

Graphical Abstract
More than 60 kinase inhibitors have been approved by the FDA, with new approvals each year for oncology and other indications.(1, 2) The epidermal growth factor receptor (EGFR) is one of the most intensively studied kinases, with three generations of targeted therapeutic drugs for the treatment of lung cancer.(3) Non small-cell lung cancer (NSCLC) patients with the L858R and del19 activating EGFR mutations are responsive to the quinazolines gefitinib and erlotinib. However, resistance to these agents typically emerges after a year, primarily through the T790M gatekeeper mutation, which enhances affinity for ATP and renders the inhibitors less able to compete.(4) Second-generation EGFR inhibitors such as afatinib and dacomitinib attempted to address this through covalent bond formation with a cysteine adjacent to the ATP site, but activity against wild-type (wt) EGFR led to gastrointestinal toxicity and a limited therapeutic index for these agents in the clinic.
The third-generation covalent inhibitor osimertinib was developed from a series of pyrimidines with selectivity for oncogenic EGFR mutants over wild-type (wt) EGFR.(5, 6) The combination of efficacy and improved tolerability led to initial FDA approval in 2015 for T790M-positive patients, and the drug progressed to first line therapy for metastatic NSCLC in 2018. As with other kinase inhibitors, however, resistance emerges, and in common with other covalent inhibitors, this often involves the cysteine residue critical to the molecule’s activity. In patients treated with both osimertinib and a first-generation inhibitor, C797S is the most frequently observed mutation, and there are currently no targeted therapies for patients who have progressed to EGFRL858R/T790M/C797S status. Other mechanisms of resistance to osimertinib include the activation of the MEK signaling pathway, and amplification of wt EGFR.(7) Most kinase inhibitors target the ATP site, and while there are some highly selective compounds, achieving selectivity across the kinome can be a challenge. Activity against two or more kinases may be beneficial in some cases, but non-selective inhibitors may have limited tolerability and be unsuitable for combination with other therapies. Allosteric inhibitors which bind outside the ATP pocket can be exquisitely selective, have greater potential for use in combination with other therapies,(8) and may delay the onset of resistance in earlier lines of therapy when combined with existing agents.
EAI045 (Fig. 1) was identified as an allosteric EGFR inhibitor following a HTS campaign to find non-ATP competitive compounds with selectivity for the EGFRL858R/T790M mutant over wt EGFR.(9) It binds to the inactive conformation of EGFR in a pocket adjacent to the ATP site. Upon activation through ligand binding, EGFR forms asymmetrical dimers in which the allosteric site is less accessible in the activator subunit. This blocks the binding of EAI045, and as a single agent it has minimal cellular and in vivo activity. The addition of cetuximab prevents dimer formation, and the combination with EAI045 demonstrated good activity in EGFRL858R/T790M models. Optimization of the isoindolinone series identified JBJ-04-125-02 (Fig. 1), with a phenyl piperazine substituent extending into a channel alongside the α-helix. The enhanced potency of JBJ-04-125-02 enables it to achieve good single-agent activity in a Ba/F3 EGFRL858R/T790M cell antiproliferative assay.(10) In mice, the compound has low in vivo clearance and a high volume of distribution, but poor oral bioavailability (Table 1). Despite its disappointing oral PK profile, efficacy was observed in an H1975 model upon daily oral dosing at 100 mg/kg. Additionally, the allosteric inhibitors co-bind with osimertinib,(11) and the combination with JBJ-04-125-02 delivered enhanced effects relative to the individual agents in a set of in vitro and in vivo studies.(10)
Fig. 1 –

structures of EAI045 and optimized isoindolinones
Table 1.
EGFRL858R/T790M enzyme and Ba/F3 cell data, and mouse in vivo PK (3 mg/kg IV, 20 mg/kg PO) for isoindolinones
| Cpd | Enzyme IC50 (nM) |
Cell IC50 (μM) |
Cell IC50 (μM)+cetux |
IV Cl (mL/min/kg) |
IV T1/2 (hr) |
Vss (L/kg) |
F % |
Auc 8hr (ng*hr/mL) |
|---|---|---|---|---|---|---|---|---|
| 1, EAI045 | 3.3 | > 10.0 | 0.221 | - | - | - | - | - |
| 2, JBJ-04-125-02 | 0.3 | 0.61 | 0.021 | 5.0 | 3.0 | 0.6 | 3 | 2523 |
| 3 | 0.3 | 0.49 | 0.006 | 10.3 | 2.8 | 1.8 | 2 | 1351 |
| 4, JBJ-09-063 | 0.1 | 0.05 | 0.006 | 15.7 | 2.3 | 2.5 | 15 | 2398 |
| 5 | 0.2 | 1.07 | 0.020 | 13.9 | 3.1 | 2.4 | 4 | 1002 |
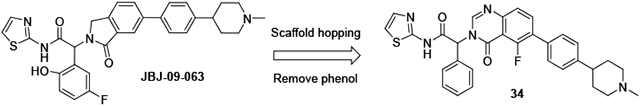
We attempted to improve permeability and bioavailability by reducing the number of H-bond donors in the molecule, and prepared N-methyl piperazine 3 and N-methyl piperidine JBJ-09-063. These changes did not significantly impact oral exposure, but the enhanced potency of JBJ-09-063 supported its use as an improved in vivo tool compound (Table 1), and JBJ-09-063 achieved tumor regression in osimertinib-resistant triple mutant (EGFRL858R/T790M/C797S) models upon daily oral dosing at 50 mg/kg.(12) At the same dose, minimal effects were observed with JBJ-09-063 in an A431 (wt EGFR) efficacy model. The removal of the phenol to give compound 5 led to a reduction in potency, without any improvement in oral bioavailability (Table 1).
Our screening cascade consisted of an EGFRL858R/T790M enzyme assay,(13) and a Ba/F3 cellular proliferation assay with two readouts, one measuring compound activity as a single agent, and one with the addition of cetuximab to prevent the formation of EGFR dimers.(13) Few of the early project compounds showed good cellular potency as single agents, and the second readout generated useful cell SAR for less potent examples. The objective was an agent for the treatment of osimertinib-resistant NSCLC, but we observed an excellent correlation between the EGFRL858R/T790M and EGFRL858R/T790M/C797S mutants in both enzyme and Ba/F3 cell assays, and we retained the EGFRL858R/T790M assays as our primary screens to provide consistency with historic project data. Ba/F3 EGFRL858R/T790M/C797S cell data was collected later for selected compounds, with this assay showing increased sensitivity to the allosteric inhibitors. Enzyme IC50 data correlated with the cellular data for cetuximab co-treatment but less so for compounds dosed as a single agent. We attributed this to asymmetric EGFR dimer formation in the cell, where one allosteric site is less accessible, and we observed an enzyme IC50 threshold of about 1 nM to achieve single-agent cell potency of <1 μM in the EGFRL858R/T790M assay.
JBJ-09-063 was sufficiently stable to deliver good efficacy upon oral dosing, but under certain conditions it underwent hydrolysis to the corresponding acid, presumably via intramolecular lactone formation. To develop allosteric EGFR inhibitors for osimertinib-resistant NSCLC, we wanted to avoid the potential chemical instability present in compounds that incorporated both the thiazole amide and phenol groups. The SAR around the thiazole was very tight, with little tolerance for even small substituents or replacements other than 2-pyridine. Another group reported an attempt to mitigate the hydrolysis risk by replacing the thiazole amide of EAI045 with a bicyclic quinazolinone group, but this led to a dramatic reduction in potency.(14) A patent application from the same group disclosed a series of isoindolinones in which the thiazole amide was replaced by indoles and benzimidazoles, with more promising activity data.(15)
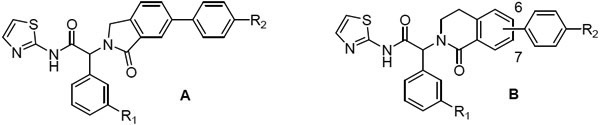
At the phenyl position, the 5-fluoro-2-phenol is preferred, but moderate potency is retained with an unsubstituted phenyl (6-7) or 3-fluorophenyl ring (5, 8, Table 2). As part of our efforts to develop chemically stable EGFR inhibitors, we removed the phenol moiety, and sought to improve the overall potency and PK profile through modifications elsewhere. Extensive exploration of the right-hand side of the isoindolinone series had culminated in the exquisite potency of JBJ-09-063, and we instead focused on replacements of the isoindolinone core. We acknowledged that new scaffolds might require re-optimization of the right-hand side, but initially incorporated N-methyl piperazine and N-methyl piperidine to deliver matched pairs with the isoindolinone series. We introduced the aryl amine at both the 6 and 7 positions of the dihydroisoquinolinone to determine if there was any preference for the positioning of this substituent in the channel revealed by our previous structural work.
Table 2.
EGFRL858R/T790M enzyme and Ba/F3 cell activity of isoindolinones (A) and dihydroisoquinolinones (B)
| |||||||
|---|---|---|---|---|---|---|---|
| Cpd | Core | R1 | R2 | Enzyme IC50 (nM) |
L858R/T790M Cell IC50 (μM) |
L858R/T790M Cetux co-dosing Cell IC50 (μM) |
L858R/T790M /C797S Cell IC50 (μM) |
| 6 | A | H | N-Me piperazine | 0.9 | 2.56 | 0.043 | - |
| 7 | A | H | N-Me piperidine | 0.9 | 1.00 | 0.020 | 0.179 |
| 8 | A | F | N-Me piperazine | 1.8 | 1.99 | 0.044 | - |
| 5 | A | F | N-Me piperidine | 0.2 | 1.07 | 0.020 | 0.152 |
| 9 | B | F | 6, N-Me piperazine | 1.1 | 3.66 | 0.801 | - |
| 10 | B | F | 6, N-Me piperidine | 2.2 | 1.99 | 0.246 | - |
| 11 | B | F | 7, N-Me piperazine | 6.8 | 1.60 | 0.126 | - |
| 12 | B | F | 7, N-Me piperidine | 3.2 | 1.47 | 0.099 | - |
We first expanded the lactam ring of the isoindolinone to generate dihydroisoquinolinones, incorporating the 3-F-phenyl substituent to give compounds 9-12 as the matched pairs of isoindolinones 5 and 8 (Table 2). Compounds were prepared according to Scheme 1. The lactones 13 were treated with POCl3 to give the corresponding chloroethyl acid chlorides, to which the 3-F phenylglycine ester was added to form the amides. The amides were cyclized to the bicyclic lactams 14 by treatment with potassium t-butoxide in refluxing dioxane. Ester hydrolysis and coupling of the acid with 2-aminothiazole gave the amides 15, with Suzuki couplings with the phenylpiperazine or phenylpiperidine boronate to generate the final compounds 9-12. Disappointingly, these compounds showed reduced enzyme and cellular potency relative to the corresponding isoindolinones regardless of the regiochemistry of the substituent (e.g. 10 or 12 v 5). Consistent with the isoindolinone pair 3 and JBJ-09-063, the piperidine example was usually more potent than the corresponding piperazine in both the enzyme and cellular assays.
Scheme 1 –

synthesis of dihydroisoquinolinones 9-12. Reagents and conditions: a) PCl5, POCl3, reflux; b) ethyl 2-amino-2-(3-fluorophenyl)acetate, iPr2NEt, THF, 0°C, then KOtBu, dioxane, 110°C, 23-35% over two steps; c) LiOH.H2O, THF, MeOH, 76-97%; d) 2-aminothiazole, iPr2NEt, HATU, DMF, 55°C, 38-73%; e) aryl amine pinacol boronate, Pd(dppf)Cl2, Na2CO3, 4:1 dioxane:water, 105°C, 49-59%.
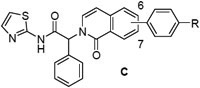
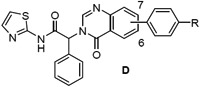
We next prepared examples 16-27 in the isoquinolinone, quinazolinone and phthalazinone series (Table 3). Alkylation of the bicyclic cores 28 with methyl 2-bromo-2-phenylacetate gave the bromoesters 29, followed by chemistry similar to that described for the dihydroisoquinolinone scaffold (Scheme 2).(13) As before, examples with a piperidine were more potent than the corresponding piperazines. For the isoquinolinone scaffold, we observed a slight preference for 6-substituents across the set of matched pairs (16 v 18 and 17 v 19). For the quinazolinones, substituents at the 6-position were clearly favored (22 v 20, 23 v 21). In the numbering system for these bicyclic series, the 6-position is on the same side of the core as the carbonyl for the quinazolinones, but the opposite side for the isoquinolinones and phthalazinones. Compound 23 was the most potent quinazolinone, with an IC50 of 0.2 nM, and a cell EC50 of 0.72 μM and 0.05 μM as a single agent in our Ba/F3 EGFRL858R/T790M and EGFRL858R/T790M/C797S assays respectively. The phthalazinones 24-27 were less potent across both the enzyme and cell assays.
Table 3.
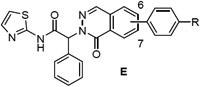
EGFRL858R/T790M enzyme and Ba/F3 cell activity of isoquinolinones (C), quinazolinones (D) and phthalazinones (E) 16-27
|
|
|
||||
|---|---|---|---|---|---|---|
| Cpd | Core | R | Enzyme IC50 (nM) |
L858R/T790M Cell IC50 (μM) |
L858R/T790M Cetux co-dosing Cell IC50 (μM) |
L858R/T790M /C797S Cell IC50 (μM) |
| 7 | 0.9 | 1.00 | 0.020 | 0.18 | ||
| 16 | C | 6, N-Me piperazine | 3.4 | 1.27 | 0.062 | - |
| 17 | C | 6, N-Me piperidine | 1.2 | 1.28 | 0.024 | 0.50 |
| 18 | C | 7, N-Me piperazine | 5.0 | 1.85 | 0.044 | - |
| 19 | C | 7, N-Me piperidine | 1.8 | 1.50 | 0.070 | 0.38 |
| 20 | D | 7, N-Me piperidine | 3.1 | 2.00 | 0.375 | - |
| 21 | D | 7, N-Me piperidine | 5.8 | 2.43 | 0.225 | 0.90 |
| 22 | D | 6, N-Me piperidine | 0.9 | 2.32 | 0.021 | - |
| 23 | D | 6, N-Me piperidine | 0.2 | 0.78 | 0.007 | 0.05 |
| 24 | E | 6, N-Me piperidine | 17.8 | 3.46 | 1.67 | - |
| 25 | E | 6, N-Me piperidine | 9.7 | 1.24 | 0.538 | 0.99 |
| 26 | E | 7, N-Me piperidine | 10.3 | 2.16 | 0.380 | - |
| 27 | E | 7, N-Me piperidine | 4.0 | 1.54 | 0.207 | 0.44 |
Scheme 2 –

synthesis of compounds 16-27. Reagents and conditions: a) methyl 2-bromo-2-phenylacetate, Cs2CO3, DMF, 50°C, 55-79%; b) LiOH, H2O, MeOH, THF, 27-99%; c) 2-aminothiazole, iPr2NEt, HATU, DMF, 60°C, 56-85%; d) aryl amine pinacol boronate, Pd(dppf)Cl2, Na2CO3, 4:1 dioxane:water, 100°C, 24-70%.
Examples from both the isoquinolinone and quinazolinone series showed promising activity, comparable and in some cases better than their isoindolinone matched pairs (6, 7, Table 2). We were conscious of the high lipophilicity of the series and prepared examples with an additional nitrogen in the core (31-32, Table 4). Inspired by the dramatic potency enhancement achieved with a fluorine scanning approach on a similar scaffold in a series of Bruton’s tyrosine kinase (BTK) inhibitors,(16) we also introduced fluorine at different positions of the quinazolinone (33-35). We prioritized compounds with the phenyl piperidine substituent at the 6-position of these scaffolds, as this regiochemistry had been preferred in the initial quinazolinone examples.
Table 4.
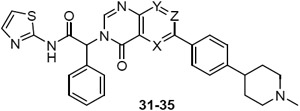
EGFRL858R/T790M enzyme and cell potency of azaquinazolinones 31-32 and F-quinazolinones 33-35
| |||||||
|---|---|---|---|---|---|---|---|
| Cpd | X | Y | Z | Enzyme IC50 (nM) |
L858R/T790M Cell IC50 (μM) |
L858R/T790M Cetux co-dosing Cell IC50 (μM) |
L858R/T790M /C797S Cell IC50 (μM) |
| 7 | 0.9 | 1.00 | 0.020 | 0.18 | |||
| 31 | N | CH | CH | 0.6 | 1.71 | 0.016 | 0.09 |
| 32 | CH | N | CH | 1.9 | 1.14 | 0.023 | 0.31 |
| 33 | CH | CF | CH | 0.6 | 0.81 | 0.010 | 0.15 |
| 34 | CF | CH | CH | 0.1 | 0.32 | 0.003 | 0.02 |
| 35 | CH | CH | CF | 1.4 | 3.00 | 0.073 | 0.28 |
These series could not be readily accessed by the synthetic route of Scheme 2, as the additional nitrogen atom or fluorine substituent appeared to render the alkylated cores unstable to the basic conditions used in the ester hydrolysis step. Azaquinazolinones 31 and 32 were instead prepared from 6-bromopyrido[3,2-d]pyrimidin-4(3H)-one and 6-bromopyrido[2,3-d]pyrimidin-4(3H)-one respectively by alkylation with methyl 2-bromo-2-phenylacetate to give the esters 37 (Scheme 3). This was followed by Suzuki couplings to install the phenyl piperidine. This reaction could be stopped to give the corresponding esters, but prolonged heating resulted in hydrolysis to the acids 38, which were coupled with 2-aminothiazole to generate the target compounds 31 and 32.
Scheme 3 –

synthesis of compounds 31-35. Reagents and conditions: a) methyl 2-bromo-2-phenylacetate, Cs2CO3, DMF, 50°C, 24-60%; b) 1-methyl-4-(4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)phenyl) piperidine, Pd(dppf)Cl2. Na2CO3, 4:1 dioxan:water, 100°C, 11-59%; c) 2-aminothiazole, iPr2NEt, HATU, DMF, 60°C, 9-48%; d) LiOH, water, 100°C, 88%; e) methyl 2-amino-2-phenylacetate, iPr2NEt, HATU, DMF, 72%; f) (EtO)3CH, reflux, 79%; g) TFA, DCM; h) 42, HATU, DBU, CH3CN, 7%.
8-F-Quinazolinone 33 was prepared by similar chemistry, from the commercially available starting material 6-bromo-8-fluoroquinazolin-4(3H)-one (36, X=CH, Y = CF). For 34, the nitrile of 6-amino-3-bromo-2-fluorobenzonitrile 39 was hydrolyzed to the acid, followed by an amide coupling with methyl 2-amino-2-phenylacetate to give 40. Cyclization to the 5-F quinazolinone was achieved by heating 40 in neat triethylorthoformate, with the usual Suzuki and amide couplings to complete the synthesis.
The 7-F quinazolinone 35 was synthesized by a different route (Scheme 3). Amino amide 42 was prepared from Boc-protected 2-aminophenylacetic acid 41. A Suzuki coupling installed the phenyl piperidine onto 6-bromo-7-fluoroquinazolin-4(3H)-one 43 to give 44, and this was coupled with amine 42 via HATU activation(17) to give 35.
The 5-F quinazolinone 34 was the most potent of this set of compounds and showed a significant improvement in potency relative to the corresponding isoindolinone 7, with an EGFRL858R/T790M IC50 of 0.1 nM, and cell EC50s of 0.32 μM and 0.02 μM in our Ba/F3 EGFRL858R/T790M and EGFRL858R/T790M/C797S assays respectively (Table 4).
We obtained a co-crystal structure of 34 binding to EGFRT790M/V948R. The V948 residue is distant from the binding site and was introduced to prevent EGFR asymmetric dimer formation during crystallization. The structure confirmed that the quinazolinone series binds in essentially the same manner as the isoindolinones,(10, 12) though lacking the hydrogen bond of the phenolic OH to the backbone of F856. The amide NH forms a key H-bond interaction with D855, and the quinazolinone core positions the phenyl piperidine substituent into the groove adjacent to the α-C-helix to enable the piperidine nitrogen to interact with acidic residue E758.
We obtained in vivo rat PK (1 mg/kg IV, 10 mg/kg PO) for 5-F-quinazolinone 34. It was found to have moderate IV Cl (30 mL/min/kg), T1/2 of 3.5 hours and oral bioavailability of 24%. Compound 34 was profiled in an H1975 tumor efficacy model (EGFRL858R/T790M) and achieved tumor regression upon oral dosing at both the 25 mg/kg and 50 mg/kg doses administered once daily, with slower re-growth post-treatment at the higher dose. The compound was well-tolerated at both doses.
In conclusion, we pursued a scaffold-hopping approach to identify new series of allosteric mutant-selective EGFR inhibitors, with good activity against the EGFRL858R/T790M and EGFRL858R/T790M/C797S mutants. The 5-fluoroquinazolinone scaffold delivered a compound with sufficient potency to achieve good in vivo efficacy without the need for the fluorophenol group, mitigating the potential chemical instability risk. Our further efforts to develop an agent for the treatment of osimertinib-resistant NSCLC will be reported in due course.
Supplementary Material
Fig. 2 –
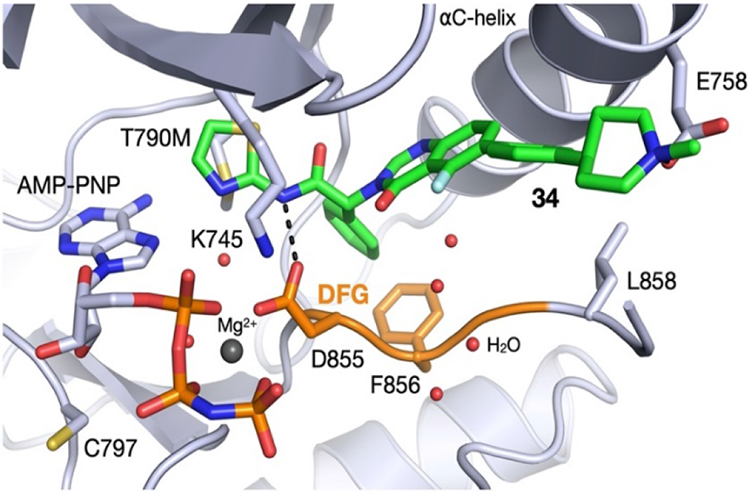
Co-crystal structure of EGFRT790M/V948R bound to 34 and AMP-PNP (2.3 Å, PDB 7LTX)
Fig. 3 –
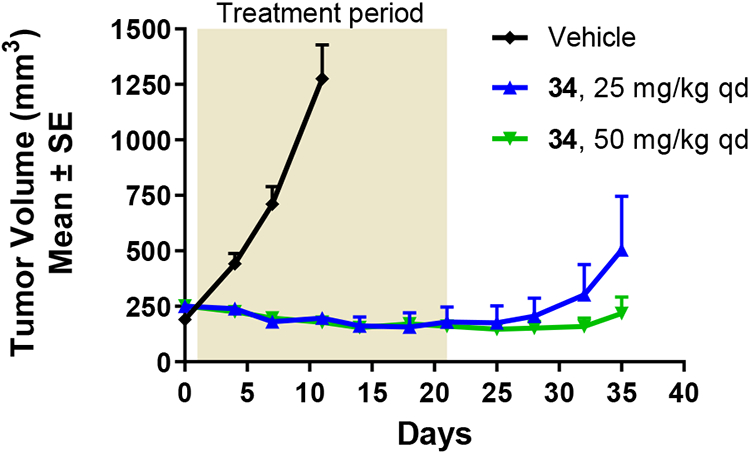
H1975 efficacy data for compound 34.
Acknowledgments
We thank Takeda Pharmaceuticals for funding support and scientific discussions. The rat PK study was conducted at WuXi AppTec. T.S.B is supported by a Ruth L. Kirschstein National Research Service Award (1F32CA247198-01). The crystallography work is based upon research conducted at the Northeastern Collaborative Access Team beamlines (P30 GM124165, P41 GM103403) utilizing resources of the Advanced Photon Source at the Argonne National Laboratory (DE-AC02-06CH11357).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Supplementary data
Supplementary data to this article can be found online at:
Declaration of interests
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
References and notes
- 1.Roskoski R Jr. Properties of FDA-approved small molecule protein kinase inhibitors: A 2021 update. Pharmacol Res. 2021;165:105463. doi: 10.1016/j.phrs.2021.105463. [DOI] [PubMed] [Google Scholar]
- 2.Xie Z, Yang X, Duan Y, Han J, Liao C. Small-Molecule Kinase Inhibitors for the Treatment of Nononcologic Diseases. J Med Chem. 2021;64(3):1283–345. doi: 10.1021/acs.jmedchem.0c01511. [DOI] [PubMed] [Google Scholar]
- 3.Ayati A, Moghimi S, Salarinejad S, Safavi M, Pouramiri B, Foroumadi A. A review on progression of epidermal growth factor receptor (EGFR) inhibitors as an efficient approach in cancer targeted therapy. Bioorg Chem. 2020;99:103811. doi: 10.1016/j.bioorg.2020.103811. [DOI] [PubMed] [Google Scholar]
- 4.Yun CH, Mengwasser KE, Toms AV, Woo MS, Greulich H, Wong KK, et al. The T790M mutation in EGFR kinase causes drug resistance by increasing the affinity for ATP. Proc Natl Acad Sci U S A. 2008;105(6):2070–5. doi: 10.1073/pnas.0709662105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ward RA, Anderton MJ, Ashton S, Bethel PA, Box M, Butterworth S, et al. Structure- and reactivity-based development of covalent inhibitors of the activating and gatekeeper mutant forms of the epidermal growth factor receptor (EGFR). J Med Chem. 2013;56(17):7025–48. doi: 10.1021/jm400822z. [DOI] [PubMed] [Google Scholar]
- 6.Finlay MR, Anderton M, Ashton S, Ballard P, Bethel PA, Box MR, et al. Discovery of a potent and selective EGFR inhibitor (AZD9291) of both sensitizing and T790M resistance mutations that spares the wild type form of the receptor. J Med Chem. 2014;57(20):8249–67. doi: 10.1021/jm500973a. [DOI] [PubMed] [Google Scholar]
- 7.Schmid S, Li JJN, Leighl NB. Mechanisms of osimertinib resistance and emerging treatment options. Lung Cancer. 2020;147:123–9. doi: 10.1016/j.lungcan.2020.07.014. [DOI] [PubMed] [Google Scholar]
- 8.Lu X, Smaill JB, Ding K. New Promise and Opportunities for Allosteric Kinase Inhibitors. Angew Chem Int Ed Engl. 2020;59(33):13764–76. doi: 10.1002/anie.201914525. [DOI] [PubMed] [Google Scholar]
- 9.Jia Y, Yun CH, Park E, Ercan D, Manuia M, Juarez J, et al. Overcoming EGFR(T790M) and EGFR(C797S) resistance with mutant-selective allosteric inhibitors. Nature. 2016;534(7605):129–32. doi: 10.1038/nature17960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.To C, Jang J, Chen T, Park E, Mushajiang M, De Clercq DJH, et al. Single and Dual Targeting of Mutant EGFR with an Allosteric Inhibitor. Cancer Discov. 2019;9(7):926–43. doi: 10.1158/2159-8290.CD-18-0903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Niggenaber J, Heyden L, Grabe T, Muller MP, Lategahn J, Rauh D. Complex Crystal Structures of EGFR with Third-Generation Kinase Inhibitors and Simultaneously Bound Allosteric Ligands. ACS Med Chem Lett. 2020;11(12):2484–90. doi: 10.1021/acsmedchemlett.0c00472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.To C, et al. , Molecular basis for cooperative binding and synergy of ATP-site and allosteric EGFR inhibitors. Nature Cancer. 2022, accepted for publication. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gray NS, Scott DA, Gero T, Eck M, Heppner D, Beyett T, et al. Nitrogen containing heterocycles as allosteric EGFR inhibitors and preparation and methods of use thereof. PCT Int. Appl. WO2020257632.
- 14.Lee S, Kim J, Duggirala KB, Go A, Shin I, Cho BC, et al. Allosteric Inhibitor TREA-0236 Containing Non-hydrolysable Quinazoline-4-one for EGFR T790M/C797S Mutants Inhibition. Bulletin of the Korean Chemical Society. 2018;39(7):895–8. doi: 10.1002/bkcs.11491. [DOI] [Google Scholar]
- 15.Lee K, Choi G, Lee S, Kim J, Cho B, Park CW, Yun J. Isoindolin-1-one derivative, method for preparing same, and pharmaceutical composition comprising same as effective component for preventing or treating cancer. PCT Int. Appl WO2020036386.
- 16.Lou Y, Sweeney ZK, Kuglstatter A, Davis D, Goldstein DM, Han X, et al. Finding the perfect spot for fluorine: improving potency up to 40-fold during a rational fluorine scan of a Bruton's Tyrosine Kinase (BTK) inhibitor scaffold. Bioorg Med Chem Lett. 2015;25(2):367–71. doi: 10.1016/j.bmcl.2014.11.030. [DOI] [PubMed] [Google Scholar]
- 17.Xiao Z, Yang MG, Li P, Carter PH. Synthesis of 3-substituted-4(3H)-quinazolinones via HATU-mediated coupling of 4-hydroxyquinazolines with amines. Org Lett. 2009; 11(6):1421–4. doi: 10.1021/o1802946p. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.