

Abstract
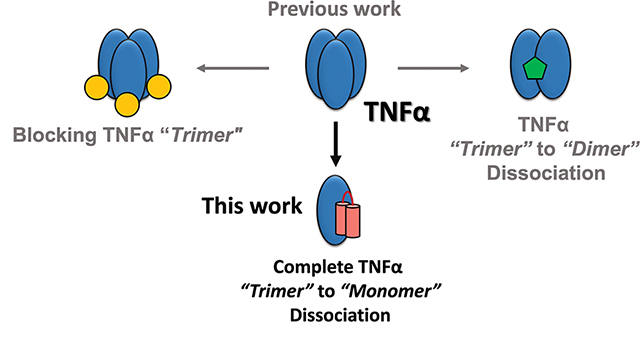
Aberrant tumor necrosis factor-α (TNFα) signaling is associated with many inflammatory diseases. The homotrimeric quaternary structure of TNFα is essential for receptor recognition and signal transduction. Previously, we described an engineered α/β-peptide inhibitor that potently suppresses TNFα activity and resists proteolysis. Here we present structural evidence that both the α/β-peptide inhibitor and an all-α analogue bind to a monomeric form of TNFα. Calorimetry data support a 1:1 inhibitor:TNFα stoichiometry in solution. In contrast, previous co-crystal structures involving peptide or small-molecule inhibitors have shown the antagonists engaging a TNFα dimer. The structural data reveal why our inhibitors favor monomeric TNFα. Previous efforts to block TNFα-induced cell death with peptide inhibitors revealed that surfactant additives to the assay conditions cause a more rapid manifestation of inhibitory activity than is observed in the absence of additives. We attributed this effect to a loose association surfactant-TNFα association that lowers the barrier to trimer dissociation. Here, we used the new structural data to design peptide inhibitors bearing a surfactant-inspired appendage intended to facilitate TNFα trimer dissociation. The appendage modified the time course of protection from cell death.
Graphical Abstract
INTRODUCTION
Excessive signaling mediated by cytokines, growth factors and other proteins is associated with many human diseases.1,2 These processes require that the soluble “message” protein engage with cell-surface receptors, and agents that block this engagement can be effective as drugs.3 Because contact between signaling proteins and their receptors usually involves large surfaces on each partner, therapeutically useful antagonists in current clinical use are themselves proteins, most commonly engineered antibodies.4,5 Antagonists of lower molecular weight would be appealing because antibodies are challenging to produce and store, and sustained use of engineered proteins can lead to adverse immunological responses.6,7 Efforts to inhibit protein-protein interactions with small molecules, however, have seldom led to clinical success, presumably because most small molecules cannot bind tightly enough to a target protein surface to prevent engagement of a much larger partner protein.3,8,9 This challenge has inspired many creative approaches based on peptides and related oligomers.10–24
Aberrant tumor necrosis factor-α (TNFα) signaling occurs in many inflammatory diseases, and engineered proteins that inhibit TNFα binding to cognate receptors have been widely adopted in human medicine.1,4 TNF superfamily proteins function as non-covalent homotrimers; protein drugs that block signaling act by binding to the trimeric cytokine in a manner that prevents association with cell-surface receptors.25,26 An alternative mechanism of antagonism would be to disrupt the trimeric TNFα quaternary structure, which is required for signal transduction. The small molecule SPD304, a modest inhibitor of TNFα signaling in cell-based assays, was originally proposed to function in this way.27 A co-crystal structure of SPD304 and TNFα revealed a dimeric form of the protein, with the small molecule engaging a surface exposed by the absence of the third protomer.27 However, a comparable co-crystal structure was subsequently reported for another small molecule inhibitor, and in this case several copies of the inhibitor could be resolved in association with the TNFα dimer.28 Retrospective analysis suggested a similar possibility for the structure containing SPD304. Other studies indicate that inhibition of TNFα signaling by SPD304 does not depend on trimer dissociation.29 A series of benzimidazole derivatives has recently been shown to block TNFα signaling by binding to the trimer and stabilizing an unsymmetrical quaternary structure that cannot properly engage with cell-surface receptors; the impressive qualities of these compounds include oral activity in mice.30,31 Luzi et al. have reported a chemically crosslinked 19-mer peptide, designated M21, that inhibits TNFα signaling by stabilizing a dimeric form, as established by a co-crystal structure.32
We recently described a 29-mer peptide (1; Figure 1a) that potently inhibits TNFα signal transduction, an activity that was correlated with disruption of the native trimeric quaternary structure.33 This inhibitor contains six non-proteinogenic residues, three derived from β-amino acids and three from 2-aminoisobutyric acid (Aib) (Figure 1b), and displays considerable resistance to proteolytic degradation.34 Here we demonstrate that α/β-peptide 1 stabilizes a monomeric form of TNFα. To our knowledge, this work provides the first demonstration of complete dissociation of the trimeric TNFα to a monomeric form. High-resolution structural data elucidate the basis of this unique inhibitory mode of action.
Figure 1.
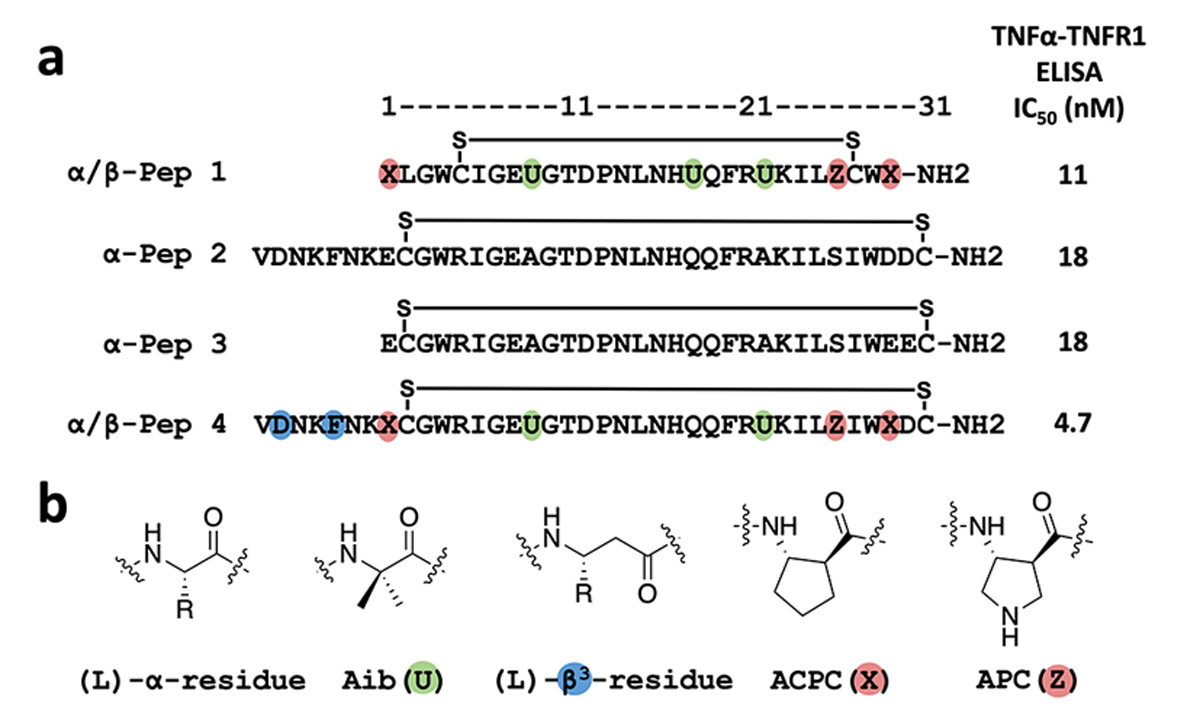
(a) Sequences of TNFα peptide inhibitors (1–4) used in this study. The two cysteines are engaged in the intramolecular disulfide bond. Unnatural amino acids are indicated by colored ovals. (b) Structures of a generic (L)-α-residue, an Aib residue (U) (green), a (L)-β3-residue (blue), a cyclic β-residue ACPC residue (X) (red), and a cyclic β-residue APC residue (Z) (red).
RESULTS AND DISCUSSION
α/β-Peptide 1 stabilizes a monomeric form of TNFα.
The design of α/β-peptide 1 was based on a 59-mer polypeptide (Z-TNFα, Figure S1) that had been selected and optimized by Jonsson et al. via phage display for binding to TNFα.35 This 59-mer has a Z domain scaffold, which adopts a three-helix bundle tertiary structure; the TNFα-binding surface is displayed by two of the three helices.7,36,37 Our previous development of 1 involved eliminating the third helix, stabilizing the necessary helix-loop-helix conformation with a disulfide, and introducing nonproteinogenic residues, among other modifications.33,35–42 Size-exclusion chromatography (SEC) data showed that 1 caused dissociation of the trimeric state,33 but we could not determine from this analysis whether a dimer or monomer was formed.
To elucidate the interaction of 1 with TNFα, we determined a cocrystal structure, which reveals a 1:1 ratio of α/β-peptide 1 and TNFα monomer (Figure 2). The crystallization solution contained 0.8 M sodium potassium tartrate, 0.1 M Tris (pH 8.5), and 4% ethylene glycol. (No surfactant was included.) The asymmetric unit contains eight independent copies of each component. The TNFα monomers are arranged in an octameric ring, with the α/β-peptides engaged by surfaces oriented toward the center of the ring (Figure 2a). All eight TNFα-peptide copies are very similar structurally (Figure S2a, Table S1). Based on data described below and the well-established significance of the trimeric TNFα quaternary structure, we hypothesize that the octameric arrangement has no biological significance and is a consequence of crystal packing forces.
Figure 2.
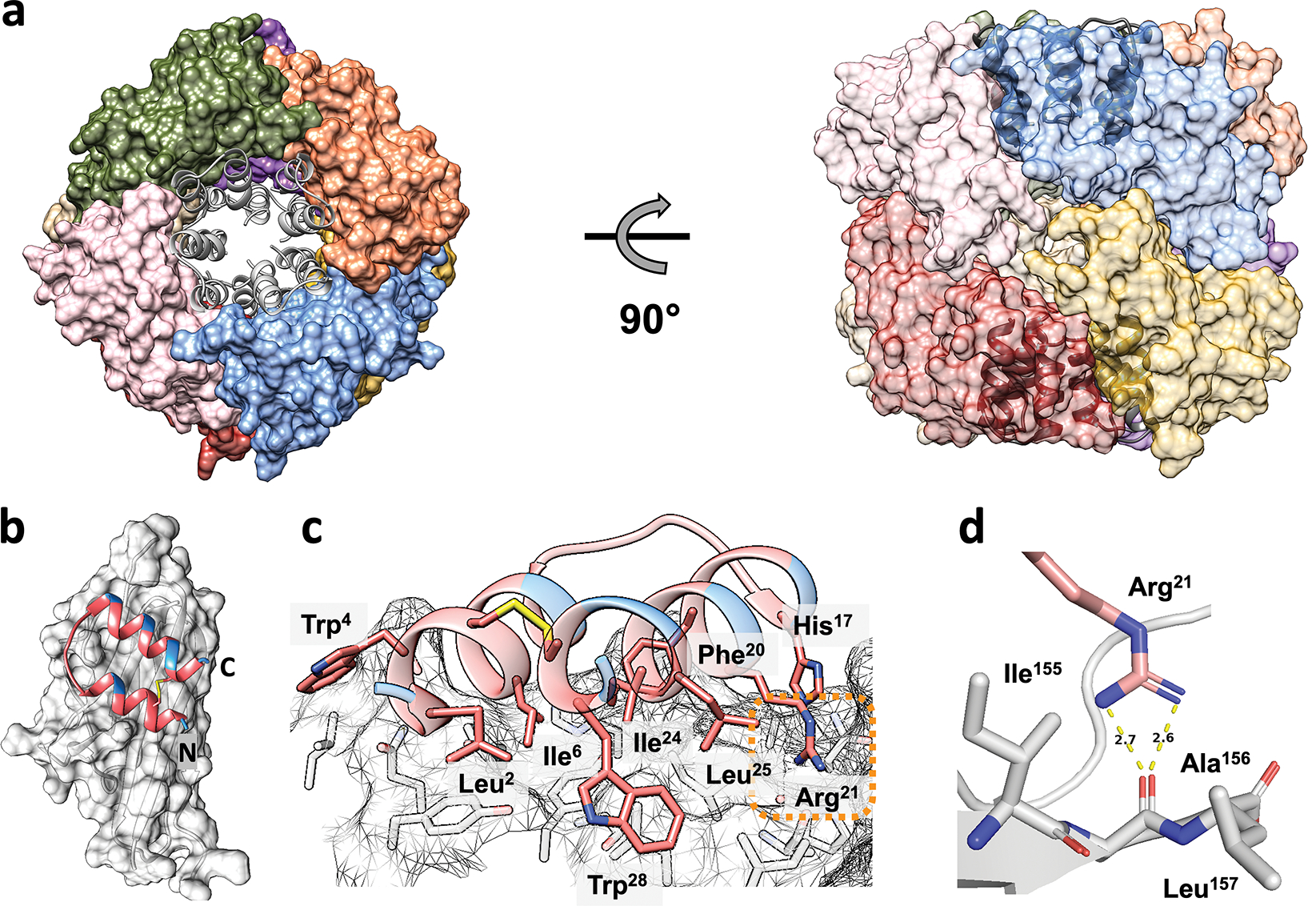
Crystal structure of α/β-peptide 1 bound to TNFα with a 1:1 stoichiometry (PDB ID: 7TA6). (a) Top (left) and side (right) views of the asymmetric unit, which contains eight copies α/β-peptide 1 (grey) and eight copies of TNFα monomer (colored). (b) α/β-Peptide 1 (pink, with unnatural residues in blue and intramolecular disulfide in yellow) bound to TNFα monomer (light grey). The N- and C- termini of α/β-Peptide 1 are labeled. (c) Close-up showing side chains of the α/β-peptide 1 that contact the TNFα surface. (d) The side chain of residue Arg21 on α/β-peptide 1 interacts with the backbone of Ala156 on TNFα through hydrogen bonding.
In the crystal structure, α/β-peptide 1 is bound to a TNFα surface that would pack against neighboring monomers in the native trimeric quaternary assembly (Figure 2b, 3a). The side chains of His17 and Phe20 on α/β-peptide 1 are oriented toward complementary TNFα surface cavities. The side chains of Leu2, Ile6, Arg21, Ile24, and Leu25 make other close contacts with the TNFα monomer surface (Figure 2c). The side chain guanidinium group of Arg21 forms hydrogen bonds with the main chain carbonyl of TNFα residue Ala156 (Figure 2d). As intended in the original design of 1, all of the β- and Aib-substitution sites are oriented toward solvent rather than toward the TNFα protomer surface (Figure 2b,c). The loop of the helix-loop-helix structure (Figure 2c) does not directly interact with the TNFα surface, as expected from our previous design. This structure thus validates the design strategy that produced α/β-peptide 1, which sought to incorporate non-proteinogenic residues to protect the peptide from proteolytic degradation without affecting the molecular surface required for engagement of TNFα.33,37
Figure 3.
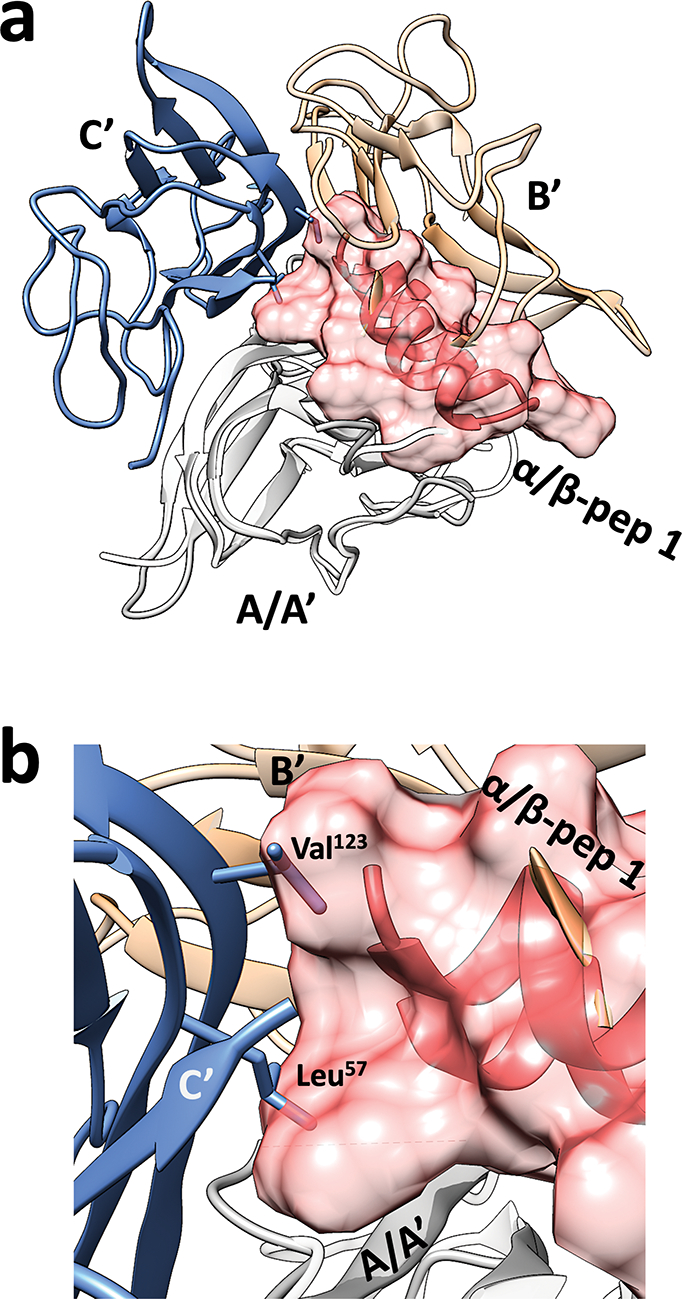
Overlay of a representative TNFα monomer and the bound α/β-peptide 1 from the TNFα+1 crystal structure with the native TNFα trimer crystal structure (PDB ID: 1TNF, denoted as A’, B’ and C’). (a) The TNFα monomer (A; dark grey ribbons) bound to 1 is aligned to TNFα monomer (A’; light gray ribbons) from the native trimer. The α/β-peptide 1 is shown as a pink volume, and the other two TNFα monomers (B’ and C’) from the native homotrimer are shown as blue and yellow ribbons. This overlay shows that binding to α/β-peptide 1 prevents association of a TNFα monomer with either of the other subunits that would constitute a native trimer. (b) Close-up showing that the surface of α/β-peptide 1 bound to the TNFα monomer (A; dark gray) occupies space that would be occupied by key interface side chains from the TNFα monomer in blue in the native homotrimer.
Trimer-to-monomer dissociation of TNFα has not been previously demonstrated, despite multiple efforts to inhibit TNFα signaling with small molecules and peptides.27,28,31–33 To understand how α/β-peptide 1 could stabilize a monomeric form of TNFα, we overlaid a representative TNFα-peptide pair from our new co-crystal structure with the native TNFα trimer structure (Figure 3a). In this overlay, the TNFα monomer from our structure was aligned with one of the monomers from the TNFα trimer (gray A and A’ in Figure 3a) with an average RMSD of 0.54 Å (Table S2). This structural similarity supports our hypothesis that 1 inhibits TNFα-mediated signaling by disrupting the trimeric quaternary structure rather than distorting monomer tertiary structure.
The structural overlay shows that the bulk of α/β-peptide 1 occupies a surface on the TNFα monomer that would engage one of the two neighbors in the trimer (yellow B’ in Figure 3a). The N- and C-termini of 1, however, extend onto a surface required to engage the other neighbor in the trimer (blue C’ in Figure 3a). In the native TNFα trimer, β-strands formed by segments 55–67 and 112–123 mediate contacts between subunits.25 Key residues from these strands, Leu57 and Val123, contribute side chains that are buried in the core of the native trimer (Figure S3b,c). The overlay in Figure 3b shows that these two side chains from the TNFα monomer in blue clash with the volume of α/β-peptide 1 bound to the TNFα monomer in gray. In contrast, the co-crystal structure of bicyclic peptide M21 engaged with a TNFα dimer32 shows that inter-subunit contacts mediated by the side chains of Leu57 and Val123 can be maintained when M21 is bound (Figure S4). Therefore, we hypothesize that the slightly larger size of 1 relative to M21 is responsible for the unique trimer-to-monomer TNFα dissociation achieved by the α/β-peptide. This hypothesis is consistent with the observation of dimeric TNFα in co-crystal structures with small molecules SPD304 and JNJ525, even though multiple small molecules appear to associate with the dimer in each case.27,28
α-Peptide 3 stabilizes a monomeric form of TNFα.
Because the features observed in a single crystal structure might not correspond to behavior in solution, we sought to co-crystallize TNFα with other peptide antagonists we had identified during the development of α/β-peptide 1 (α-peptides 2 and 3 in Figure 1 and α/β-peptides 4–8 in Figure S1).33 Most efforts failed, but co-crystallization of 3 with TNFα yielded a high-resolution structure. (Peptide 3 and α/β-peptide 1 are comparable in their ability to inhibit binding of TNFα to TNF receptor 1 (Figure S1, S5).) Peptide 3 co-crystallized with TNFα from a solution containing 1.5 M (NH4)2SO4, 0.1 M Tris (pH 8.5), and 12% glycerol. (No surfactant was included.) This crystal contained a 1:1 ratio of 3 and the TNFα protomer, matching the stoichiometry observed for the crystal containing α/β-peptide 1 and TNFα. The asymmetric unit contains two TNFα protomers, each bound to a molecule of 3. The relative orientations of these two TNFα protomers are quite distinct from the parallel packing in the native trimer (Figure S6a vs Figure S3a). Despite the considerable difference in the asymmetric units of the TNFα+1 and TNFα+3 structures, the interface between α-peptide 3 and the TNFα monomer is very similar to the interface between α/β-peptide 1 and the TNFα monomer (Figure S6a, Figure S2c and Table S1).
The mechanism by which α-peptide 3 induces trimer-to-monomer disruption of TNFα appears to be similar to that described above for α/β-peptide 1. Overlaying the TNFα+3 structure with the native TNFα trimer (Figure 4) reveals that the termini of 3 occupy the region that is required by Leu57 and Val123 from a neighboring protomer in the native trimer. This portion of 3 further extends into the region occupied by the Tyr59 and Tyr119 side chains of the neighboring protomer in the native trimer (Figure 4b). Overall, the co-crystal structure containing TNFα and inhibitor 3 supports our mechanistic explanation for the unique ability of helix-loop-helix peptides to induce TNFα trimer disassembly to an inactive monomeric form and thereby block signaling mediated by this cytokine.
Figure 4.
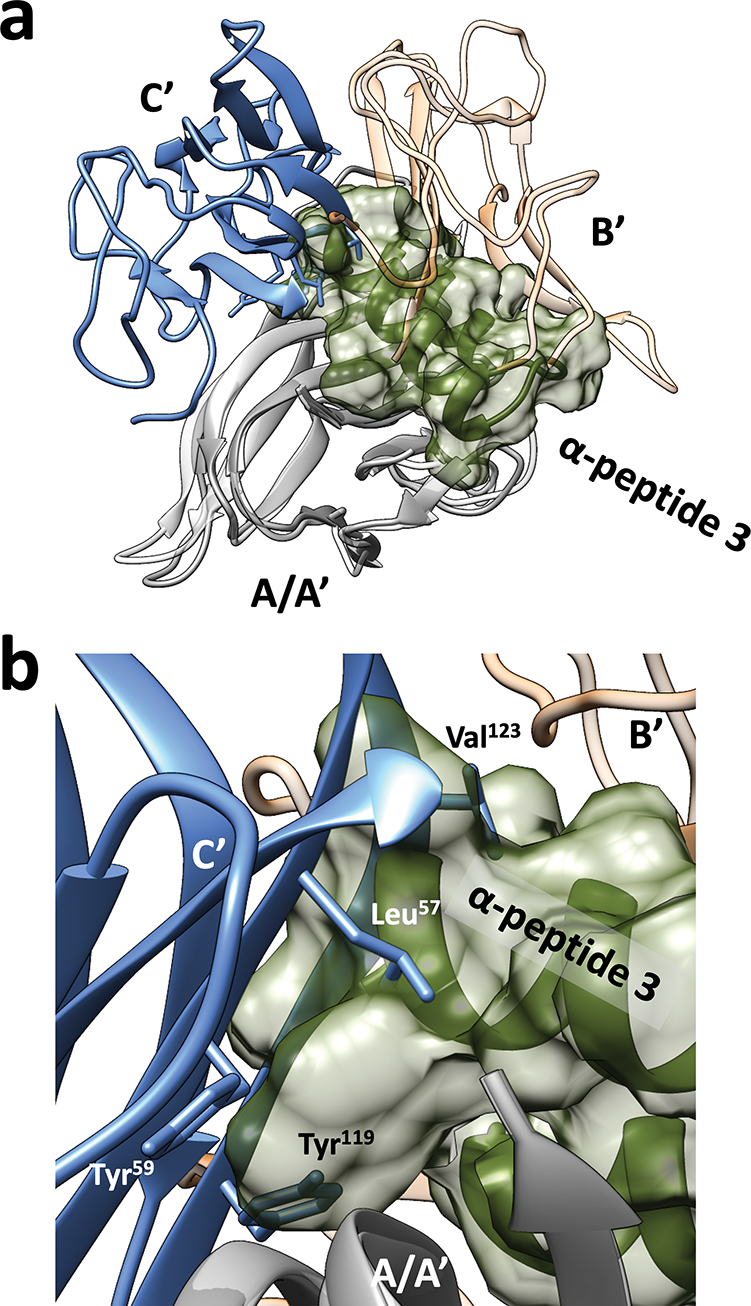
(a) Overlay one TNFα and α-peptide 3 pair in TNFα+3 crystal structure (PDB ID: 7TA3) with the native TNFα trimer crystal structure (PDB ID: 1TNF) as in Figure 3. The α-peptide 3 (green volume) prevents association of the TNFα monomer with either of the other subunits (B’ and C’) that would constitute a native trimer. (b) Zoom in showing how the N- and C-termini of 3 occupy space that would be occupied by key interface side chains from the TNFα monomer in blue in the native homotrimer.
Evidence for 1:1 binding stoichiometry in solution.
Our previous SEC analysis of interactions between TNFα and peptide inhibitors showed disruption of the native cytokine trimer but could not distinguish between dimer and monomer as the resulting form.33 Additional SEC data that support this conclusion are provided in Figure S8. To determine whether the stoichiometries of the TNFα-inhibitor complexes observed in the crystal structures correspond to behavior in solution, we used isothermal titration calorimetry (ITC) to characterize the interaction between the α-peptide inhibitor 3 with TNFα (Figure 5). Each addition of peptide solution caused an instantaneous heat absorption followed rapidly by heat release. Control ITC studies (Figure S7) showed that the initial endothermic effect is not related to TNFα and arises instead from peptide dilution. The exothermic effect results from interaction between peptide 3 and TNFα. Analysis of the ITC data revealed a 1:1 binding stoichiometry (N = 0.93 ± 0.09), and an apparent KD of 2.1 ± 0.5 μM. Very similar results were obtained with α-peptide 2 (N = 0.93 ± 0.04; KD of 3.3 ± 0.2 μM).
Figure 5.
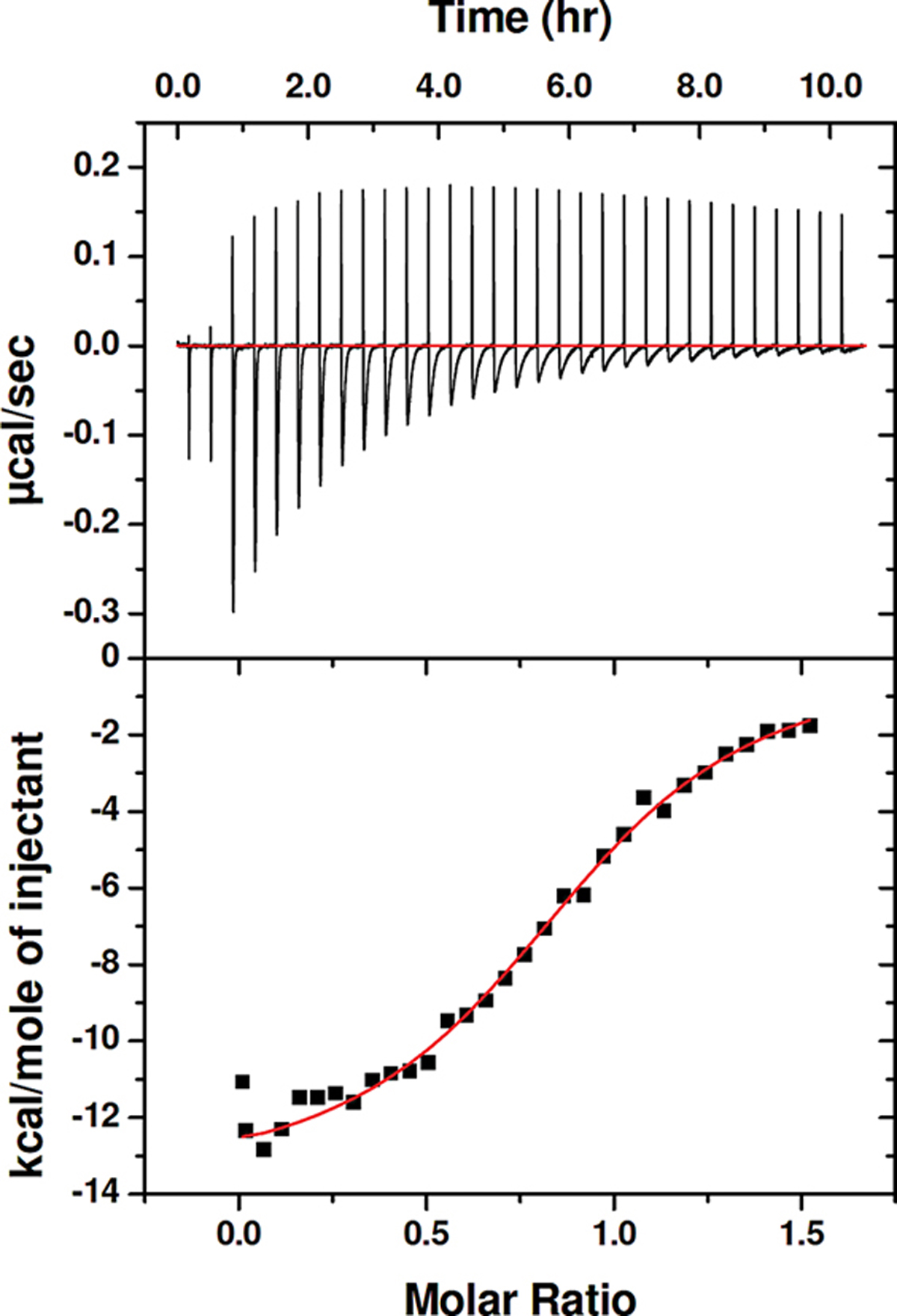
An isothermal titration calorimetry (ITC) experiment for addition of α-peptide 3 to TNFα in PBS with 0.05% TWEEN-20. The binding affinity (KD) and stoichiometry (N) values were calculated from three independent experiments.
The 1:1 stoichiometry indicated by the ITC analysis is consistent with a final complex in which one molecule of peptide binds to one TNFα protomer, or a complex in which three molecules of peptide bind to the native TNFα trimer. However, our SEC data demonstrating the disruption of the TNFα trimer by 2 are provided in Figure S8, and since the crystal structures of TNFα+3 and TNFα+1 show single peptide molecules bound to TNFα protomer, we conclude that the complex formed in solution involves a single molecule of peptide bound to a TNFα protomer. The exotherm that results from formation of this complex must include contributions from dissociation of the TNFα trimer and from binding of the inhibitor to monomeric TNFα.
Evaluation of hydrophobically modified peptides for inhibition of TNFα-induced cell toxicity.
We previously showed that α/β-peptide 1 can interfere with TNFα-mediated killing of WEHI-13VAR cells.33 TNFα-induced cell death results from binding of the cytokine to cell-surface receptors; therefore, inhibitory efficacy in this cellular assay is a measure of a peptide’s ability to block TNFα-receptor interaction. We found that if the antagonist peptide and TNFα were pre-incubated together for one hour before being added to the cells, then the peptide had no effect, and the toxic effects of TNFα were fully manifested. However, if a low concentration of the surfactant TWEEN20 was included during the pre-incubation, then significant inhibition of TNFα-induced cell killing was observed with low concentrations of the antagonist (IC50 for 1 < 10 nM).
Based on these and related findings,33 and a reconsideration of results from other groups,28,29,32,43,44 we concluded that peptide inhibitors such as 1 must overcome a significant kinetic barrier to dissociation of the TNFα trimer in order to block cell toxicity by stabilizing an inactive and dissociated form of the cytokine. Our data suggested that TWEEN20 can interact with TNFα in a way that facilitates but does not induce trimer dissociation. This interaction was attributed to the hydrophobic moiety within TWEEN20, a dodecanoyl group (Figure 6a); similar behavior was observed with other amphiphilic or hydrophobic additives, including the small molecule SPD304.33
Figure 6.

(a) Chemical structure of TWEEN 20. (b) Chemical structure of oligo-ethylene glycol (PEG16) and an N-terminal Lys residue bearing two dodecanoyl groups (C12)2 on the α and ε nitrogens. (c) A depiction of the lipid-modified peptide (C12)2-PEG16-1 where peptide 1 residues are indicated by circles.
These prior results led us to hypothesize that a peptide antagonist such as 1 might display more rapid inhibition of TNFα signaling if a hydrophobic unit were appended. This hydrophobic unit would be intended to lower the barrier to TNFα trimer dissociation and thereby facilitate capture of a TNFα protomer by the linked peptide inhibitor. The co-crystal structures described above show that the N-termini of α/β-peptide 1 and α-peptide 3 do not make direct contact with the TNFα monomer surface (Figure 2b, S6b), which suggested that N-terminal modification of these peptide antagonists might be tolerated with minimal loss of antagonist activity.
We tested our hypothesis with a design featuring an N-terminal Lys residue bearing dodecanoyl groups on both the α and ε nitrogens (Figure 6b). A flexible oligo-ethylene glycol spacer was placed between this modified Lys residue and the N-terminus of the antagonist segment (Figure 6b). The resulting peptide, (C12)2-PEG16-1 (Figure 6c), was compared with 1 itself in the ELISA assay for inhibition of TNFα binding to TNFR1 (Table S3; Figure S9). These measurements were carried out in the presence of 0.05% TWEEN20, to avoid complications from non-specific binding to ELISA plate or reagents. The comparison revealed that the inhibitory potency of (C12)2-PEG16-1 was substantially diminished relative to that of 1 itself: IC50 for (C12)2-PEG16-1 was ~44-fold higher than IC50 for 1.
The difference in ELISA results for 1 vs. (C12)2-PEG16-1, in conjunction with the crystal structures of 1 and 3 bound to TNFα, led us to conclude that the large N-terminal appendage might be too close to the peptide surface that engages the TNFα protomer to allow optimal binding of (C12)2-PEG16-1. We therefore extended the hydrophobic modification strategy to longer α/β-peptide 4 (Figure 1), which is an analogue of α-peptide 2. In unmodified form, 4 is slightly more effective than 1 or 2 in blocking TNFα-TNFR1 association, as indicated by ELISA (Table S3; Figure S9). We hypothesized that lengthening of the N-terminal region (4 vs. 1) might diminish interference of the hydrophobic appendage with binding to TNFα. Comparison of 4 vs. (C12)2-PEG16-4 via ELISA, in the presence of 0.05% TWEEN20, supported this structure-based hypothesis: IC50 for (C12)2-PEG16-4 was only ~7-fold higher than IC50 for 4 (Table S3; Figure S9).
To ask how the hydrophobic appendage affects the protection of cells from TNFα-induced toxicity, we compared 4 and (C12)2-PEG16-4 in a time course assay with WEHI-VAR13 cells in the absence of TWEEN20 (Figure 7). Almost no cells survived when 0.08 ng/mL TNFα was added without any antagonist; this condition was used to define 0% viability. Cell survival in the absence of TNFα was used to define 100% viability. These experiments explored two parameters: (1) the concentration of the antagonist (4 or (C12)2-PEG16-4) with which TNFα was pre-incubated, before addition to the cells; and (2) the length of the pre-incubation period, which varied between 1 hr and 24 hr at 37°C.
Figure 7.
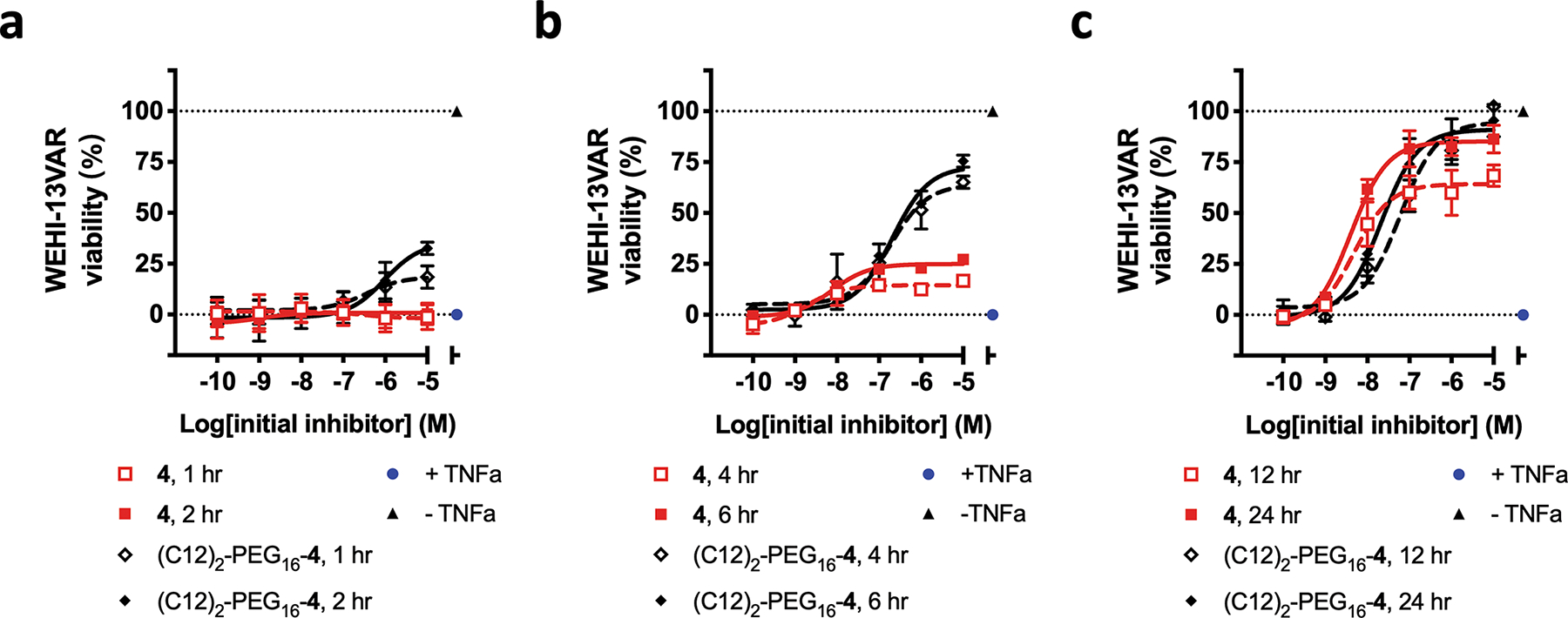
Protection of WEHI-13VAR cells from TNFα-induced toxicity, by α/β-peptide 4 and a hydrophobically modified analogue, (C12)2-PEG16-4. These studies show the effects of two variables: concentrations of the peptide antagonist (4 or (C12)2-PEG16-4), and the pre-incubation times of the peptide antagonist with TNFα before the mixture was added to the cells ((a) 1 or 2 hr; (b) 4 or 6 hr, (c) 12 or 24 hr). “+TNFα”: TNFα + DMSO; “−TNFα”: PBS + DMSO. All pre-incubation steps were conducted at 37°C in PBS. No amphiphilic additive (e.g., TWEEN20) was used at any point.
The cell survival assays showed that the hydrophobic modification influences the time course over which the α/β-peptide antagonist manifests its protective effect. After 2 hr pre-incubation of TNFα and antagonist 4, no protection was detected even at the highest α/β-peptide concentration (10 μM initial concentration, which was diluted 350-fold upon addition to cells; see SI sec II 7c for details). In contrast, at this time point 10 μM (C12)2-PEG16-4 led to survival of ~25% of the cells (Figure 7a). The maximum level of cell survival achieved with (C12)2-PEG16-4 after 2 hr pre-incubation exceeded that achieved with unmodified 4 up to 6 hr of pre-incubation (Figure 7b). At 12 hr, 10 μM (C12)2-PEG16-4 protected nearly all cells from TNFα-induced death, while only ~2/3 cells survived with 10 μM 4 (Figure 7c). After 24 hr pre-incubation, however, both 4 and the hydrophobically modified analogue protected nearly all cells from the toxic effects of TNFα (Figure 7c). The dose-response data obtained after 24 hr pre-incubation suggested that the IC50 for 4 (4.0 nM) is approximately 5-fold higher than the IC50 for (C12)2-PEG16-4 (Table S4). This modest difference in the abilities of 4 and (C12)2-PEG16-4 to block TNFα-induced cell death is very similar to the difference in their abilities to inhibit TNFα-TNFR1 interaction as measured by ELISA (Table S3).
The cell assay data in Figure 7 support our design hypothesis that the two portions of hybrid molecule (C12)2-PEG16-4 can work synergistically to affect the time course over which protection of WEHI VAR13 cells from TNFα-induced toxicity is manifested. We propose that the dodecanoyl segments of this inhibitor associate with the TNFα trimer in a manner that loosens the quaternary structure and facilitates full-fledged trimer disruption mediated by the α/β-peptide portion. These observations raise interesting questions about the effects of hydrophobically modified TNFα antagonists in vivo, where the overall effect is likely to result from an interplay among thermodynamic and kinetic factors.
CONCLUSIONS
We have shown that α/β-peptide 1, a proteolysis-resistant 29-mer, disrupts the native trimeric quaternary structure of TNFα and stabilizes a monomeric form. This disruptive activity presumably explains how 1 protects cells from TNFα-induced death.33 Previous structural studies have shown that small molecules, apparently acting in clusters, and a cross-linked 19-mer peptide can induce trimer-to-dimer dissociation of TNFα.27,28,32 Our structural analysis of the complex formed by α/β-peptide 1 and monomeric TNFα and of the analogous complex formed by α-peptide 3 reveals that these peptides occupy a moderately larger surface area on a TNFα protomer relative to the other inhibitors (Figure S10), which presumably explains the high potencies of 1 and 3 and the trimer-to-monomer dissociation induced by these inhibitors. Comparisons between the structure of α/β-peptide 1 and α-peptide 3 bound to TNFα show how strategic replacement of proteinogenic residues by β residues or Aib at non-contact sites can preserve a broad and specific binding surface displayed by a mini-protein tertiary structure while conferring resistance to proteolysis.
Dissociation of the native TNFα trimer by our peptides or other smaller agents requires that the antagonist overcome a substantial kinetic barrier.28,29,32,33 We previously discovered that additives such as TWEEN20 or SPD304 shorten the time course of antagonism manifested by peptides such as 1. The additive apparently lowers the barrier to TNFα trimer dissociation.33 The two co-crystal structures reported here suggested that appending a lipophilic group to the N-terminus of an α/β-peptide antagonist might improve the time course of cell rescue from TNFα-induced death. The anticipated improvement was observed for (C12)2-PEG16-4, relative to the unmodified α/β-peptide, 4.
These studies provide a basis for future efforts to block aberrant oligomeric cytokine signaling in vivo with backbone-modified peptides that bear a hydrophobic appendage and resist proteolysis. Such molecules might ultimately be useful alternatives to the engineered antibodies and receptor fragments that are currently used to treat inflammatory diseases mediated by TNFα. Peptides that bear a hydrophobic appendage and activate the glucagon-like peptide-1 receptor (GLP-1R), such as liraglutide and semaglutide, are widely used to treat type 2 diabetes.45 In these cases, the appendage apparently promotes peptide binding to albumin in the bloodstream, which hinders peptide excretion. Attaching a lipid moiety enhances the potency of peptide inhibitors of enveloped virus infection.46 The lipid is believed to localize such peptides in the cell membrane and thereby enhance their potency in blocking the function of viral fusion proteins. Pharmacokinetic benefits, such as those seen with GLP-1R agonists, and/or localization benefits observed for antiviral lipopeptides, might be manifested in vivo for an agent such as (C12)2-PEG16-4.
Supplementary Material
ACKNOWLEDGMENT
This work was supported in part by the National Institutes of Health grant R01 GM056414. This research used resources of the Advanced Photon Source, a U.S. Department of Energy (DOE) Office of Science User Facility operated for the DOE Office of Science by Argonne National Laboratory under Contract No. DE-AC02-06CH11357. Use of the LS-CAT Sector 21 was supported by the Michigan Economic Development Corporation and the Michigan Technology Tri-Corridor (Grant 085P1000817). GM/CA@APS has been funded by the National Cancer Institute (ACB-12002) and the National Institute of General Medical Sciences (AGM-12006, P30GM138396).
Footnotes
The authors declare the following competing financial interest(s): S.H.G. is a co-founder of Longevity Biotech, Inc., which is pursuing biomedical applications of α/β-peptides.
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website.
Materials and instrumentations (Section I), method descriptions (Section II), additional figures and tables (Section III), and characterization data for all synthesized peptides (Section IV) (PDF).
PDB Accession Codes: 7TA3, 7TA6
REFERENCES
- (1).Brenner D; Blaser H; Mak TW Regulation of Tumour Necrosis Factor Signalling: Live or Let Die. Nature Reviews Immunology. 2015, pp 362–374. 10.1038/nri3834. [DOI] [PubMed] [Google Scholar]
- (2).Ivashkiv LB IFNγ: Signalling, Epigenetics and Roles in Immunity, Metabolism, Disease and Cancer Immunotherapy. Nature Reviews Immunology. 2018, pp 545–558. 10.1038/s41577-018-0029-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Attwood MM; Jonsson J; Rask-Andersen M; Schiöth HB Soluble Ligands as Drug Targets. Nature Reviews Drug Discovery. 2020, pp 695–710. 10.1038/s41573-020-0078-4. [DOI] [PubMed] [Google Scholar]
- (4).Sedger LM; McDermott MF TNF and TNF-Receptors: From Mediators of Cell Death and Inflammation to Therapeutic Giants - Past, Present and Future. Cytokine and Growth Factor Reviews. 2014, pp 453–472. 10.1016/j.cytogfr.2014.07.016. [DOI] [PubMed] [Google Scholar]
- (5).Mitoma H; Horiuchi T; Tsukamoto H; Ueda N Molecular Mechanisms of Action of Anti-TNF-α Agents – Comparison among Therapeutic TNF-α Antagonists. Cytokine 2018, 101, 56–63. 10.1016/j.cyto.2016.08.014. [DOI] [PubMed] [Google Scholar]
- (6).Biancheri P; Brezski RJ; Di Sabatino A; Greenplate AR; Soring KL; Corazza GR; Kok KB; Rovedatti L; Vossenkämper A; Ahmad N; et al. Proteolytic Cleavage and Loss of Function of Biologic Agents That Neutralize Tumor Necrosis Factor in the Mucosa of Patients with Inflammatory Bowel Disease. Gastroenterology 2015, 149 (6), 1564–1574.e3. 10.1053/j.gastro.2015.07.002. [DOI] [PubMed] [Google Scholar]
- (7).Ståhl S; Gräslund T; Eriksson Karlström A; Frejd FY; Nygren PÅ; Löfblom J Affibody Molecules in Biotechnological and Medical Applications. Trends Biotechnol. 2017, 35 (8), 691–712. 10.1016/j.tibtech.2017.04.007. [DOI] [PubMed] [Google Scholar]
- (8).Arkin MMR; Wells JA Small-Molecule Inhibitors of Protein-Protein Interactions: Progressing towards the Dream. Nat. Rev. Drug Discov. 2004, 3 (4), 301–317. 10.1038/nrd1343. [DOI] [PubMed] [Google Scholar]
- (9).Scott DE; Bayly AR; Abell C; Skidmore J Small Molecules, Big Targets: Drug Discovery Faces the Protein-Protein Interaction Challenge. Nat. Rev. Drug Discov. 2016, 15 (8), 533–550. 10.1038/nrd.2016.29. [DOI] [PubMed] [Google Scholar]
- (10).Shi Y; Sang P; Lu J; Higbee P; Chen L; Yang L; Odom T; Daughdrill G; Chen J; Cai J Rational Design of Right-Handed Heterogeneous Peptidomimetics as Inhibitors of Protein-Protein Interactions. J. Med. Chem. 2020, 63 (21), 13187–13196. 10.1021/acs.jmedchem.0c01638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Sang P; Zhang M; Shi Y; Li C; Abdulkadir S; Li Q; Ji H; Cai J Inhibition of β-Catenin/B Cell Lymphoma 9 Protein–protein Interaction Using α-Helix–Mimicking Sulfono-γ-AApeptide Inhibitors. Proc. Natl. Acad. Sci. U. S. A. 2019, 166 (22), 10757–10762. 10.1073/pnas.1819663116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Merritt HI; Sawyer N; Arora PS Bent into Shape: Folded Peptides to Mimic Protein Structure and Modulate Protein Function. Pept. Sci. 2020, 112 (1), e24145. 10.1002/pep2.24145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Østergaard S; Kofoed J; Paulsson JF; Madsen KG; Jorgensen R; Wulff BS Design of Y 2 Receptor Selective and Proteolytically Stable PYY 3–36 Analogues. J. Med. Chem. 2018, 61 (23), 10519–10530. 10.1021/acs.jmedchem.8b01046. [DOI] [PubMed] [Google Scholar]
- (14).Berlicki Ł; Kaske M; Gutiérrez-Abad R; Bernhardt G; Illa O; Ortuño RM; Cabrele C; Buschauer A; Reiser O Replacement of Thr32 and Gln34 in the C -Terminal Neuropeptide y Fragment 25–36 by Cis -Cyclobutane and Cis - Cyclopentane β-Amino Acids Shifts Selectivity toward the Y4 Receptor. J. Med. Chem. 2013, 56 (21), 8422–8431. 10.1021/jm4008505. [DOI] [PubMed] [Google Scholar]
- (15).Tošovská P; Arora PS Oligooxopiperazines as Nonpeptidic α-Helix Mimetics. Org. Lett. 2010, 12 (7), 1588–1591. 10.1021/ol1003143. [DOI] [PubMed] [Google Scholar]
- (16).Horne WS; Grossmann TN Proteomimetics as Protein-Inspired Scaffolds with Defined Tertiary Folding Patterns. Nat. Chem. 2020, 12 (4), 331–337. 10.1038/s41557-020-0420-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Wendt M; Bellavita R; Gerber A; Efrém NL; van Ramshorst T; Pearce NM; Davey PRJ; Everard I; Vazquez-Chantada M; Chiarparin E; et al. Bicyclic β-Sheet Mimetics That Target the Transcriptional Coactivator β-Catenin and Inhibit Wnt Signaling. Angew. Chemie - Int. Ed. 2021, 60 (25), 13937–13944. 10.1002/anie.202102082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Adihou H; Gopalakrishnan R; Förster T; Guéret SM; Gasper R; Geschwindner S; Carrillo García C; Karatas H; Pobbati AV; Vazquez-Chantada M; et al. A Protein Tertiary Structure Mimetic Modulator of the Hippo Signalling Pathway. Nat. Commun. 2020, 11 (1), 5425. 10.1038/s41467-020-19224-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Cussol L; Mauran-Ambrosino L; Buratto J; Belorusova AY; Neuville M; Osz J; Fribourg S; Fremaux J; Dolain C; Goudreau SR; et al. Structural Basis for α-Helix Mimicry and Inhibition of Protein–Protein Interactions with Oligourea Foldamers. Angew. Chemie - Int. Ed. 2021, 60 (5), 2296–2303. 10.1002/anie.202008992. [DOI] [PubMed] [Google Scholar]
- (20).Mbianda J; Bakail M; André C; Moal G; Perrin ME; Pinna G; Guerois R; Becher F; Legrand P; Traoré S; et al. Optimal Anchoring of a Foldamer Inhibitor of ASF1 Histone Chaperone through Backbone Plasticity. Sci. Adv. 2021, 7 (12), eabd9153. 10.1126/sciadv.abd9153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Hegedüs Z; Hóbor F; Shoemark DK; Celis S; Lian LY; Trinh CH; Sessions RB; Edwards TA; Wilson AJ Identification of β-Strand Mediated Protein-Protein Interaction Inhibitors Using Ligand-Directed Fragment Ligation. Chem. Sci. 2021, 12 (6), 2286–2293. 10.1039/d0sc05694d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Hetherington K; Hegedus Z; Edwards TA; Sessions RB; Nelson A; Wilson AJ Stapled Peptides as HIF-1α/P300 Inhibitors: Helicity Enhancement in the Bound State Increases Inhibitory Potency. Chem. - A Eur. J. 2020, 26 (34), 7638–7646. 10.1002/chem.202000417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Hong SH; Yoo DY; Conway L; Richards-Corke KC; Parker CG; Arora PS A Sos Proteomimetic as a Pan-Ras Inhibitor. Proc. Natl. Acad. Sci. U. S. A. 2021, 118 (18), 1–11. 10.1073/pnas.2101027118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Zwillinger M; Reddy PS; Wicher B; Mandal PK; Csékei M; Fischer L; Kotschy A; Huc I Aromatic Foldamer Helices as α-Helix Extended Surface Mimetics. Chem. - A Eur. J. 2020, 26 (72), 17366–17370. 10.1002/chem.202004064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Eck MJ; Sprang SR The Structure of Tumor Necrosis Factor-α at 2.6 Å Resolution. Implications for Receptor Binding. J. Biol. Chem. 1989, 264 (29), 17595–17605. 10.2210/pdb1tnf/pdb. [DOI] [PubMed] [Google Scholar]
- (26).Liang S; Dai J; Hou S; Su L; Zhang D; Guo H; Hu S; Wang H; Rao Z; Guo Y; et al. Structural Basis for Treating Tumor Necrosis Factor α (TNFα)-Associated Diseases with the Therapeutic Antibody Infliximab. J. Biol. Chem. 2013, 288 (19), 13799–13807. 10.1074/jbc.M112.433961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).He MM; Smith AS; Oslob JD; Flanagan WM; Braisted AC; Whitty A; Cancilla MT; Wang J; Lugovskoy AA; Yoburn JC; et al. Medicine: Small-Molecule Inhibition of TNF-α. Science (80-. ). 2005, 310 (5750), 1022–1025. 10.1126/science.1116304. [DOI] [PubMed] [Google Scholar]
- (28).Blevitt JM; Hack MD; Herman KL; Jackson PF; Krawczuk PJ; Lebsack AD; Liu AX; Mirzadegan T; Nelen MI; Patrick AN; et al. Structural Basis of Small-Molecule Aggregate Induced Inhibition of a Protein-Protein Interaction. J. Med. Chem. 2017, 60 (8), 3511–3517. 10.1021/acs.jmedchem.6b01836. [DOI] [PubMed] [Google Scholar]
- (29).Hofmann D; Salmon L; Wider G Activity of Tumor Necrosis Factor α Is Modulated by Dynamic Conformational Rearrangements. J. Am. Chem. Soc. 2018, 140 (1), 167–175. 10.1021/jacs.7b05050. [DOI] [PubMed] [Google Scholar]
- (30).McMillan D; Martinez-Fleites C; Porter J; Fox D; Davis R; Mori P; Ceska T; Carrington B; Lawson A; Bourne T; et al. Structural Insights into the Disruption of TNF-TNFR1 Signalling by Small Molecules Stabilising a Distorted TNF. Nat. Commun. 2021, 12 (1), 582. 10.1038/s41467-020-20828-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).O’Connell J; Porter J; Kroeplien B; Norman T; Rapecki S; Davis R; McMillan D; Arakaki T; Burgin A; Fox D; et al. Small Molecules That Inhibit TNF Signalling by Stabilising an Asymmetric Form of the Trimer. Nat. Commun. 2019, 10 (1), 5795. 10.1038/s41467-019-13616-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).Luzi S; Kondo Y; Bernard E; Stadler LKJ; Vaysburd M; Winter G; Holliger P Subunit Disassembly and Inhibition of TNFα by a Semi-Synthetic Bicyclic Peptide. Protein Eng. Des. Sel. 2015, 28 (2), 45–52. 10.1093/protein/gzu055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).Checco JW; Eddinger GA; Rettko NJ; Chartier AR; Gellman SH Tumor Necrosis Factor-α Trimer Disassembly and Inactivation via Peptide-Small Molecule Synergy. ACS Chem. Biol. 2020, 15 (8), 2116–2124. 10.1021/acschembio.0c00313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).Steer DL; Lew RA; Perlmutter P; Smith AI; Aguilar MI The Use of β-Amino Acids in the Design of Protease and Peptidase Inhibitors. Lett. Pept. Sci. 2001, 8 (3–5), 241–246. 10.1023/A:1016237415473. [DOI] [Google Scholar]
- (35).Jonsson A; Wållberg H; Herne N; Ståhl S; Frejd FY Generation of Tumour-Necrosis-Factor-α-Specific Affibody Molecules Capable of Blocking Receptor Binding in Vitro. Biotechnol. Appl. Biochem. 2009, 54 (2), 93–103. 10.1042/ba20090085. [DOI] [PubMed] [Google Scholar]
- (36).Löfblom J; Feldwisch J; Tolmachev V; Carlsson J; Ståhl S; Frejd FY Affibody Molecules: Engineered Proteins for Therapeutic, Diagnostic and Biotechnological Applications. FEBS Letters. 2010, pp 2670–2680. 10.1016/j.febslet.2010.04.014. [DOI] [PubMed] [Google Scholar]
- (37).Checco JW; Kreitler DF; Thomas NC; Belair DG; Rettko NJ; Murphy WL; Forest KT; Gellman SH Targeting Diverse Protein–Protein Interaction Interfaces with α/β-Peptides Derived from the Z-Domain Scaffold. Proc. Natl. Acad. Sci. 2015, 112 (15), 4552–4557. 10.1073/pnas.1420380112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (38).Checco JW; Gellman SH Iterative Nonproteinogenic Residue Incorporation Yields α/β-Peptides with a Helix–Loop–Helix Tertiary Structure and High Affinity for VEGF. ChemBioChem 2017, 18 (3), 291–299. 10.1002/cbic.201600545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (39).Buck E; Wells JA Disulfide Trapping to Localize Small-Molecule Agonists and Antagonists for a G Protein-Coupled Receptor. Proc. Natl. Acad. Sci. 2005, 102 (8), 2719–2724. 10.1073/PNAS.0500016102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (40).Horne WS; Johnson LM; Ketas TJ; Klasse PJ; Lu M; Moore JP; Gellman SH Structural and Biological Mimicry of Protein Surface Recognition by α/β-Peptide Foldamers. Proc. Natl. Acad. Sci. U. S. A. 2009, 106 (35), 14751–14756. 10.1073/pnas.0902663106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (41).Braisted AC; Wells JA Minimizing a Binding Domain from Protein A. Proc. Natl. Acad. Sci. U. S. A. 1996, 93 (12), 5688–5692. 10.1073/pnas.93.12.5688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (42).Erlanson DA; Wells JA; Braisted AC Tethering: Fragment-Based Drug Discovery. Annual Review of Biophysics and Biomolecular Structure. 2004, pp 199–223. 10.1146/annurev.biophys.33.110502.140409. [DOI] [PubMed] [Google Scholar]
- (43).Poiesi C; Albertini A; Ghielmi S; Cassani G; Corti A Kinetic Analysis of TNF-α Oligomer-Monomer Transition by Surface Plasmon Resonance and Immunochemical Methods. Cytokine 1993, 5 (6), 539–545. 10.1016/S1043-4666(05)80002-X. [DOI] [PubMed] [Google Scholar]
- (44).Van Schie KA; Ooijevaar-De Heer P; Dijk L; Kruithof S; Wolbink G; Rispens T Therapeutic TNF Inhibitors Can Differentially Stabilize Trimeric TNF by Inhibiting Monomer Exchange. Sci. Rep. 2016, 6 (1), 32747. 10.1038/srep32747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (45).Lau J; Bloch P; Schäffer L; Pettersson I; Spetzler J; Kofoed J; Madsen K; Knudsen LB; McGuire J; Steensgaard DB; et al. Discovery of the Once-Weekly Glucagon-Like Peptide-1 (GLP-1) Analogue Semaglutide. J. Med. Chem. 2015, 58 (18), 7370–7380. 10.1021/acs.jmedchem.5b00726. [DOI] [PubMed] [Google Scholar]
- (46).Porotto M; Rockx B; Yokoyama CC; Talekar A; DeVito I; Palermo LM; Liu J; Cortese R; Lu M; Feldmann H; et al. Inhibition of Nipah Virus Infection In Vivo: Targeting an Early Stage of Paramyxovirus Fusion Activation during Viral Entry. PLoS Pathog. 2010, 6 (10), e1001168. 10.1371/journal.ppat.1001168. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.