heteromultimers with other cation channels is a potentially important mechanism for controlling TRPV4 channel activity. Other proteins that have been shown to alter TRPV4 channel activity include calmodulin, protein kinases, and structural proteins, particularly A-kinase anchoring protein 150 (AKAP150) and caveolin-1 (Cav-1) (Fig. 1).
TRPV4 channels have a calmodulin-binding site on the C-terminal, which enables the biphasic regulation of channel activity by Ca2+. Lower concentrations of intracellular Ca2+ potentiate the activity of the channels, whereas higher concentrations inhibit channel activity (Strotmann, Schultz, & Plant, 2003). Similarly, studies in expression systems provided the first evidence that TRPV4 channel activity is potentiated by protein kinase A (PKA) or protein kinase C (PKC)-dependent channel phosphorylation (Cao et al., 2018; Fan, Zhang, & McNaughton, 2009). PKA and PKC appear to act on distinct sites on TRPV4 channels to potentiate its activity. Notably, AKAP150, which can anchor PKC and PKA, was found to promote TRPV4 channel phosphorylation by PKC and PKA in expression systems. In this regard, AKAP150 was localized at MEPs in native endothelium, where it enhanced PKC-dependent activation of TRPV4 channels (Sonkusare et al., 2014). Moreover, the classical endothelial muscarinic receptor agonist acetylcholine activated endothelial AKAP150-PKC-TRPV4 channel signaling at MEPs to cause vasodilation. TRPV4 channels at MEPs open in a coupled manner, indicating simultaneous openings of multiple channels in a cluster. Coupled openings of TRPV4 channels amplify the Ca2+ influx through a small number of channels, making it possible for one or two open channels to have a functional effect. Coupled openings of TRPV4 channels required the presence of AKAP150, as indicated by the loss of coupling in the arteries from AKAP150−/− mice (Sonkusare et al., 2014). Interestingly, the coupled openings of TRPV4 channels were not dependent on PKC, indicating that AKAP150 can enhance TRPV4 channel activity in PKC-dependent and -independent manner. Whether the PKC-independent component of AKAP150-regulation of TRPV4 channel activity involves PKA and/or channel regulation via direct binding of AKAP150 to TRPV4 channels remains unknown.
Cav-1 is an essential structural protein of the membrane invaginations called caveolae, which serve to localize key signaling elements (Fridolfsson, Roth, Insel, & Patel, 2014; Minshall, Sessa, Stan, Anderson, & Malik, 2003). There is no direct evidence supporting the regulation of TRPV4 channel activity by Cav-1, although Cav-1 has been shown to associate with TRPV4 channels in cultured ECs. Cav-1 co-immunoprecipitated with TRPV4 channels in cultured ECs, and endothelium-dependent vasodilation in response to TRPV4 channel activator was decreased in conduit arteries from Cav-1−/− mice (Saliez et al., 2008). A recent study showed that endothelial Cav-1 anchoring of PKC increases TRPV4 channel activity and promotes endothelium-dependent vasodilation in pulmonary resistance arteries (Marziano, Hong, Cope, Ottolini, & Sonkusare, 2019). The uterine arteries from rats showed a higher contribution of endothelial TRPV4 channels in controlling vasodilation during pregnancy, an effect that was attributed to a higher expression of TRPV4 channels and their association with caveolae (Senadheera et al., 2013). Thus, multiple studies suggest a potential regulation of TRPV4 channel activity by Cav-1, although further studies are needed to understand the precise linkages in endothelial Cav-1-TRPV4 channel signaling.
Protein kinase G (PKG), a cGMP-dependent protein kinase, has also been shown to regulate endothelial TRPV4 channel activity. PKG inhibited flow-induced TRPV4-TRPC1 channel-dependent Ca2+ influx in ECs, and flow-mediated vasodilation (Ma et al., 2010). Also, elevated intravascular pressure in pulmonary microvessels induced endothelial TRPV4 channel-dependent Ca2+ influx that increased vascular permeability, but concomitantly activated a negative-feedback mechanism involving cGMP-dependent attenuation of the TRPV4 channel activity (Yin et al., 2008). Recently, Marziano et al. demonstrated that TRPV4 channel-dependent release of NO leads to vasodilation of pulmonary resistance arteries and activation of endothelial guanylyl cyclase (GC)-PKG-dependent negative-feedback loop that inhibits the coupled openings of TRPV4 channels (Marziano et al., 2017), thereby limiting Ca2+ influx through the channel. Whether PKG also requires anchoring by a scaffolding protein to modulate TRPV4 channel activity has not been tested.
5. Physiological stimuli that activate endothelial TRPV4 channels
Recent studies in cell type-specific knockout mice have immensely improved our understanding of the physiological roles of TRPV4 channels from individual cell types. Endothelium-specific TRPV4 knockout mouse has not been generated; thus, the precise physiological functions of endothelial TRPV4 channels remain unclear. Using highly selective and potent pharmacological tools (Table 1) and global knockout mice, numerous physiological stimuli have been shown to alter the activity of endothelial TRPV4 channels (Fig. 2) (Caires et al., 2017; Darby, Grace, Baratchi, & McIntyre, 2016; Meng, Zhao, Xie, Han, & Ji, 2018), as discussed below.
Table 1.
Pharmacological agonists and antagonists of TRPV4 channels.
Fig. 2.
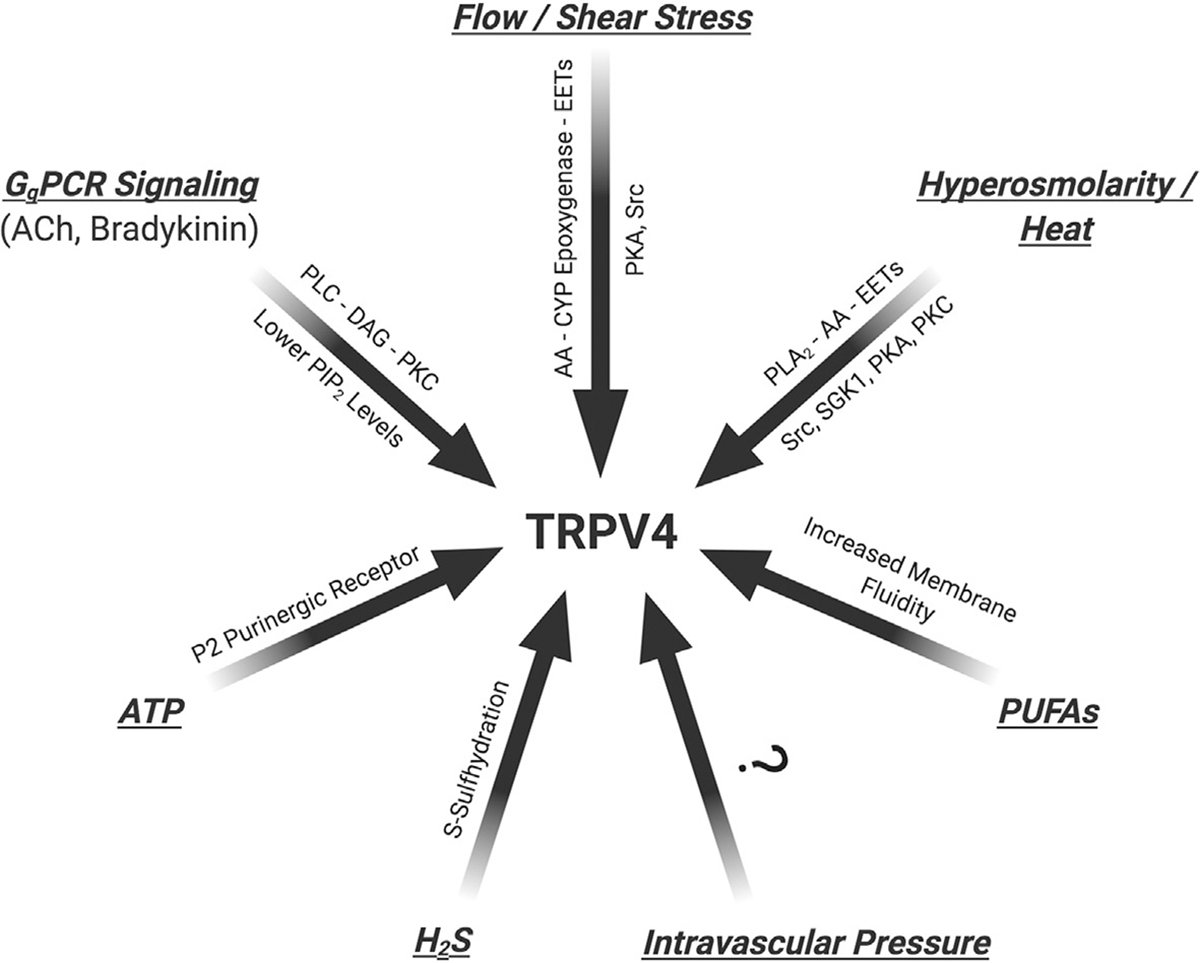
Physiological stimuli and cellular regulators of endothelial TRPV4 channel activity. TRPV4, transient receptor potential vanilloid 4; GqPCR, Gq protein-coupled receptors; ATP, adenosine triphosphate; CYP, cytochrome P450; Src, Src family kinases; PKA, protein kinase A; PKC, protein kinase C; SGK-1, serine/threonine kinase-1; EET, epoxyeicosatrienoic acid; PLA2, phospholipase A2; PLC, phospholipase C; DAG, diacylglycerol; PUFA, ω-polyunsaturated fatty acids; H2S, hydrogen sulfide; ACh, acetylcholine.
5.1. Mechanical stimuli (flow/shear stress/intravascular pressure)
Nikolaev et al. recently reported that TRPV4 channels are insensitive to mechanical stretch (Nikolaev et al., 2019). While the direct mechanosensing properties of TRPV4 channels remain in doubt, it is clear that mechano-activated signaling pathways can increase the activity of TRPV4 channels. Many studies, using genetic and pharmacological approaches, provide evidence that TRPV4 channels play an essential role in shear stress-induced activation of endothelial Ca2+ signaling. Shear stress-induced vasodilation of rat carotid artery and arteria gracilis was inhibited by non-selective TRPV4 channel inhibitor ruthenium red (Kohler et al., 2006). Also, shear stress- and flow/reperfusion-induced vasodilation was significantly attenuated in carotid arteries from TRPV4−/− mice (Hartmannsgruber et al., 2007). Moreover, increased luminal flow dilated carotid artery via EET-endothelium derived hyperpolarization (EDH) pathway, a response that was blunted in the arteries from TRPV4−/− mice. Interestingly, endothelial TRPV4 channels translocated from perinuclear area to the cell membrane in response to increased shear stress (Loot et al., 2008). In mouse mesenteric arteries, shear stress elicited a rapid Ca2+ influx through endothelial TRPV4 channels and caused vasodilation via eNOS and EDH-dependent mechanisms (Mendoza et al., 2010).
Mechanical stretch was also shown to activate endothelial TRPV4 channels to induce capillary cell reorientation via phosphatidylinositol 3-kinase-dependent stimulation of integrin-integrin signaling (Thodeti et al., 2009). Additionally, Bagher et al. demonstrated that a decrease in intravascular pressure increases Ca2+ influx through endothelial TRPV4 channels at MEPs in cremaster arterioles (Bagher et al., 2012). Thus, evidence in the literature clearly supports the concept that endothelial TRPV4 channels are activated by mechanical stimuli, including shear stress, and changes in intravascular pressure.
5.2. Changes in osmolarity and temperature
Early studies of TRPV4 channels revealed that the channel could be activated by increases in temperature or by changes in osmolarity that alter the cell size. Strotmann, Harteneck, Nunnenmacher, Schultz, and Plant (2000) demonstrated a rapid increase in TRPV4 channel activity and intracellular Ca2+ concentration with small decrease in extracellular osmolarity. Similarly, Vriens et al. showed that hypotonic extracellular solution activates phospholipase A2 (PLA2)-dependent formation of arachidonic acid and EETs, which are purported TRPV4 channel activators (Vriens et al., 2004). Intriguingly, a heat ramp to ~40 °C increased the inward current through TRPV4 channels and intracellular Ca2+ concentration in freshly isolated mouse aorta ECs (Watanabe, Vriens, et al., 2002) supporting the idea that hyperthermia may activate endothelial TRPV4 channels to cause vasodilation and increase blood flow. Whether temperature- or osmolarity-induced activation of endothelial TRPV4 channels contributes to vasodilation has not been elucidated. Studies in other cell types suggest that PKA, PKC, Src family kinases, and serine/threonine kinase-1 sensitize TRPV4 channels to activation by heat or mechanical stimuli (Fig. 2) (Darby et al., 2016). However, TRPV4 channel sensitization by PKA or Src family kinases has not been verified in ECs.
5.3. G-protein-coupled receptor (GPCR) activation
GPCR agonists are one of the best-known endogenous activators of endothelial TRPV4 channels. Gq protein-coupled receptor (GqPCR) signaling has been associated with endothelial TRPV4 channel activation via two distinct mechanisms: (1) PLC-diacylglycerol (DAG)-PKC-dependent activation of TRPV4 channels in resistance arteries (Sonkusare et al., 2014); and (2) depletion of a negative regulator of TRPV4 channel activity, phosphatidylinositol 4,5-bisphosphate (PIP2), in capillary endothelium (Harraz, Longden, Hill-Eubanks, & Nelson, 2018). Sonkusare et al. showed that the classical Gq-coupled muscarinic receptor agonist, acetylcholine, activated TRPV4 channels at MEPs via PLC-DAG-PKC signaling (Sonkusare et al., 2012, 2014). Bradykinin, an inflammatory mediator that acts on Gq-coupled bradykinin receptors, similarly stimulated the activity of TRPV4 channels at MEPs (Sonkusare et al., 2014). Anchoring of PKC by AKAP150 at MEPs was found to be essential for acetylcholine- or bradykinin-induced TRPV4 channel activation. Interestingly, Marziano et al. recently showed that endogenous purinergic receptor agonist adenosine triphosphate (ATP) dilates pulmonary resistance arteries via activation of endothelial TRPV4-eNOS-NO signaling (Marziano et al., 2017). This effect was found to be dependent on P2 purinergic receptors (Marziano et al., 2017). Pulmonary endothelium is known to express P2Y1 and P2Y2 receptor subtypes, both Gq-coupled receptors (Zemskov, Lucas, Verin, & Umapathy, 2011). Thus, it is conceivable that ATP activates TRPV4 channels in pulmonary endothelium via activation of one or both of these receptors.
Patch clamp studies in freshly isolated mouse brain capillary ECs indicated that GqPCR activation increases TRPV4 channel currents mainly via PLC-mediated hydrolysis of plasma membrane PIP2, a known inhibitor of TRPV4 channel activity (Harraz et al., 2018). It can be speculated that GqPCR agonists activate endothelial TRPV4 channels via distinct signaling mechanisms in arterial and capillary ECs. Capillaries are characterized by the absence of SMCs, and therefore, MEPs. Thus, the signaling microdomains that enable GqPCR-TRPV4 channel signaling in arterial endothelium could be absent from capillary endothelium. The absence of IK/SK channel currents in capillary endothelium further supports the differences in ion channel signaling networks between arterial and capillary endothelium (Longden et al., 2017).
Studies in expression systems show that TRPV4 channels can also be phosphorylated and activated by PKA, a phenomenon that is potentiated by AKAP150 (Cao et al., 2018; Fan et al., 2009). Whether PKA can activate endothelial TRPV4 channels is not known. PKA is activated by Gs-coupled receptor agonists, resulting in adenyl cyclase (AC)-cAMP-PKA signaling. Two main Gs-coupled receptors that are expressed on EC membrane include histamine and prostacyclin receptors (Alfranca, Iniguez, Fresno, & Redondo, 2006; Hekimian, Cote, Van Sande, & Boeynaems, 1992; Heltianu, Simionescu, & Simionescu, 1982). Whether endothelial histamine or prostacyclin receptors can activate TRPV4 channels is not known.
6. Cellular regulators of endothelial TRPV4 channel activity
6.1. Inositol 1,4,5-trisphosphate (IP3)
A recent study by Takahashi et al. reported an IP3 binding domain on TRPV4 channels and showed that binding of IP3 to this domain increases the activity of TRPV4 channels (Takahashi et al., 2014). To this effect, Hong et al. recently showed that IP3 generated in SMCs by the activation of SMC α1-adrenergic receptors or thromboxane A2 receptors could travel to ECs via MEGJs, and activate endothelial TRPV4 channels. While the activation of α1-adrenergic receptors or thromboxane A2 receptors causes vasoconstriction, the IP3-dependent activation of endothelial TRPV4 channels serves as a negative-feedback mechanism that limits the vasoconstriction (Hong et al., 2018). Garland et al. suggested that increased intracellular Ca2+ from SMCs can cross MEGJs and directly activate endothelial IK and SK channels, thereby limiting SMC contraction. It was also proposed that the type of SMC stimulus and the extent of stimulation may determine the negative regulatory mechanism that is activated (Hong et al., 2018). The activity of IP3Rs is known to be regulated by intracellular Ca2+; indeed, Ca2+ is essentially a ligand for IP3Rs. In this context, Heathcote et al. recently demonstrated that Ca2+ influx through endothelial TRPV4 channels activates Ca2+-induced Ca2+ release through IP3R ion channels, thus amplifying the Ca2+ response that underlies a large-scale endothelial communication system (Heathcote et al., 2019).
6.2. Hydrogen sulfide (H2S)
H2S, a gaseous mediator produced by ECs, was shown to cause vasodilation via endothelial TRPV4 channel activation in phenylephrine-constricted mesenteric arteries (Naik, Osmond, Walker, & Kanagy, 2016). H2S also increased the activity of TRPV4 channel-dependent Ca2+ signals in ECs of pressurized mesenteric arteries. Interestingly, TRPV4 channel-dependent Ca2+ signaling resulted in the activation of endothelial big conductance, Ca2+-activated K+ (BK) channels. TRPV4 channel activator increased the BK channel currents in isolated ECs, and BK channel inhibitor abolished H2S-induced vasodilation (Naik et al., 2016). While SMC TRPV4 channels have been linked with BK channel activation in the past (Earley et al., 2005), this was the first report on TRPV4-BK channel signaling in ECs. The mechanistic determinants of preferential TRPV4-BK or TRPV4-IK/SK channel signaling linkages will be an exciting topic for future investigations.
6.3. Metabolites of arachidonic acid
Mechanical stimuli are known to activate cytochrome P450 (CYP) epoxygenases and EET formation in ECs. EETs released in response to mechanical stimuli can regulate intracellular Ca2+ levels and vascular resistance. Increased flow/shear stress elicited vasodilation via CYP epoxygenase-dependent activation of endothelial TRPV4 channels in mouse carotid arteries (Loot et al., 2008). Marrelli et al. showed that PLA2, via its main product, arachidonic acid (AA), increases endothelial TRPV4 channel activity and endothelial Ca2+ levels in cerebral arteries (Marrelli et al., 2007; Watanabe et al., 2003). Vriens et al. further showed that CYP epoxygenase metabolizes AA to biologically active EETs that increase the activity of endothelial TRPV4 channels (Vriens et al., 2005). Intriguingly, in human coronary arteries, the administration of AA caused EC membrane hyperpolarization and direct activation of TRPV4 channel-dependent Ca2+ entry (Cao et al., 2018; Zheng et al., 2013). This phenomenon also required basal phosphorylation of endothelial TRPV4 channels by PKA. Thus, the CYP epoxygenase-AA-EETs signaling axis appears to play an essential role in increasing the activity of endothelial TRPV4 channels and endothelium-dependent vasodilation (Fig. 2). It is important to note that not all EETs can activate TRPV4 channels. While 5,6-EET, 8,9-EET, and 11,12-EET activated TRPV4 channels in expression systems and in ECs, 14,15-EET had no effect on TRPV4 channel activity (Sonkusare et al., 2014; Thorneloe et al., 2012; Vriens et al., 2005; Watanabe et al., 2003; Zheng et al., 2013).
6.4. ω-3 Polyunsaturated fatty acids (PUFAs)
PUFAs from fish oil have been shown to have a protective effect on vascular function. Caires et al. recently demonstrated that ω-3 fatty acid metabolites (eicosapentaenoic acid, EPA; 17, 18-epoxyeicosatetraenoic acid, 17, 18-EEQ) contribute to the regulation of endothelial TRPV4 channel activity in C. elegans and provided evidence that PUFAs enhance TRPV4 channel activity in human microvascular endothelial cells by modifying the membrane fluidity and bending stiffness without altering channel expression or trafficking (Caires et al., 2017). These results reinforce the concept that lipid metabolites are crucial modulators of endothelial TRPV4 channel activity.
7. Heterogeneity in physiological regulators and downstream targets of endothelial TRPV4 channels
TRPV4 channel-dependent endothelial Ca2+ signaling mechanisms have been elucidated in several vascular beds. There appears to be considerable heterogeneity among different vascular beds regarding the molecular mechanisms that regulate TRPV4 channel activity. For example, the activity of endothelial TRPV4 channels is inhibited by a NO-GC-PKG-dependent negative-feedback pathway in pulmonary arteries. Still, such a mechanism is not observed in mesenteric arteries (Marziano et al., 2017). AKAP150 increases the activity of TRPV4 channels at MEPs in mesenteric arteries (Sonkusare et al., 2014); however, AKAP150 is not present in pulmonary arteries (Marziano et al., 2017). Physiological activators of TRPV4 channels also appear to be different in different vascular beds. For example, ATP can activate endothelial TRPV4 channels in pulmonary arteries but not in mesenteric arteries (Marziano et al., 2017) (Fig. 1). Moreover, acetylcholine and bradykinin activated endothelial TRPV4 channels in mesenteric arteries but not in pulmonary arteries. Additionally, endothelial TRPV4 channels appear to couple with distinct downstream signaling targets in different vascular beds. In systemic resistance arteries, TRPV4 channels exclusively couple with IK/SK channels (Bagher et al., 2012; Sonkusare et al., 2012), whereas in pulmonary resistance arteries, they couple predominantly with eNOS (Marziano et al., 2017). The molecular mechanisms that are responsible for the heterogeneity in regulatory mechanisms and downstream targets of endothelial TRPV4 channels remain unclear, and further research in this area may provide exciting insights on how differential TRPV4 channel regulation contributes to endothelial cell heterogeneity. Table 2 summarizes TRPV4 channel-dependent vasodilatory mechanisms in different vascular beds.
Table 2.
Summary of endothelial TRPV4 channel-dependent vasodilatory mechanisms in different vascular beds.
| Vascular bed | Species | Endothelial TRPV4 channel-dependent mechanism | References |
|---|---|---|---|
| Cerebral arteries | Rat, mouse, human | ✓ PLA2 activation increases endothelial Ca2+ influx through TRPV4 channels ✓ acetylcholine-dependent vasodilation via TRPV4 channels ✓ GqPCR signaling activates endothelial TRPV4 channels over Kir2.1 channels |
Harraz et al. (2018), Hatano, Suzuki, Itoh, and Muraki (2013), Marrelli et al. (2007), Zhang et al. (2013) |
| Retinal artery | Bovine | ✓ TRPV4 channel control intracellular Ca2+ concentration in cultured retinal microvascular ECs | Monaghan et al. (2015) |
| Carotid artery | Rat, mouse | ✓ TRPV4 channel activation triggers NO- and EDH-dependent vasodilation ✓ Dynamic TRPV4/SK channel complexes in caveolae of ECs promote flow-mediated vasodilation ✓ Flow-induced endothelium-dependent vasodilation via activation of cytochrome P450 epoxygenase-EETs-TRPV4 channel signaling |
Goedicke-Fritz et al. (2015), Kohler et al. (2006), Loot et al. (2008) |
| Arteria gracilis | Rat | ✓ TRPV4 channel activation triggers NO- and EDH-dependent vasodilation ✓ Acetylcholine activates TRPV4 channel-mediated Ca2+ signals ✓ TRPV4-SK/IK channel signaling ✓ Co-localization of Cav-1, TRPV4, and BK channels promotes vasodilation |
Kohler et al. (2006), Naik and Walker (2018) |
| Microvascular ECs | Human | ✓ Caveolar association of Cav-1, TRPV4 and SK channels in microvascular ECs ✓ Shear stress induces a partial de novo colocalization of IK channels with Cav-1 and TRPV4 channels ✓ PUFAs enhance TRPV4 channel activity by modifying cell membrane fluidity and bending stiffness |
Caires et al. (2017), Goedicke-Fritz et al. (2015) |
| Feed artery | Human | ✓ TRPV4 channels attenuate α1-adrenergic vasoconstriction | Gifford et al. (2014) |
| Coronary artery | Human | ✓ Flow-induced activation of endothelial TRPV4 channels and mitochondrial reactive oxygen species production ✓ Arachidonic acid-activation of TRPV4 channels via PKA-mediated phosphorylation |
Bubolz et al. (2012), Cao et al. (2018), Zheng et al. (2013) |
| Pulmonary artery | Rat, mouse | ✓ Endothelium-dependent vasodilation mainly through TRPV4-eNOS signaling ✓ NO release activates endothelial GC-PKG-dependent negative-feedback pathway that limits TRPV4 channel activity ✓ ATP dilates pulmonary arteries mainly via TRPV4-eNOS signalings ✓ Endothelial Cav-1-PKC-TRPV4 channel signaling promotes vasodilation |
Marziano et al. (2017), Marziano et al. (2019), Sukumaran et al. (2013) |
| Mesenteric artery | Mouse, rat | ✓ EDH-dependent dilation in resistance arteries, EDH- and NO-dependent dilation in conduit arteries ✓ 11,12-EET induces TRPV4 channel activation and vasodilation ✓ Shear stress dilates via NO- and EDH-dependent mechanisms ✓ PKCα-TRPV4 channel signaling mediates vasodilation to acetylcholine ✓ Cooperative TRPV4 channel openings increase endothelial Ca2+ influx and cause vasodilation through IK and SK channels ✓ TRPV4 channels colocalize with Cav-1 ✓ PKC-AKAP150-TRPV4 channel signaling at MEPs underlies vasodilation ✓ Endothelial TRPV4-C1-P2 complex mediates flow-induced Ca2+ entry |
Adapala et al. (2011), Du et al. (2014), Earley et al. (2009), Ma et al. (2013), Mendoza et al. (2010), Rath et al. (2012), Seki et al. (2017), Sonkusare et al. (2012) |
| Uterine radial artery | Rat | ✓ The number of endothelial TRPV4 channels and caveolae is increased in pregnancy | Senadheera et al. (2013) |
| Bladder vessel | Rat, mouse | ✓ Endothelial TRPV4 channels are expressed | Gevaert et al. (2007) |
| Cremaster artery | Rat | ✓ Endothelial TRPV4 channels increase Ca2+ signal frequency at MEPs in response to low intraluminal pressure | Bagher et al. (2012) |
8. Alterations in endothelial TRPV4 channel activity under non-physiological or disease conditions
Brachyolmia, a heterogeneous group of skeletal dysplasias, was the first human disease to be associated with abnormal TRPV4 channel activity (Rock et al., 2008). Several autosomal-dominant skeletal dysplasias and neuromuscular disorders have since been associated with altered TRPV4 channel activity (Dai et al., 2010; White et al., 2016). Recent studies in animal models and patients demonstrate a vital role for altered endothelial TRPV4 channel activity in the pathogenesis of cardiovascular diseases. Depending on the disease condition and vascular bed under consideration, the activity of endothelial TRPV4 channels is increased or decreased in vascular disorders.
8.1. Endothelial TRPV4 channel activity in pulmonary vascular disease
Pulmonary hypertension (PH) is characterized by elevated pulmonary arterial pressure (>25mmHg), pulmonary vasoconstriction, and pulmonary vascular remodeling. Abnormal endothelial TRPV4 channel activity has been implicated in the pathogenesis of PH. Marziano et al. demonstrated that TRPV4 channels are a crucial pathway for endothelial Ca2+ influx and vasodilation in small pulmonary arteries that control pulmonary vascular resistance (Marziano et al., 2017). In PH, attenuated Ca2+ influx through endothelial TRPV4 channels resulted in impaired vasodilation of pulmonary resistance arteries and an increase in pulmonary arterial pressure. On the contrary, studies by Suresh et al. in a rat model of pulmonary arterial hypertension (PAH) showed that Ca2+ influx through TRPV4 channels is increased in cultured microvascular endothelial cells (MVECs) in PAH (Suresh et al., 2018). Moreover, studies in cultured ECs showed that the increase in TRPV4 channel activity was due to an elevated mitochondrial production of hydrogen peroxide in PAH. The differences in the results in small pulmonary arteries and MVECs could be attributed to differential regulation of TRPV4 channel activity in ECs from small arteries vs capillaries or differences in conditions (culture conditions vs native cells). Nevertheless, these results show that PH is associated with abnormal Ca2+ influx through endothelial TRPV4 channels. Therefore, the mechanisms that drive the abnormal TRPV4 channel activity in PH could potentially be targeted for therapeutic benefit.
Endothelial TRPV4 channels are critical controllers of lung capillary permeability, and elevated TRPV4 channel activity is associated with increased capillary permeability. In heart failure patients and animal models, TRPV4 channel expression in pulmonary vasculature was increased. Moreover, administering TRPV4 channel inhibitor (GSK2193874) prevented the increased vascular permeability and resultant pulmonary edema in an experimental model of elevated pulmonary venous pressure, and in a mouse model of myocardial infarction (Thorneloe et al., 2012). On the contrary, TRPV4 channel activator (GSK1016790A) decreased right ventricular and systemic pressures and led to a pulmonary circulatory collapse. In another study, the TRPV4 channel activator stimulated IK channels and eNOS, increased vascular permeability, and caused lung hemorrhage (Pankey, Zsombok, Lasker, & Kadowitz, 2014; Wandall-Frostholm et al., 2015). A study in isolated mouse lungs showed CYP epoxygenase-EET-dependent modulation of Ca2+ entry through TRPV4 channels in response to high pulmonary vascular pressure (Jian, King, Al-Mehdi, Liedtke, & Townsley, 2008). Yin et al. showed that pressure-induced Ca2+ entry through endothelial TRPV4 channels not only increased lung vascular permeability but also concomitantly activated a NO-cGMP-dependent negative-feedback loop, preserving the pulmonary vascular barrier (Yin et al., 2008). These studies prove that endothelial TRPV4 channels play a central role in the regulation of capillary permeability, particularly in the lung, and that higher TRPV4 channel activity increases capillary permeability.
8.2. Endothelial TRPV4 channel activity in hypertension
Systemic hypertension is characterized by elevated systolic/diastolic blood pressure (>130/80mmHg). Endothelial dysfunction and increased contractility of resistance arteries are hallmarks of hypertension. In some of the earliest studies on the role of TRPV4 channels in cardiovascular disease, Earley et al. found that NOS inhibitor-induced hypertension was augmented in global TRPV4−/− mice. The authors proposed that endothelial TRPV4 channel-dependent vasodilation opposes hypertensive effects of the NOS inhibitor (Earley et al., 2009). Furthermore, in an angiotensin II-infused mouse model of hypertension, endothelial TRPV4 channel activity and acetylcholine stimulation of TRPV4 channels were impaired exclusively at MEPs. This abnormality in TRPV4 channel activity was attributed to lower AKAP150 levels in hypertension, which resulted in a loss of coupled openings of TRPV4 channel and acetylcholine-PKC activation of the channel, thereby lowering Ca2+ influx at MEPs (Sonkusare et al., 2014). High-fat diet-induced obesity was also associated with a decrease in TRPV4 channel activity at MEPs (Hong, Cope, Marziano, & Sonkusare, 2016a). It is not known whether the mechanisms for reduced TRPV4 channel activity are similar between angiotensin II-induced hypertension and obesity. Neither angiotensin II-induced hypertension nor obesity was associated with a loss of function of IK/SK channels (Hong et al., 2016a; Sonkusare et al., 2014). However, in mesenteric arteries from stroke-prone spontaneously hypertensive rats, the activity of endothelial TRPV4 and SK channels and EDH-mediated vasodilation were attenuated (Seki et al., 2017).
In a mouse model of salt-induced hypertension, hypercontractility of aorta was linked to TRPV4 channel-mediated increase in endothelial Ca2+ and subsequent activation of PLA2, which increased the formation of prostaglandin F2α (PGF2α) by endothelial COX2 (Zhang et al., 2018). Intriguingly, a study by Gao et al. in salt-induced hypertensive rats found enhanced TRPV4 channel expression in mesenteric arteries and higher TRPV4 channel-mediated vasodilation. These results suggested that a compensatory TRPV4 channel activation may prevent impaired vasodilation in salt-induced hypertension (Gao, Sui, Garavito, Worden, & Wang, 2009).
8.3. Endothelial TRPV4 channel activity in other vascular disorders
Endothelial TRPV4 channels also played a vital role in mediating vasodilation of cerebral arteries (Zhang et al., 2013). The activity of endothelial TRPV4 channels in cerebral arteries was decreased in a rat model of cerebrovascular pathology in Alzheimer’s disease (Zhang et al., 2013). TRPV4 channel-mediated vasodilation was restored by superoxide dismutase or catalase in this model (Zhang et al., 2013), indicating that redox signaling impairs endothelial TRPV4 channel activity.
Endothelial TRPV4-SK channel signaling was impaired in streptozotocin-induced diabetic rats, possibly contributing to endothelial dysfunction in this model of type 1 diabetes (Ma et al., 2013). Moreover, the expression and activity of TRPV4 channels in retinal vascular ECs were attenuated following the exposure to high glucose (Monaghan et al., 2015), suggesting that hyperglycemia-induced TRPV4 channel dysfunction may contribute to diabetic retinopathy. Rath et al. showed that TRPV4 channels promote NO signaling and contribute to hypoxia-induced vasodilation of systemic arteries under ischemic conditions (Rath et al., 2012). Furthermore, TRPV4 channel inhibition restored high temperature-induced decline in vasoconstriction to phenylephrine in human skeletal muscle feed artery, suggesting that increased body temperature recruits endothelial TRPV4 channels to elicit a sympatholysis-like inhibition of α1 adrenergic receptor-mediated vasoconstriction (Gifford et al., 2014). These results provide further evidence that TRPV4 channels are important for endothelial homeostasis, and impaired TRPV4 channel activity contributes to endothelial dysfunction in vascular disorders.
9. Summary
TRPV4 channels are a major Ca2+ influx pathway that controls endothelial Ca2+ signaling mechanisms and promotes endothelium-dependent vasodilation. There appears to be substantial heterogeneity in physiological regulators and downstream targets of TRPV4 channels. Many physiological stimuli (shear stress, stretch, osmolarity, temperature, GPCR signaling, H2S, lipid metabolites) can activate endothelial TRPV4 channels to increase Ca2+ influx in ECs. Among all TRP channels, TRPV4 channels have some of the best (specific and potent) pharmacological activators and inhibitors available for reliably studying the channel activity (Table 1). Patch clamp electrophysiology has traditionally been used to study the ionic currents through TRPV4 channels in native ECs. Recent studies provide evidence that TRPV4 channel activity can be studied by optically recording Ca2+ influx through individual TRPV4 channels in the intact endothelium under physiological conditions. Multiple studies suggest that endothelial TRPV4 channel activity is impaired in vascular disorders, including systemic hypertension, diabetes, Alzheimer’s disease, obesity, and pulmonary hypertension. The activity of TRPV4 channels in capillary endothelium appears to be increased in diseases characterized by elevated capillary permeability, particularly in the lung.
10. Perspectives and future directions
The role of endothelial TRPV4 channels in endothelium-dependent vasodilation is well-established. However, the physiological significance of TRPV4 channel-mediated vasodilation remains elusive. Global TRPV4−/− mice lack a distinct vascular phenotype, which could be attributed to compensatory upregulation of another Ca2+ entry pathway. Future studies in endothelium-specific TRPV4 knockout mice are needed for a more definitive assessment of the physiological roles of endothelial TRPV4 channels. Moreover, endothelium-specific knockout mice for proteins that control TRPV4 channel activity (AKAP150 or Cav-1, for example) will provide further insights into the physiological importance of endothelial TRPV4 channel regulation. If endothelium-dependent vasodilation lowers vascular resistance and blood pressure, then endothelium-specific knockout mice will be expected to show a higher resting blood pressure. The use of endothelium-specific knockout mice may also lead to discoveries on physiological stimuli that activate endothelial TRPV4 channels. The loss of endothelial function is a significant problem in almost all cardiovascular disorders. Understanding the mechanisms that lead to TRPV4 channel dysfunction may result in novel therapeutic strategies to restore endothelial Ca2+ signaling and vasodilation in disease conditions. A variety of physiological stimuli, regulators, and downstream targets couple with TRPV4 channels in different vascular beds. Understanding the molecular mechanisms for the differential regulation and targets of TRPV4 channel activity may provide crucial insights into molecular mechanisms that underlie endothelial cell heterogeneity. Different vascular beds are exposed to distinct physiological environments (intravascular pressure, flow, the concentration of dissolved oxygen, humoral mediators). Whether the unique physiological conditions experienced by ECs in a vascular bed determine the vasodilatory signaling linkages in that vascular bed remains unexplored. The majority of signaling elements for TRPV4 channel-dependent vasodilation are known to be concentrated at MEPs. Understanding the mechanisms for translocation of these signaling elements to MEPs will be an exciting topic for future investigations. Finally, substantial sex differences in TRPV4 channel activity have been shown in SMCs. Therefore, potential sex differences in endothelial TRPV4 channel activity and their physiological relevance need further examination.
Acknowledgment
This work was supported by grants from the NIH (HL142808, HL146914, and HL147555) to S.K.S.
References
- Adapala RK, Talasila PK, Bratz IN, Zhang DX, Suzuki M, Meszaros JG, et al. (2011). PKCalpha mediates acetylcholine-induced activation of TRPV4-dependent calcium influx in endothelial cells. American Journal of Physiology. Heart and Circulatory Physiology, 301(3), H757–H765. 10.1152/ajpheart.00142.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alfranca A, Iniguez MA, Fresno M, & Redondo JM (2006). Prostanoid signal transduction and gene expression in the endothelium: Role in cardiovascular diseases. Cardiovascular Research, 70(3), 446–456. 10.1016/j.cardiores.2005.12.020. [DOI] [PubMed] [Google Scholar]
- Bagher P, Beleznai T, Kansui Y, Mitchell R, Garland CJ, & Dora KA (2012). Low intravascular pressure activates endothelial cell TRPV4 channels, local Ca2+ events, and IKCa channels, reducing arteriolar tone. Proceedings of the National Academy of Sciences of the United States of America, 109(44), 18174–18179. 10.1073/pnas.1211946109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bubolz AH, Mendoza SA, Zheng X, Zinkevich NS, Li R, Gutterman DD, et al. (2012). Activation of endothelial TRPV4 channels mediates flow-induced dilation in human coronary arterioles: Role of Ca2+ entry and mitochondrial ROS signaling. American Journal of Physiology. Heart and Circulatory Physiology, 302(3), H634–H642. 10.1152/ajpheart.00717.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caires R, Sierra-Valdez FJ, Millet JRM, Herwig JD, Roan E, Vasquez V, et al. (2017). Omega-3 fatty acids modulate TRPV4 function through plasma membrane remodeling. Cell Reports, 21(1), 246–258. 10.1016/j.celrep.2017.09.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao S, Anishkin A, Zinkevich NS, Nishijima Y, Korishettar A, Wang Z, et al. (2018). Transient receptor potential vanilloid 4 (TRPV4) activation by arachidonic acid requires protein kinase A-mediated phosphorylation. The Journal of Biological Chemistry, 293(14), 5307–5322. 10.1074/jbc.M117.811075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clapham DE, Montell C, Schultz G, & Julius D (2003). International union of pharmacology. XLIII. Compendium of voltage-gated ion channels: Transient receptor potential channels. Pharmacological Reviews, 55(4), 591–596. 10.1124/pr.55.4.6. [DOI] [PubMed] [Google Scholar]
- Dai J, Cho TJ, Unger S, Lausch E, Nishimura G, Kim OH, et al. (2010). TRPV4-pathy, a novel channelopathy affecting diverse systems. Journal of Human Genetics, 55(7), 400–402. 10.1038/jhg.2010.37. [DOI] [PubMed] [Google Scholar]
- Darby WG, Grace MS, Baratchi S, & McIntyre P (2016). Modulation of TRPV4 by diverse mechanisms. The International Journal of Biochemistry & Cell Biology, 78, 217–228. 10.1016/j.biocel.2016.07.012. [DOI] [PubMed] [Google Scholar]
- Davies PF (1995). Flow-mediated endothelial mechanotransduction. Physiological Reviews, 75(3), 519–560. 10.1152/physrev.1995.75.3.519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davies PF (2009). Hemodynamic shear stress and the endothelium in cardiovascular pathophysiology. Nature Clinical Practice. Cardiovascular Medicine, 6(1), 16–26. 10.1038/ncpcardio1397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis MJ, Hill MA, & Kuo L (2008). Local regulation of microvascular perfusion. In Tuma RF, Duran WN, & Ley K (Eds.), Handbook of physiology: The cardiovascular system, microcirculation (section 2) (2nd ed., pp. 161–284). San Diego, CA: Academic Press. [Google Scholar]
- Demuro A, & Parker I (2005). “Optical patch-clamping”: Single-channel recording by imaging Ca2+ flux through individual muscle acetylcholine receptor channels. The Journal of General Physiology, 126(3), 179–192. 10.1085/jgp.200509331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deng Z, Paknejad N, Maksaev G, Sala-Rabanal M, Nichols CG, Hite RK, et al. (2018). Cryo-EM and X-ray structures of TRPV4 reveal insight into ion permeation and gating mechanisms. Nature Structural & Molecular Biology, 25, 252–260. 10.1038/s41594-018-0037-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Du J, Ma X, Shen B, Huang Y, Birnbaumer L, & Yao X (2014). TRPV4, TRPC1, and TRPP2 assemble to form a flow-sensitive heteromeric channel. The FASEB Journal, 28(11), 4677–4685. 10.1096/fj.14-251652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dunn KM, Hill-Eubanks DC, Liedtke WB, & Nelson MT (2013). TRPV4 channels stimulate Ca2+-induced Ca2+ release in astrocytic endfeet and amplify neurovascular coupling responses. Proceedings of the National Academy of Sciences of the United States of America, 110(15), 6157–6162. 10.1073/pnas.1216514110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Earley S, & Brayden JE (2015). Transient receptor potential channels in the vasculature. Physiological Reviews, 95(2), 645–690. 10.1152/physrev.00026.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Earley S, Heppner TJ, Nelson MT, & Brayden JE (2005). TRPV4 forms a novel Ca2+ signaling complex with ryanodine receptors and BKCa channels. Circulation Research, 97(12), 1270–1279. 10.1161/01.RES.0000194321.60300.d6. [DOI] [PubMed] [Google Scholar]
- Earley S, Pauyo T, Drapp R, Tavares MJ, Liedtke W, & Brayden JE (2009). TRPV4-dependent dilation of peripheral resistance arteries influences arterial pressure. American Journal of Physiology. Heart and Circulatory Physiology, 297(3), H1096–H1102. 10.1152/ajpheart.00241.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ellinsworth DC, Sandow SL, Shukla N, Liu Y, Jeremy JY, & Gutterman DD (2016). Endothelium-derived hyperpolarization and coronary vasodilation: Diverse and integrated roles of epoxyeicosatrienoic acids, hydrogen peroxide, and gap junctions. Microcirculation, 23(1), 15–32. 10.1111/micc.12255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Everaerts W, Zhen X, Ghosh D, Vriens J, Gevaert T, Gilbert JP, et al. (2010). Inhibition of the cation channel TRPV4 improves bladder function in mice and rats with cyclophosphamide-induced cystitis. Proceedings of the National Academy of Sciences of the United States of America, 107(44), 19084–19089. 10.1073/pnas.1005333107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Falcone JC, Kuo L, & Meininger GA (1993). Endothelial cell calcium increases during flow-induced dilation in isolated arterioles. The American Journal of Physiology, 264(2 Pt. 2), H653–H659. 10.1152/ajpheart.1993.264.2.H653. [DOI] [PubMed] [Google Scholar]
- Fan HC, Zhang X, & McNaughton PA (2009). Activation of the TRPV4 ion channel is enhanced by phosphorylation. The Journal of Biological Chemistry, 284(41), 27884–27891. 10.1074/jbc.M109.028803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fridolfsson HN, Roth DM, Insel PA, & Patel HH (2014). Regulation of intracellular signaling and function by caveolin. The FASEB Journal, 28(9), 3823–3831. 10.1096/fj.14-252320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao F, Sui D, Garavito RM, Worden RM, & Wang DH (2009). Salt intake augments hypotensive effects of transient receptor potential vanilloid 4: Functional significance and implication. Hypertension, 53(2), 228–235. 10.1161/HYPERTENSIONAHA.108.117499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gevaert T, Vriens J, Segal A, Everaerts W, Roskams T, Talavera K, et al. (2007). Deletion of the transient receptor potential cation channel TRPV4 impairs murine bladder voiding. The Journal of Clinical Investigation, 117(11), 3453–3462. 10.1172/JCI31766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gifford JR, Ives SJ, Park SY, Andtbacka RH, Hyngstrom JR, Mueller MT, et al. (2014). Alpha1- and alpha2-adrenergic responsiveness in human skeletal muscle feed arteries: The role of TRPV ion channels in heat-induced sympatholysis. American Journal of Physiology. Heart and Circulatory Physiology, 307(9), H1288–H1297. 10.1152/ajpheart.00068.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goedicke-Fritz S, Kaistha A, Kacik M, Markert S, Hofmeister A, Busch C, et al. (2015). Evidence for functional and dynamic microcompartmentation of Cav-1/TRPV4/K(Ca) in caveolae of endothelial cells. European Journal of Cell Biology, 94(7–9), 391–400. 10.1016/j.ejcb.2015.06.002. [DOI] [PubMed] [Google Scholar]
- Harraz OF, Longden TA, Hill-Eubanks D, & Nelson MT (2018). PIP2 depletion promotes TRPV4 channel activity in mouse brain capillary endothelial cells. eLife, 7, e38689. 10.7554/eLife.38689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harteneck C, & Schultz G (2007). TRPV4 and TRPM3 as volume-regulated cation channels. In Liedtke W & Heller S (Eds.), TRP ion channel function in sensory transduction and cellular signaling cascades. Boca Raton, FL: CRC Press/Taylor & Francis. [Google Scholar]
- Hartmannsgruber V, Heyken WT, Kacik M, Kaistha A, Grgic I, Harteneck C, et al. (2007). Arterial response to shear stress critically depends on endothelial TRPV4 expression. PLoS One, 2(9), e827. 10.1371/journal.pone.0000827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hatano N, Suzuki H, Itoh Y, & Muraki K (2013). TRPV4 partially participates in proliferation of human brain capillary endothelial cells. Life Sciences, 92(4–5), 317–324. 10.1016/j.lfs.2013.01.002. [DOI] [PubMed] [Google Scholar]
- Heathcote HR, Lee MD, Zhang X, Saunter CD, Wilson C, & McCarron JG (2019). Endothelial TRPV4 channels modulate vascular tone by Ca2+-induced Ca2+ release at inositol 1,4,5-trisphosphate receptors. British Journal of Pharmacology, 176(17), 3297–3317. 10.1111/bph.14762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hekimian G, Cote S, Van Sande J, & Boeynaems JM (1992). H2 receptor-mediated responses of aortic endothelial cells to histamine. The American Journal of Physiology, 262(1 Pt. 2), H220–H224. 10.1152/ajpheart.1992.262.1.H220. [DOI] [PubMed] [Google Scholar]
- Heltianu C, Simionescu M, & Simionescu N (1982). Histamine receptors of the microvascular endothelium revealed in situ with a histamine-ferritin conjugate: Characteristic high-affinity binding sites in venules. The Journal of Cell Biology, 93(2), 357–364. 10.1083/jcb.93.2.357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hong K, Cope EL, DeLalio LJ, Marziano C, Isakson BE, & Sonkusare SK (2018). TRPV4 (transient receptor potential vanilloid 4) channel-dependent negative feedback mechanism regulates Gq protein-coupled receptor-induced vasoconstriction. Arteriosclerosis, Thrombosis, and Vascular Biology, 38(3), 542–554. 10.1161/ATVBAHA.117.310038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hong K, Cope EL, Marziano C, & Sonkusare SK (2016a). Loss of local Ca2+ signaling networks in the endothelium in diet induced obesity. The FASEB Journal, 30(1), 1.26733108 [Google Scholar]
- Hong K, Cope EL, Marziano C, & Sonkusare SK (2016b). Smooth muscle α1-adrenergic receptor activation initiates a negative feedback mechanism that involves Ca2+ influx through endothelial TRPV4 channels. The FASEB Journal, 31(1), 1. [Google Scholar]
- Huh D, Leslie DC, Matthews BD, Fraser JP, Jurek S, Hamilton GA, et al. (2012). A human disease model of drug toxicity-induced pulmonary edema in a lung-on-a-chip microdevice. Science Translational Medicine, 4(159). 10.1126/scitranslmed.3004249.159ra147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jian MY, King JA, Al-Mehdi AB, Liedtke W, & Townsley MI (2008). High vascular pressure-induced lung injury requires P450 epoxygenase-dependent activation of TRPV4. American Journal of Respiratory Cell and Molecular Biology, 38(4), 386–392. 10.1165/rcmb.2007-0192OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones JL, Peana D, Veteto AB, Lambert MD, Nourian Z, Karasseva NG, et al. (2019). TRPV4 increases cardiomyocyte calcium cycling and contractility yet contributes to damage in the aged heart following hypoosmotic stress. Cardiovascular Research, 115(1), 46–56. 10.1093/cvr/cvy156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kohler R, Heyken WT, Heinau P, Schubert R, Si H, Kacik M, et al. (2006). Evidence for a functional role of endothelial transient receptor potential V4 in shear stress-induced vasodilatation. Arteriosclerosis, Thrombosis, and Vascular Biology, 26(7), 1495–1502. 10.1161/01.ATV.0000225698.36212.6a. [DOI] [PubMed] [Google Scholar]
- Longden TA, Dabertrand F, Koide M, Gonzales AL, Tykocki NR, Brayden JE, et al. (2017). Capillary K+-sensing initiates retrograde hyperpolarization to increase local cerebral blood flow. Nature Neuroscience, 20(5), 717–726. 10.1038/nn.4533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loot AE, Popp R, Fisslthaler B, Vriens J, Nilius B, & Fleming I (2008). Role of cytochrome P450-dependent transient receptor potential V4 activation in flow-induced vasodilatation. Cardiovascular Research, 80(3), 445–452. 10.1093/cvr/cvn207. [DOI] [PubMed] [Google Scholar]
- Ma X, Du J, Zhang P, Deng J, Liu J, Lam FF, et al. (2013). Functional role of TRPV4-KCa2.3 signaling in vascular endothelial cells in normal and streptozotocin-induced diabetic rats. Hypertension, 62(1), 134–139. 10.1161/HYPERTENSIONAHA.113.01500. [DOI] [PubMed] [Google Scholar]
- Ma X, Qiu S, Luo J, Ma Y, Ngai CY, Shen B, et al. (2010). Functional role of vanilloid transient receptor potential 4-canonical transient receptor potential 1 complex in flow-induced Ca2+ influx. Arteriosclerosis, Thrombosis, and Vascular Biology, 30(4), 851–858. 10.1161/ATVBAHA.109.196584. [DOI] [PubMed] [Google Scholar]
- Marrelli SP, O’Neil RG, Brown RC, & Bryan RM Jr. (2007). PLA2 and TRPV4 channels regulate endothelial calcium in cerebral arteries. American Journal of Physiology. Heart and Circulatory Physiology, 292(3), H1390–H1397. 10.1152/ajpheart.01006.2006. [DOI] [PubMed] [Google Scholar]
- Marziano C, Hong K, Cope EL, Kotlikoff MI, Isakson BE, & Sonkusare SK (2017). Nitric oxide-dependent feedback loop regulates transient receptor potential vanilloid 4 (TRPV4) channel cooperativity and endothelial function in small pulmonary arteries. Journal of the American Heart Association, 6(12). e007157. 10.1161/JAHA.117.007157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marziano C, Hong K, Cope EL, Ottolini M, & Sonkusare SK (2019). Peroxynitrite impairs caveolin-1-TRPV4 channel signaling in pulmonary hypertension. The FASEB Journal, 33(1), 1.30593122 [Google Scholar]
- Mendoza SA, Fang J, Gutterman DD, Wilcox DA, Bubolz AH, Li R, et al. (2010). TRPV4-mediated endothelial Ca2+ influx and vasodilation in response to shear stress. American Journal of Physiology. Heart and Circulatory Physiology, 298(2), H466–H476. 10.1152/ajpheart.00854.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meng G, Zhao S, Xie L, Han Y, & Ji Y (2018). Protein S-sulfhydration by hydrogen sulfide in cardiovascular system. British Journal of Pharmacology, 175(8), 1146–1156. 10.1111/bph.13825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mercado J, Baylie R, Navedo MF, Yuan C, Scott JD, Nelson MT, et al. (2014). Local control of TRPV4 channels by AKAP150-targeted PKC in arterial smooth muscle. The Journal of General Physiology, 143(5), 559–575. 10.1085/jgp.201311050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minshall RD, Sessa WC, Stan RV, Anderson RG, & Malik AB (2003). Caveolin regulation of endothelial function. American Journal of Physiology. Lung Cellular and Molecular Physiology, 285(6), L1179–L1183. 10.1152/ajplung.00242.2003. [DOI] [PubMed] [Google Scholar]
- Monaghan K, McNaughten J, McGahon MK, Kelly C, Kyle D, Yong PH, et al. (2015). Hyperglycemia and diabetes downregulate the functional expression of TRPV4 channels in retinal microvascular endothelium. PLoS One, 10(6). e0128359. 10.1371/journal.pone.0128359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naik JS, Osmond JM, Walker BR, & Kanagy NL (2016). Hydrogen sulfideinduced vasodilation mediated by endothelial TRPV4 channels. American Journal of Physiology. Heart and Circulatory Physiology, 311(6), H1437–H1444. 10.1152/ajpheart.00465.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naik JS, & Walker BR (2018). Endothelial-dependent dilation following chronic hypoxia involves TRPV4-mediated activation of endothelial BK channels. Pflügers Archiv, 470(4), 633–648. 10.1007/s00424-018-2112-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nikolaev YA, Cox CD, Ridone P, Rohde PR, Cordero-Morales JF, Vasquez V, et al. (2019). Mammalian TRP ion channels are insensitive to membrane stretch. Journal of Cell Science, 132(23). 10.1242/jcs.238360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nilius B, & Voets T (2013). The puzzle of TRPV4 channelopathies. EMBO Reports, 14, 152–163. 10.1038/embor.2012.219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ottolini M, Hong K, & Sonkusare SK (2019). Calcium signals that determine vascular resistance. Wiley Interdisciplinary Reviews. Systems Biology and Medicine, 11, e1448. 10.1002/wsbm.1448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pankey EA, Zsombok A, Lasker GF, & Kadowitz PJ (2014). Analysis of responses to the TRPV4 agonist GSK1016790A in the pulmonary vascular bed of the intact-chest rat. American Journal of Physiology. Heart and Circulatory Physiology, 306(1), H33–H40. 10.1152/ajpheart.00303.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phuong TTT, Redmon SN, Yarishkin O, Winter JM, Li DY, & Krizaj D (2017). Calcium influx through TRPV4 channels modulates the adherens contacts between retinal microvascular endothelial cells. The Journal of Physiology, 595(22), 6869–6885. 10.1113/JP275052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rath G, Saliez J, Behets G, Romero-Perez M, Leon-Gomez E, Bouzin C, et al. (2012). Vascular hypoxic preconditioning relies on TRPV4-dependent calcium influx and proper intercellular gap junctions communication. Arteriosclerosis, Thrombosis, and Vascular Biology, 32(9), 2241–2249. 10.1161/ATVBAHA.112.252783. [DOI] [PubMed] [Google Scholar]
- Rock MJ, Prenen J, Funari VA, Funari TL, Merriman B, Nelson SF, et al. (2008). Gain-of-function mutations in TRPV4 cause autosomal dominant brachyolmia. Nature Genetics, 40(8), 999–1003. 10.1038/ng.166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saliez J, Bouzin C, Rath G, Ghisdal P, Desjardins F, Rezzani R, et al. (2008). Role of caveolar compartmentation in endothelium-derived hyperpolarizing factor-mediated relaxation: Ca2+ signals and gap junction function are regulated by caveolin in endothelial cells. Circulation, 117(8), 1065–1074. 10.1161/CIRCULATIONAHA.107.731679. [DOI] [PubMed] [Google Scholar]
- Seki T, Goto K, Kiyohara K, Kansui Y, Murakami N, Haga Y, et al. (2017). Downregulation of endothelial transient receptor potential vanilloid type 4 channel and small-conductance of Ca2+-activated K+ channels underpins impaired endothelium-dependent hyperpolarization in hypertension. Hypertension, 69(1), 143–153. 10.1161/HYPERTENSIONAHA.116.07110. [DOI] [PubMed] [Google Scholar]
- Senadheera S, Bertrand PP, Grayson TH, Leader L, Murphy TV, & Sandow SL (2013). Pregnancy-induced remodelling and enhanced endothelium-derived hyperpolarization-type vasodilator activity in rat uterine radial artery: Transient receptor potential vanilloid type 4 channels, caveolae and myoendothelial gap junctions. Journal of Anatomy, 223(6), 677–686. 10.1111/joa.12127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shibasaki K, Ikenaka K, Tamalu F, Tominaga M, & Ishizaki Y (2014). A novel subtype of astrocytes expressing TRPV4 (transient receptor potential vanilloid 4) regulates neuronal excitability via release of gliotransmitters. The Journal of Biological Chemistry, 289(21), 14470–14480. 10.1074/jbc.M114.557132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sonkusare SK, Bonev AD, Ledoux J, Liedtke W, Kotlikoff MI, Heppner TJ, et al. (2012). Elementary Ca2+ signals through endothelial TRPV4 channels regulate vascular function. Science, 336(6081), 597–601. 10.1126/science.1216283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sonkusare SK, Dalsgaard T, Bonev AD, Hill-Eubanks DC, Kotlikoff MI, Scott JD, et al. (2014). AKAP150-dependent cooperative TRPV4 channel gating is central to endothelium-dependent vasodilation and is disrupted in hypertension. Science Signaling, 7(333), ra66. 10.1126/scisignal.2005052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strotmann R, Harteneck C, Nunnenmacher K, Schultz G, & Plant TD (2000). OTRPC4, a nonselective cation channel that confers sensitivity to extracellular osmolarity. Nature Cell Biology, 2(10), 695–702. 10.1038/35036318. [DOI] [PubMed] [Google Scholar]
- Strotmann R, Schultz G, & Plant TD (2003). Ca2+-dependent potentiation of the nonselective cation channel TRPV4 is mediated by a C-terminal calmodulin binding site. The Journal of Biological Chemistry, 278(29), 26541–26549. 10.1074/jbc.M302590200. [DOI] [PubMed] [Google Scholar]
- Sukumaran SV, Singh TU, Parida S, Narasimha Reddy Ch E, Thangamalai R, Kandasamy K, et al. (2013). TRPV4 channel activation leads to endothelium-dependent relaxation mediated by nitric oxide and endothelium-derived hyperpolarizing factor in rat pulmonary artery. Pharmacological Research, 78, 18–27. 10.1016/j.phrs.2013.09.005. [DOI] [PubMed] [Google Scholar]
- Sullivan MN, Francis M, Pitts NL, Taylor MS, & Earley S (2012). Optical recording reveals novel properties of GSK1016790A-induced vanilloid transient receptor potential channel TRPV4 activity in primary human endothelial cells. Molecular Pharmacology, 82(3), 464–472. 10.1124/mol.112.078584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suresh K, Servinsky L, Jiang H, Bigham Z, Yun X, Kliment C, et al. (2018). Reactive oxygen species induced Ca2+ influx via TRPV4 and microvascular endothelial dysfunction in the SU5416/hypoxia model of pulmonary arterial hypertension. American Journal of Physiology. Lung Cellular and Molecular Physiology, 314(5), L893–L907. 10.1152/ajplung.00430.2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tajada S, Moreno CM, O’Dwyer S, Woods S, Sato D, Navedo MF, et al. (2017). Distance constraints on activation of TRPV4 channels by AKAP150-bound PKCalpha in arterial myocytes. The Journal of General Physiology, 149(6), 639–659. 10.1085/jgp.201611709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahashi N, Hamada-Nakahara S, Itoh Y, Takemura K, Shimada A, Ueda Y, et al. (2014). TRPV4 channel activity is modulated by direct interaction of the ankyrin domain to PI(4,5)P(2). Nature Communications, 5, 4994. 10.1038/ncomms5994. [DOI] [PubMed] [Google Scholar]
- Thodeti CK, Matthews B, Ravi A, Mammoto A, Ghosh K, Bracha AL, et al. (2009). TRPV4 channels mediate cyclic strain-induced endothelial cell reorientation through integrin-to-integrin signaling. Circulation Research, 104(9), 1123–1130. 10.1161/CIRCRESAHA.108.192930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thorneloe KS, Cheung M, Bao W, Alsaid H, Lenhard S, Jian MY, et al. (2012). An orally active TRPV4 channel blocker prevents and resolves pulmonary edema induced by heart failure. Science Translational Medicine, 4(159), 159ra148. 10.1126/scitranslmed.3004276. [DOI] [PubMed] [Google Scholar]
- Thorneloe KS, Sulpizio AC, Lin Z, Figueroa DJ, Clouse AK, McCafferty GP, et al. (2008). N-((1S)-1-{[4-((2S)-2-{[(2,4-dichlorophenyl)sulfonyl]amino}-3-hydroxypropanoyl)-1-piperazinyl]carbonyl}-3-methylbutyl)-1-benzothiophene-2-carboxamide (GSK1016790A), a novel and potent transient receptor potential vanilloid 4 channel agonist induces urinary bladder contraction and hyperactivity: Part I. The Journal of Pharmacology and Experimental Therapeutics, 326(2), 432–442. 10.1124/jpet.108.139295. [DOI] [PubMed] [Google Scholar]
- Vincent F, Acevedo A, Nguyen MT, Dourado M, DeFalco J, Gustafson A, et al. (2009). Identification and characterization of novel TRPV4 modulators. Biochemical and Biophysical Research Communications, 389(3), 490–494. 10.1016/j.bbrc.2009.09.007. [DOI] [PubMed] [Google Scholar]
- Vriens J, Owsianik G, Fisslthaler B, Suzuki M, Janssens A, Voets T, et al. (2005). Modulation of the Ca2+ permeable cation channel TRPV4 by cytochrome P450 epoxygenases in vascular endothelium. Circulation Research, 97(9), 908–915. 10.1161/01.RES.0000187474.47805.30. [DOI] [PubMed] [Google Scholar]
- Vriens J, Watanabe H, Janssens A, Droogmans G, Voets T, & Nilius B (2004). Cell swelling, heat, and chemical agonists use distinct pathways for the activation of the cation channel TRPV4. Proceedings of the National Academy of Sciences of the United States of America, 101(1), 396–401. 10.1073/pnas.0303329101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wandall-Frostholm C, Dalsgaard T, Bajoriunas V, Olivan-Viguera A, Sadda V, Beck L, et al. (2015). Genetic deficit of KCa3.1 channels protects against pulmonary circulatory collapse induced by TRPV4 channel activation. British Journal of Pharmacology, 172(18), 4493–4505. 10.1111/bph.13234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watanabe H, Davis JB, Smart D, Jerman JC, Smith GD, Hayes P, et al. (2002). Activation of TRPV4 channels (hVRL-2/mTRP12) by phorbol derivatives. The Journal of Biological Chemistry, 277(16), 13569–13577. 10.1074/jbc.M200062200. [DOI] [PubMed] [Google Scholar]
- Watanabe H, Vriens J, Prenen J, Droogmans G, Voets T, & Nilius B (2003). Anandamide and arachidonic acid use epoxyeicosatrienoic acids to activate TRPV4 channels. Nature, 424(6947), 434–438. 10.1038/nature01807. [DOI] [PubMed] [Google Scholar]
- Watanabe H, Vriens J, Suh SH, Benham CD, Droogmans G, & Nilius B (2002). Heat-evoked activation of TRPV4 channels in a HEK293 cell expression system and in native mouse aorta endothelial cells. The Journal of Biological Chemistry, 277(49), 47044–47051. 10.1074/jbc.M208277200. [DOI] [PubMed] [Google Scholar]
- White JP, Cibelli M, Urban L, Nilius B, McGeown JG, & Nagy I (2016). TRPV4: Molecular conductor of a diverse orchestra. Physiological Reviews, 96(3), 911–973. 10.1152/physrev.00016.2015. [DOI] [PubMed] [Google Scholar]
- Wu S, Jian MY, Xu YC, Zhou C, Al-Mehdi AB, Liedtke W, et al. (2009). Ca2+ entry via alpha1G and TRPV4 channels differentially regulates surface expression of P-selectin and barrier integrity in pulmonary capillary endothelium. American Journal of Physiology. Lung Cellular and Molecular Physiology, 297(4), L650–L657. 10.1152/ajplung.00015.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yin J, Hoffmann J, Kaestle SM, Neye N, Wang L, Baeurle J, et al. (2008). Negative-feedback loop attenuates hydrostatic lung edema via a cGMP-dependent regulation of transient receptor potential vanilloid 4. Circulation Research, 102(8), 966–974. 10.1161/CIRCRESAHA.107.168724. [DOI] [PubMed] [Google Scholar]
- Zemskov E, Lucas R, Verin AD, & Umapathy NS (2011). P2Y receptors as regulators of lung endothelial barrier integrity. Journal of Cardiovascular Disease Research, 2(1), 14–22. 10.4103/0975-3583.78582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang DX, Mendoza SA, Bubolz AH, Mizuno A, Ge ZD, Li R, et al. (2009). Transient receptor potential vanilloid type 4-deficient mice exhibit impaired endothelium-dependent relaxation induced by acetylcholine in vitro and in vivo. Hypertension, 53(3), 532–538. 10.1161/HYPERTENSIONAHA.108.127100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang L, Papadopoulos P, & Hamel E (2013). Endothelial TRPV4 channels mediate dilation of cerebral arteries: Impairment and recovery in cerebrovascular pathologies related to Alzheimer’s disease. British Journal of Pharmacology, 170(3), 661–670. 10.1111/bph.12315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang P, Sun C, Li H, Tang C, Kan H, Yang Z, et al. (2018). TRPV4 (transient receptor potential vanilloid 4) mediates endothelium-dependent contractions in the aortas of hypertensive mice. Hypertension, 71(1), 134–142. 10.1161/HYPERTENSIONAHA.117.09767. [DOI] [PubMed] [Google Scholar]
- Zheng X, Zinkevich NS, Gebremedhin D, Gauthier KM, Nishijima Y, Fang J, et al. (2013). Arachidonic acid-induced dilation in human coronary arterioles: Convergence of signaling mechanisms on endothelial TRPV4-mediated Ca2+ entry. Journal of the American Heart Association, 2(3), e000080. 10.1161/JAHA.113.000080. [DOI] [PMC free article] [PubMed] [Google Scholar]