

Abstract
A series of tetrahydropyrimidinyl-substituted benzimidazoles attached to various aliphatic or aromatic residues via phenoxymethylene were synthesised to investigate their antibacterial activities against selected Gram-positive and Gram-negative bacteria. The influence of the type of substituent at the C-3 and C-4 positions of the phenoxymethylene linker on the antibacterial activity was observed, showing that the aromatic moiety improved the antibacterial potency. Of all the evaluated compounds, benzoyl-substituted benzimidazole derivative 15a was the most active compound, particularly against the Gram-negative pathogens E. coli (MIC = 1 μg mL−1) and M. catarrhalis (MIC = 2 μg mL−1). Compound 15a also exhibited the most promising antibacterial activity against sensitive and resistant strains of S. pyogenes (MIC = 2 μg mL−1). Significant stabilization effects and positive induced CD bands strongly support the binding of the most biologically active benzimidazoles inside the minor grooves of AT-rich DNA, in line with docking studies. The predicted physico-chemical and ADME properties lie within drug-like space except for low membrane permeability, which needs further optimization. Our findings encourage further development of novel structurally related 5(6)-tetrahydropyrimidinyl substituted benzimidazoles in order to optimize their antibacterial effect against common respiratory pathogens.
The most active benzimidazole 15a against E. coli, M. catarrhalis and both sensitive and resistant strains of S. pyogenes showed preference toward AT-rich sites and minor groove binding mode, which was in line with docking studies.
1. Introduction
Bacterial infections are an increasing public health problem in hospitals and in the community, which accounts for the main cause of morbidity and mortality worldwide. The value of antibiotics, as effective treatment against bacterial infections, is immeasurable for human health.1,2 However, the increasing prevalence of hospital and community-acquired infections caused by multidrug-resistant (MDR) bacterial pathogens limits the usefulness of currently available antibiotic therapies.3–5 Antimicrobial resistance among Gram-positive bacteria, particularly Staphylococcus aureus, Enterococcus faecium, and Streptococcus pneumoniae, poses a serious public health threat.6 Among the Gram-negative bacterial pathogens, Escherichia coli, Klebsiella pneumoniae, Pseudomonas aeruginosa, and Acinetobacter baumannii are the most common to develop antibiotic resistance.7,8 The low permeability of the outer membrane in Gram-negative bacteria, the presence of efflux pumps, the production of degrading enzymes, and the modification of drug targets are the main mechanisms used by bacteria to resist the action of antibiotics.9Escherichia coli accounts for nearly 80% of community-acquired and 50% of hospital-acquired urinary tract infections (UTI).10 After being considered a harmless inhabitant of the upper respiratory tract for a long time, in recent years, Moraxella catarrhalis has been established as an emerging unencapsulated Gram-negative mucosal pathogen. Moraxella catarrhalis is a human-restricted pathogen responsible for respiratory tract infections such as childhood otitis media (OM) and exacerbations of chronic obstructive pulmonary disease (COPD) in adults.11–13 It is the third most common cause of OM after Haemophilus influenzae and Streptococcus pneumoniae, is responsible for up to 20% of the cases and is the second most common cause of COPD, making it one of the primary medical issues related to bacteria.14 To date, most therapeutic guidelines recommend antibiotics for the treatment of OM cases. However, excessive and improper use of antibiotics led to the development of antibacterial resistance worldwide during the last few decades, suggesting that the incidence of these infections may continue to rise.2 Antimicrobial resistance is genetically determined and can arise both from mutations in the pre-existing bacterial genome and from the uptake of extra-chromosomal genetic elements via horizontal gene transfer.15,16
Among the important pharmacophores, the azole framework is still considered quite a significant structure for design of compounds with antibacterial,17–20 antitumor,21,22 antiviral,23 antitrypanosomal17,24,25 and anti-inflammatory26 activities. From benzazole compounds, benzimidazoles and their derivatives have demonstrated a wide array of pharmacological activities depending on the substitution pattern of the benzimidazole ring.27–29 Structural modifications at the 2, 5 and 6 positions on the benzimidazole scaffold have provided a wide spectrum of compound classes as well as the scope for the enhancement of activity.23,30,31
Based on the comparative analysis of structures of various substituted benzimidazole derivatives as antimicrobial agents, it was noted that a bulky aromatic or heteroaromatic moiety and electron-rich group is required at either N-1 or C-2 through a linker of appropriate length.32,33 Benzimidazoles are purinomimetics that have the potential to readily interact with the various biopolymers found in living systems. As competitive inhibitors, benzimidazoles can replace purine, which blocks the biosynthesis of nucleic acids and proteins in the bacterial cell wall, thereby inhibiting the growth of bacteria.34,35
Importantly, benzimidazole derivatives have been shown to interact with DNA and were also used in the medicinal field where the primary target was found to be DNA and DNA associated processes.25,36–43
From the series of Hoechst dye analogues, I, Hoechst 33258, (Fig. 1) has shown notable antibacterial activity against methicillin resistant S. aureus (MRSA) and vancomycin resistant enterococci (VRE).44 By simply changing the configuration of the binding of the two benzimidazole units from head to tail to symmetric tail to tail, ridinilazole (II, SMT-19969) has been developed. Ridinilazole has been recently approved for the treatment of Clostridium difficile infection and prevention of recurrent disease, and found a propensity to bind only AT-rich DNA.45–48 Moreover, from dicationic bis-benzimidazoles, III, DB-325 with two benzimidazole units connected by a symmetric diaryl spacer was found to bind to the minor groove of DNA and showed potent anti-MRSA and anti-VRE activity.49 Triazole-bearing 2-aromatic benzimidazole amidines IV and V (Table 1) showed binding affinities to ctDNA, with the non-substituted amidine IV inhibiting the growth of Gram-negative extended spectrum beta-lactamase (ESBL) producing E. coli, and isopropyl-substituted analogue V showing strong inhibitory activity against Gram-positive MRSA strain.17
Fig. 1. Structures of some benzimidazole-based compounds with antibacterial activity.

In vitro antibacterial activity of novel benzimidazoles 12a–19a, 12b–18b, 12c, 13c, 15c–19c, 21a, 23a–24a and benzothiazole derivatives 21b, 23b and 22c–24c against Gram-positive and Gram-negative bacteria.

| |||||||
|---|---|---|---|---|---|---|---|
| Compd | X | R1 | R2 | S. aureus | E. faecalis | MICa (μg mL−1) | E. coli AcrAB− |
| M. catarrhalis | |||||||
| 12a | NH | H |

|
>128 | >128 | 8 | 16 |
| 12b | NH | F | >128 | >128 | 32 | 32 | |
| 12c | NH | OCH3 | >128 | >128 | 8 | 16 | |
| 13a | NH | H |
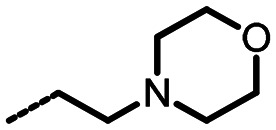
|
>128 | >128 | 32 | 64 |
| 13b | NH | F | >128 | >128 | 32 | 64 | |
| 13c | NH | OCH3 | >128 | >128 | 16 | 64 | |
| 14a | NH | H |

|
>128 | >128 | 32 | 32 |
| 14b | NH | F | >128 | >128 | 128 | 64 | |
| 15a | NH | H |

|
32 | 32 | 2 | 1 |
| 15b | NH | F | 32 | 32 | 2 | 4 | |
| 15c | NH | OCH3 | 64 | 64 | 2 | 4 | |
| 16a | NH | H |

|
128 | >128 | 4 | 16 |
| 16b | NH | F | 128 | >128 | 4 | 32 | |
| 16c | NH | OCH3 | 128 | >128 | 4 | 16 | |
| 17a | NH | H |

|
>128 | >128 | 2 | 16 |
| 17b | NH | F | >128 | >128 | 8 | 32 | |
| 17c | NH | OCH3 | >128 | >128 | 8 | 64 | |
| 18a | NH | H |

|
128 | 128 | 8 | 16 |
| 18b | NH | F | 128 | >128 | 16 | 32 | |
| 18c | NH | OCH3 | >128 | 64 | 8 | 16 | |
| 19a | NH | H |

|
>128 | >128 | 32 | 32 |
| 19c | NH | OCH3 | >128 | >128 | 128 | 32 | |
| 21a | S | H |

|
128 | >128 | 8 | 32 |
| 21b | S | F | 128 | 128 | 4 | 16 | |
| 22c | S | OCH3 |

|
>128 | >128 | 32 | 128 |
| 23a | S | H |

|
>128 | >128 | 32 | >128 |
| 23b | S | F | 128 | >128 | >128 | >128 | |
| 23c | S | OCH3 | >128 | >128 | 32 | 128 | |
| 24a | S | H |

|
128 | 128 | 4 | 32 |
| 24c | S | OCH3 | >128 | 128 | 16 | 64 | |
| AZM b | 2 | 0.125 | 8 | 0.5 | |||
Minimal inhibitory concentrations.
Azithromycin.
Based on the above-mentioned and in continuation of our recent work on the development of aromatic benzimidazole amidines as antibacterial agents, we have synthesised new chemical entities containing a positively charged 3,4,5,6-tetrahydropyrimidine moiety at the 5(6)-position of the benzimidazole core and different aliphatic and aromatic units at the phenoxymethylene attached to C-2 of the benzimidazole. The activities of novel 5(6)-amidino benzimidazoles 12a–19a, 12b–18b, 12c, 13c and 15c–19c against Gram-positive and Gram-negative bacteria were evaluated. Given the importance of benzothiazoles in the area of drug discovery50–53 and to evaluate their influence on antibacterial activity compared to benzimidazole pharmacophores, selected benzothiazole analogues of benzimidazoles with the best activity were additionally synthesised. Compounds with the most potent antibacterial activity were further investigated in terms of their DNA binding affinities by UV-vis and CD spectroscopy, as well as thermal denaturation experiments to elucidate the type of interaction with DNA.
2. Results and discussion
2.1. Chemistry
A focused library of 22 amidino-substituted benzimidazole derivatives 12a–19a, 12b–18b, 12c, 13c and 15c–19c was synthesised by cyclocondensation from 4-(tetrahydropyrimidin-2-yl)-benzene-1,2-diamine 11 and corresponding benzaldehyde intermediates as shown in Scheme 2. The 4-(tetrahydropyrimidin-2-yl)-benzene-1,2-diamine 11 was prepared by the acidic Pinner reaction from the corresponding nitrile-substituted precursor as previously reported in the literature.54 Key intermediates for the synthesis of 1,2,3-triazole-substituted benzimidazole derivatives (8a–8c, 9a–9c and 10a–10c) were prepared by regioselective copper(i) catalysed cycloaddition from O-propargylated benzaldehydes (2a–2c) and corresponding azides (Scheme 1).
Scheme 2. Reagents and conditions: (i) 4-amidinobenzene-1,2-diamine 11, NaHSO3, EtOH, reflux, 8 h, corresponding amidine free bases, EtOH/HCl(g), 24 h, rt.

Scheme 1. Reagents and conditions: (i) RCl/RBr, CH3CN, K2CO3, reflux, 8 h; (ii) CuI, TMSN3, DMF : H2O = 9 : 1, reflux, 6 h; (iii) benzyl chloride, NaN3, Et3N, 30 min, rt, Cu(OAc)2, t-BuOH : H2O = 1 : 1, 24 h; (iv) 4-(2-azidoethyl)morpholine, Cu(OAc)2, MeOH, reflux, 24 h.

With the aim of assessing the influence of the type of right-hand unit on antibacterial activity, diethylaminoethyl (12a–12c), ethynylmorpholine (13a–13c), morpholinoyl (14a–14b), benzoyl (15a–15c) and picolyl (16a–16c) substituents were introduced to the phenoxymethylene linker of the amidino-substituted benzimidazoles through O-alkylation of the 4-hydroxybenzaldehydes 1a–1c with the corresponding halides and K2CO3 which afforded the benzaldehyde precursors 2a–7a, 2b–7b and 2c–7c (Scheme 1), which were converted to target 2-aryl-5(6)-(tetrahydropyrimidin-2-yl)-substituted benzimidazole (12a–16a, 12b–13b, 15b–16b and 12c–16c) hybrids with NaHSO3 as an oxidative reagent (Scheme 2). To assess the influence of the triazole moiety on the antibacterial activity, benzaldehyde precursors with non-substituted triazole 8a–8c were prepared with copper(i) iodide and trimethylsilylazide (TMSN3), while 1-benzyl-1,2,3-triazole benzaldehydes 9a–9c were synthesised in good to excellent yields (66–97%) in a one-pot click reaction using Cu(ii) acetate, as a catalyst. Additionally, 1,3-dipolar cycloaddition of 2-(4-morpholino)ethylazide and terminal alkynes (2a–2c) yielded the corresponding 1-ethylmorpholino-1,2,3-triazole benzaldehyde precursors 10a–10c. Condensation of 4-(1,2,3-triazol-1-yl)benzaldehyde derivatives 8a–8c, 9a–9c and 10a–10c with 4-(tetrahydropyrimidin-2-yl)-benzene-1,2-diamine 11 and NaHSO3 afforded the target amidino-substituted benzimidazoles 17a–17c, 18a–18c, 19a and 19c.
To evaluate the influence of benzothiazole bioisosteres on antibacterial activity, a series of target 6-(tetrahydropyrimidinyl)benzothiazole analogues 21a, 21b, 22c, 23a–23c, 24a, and 24c were synthesised according to the procedure depicted in Scheme 3.
Scheme 3. Reagents and conditions: (i) HOAc, reflux, 3 h, then H2O/NaOH, pH 12, corresponding amidine free bases, EtOH/HCl(g), 24 h, rt.

Amidino-substituted 2-aminothiophenole 20 was prepared from 6-cyanobenzothiazole by the Pinner method, as previously reported.54 Condensation of amidino-substituted 2-aminothiophenole 20 with corresponding benzaldehyde precursors (6a–9a, 6b–9b, 6c–9c) was carried out in acetic acid, followed by a simple acid–base reaction step to afford the targeted amidino-substituted benzothiazole hydrochlorides.
2.2. Antibacterial profiling
The synthesised 2-substituted benzimidazoles 12a–19a, 12b–18b, 12c, 13c, 15c–19c and benzothiazole analogues 21a, 21b, 22c, 23a–23c, 24a, and 24c were profiled against Gram-positive bacteria including Staphylococcus aureus (ATCC 29213) and Enterococcus faecalis (ATCC29212), and Gram-negative bacteria including Moraxella catarrhalis (ATCC 23246) and hypersensitive Escherichia coli AcrAB− strain. This strain of E. coli has inactivated AcrAB–TolC multidrug-resistance efflux pump, responsible for active extrusion of metabolic waste, toxic compounds, dyes and a wide range of antibiotics from the bacterial cell.55 The antibiotic azithromycin (AZM) was used as a standard control. The minimal inhibitory concentrations (MICs) of the newly synthesised compounds and azithromycin against the above-mentioned bacterial strains are presented in Tables 1 and 2.
In vitro antibacterial activity of selected compounds against Gram-positive streptococci strains.

| |||||||
|---|---|---|---|---|---|---|---|
| Compd | R1 | R2 | X | MICa (μg mL−1) | |||
| S. pneumoniae B0652 (eryS) | S. pneumoniae B0326 (M) | S. pyogenes B0542 (eryS) | S. pyogenes B0545 (M) | ||||
| 12a | H |

|
NH | 128 | >128 | 16 | 8 |
| 12c | OCH3 | NH | 32 | 128 | 16 | 8 | |
| 15a | H |

|
NH | 16 | 32 | 2 | 2 |
| 15b | F | NH | 64 | 64 | 4 | 4 | |
| 15c | OCH3 | NH | 32 | 64 | 2 | 4 | |
| 16a | H |

|
NH | 32 | 64 | 4 | 4 |
| 16c | OCH3 | NH | 64 | 64 | 4 | 4 | |
| 17a | H |

|
NH | 8 | 32 | 4 | 4 |
| 18a | H |

|
NH | 128 | 128 | 8 | 8 |
| 18c | OCH3 | NH | >128 | 128 | 16 | 16 | |
| 21b | F |

|
S | 32 | 128 | 32 | 128 |
| AZM b | 0.125 | 16 | 0.125 | 8 | |||
| Ampicillin | 0.25 | 0.25 | 0.25 | 0.25 | |||
Minimal inhibitory concentrations.
Azithromycin.
The results showed that the majority of tested benzimidazole compounds do not exhibit activity against Gram-positive bacteria S. aureus and E. faecalis (MICs > 128 μg mL−1), except for benzoyl-substituted benzimidazole derivatives 15a–15c, which displayed MICs of 32 and 64 μg mL−1 against both Gram-positive strains. Interestingly, all tested compounds showed growth-inhibition of Gram-negative bacteria used. Benzimidazoles 13a–13c, 14a, 14b, 19a and 19b with a morpholine moiety displayed slight activity against M. catarrhalis and E. coli (MICs = 16–128 μg mL−1). Another aliphatic diethylaminoethoxy unit in 12a–12c also decreased the growth inhibition of Gram-negative bacteria (Fig. 2). Conversely, the aromatic moiety in benzimidazoles 15a–15c, 16a–16c, 17a–17c and 18a–18c enhanced the activity, particularly towards M. catarrhalis. Among these compounds, the most active benzimidazoles containing benzoyl (15a–15c), pyridine (16a–16c) and unsubstituted triazole (17a) exhibited Moraxella catarrhalis inhibition with MICs in the range of 2–4 μg mL−1. These activities were comparable to that of the reference drug AZM. Moreover, the most prominent growth-inhibition of efflux deficient E. coli was observed for compounds 15a–15c (MICs = 1–4 μg mL−1). The results showed that benzothiazole compounds exhibited a lack of activity against Gram-positive bacteria S. aureus and E. faecalis (MICs ≥ 128 μg mL−1). All tested benzothiazole analogues showed growth-inhibition of Gram-negative bacteria, except for the benzothiazole with unsubstituted triazole 23b. The most active benzothiazoles containing benzoyl (21a and 21b) exhibited activity against M. catarrhalis with MICs in the range of 4–8 μg mL−1 and E. coli with MICs in the range of 16–32 μg mL−1, which is in accord with the results of benzimidazole analogues 15a and 15b.
Fig. 2. Insights into the SAR for antibacterial activity.

Comparing the impact of the substituent at the phenoxymethylene linker to the benzimidazole core on the antibacterial effect, it can be observed that 3-unsubstituted phenoxymethylene exhibited a somewhat better potency compared to the corresponding 3-fluoro- and 3-methoxy-substituted analogues (Fig. 2). Moreover, the obtained results revealed that the type of N-heterocycle scaffold significantly influenced the antibacterial activity. In general, benzothiazole derivatives 21a, 21b, 22c, 23a–23c, 24a and 24c displayed lower activity in comparison to their benzimidazole analogues 15a, 15b, 16c, 17a–17c, 18a and 18c (Fig. 2).
Many processes of drug disposition depend on the capability of the drug to cross membranes, hence the lipophilic character of compounds is an important parameter related to membrane permeation in the biological system.12,56 Therefore, the octanol/water partition coefficients log P being a measure of hydrophobicity/lipophilicity were calculated (Table 6). The relationship between the lipophilicity of the evaluated compounds and the antibacterial activity against Gram-negative bacteria was observed. Benzimidazoles 13a–13c, 14a, 14b, 19a and 19c with a morpholine moiety had log P < 2.5 and the lowest activity against Gram-negative bacteria, while benzimidazoles 15a–15c with benzoyl, which exhibited the best antibacterial activity against M. catarrhalis and E. coli, had higher lipophilic properties with log P values ranging from 3.10–3.46. Generally, benzothiazoles demonstrated higher log P values compared to those of corresponding benzimidazole analogues. In line with this, benzothiazoles 21a, 21b and 24a with the best antibacterial activity against M. catarrhalis had log P values ranging from 3.63–4.23.
Calculated structural, physicochemical and ADME properties.
| Compd | HBDa | HBAb | TPSAc | Molecular weight | log P | log D (pH = 7.40) | pKa | Solubility | CYP3A4d | PPBe |
|---|---|---|---|---|---|---|---|---|---|---|
| 12a | 2 | 6 | 65.54 | 391.51 | 3.22 | −0.58 | 14.07 | Soluble | 0.63 | 93.68 |
| 12b | 2 | 6 | 65.54 | 409.5 | 3.26 | −0.49 | 14.04 | Soluble | 0.75 | 92.91 |
| 12c | 2 | 7 | 74.77 | 421.54 | 2.86 | −0.95 | 14.06 | Soluble | 0.79 | 91.49 |
| 13a | 2 | 7 | 74.77 | 405.49 | 2.18 | 0.14 | 14.07 | Soluble | 0.59 | 89.31 |
| 13b | 2 | 7 | 74.77 | 423.48 | 2.19 | 0.14 | 14.04 | Soluble | 0.64 | 87.88 |
| 13c | 2 | 8 | 84.00 | 435.52 | 1.85 | −0.2 | 14.06 | Soluble | 0.61 | 86.74 |
| 14a | 2 | 8 | 91.84 | 419.48 | 1.46 | −0.55 | 14.07 | Soluble | 0.48 | 93.14 |
| 14b | 2 | 8 | 91.84 | 437.47 | 1.60 | −0.42 | 14.04 | Soluble | 0.48 | 94.77 |
| 15a | 2 | 6 | 79.37 | 410.47 | 3.26 | 1.25 | 14.07 | Insoluble | 0.51 | 99.46 |
| 15b | 2 | 6 | 79.37 | 428.46 | 3.42 | 1.40 | 14.04 | Insoluble | 0.51 | 98.71 |
| 15c | 2 | 7 | 88.6 | 440.49 | 3.10 | 1.08 | 14.06 | Insoluble | 0.65 | 98.72 |
| 16a | 2 | 6 | 75.19 | 383.45 | 2.58 | 0.57 | 14.07 | Soluble | 0.47 | 97.55 |
| 16b | 2 | 6 | 75.19 | 401.44 | 2.63 | 0.61 | 14.04 | Soluble | 0.66 | 96.96 |
| 16c | 2 | 7 | 84.42 | 413.47 | 2.67 | 0.66 | 14.06 | Soluble | 0.57 | 97.24 |
| 17a | 3 | 8 | 103.87 | 373.41 | 1.90 | −0.15 | 14.09 | Soluble | 0.17 | 91.52 |
| 17b | 3 | 8 | 103.87 | 391.4 | 2.06 | 0.00 | 14.06 | Soluble | 0.25 | 91.89 |
| 17c | 3 | 9 | 113.1 | 403.44 | 1.75 | −0.3 | 14.08 | Soluble | 0.4 | 91.07 |
| 18a | 2 | 8 | 93.01 | 463.53 | 2.88 | 0.87 | 14.07 | Soluble | 0.71 | 99.43 |
| 18b | 2 | 8 | 93.01 | 481.52 | 2.96 | 0.94 | 14.04 | Soluble | 0.68 | 98.86 |
| 18c | 2 | 9 | 102.24 | 493.56 | 3.08 | 1.07 | 14.07 | Soluble | 0.76 | 99.21 |
| 19a | 2 | 10 | 105.48 | 486.57 | 1.59 | −0.46 | 14.07 | Soluble | 0.53 | 91.27 |
| 19c | 2 | 11 | 114.71 | 516.6 | 1.58 | −0.47 | 14.07 | Soluble | 0.58 | 90.54 |
| 21a | 1 | 5 | 91.82 | 427.52 | 4.23 | 2.23 | 12.78 | Insoluble | 0.64 | 99.53 |
| 21b | 1 | 5 | 91.82 | 445.51 | 4.18 | 2.18 | 12.77 | Insoluble | 0.7 | 99.32 |
| 22c | 1 | 6 | 96.87 | 430.52 | 3.63 | 1.63 | 12.78 | Soluble | 0.67 | 97.51 |
| 23a | 2 | 7 | 116.32 | 390.46 | 2.75 | 0.71 | 12.81 | Soluble | 0.24 | 95.7 |
| 23b | 2 | 7 | 116.32 | 408.45 | 2.70 | 0.66 | 12.79 | Soluble | 0.37 | 95.32 |
| 23c | 2 | 8 | 125.55 | 420.49 | 2.71 | 0.68 | 12.8 | Soluble | 0.45 | 95.73 |
| 24a | 1 | 7 | 105.46 | 480.59 | 3.63 | 1.63 | 12.78 | Soluble | 0.69 | 99.41 |
| 24c | 1 | 8 | 114.69 | 510.61 | 3.48 | 1.48 | 12.78 | Soluble | 0.8 | 99.23 |
HBD – hydrogen bond donor.
HBA – hydrogen bond acceptor.
TPSA – total polar surface area.
Substrate probability.
PPB – plasma protein binding.
Further profiling of selected compounds 12a, 12c, 15a–15c, 16a, 16c, 17a, 18a, 18c and 21b, showing the most potent antibacterial activity, was performed against sensitive and resistant strains of S. pneumoniae and S. pyogenes. Macrolide sensitive (eryS) and resistant (M) strains for both streptococci species were tested. The M phenotype exhibits efflux pump mediated resistance, which is the most common mechanism for resistance that ensures antibiotics are exported out of the bacterial cells via membrane proteins.57 Reference antibiotics azithromycin and ampicillin were used as standard controls.
The results presented in Table 2 demonstrate that all tested compounds generally showed better activity against both S. pyogenes strains compared to those of S. pneumoniae strains. Compounds 15a and 17a were the most active against sensitive S. pneumoniae with MIC values of 16 μg mL−1 and 8 μg mL−1, respectively. Compounds 15a–15c, 16a, 16c and 17a exhibited excellent antibacterial activity against both sensitive and macrolide resistant S. pyogenes strains with MIC values of 2–4 μg mL−1. Conversely, benzothiazole 21b exhibited the lowest activity against evaluated Gram-positive resistant streptococci strains.
The cytotoxicity of selected compounds on HepG2 cells was assessed by the MTS assay.58 All tested compounds had no significant effect on cell viability with IC50 > 100 μM (Table S1, ESI†).
2.3. DNA binding study
The antibacterial activity of several drugs with a heterocyclic ring can result from their interaction with bacterial DNA and RNA.59 Blocking DNA replication and transcription or interfering with DNA processing enzymes may be responsible for the antibacterial effect.60,61 Many benzimidazole and benzothiazole compounds have been shown to have significant antibacterial activity.17,53
Based on the antibacterial activity, six benzimidazoles (15a–15c, 16a, 16c and 17a) were selected for the binding study with DNA and RNA. To evaluate the difference in nucleic acid binding between the benzimidazole and benzothiazole motif, we also selected two benzothiazoles (21a and 21b) showing the highest antibacterial activity.
Almost all the compounds were soluble in redistilled water. Only 17a was dissolved in DMSO. Measurements were done in an aqueous buffer (pH = 7, sodium cacodylate buffer, I = 0.05 mol dm−3). The UV/vis spectra (ESI†) of buffered solutions of 15a–15c, 16a, 16c, 17a, 21a and 21b were proportional to their concentrations up to c = 2 × 10−5 mol dm−3, suggesting that the studied compounds do not aggregate by intermolecular stacking under experimental conditions. Absorption maxima and corresponding molar extinction coefficients (ε) are given in Table S2 (ESI†).
The estimation of polynucleotide binding constants was investigated by fluorescence spectroscopy. Circular dichroism (CD) titrations and thermal melting (Tm) experiments were used for the determination of the binding modes (intercalation, groove binding, external binding). While the CD reports on the impact of studied compounds on the CD spectra of polynucleotides, the thermal melting provides information about the polynucleotide stabilization driven by the compounds (ΔTm value).
Interactions of compounds were studied with double-stranded (ds-) polynucleotides displaying differences in sequence composition and/or the secondary structure: calf thymus (ct) DNA with mixed base-pair composition presents a classical B-helix,62 alternating dAdT-DNA also forms the classical B-helix and rArU-RNA (ds-RNA) is characterized by an A-helical structure of wide and shallow minor grooves and deep and narrow major grooves.62
In most cases, the titration with B-helical nucleic acid structures (ctDNA and p(dAdT)2) yielded a fluorescence intensity increase of benzimidazole compounds (Fig. 3). The titration of 16a and 17a with ctDNA also showed an emission intensity increase. However, the changes were linear and too small, so the binding parameters could not be calculated from the model. In general, the fluorescence intensity change of benzothiazole analogues 21a and 21b upon titration with polynucleotides did not change considerably (ESI†). The binding affinity and stoichiometry could be determined only when 21b was titrated with p(dAdT)2 (Table 3). The addition of rArU to the buffer aqueous solution of all studied compounds has not affected the fluorescence intensity (ESI†).
Fig. 3. Changes in the fluorescence spectrum of 15a (c = 1 × 10−6 mol dm−3, λexc = 312 nm) upon titration with poly(dAdT)2 (c = 1 × 10−6–1.5 × 10−5 mol dm−3); inset: dependence of 15a fluorescence intensity at λ = 382 nm upon c(poly(dAdT)2) increase, at pH = 7.0, sodium cacodylate buffer, I = 0.05 mol dm−3.

Binding constants (log Ks)a,b and parametersc calculated from the fluorescence titrations of 15a–c, 16a, 16c, 17a, 21a and 21b with double-stranded (ds-) polynucleotides at pH = 7.0 (buffer sodium cacodylate, I = 0.05 mol dm−3).
| Compd | ctDNA | p(dAdT)2 | ||
|---|---|---|---|---|
| log Ks | I/I0c | log Ks | I/I0c | |
| 15a | 6.5 | 3.4 | 5.5 | 7.2 |
| 15b | 5.6 | 11.4 | 5.9 | 15.4 |
| 15c | 6.2 | 7.6 | 6.0 | 9.5 |
| 16a | NDd | NDd | 5.4 | 2.2 |
| 16c | 6.4 | 63.1 | 5.9 | 71.5 |
| 17a | NDd | NDd | 6.6 | 2.0 |
| 21a | NDd | NDd | 5.3 | 4.9 |
| 21b | NDd | NDd | NDd | NDd |
Accuracy of n ± 20%, consequently log Ks values vary in the same order of magnitude.
Processing of titration data by means of the Scatchard equation63 gave values of ratio n [bound compound]/[polynucleotide] from 0.1–0.4, for easier comparison all data were recalculated for fixed n = 0.2.; correlation coefficients were >0.99 for most of the calculated Ks.
I 0 – starting fluorescence intensity of 15a–c, 16a, 16c, 17a, 21a and 21b; I – fluorescence intensity of 15a–c, 16a, 16c, 17a, 21a and 21b/polynucleotide complex calculated using the Scatchard equation.
Not determined; fluorescence changes of the studied compound with polynucleotides were too small/linear for accurate calculation of binding constants.
The binding constants Ks and ratios n[bound compound]/[DNA/RNA] obtained by processing fluorimetric titration data with the Scatchard equation63,64 are summarized in Table 3. Compounds 16a, 17a, and 21a showed greater affinity towards AT-rich DNA, while 15a showed substantially higher affinity towards mixed base-pair B-helical DNA (ctDNA).
Noteworthily, the binding affinities of benzimidazoles are higher than those of their benzothiazole counterparts, for both ctDNA and AT-DNA. Due to small changes in emission intensity in titrations with RNA (rArU), it was not possible to calculate the binding constants.
To further evaluate the non-covalent binding of molecules to ds-polynucleotides, we compared the melting temperature (Tm) values65 of unbound DNA sequences with their saturated complexes (Fig. 4, Table 4 and ESI†).
Fig. 4. Melting curves of poly(dAdT)2 upon addition of 15a, 15c, 16a, 16c and 17a at ratio, r ([compound]/[polynucleotide]) = 0.3, pH = 7.0 (buffer sodium cacodylate, I = 0.05 mol dm−3).

The ΔTma values (°C) of studied ds-polynucleotides upon addition of ratio rb = 0.3 and of 15a–c, 16a, 16c, 17a, 21a and 21b at pH 7.0 (sodium cacodylate buffer, I = 0.05 mol dm−3).
| r b = 0.3 | 15a | 15b | 15c | 16a | 16c | 17a | 21a | 21b |
|---|---|---|---|---|---|---|---|---|
| ctDNA | 3.3 | 2.4 | 1.6 | 4.4 | 2.6 | 3.8 | 0 | 0 |
| Poly A–poly U | 3.7 | 1.0 | 0 | 0 | 0 | 0 | NDc | NDc |
| Poly(dA–dT)2 | 16.9 | 16.0 | 16.9 | 11.0 | 12.1 | 11.9 | 3.1 | 3.8 |
Difference between Tm value of free polynucleotide and complex with a small molecule; error in ΔTm: ±0.5 °C.
r = [compound]/[polynucleotide].
Not determined.
The influence of the benzimidazole compounds on the thermal melting of p(dAdT)2 was rather significant, while the benzothiazole counterparts 21a and 21b exhibited considerably lower ΔTm values. However, the studied compounds showed a relatively small stabilization effect of ctDNA, or in the case of benzothiazoles, no stabilization at all (Table 4). The difference in stabilization effect can be explained by ctDNA composition, which contains 58% AT and 42% GC nucleobases, and the specific affinity of the compounds for the AT sequences. With the exception of 15a and 15b, the melting temperature of the rArU did not change upon the addition of the studied compounds.
The preference for the AT sequences is an expected feature of small molecules with a preference for minor groove binding sites.66,67 The minor groove of ds-RNA is not a suitable binding site for studied compounds since it is broad and shallow compared to the deep and narrow minor groove of ds-DNA.62
The useful information on the interaction of compounds with DNA and RNA can be obtained in the CD region where compounds absorb light and polynucleotides are not CD-active,68λ > 300 nm. The studied compounds are achiral and themselves do not display CD spectra, however, upon formation of complexes with ds-polynucleotides, the mutual orientations of the small molecule and polynucleotide chiral axis could be obtained, offering useful information about modes of interaction.69,70 Furthermore, all the studied compounds have absorption bands that coincide with the CD signal of polynucleotides (≤260 nm), corresponding to the helix conformation of ctDNA, p(dAdT)2 and rArU, therefore it is difficult to evaluate the influence of the compounds on polynucleotide helicity.
All the compounds exhibited positive induced CD spectra (ICD) with ctDNA and poly(dAdT)2 in the region from 300–360 nm (Fig. 5 and ESI†). Such changes are typical for classical minor groove binders like DAPI, as well as for cationic benzimidazoles and indoles that were found to be minor groove binders.71,72 Noteworthily, a weak negative ICD signal, located around 330 nm, was also observed in titrations of rArU with 15a, 15b and 15c (ESI†).
Fig. 5. CD titration of poly(dAdT)2 (c = 2.0 × 10−5 mol dm−3) with 15a at molar ratios r = [compound]/[polynucleotide] (pH = 7.0, buffer sodium cacodylate, I = 0.05 mol dm−3).

Usually, a positive ICD band, with an intensity similar to or stronger than the CD band of DNA/RNA, strongly supports the minor groove binding to DNA.73 Weak negative ICD signals in titrations with RNA as well as reported stabilization effects suggest an intercalative way of binding. Thus, it can be concluded that the studied compounds bind to the minor groove of ctDNA and AT-DNA. This is additionally supported by thermal stabilization of ds-polynucleotides (Table 4) and the binding constants, log Ks ≈ 106 (Table 3).
2.4. Computational analysis of binding to DNA
In order to further investigate the results obtained by fluorescence and CD spectroscopy, the binding of imidazoline-substituted benzimidazoles 15a and 15b as well as benzothiazoles 21a and 21b to the DNA minor groove was investigated.
Regarding compounds with the benzimidazole core, two tautomers are possible. The stability of both tautomeric forms was explored by quantum mechanical calculations at the B3LYP-D3/6-31G+(2d,p) level including the implicit water effect. The difference between tautomers is negligible and it amounts to 0.2 kcal mol−1. Therefore, both tautomeric forms were explored in the docking exercise and the one that forms more stable interactions with the DNA is selected.
Available X-ray structures of DNA complexes with binders, structurally related to imidazoline-substituted benzimidazoles, were analysed previously25 and computational experiments were carried out using the crystal structure of the DB921-D(CGCGAATTCGCG)2 complex (pdb: 2B0K) as an example of binding to the AT rich DNA and the crystal structure of the Forkhead domain of human FOXN1 in complex with human ctDNA (6EL8) was used for selectivity investigation. Compounds 15a and 21a as well as their close analogues 15b and 21b were docked to the X-ray template of DNA as described in the Methods section (4.6. Computational methods). Both water mediated and direct binding to the minor groove of the DNA were examined. Structures of inhibitors and their interactions with DNA are given in Fig. 6.
Fig. 6. Interactions of compound 15a and 21a with DNA: a) 2D projection of interactions with 15a; b) 2D projection of interactions with 21a; c) 3D surface of 15a binding to the DNA minor groove; d) aligned binding poses for 15a and 21a.

Compounds 15a and 21a could bind directly as well as through water mediated interactions in order to compensate for the deviations from the isohelical structure. Direct binding is more favorable for the benzimidazole compounds. 15a forms three hydrogen-bonds with the DNA bases, while benzothiazole forms only one-hydrogen bond, in agreement with experimentally determined lower binding constants and melting temperatures. Dihedral angles of 40 and 61° are close to the helical pitch angle of 45° enabling optimal curvature and complementary shape for effective binding to DNA.74
In the case of water mediated binding, the carbonyl group from the benzaldehyde forms a H-bond with the dT20 residue of a DNA chain B inducing sub-optimal binding with weaker interactions of the amidine moiety and its placement partially outside of the minor groove. Although not experimentally examined for the DNA binding, compounds containing morpholine and dimethyl amino moieties are predicted to have even better binding than 15a due to additional positive charge that could interact with negatively charged phosphate groups of the DNA chains. The reason for their weaker anti-bacterial profile is probably due to the lower permeability through the bacterial cell wall.
Complexes obtained by docking were further optimized by the MMGBSA method and free energies of binding are shown in Table 5. Molecular mechanics was used to estimate the strength of interaction and a generalized Born model and solvent accessibility method describe implicitly solvent effects. Benzimidazole analogues bind with higher affinity to both AT rich and ct-DNA, while benzothiazole analogues show higher selectivity between AT-rich and ct-DNA.
Ligand ΔG(int)/kcal mol−1 with AT-rich DNA and ctDNA calculated by MM–GBSA.
| Compd | AT-rich DNA | ctDNA |
|---|---|---|
| 15a | −90.1 | −86.0 |
| 15b | −91.7 | −86.8 |
| 21a | −80.3 | −65.0 |
| 21b | −81.6 | −63.0 |
In order to explore the dynamics of water mediated ligand–DNA interactions, molecular dynamics simulations in water as an explicit solvent were performed for 100 ns at room temperature. Simulations were carried out for 15a and 21a ligands with MMGBSA optimised complex structures used as starting points. The average number of direct H-bonds during the simulation is in agreement with docking and MMGBSA predictions, while water mediated H-bonds are more dynamic and occur with lower frequency than direct H-bonds as shown in Fig. 7. Compound 21a with a benzothiazole scaffold forms a smaller number of hydrogen bonds, both direct and water mediated, than compound 15a with a benzimidazole core, in agreement with experimental data.
Fig. 7. Average number of direct and water mediated H-bonds during MD simulation for a) 15a and b) 21a ligands.

2.5. In silico PhysChem and ADME profiling
The calculated structural, physico-chemical and ADME parameters for the prepared compounds are given in Table 6. All the compounds are within the thresholds proposed by the Lipinski rule of 5, with good solubility, optimal lipophilicity, HBD and HBA. Several compounds have borderline molecular weight and a higher number of aromatic rings which correlates with the predicted high plasma protein binding. The strong basicity of the prepared compounds could be a limiting factor for membrane permeability and consequently oral absorption.
The physico-chemical space of the prepared compounds was compared with previously profiled analogues25 in order to estimate their permeability as shown in Fig. 8. Lipophilicity expressed as calculated log P values and polarity expressed as total polar surface area (TPSA) were compared for two sets. Compounds with low log P and high TPSA, 13a and 19c as well as compounds with high log P and low TPSA, 12a, 12b, 15a and 15b (green rectangles) are expected to have the best, but still borderline permeabilities. The major bottleneck to the permeability is the permanently charged amidine which is why compounds in the present study are designed to have only one amidine moiety although di-amidine compounds have shown the best anti-infective properties due to strong binding to DNA. The second amidine group is replaced by ionisable and less basic morpholine or di-methylamino groups.
Fig. 8. Physico-chemical properties of the prepared compounds; lipophilicity (log P) vs. polar surface area (TPSA). Compounds colored by permeability are from ref. 25.

Regarding metabolic stability, previous studies have shown that compounds with morpholine and di-methyl amine substituents were metabolically stable while compounds with unsubstituted terminal phenyl rings and substituted triazoles are more prone to metabolism, which is qualitatively in agreement with predicted metabolic probability shown in Table 6. Therefore, it could be expected that analogous compounds in this study will also have similar clearance. In conclusion, for the investigated chemical series, the physico-chemical and ADME properties could be optimized, with the exception of expected low membrane permeability.
3. Conclusions
Synthesis of novel amidino benzimidazoles 12a–19a, 12b–18b, 12c, 13c, 15c–19c was carried out using the method based on the condensation reaction of aryl carbaldehydes with o-phenylenediamine with NaHSO3 as an oxidative reagent.
The observed SAR among the evaluated compounds suggests that an increase of antibacterial activity is affected by the introduction of an aromatic moiety at phenoxymethylene linked to the 2-position of the benzimidazole skeleton. Moreover, benzoyl-substituted benzimidazole derivatives 15a–15c exhibited the most pronounced activity against Gram-negative pathogens M. catarrhalis and E. coli (MICs = 1–4 μg mL−1). Inhibition of Moraxella catarrhalis was comparable to that of the reference antibiotic azithromycin (MICs = 8 μg mL−1). Compounds 15a–15c showed also potent activity against both sensitive and macrolide resistant S. pyogenes strains with MIC values of 2–4 μg mL−1, highlighting 15a with an unsubstituted phenoxymethylene linker as the most promising compound.
Summarised DNA and RNA binding results (significant stabilization effects of AT-DNA, high binding affinities and strong positive ICD spectra) for 15a–15c, 16a, 16c, 17a, 21a and 21b point to their preference toward AT-rich sites and minor groove binding mode.58 Noteworthily, the binding affinities of benzimidazoles as well as stabilization effects of DNA are higher than those of their benzothiazole analogues. This is most pronounced for fluorinated benzimidazole 15b and its benzothiazole analogue 21b. Those results are in correlation with antibacterial activity as well as docking studies and suggest that AT-rich DNA sequences are potential targets of the tested compounds. The predicted physico-chemical and ADME properties are within drug-like space except for low permeability. Since various antibacterial drugs have been shown to target AT-rich sites commonly found in bacterial promoters and origins of replication as, for example, antibiotics based on distamycin that have entered clinical trials, hit molecules identified in this study are promising starting points for further chemical as well as pharmacological optimization.
4. Experimental
4.1. General
All the solvents and chemicals were purchased from Aldrich (St. Louis, MO, USA) and Acros (Geel, Belgium). Thin layer chromatography was performed on pre-coated Merck silica gel 60F-254 plates (Merck, Kenilworth, NJ, USA), while a glass column slurry-packed under gravity with 0.063–0.2 mm silica gel (Fluka, Seelze, Germany) was employed for column chromatography. Melting points of compounds were determined using the Kofler micro hot-stage. 1H and 13C NMR spectra were recorded on a Varian Gemini 300 (300 and 75 MHz) or Varian Gemini 600 (600 and 150 MHz). All data were recorded in dimethyl sulfoxide (DMSO-d6) at 298 K. Chemical shifts were referenced to the residual solvent signal of DMSO at δ 2.50 ppm for 1H and δ 39.50 ppm for 13C. Individual resonances were assigned on the basis of their chemical shifts, signal intensities, multiplicity of resonances, and H–H coupling constants. Purity was assessed by elemental analysis. Elemental composition analysis (C, H, N) of all novel compounds was within 0.4% of the calculated values.
4.2. Experimental procedures for the preparation of compounds
Precursor compounds 2a,752b,252c,763a,253b,253c,254a,254b,254c,255a,255b,255c,256a,256b,256c,257a,257b,257c,258a,259a,229b,259c,2510a,2510b,2510c,2511 (ref. 54) and 20 (ref. 54) were synthesised in accordance with procedures given in the literature.
4.2.1. General procedure for synthesis of 1H-1,2,3-triazole benzaldehydes 8a–8c
To a reaction mixture of the corresponding terminal alkyne (2a–2c) in DMF : MeOH = 9 : 1 (4.6 mL) TMSN3 (1.5 eq.) and CuI (0.1 eq.) were added. The reaction mixture was stirred for 6 h at 100 °C. The solvent was removed under reduced pressure and the crude reaction mixture was purified by column chromatography with CH2Cl2.
4-[(1H-1,2,3-Triazol-4-yl)methoxy]-3-fluorobenzaldehyde (8b)
Compound 8b was prepared using the above described procedure from 2b (600 mg, 3.37 mmol) to obtain 8b as a yellow solid (371.0 mg, 49%; m.p. 120–123 °C). 1H NMR (300 MHz, DMSO-d6) δ 15.16 (1H, s, NH), 9.88 (1H, d, J = 2.0 Hz, CHO), 7.96 (1H, s), 7.80 (1H, d, J = 8.5 Hz), 7.71 (1H, dd, J = 11.3; 1.9 Hz), 7.60 (1H, t, J = 8.3 Hz), 5.42 (2H, s, OCH2). 13C NMR (75 MHz, DMSO-d6) δ 191.31 (CHO), 152.15 (d, JCF = 247.3 Hz), 151.55, 130.44, 128.67 (d, JCF = 18.3 Hz), 115.80, 115.50, 115.48, 62.46 (OCH2).
4-[(1H-1,2,3-Triazol-4-yl)methoxy]-3-methoxybenzaldehyde (8c)
Compound 8c was prepared using the above described procedure from 2c (600 mg, 3.12 mmol) to obtain 8c as a yellow solid (410.0 mg, 55%; m.p. 142–145 °C). 1H NMR (300 MHz, DMSO-d6) δ 15.14 (1H, s, NH), 9.85 (1H, s, CHO), 8.00 (1H, s), 7.56 (1H, dd, J = 8.2, 1.7 Hz), 7.40 (2H, dd, J = 7.6, 5.0 Hz), 5.30 (2H, s, OCH2), 3.81 (3H, s, OCH3). 13C NMR (151 MHz, DMSO-d6) δ 191.38 (CHO), 149.30, 129.94, 125.76, 112.63, 109.74, 61.62 (OCH2), 55.47 (OCH3).
4.2.2. General procedure for the synthesis of 2,5(6)-disubstituted benzimidazole derivatives 12a–19a, 12b–18b, 12c, 13c, and 15c–19c
The reaction mixture of benzaldehyde derivatives (3a–10a, 3b–10b, 3c–10c), o-phenylenediamine 11 and 40% NaHSO3 (aq) was dissolved in EtOH (15 mL) and stirred under reflux for 8 h. After completion of the reaction, the mixture was evaporated to dryness and the crude residue was purified by column chromatography (CH2Cl2 : CH3OH = 4 : 1). The obtained residue was dissolved in HCl saturated EtOH (8–10 mL) and stirred for 24 h. The addition of ether resulted in precipitation of products. The solid was collected by filtration, washed with anhydrous ether, and dried under vacuum.
2-(4-(2-(Diethylamino)ethoxy)phenyl)-5(6)-(3,4,5,6-tetrahydropyrimidin-2-yl)-1H-benzimidazole dihydrochloride (12a)
Compound 12a was prepared using the above described method from 3a (150 mg, 0.68 mmol) and o-phenylenediamine 11 (129 mg, 0.68 mmol) to obtain 12a as a brown solid (74.9 mg, 25%, m.p. > 240 °C). 1H NMR (400 MHz, DMSO) δ 10.70 (1H, s, NH+), 10.17 (2H, s, CNH), 8.48 (2H, d, J = 8.7 Hz), 8.15 (1H, s), 7.91 (1H, d, J = 8.5 Hz), 7.74 (1H, d, J = 8.5 Hz), 7.31 (2H, d, J = 9.0 Hz), 4.55 (2H, t, J = 5.0 Hz, OCH2), 3.57–3.50 (8H, m, NCH2, signal overlapping with H2O signal), 3.23 (2H, qd, J = 11.8, 7.0 Hz, NCH2), 2.06–1.98 (2H, m, CH2), 1.28 (6H, t, J = 7.2 Hz, CH3). 13C NMR (75 MHz, DMSO) δ 161.35f, 159.62 (CNH), 152.56, 130.51, 124.51, 124.00, 116.03, 114.91, 63.26 (OCH2), 49.94 (NCH2), 47.44 (NCH2), 39.32 (NCH2), 18.23 (CH2), 8.92 (CH3). Anal. calcd. for C23H29N5O × 2HCl × 1.5H2O (Mr = 491.45): C 56.21, H 6.97, N 14.25; found: C 56.33, H 6.76, N 14.25.
2-(4-(2-(Diethylamino)ethoxy)-3-fluorophenyl)-5(6)-(3,4,5,6-tetrahydropyrimidin-2-yl)-1H-benzimidazole dihydrochloride (12b)
Compound 12b was prepared using the above described method from 3b (150 mg, 0.63 mmol) and o-phenylenediamine 11 (120 mg, 0.63 mmol) to obtain 12b as a white solid (60.7 mg, 21%, m.p. > 240 °C). 1H NMR (400 MHz, DMSO) δ 10.98 (1H, s, NH+), 10.18 (2H, s, CNH), 8.39 (1H, d, J = 12.2 Hz), 8.33 (1H, d, J = 8.6 Hz), 8.14 (1H, s), 7.87 (1H, d, J = 8.5 Hz), 7.71 (1H, d, J = 8.5 Hz), 7.54 (1H, t, J = 8.7 Hz), 4.64 (2H, t, J = 4.8 Hz, OCH2), 3.59 (4H, dd, J = 9.4, 4.7 Hz, NCH2), 3.53 (4H, s, NCH2), 3.27–3.21 (2H, m, CH2), 2.06–1.96 (2H, m, CH2), 1.29 (6H, t, J = 7.2 Hz, CH3). 13C NMR (101 MHz, DMSO) δ 159.75 (CNH), 151.89 (d, JCF = 245.1 Hz), 152.21, 148.83, 125.48, 123.98, 123.44, 116.03, 115.83 (d, J = 21.3 Hz), 115.34, 64.42 (OCH2), 49.85 (NCH2), 47.65 (NCH2), 18.28 (CH2), 8.96 (CH3). Anal. calcd. for C23H28FN5O × 2HCl × 2.25H2O (Mr = 522.95): C 52.82, H 6.65, N 13.39; found: C 52.73, H 6.73, N 13.24.
2-(4-(2-(Diethylamino)ethoxy)-3-methoxyphenyl)-5(6)-(3,4,5,6-tetrahydropyrimidin-2-yl)-1H-benzimidazole dihydrochloride (12c)
Compound 12c was prepared using the above described method from 3c (150 mg, 0.60 mmol) and o-phenylenediamine 11 (114 mg, 0.60 mmol) to obtain 12c as a brown solid (87.5 mg, 32%, m.p. > 240 °C). 1H NMR (400 MHz, DMSO) δ 10.97 (1H, s, NH+), 10.30 (2H, s, CNH), 8.38 (1H, s), 8.24 (1H, d, J = 8.4 Hz), 8.20 (1H, d, J = 1.1 Hz), 7.96 (1H, d, J = 8.6 Hz), 7.82 (1H, dd, J = 8.6, 1.4 Hz), 7.37 (1H, d, J = 8.6 Hz), 4.58 (2H, t, J = 4.9 Hz, OCH2), 3.98 (3H, s, OCH3), 3.58–3.50 (8H, m, NCH2, signal overlapping with H2O signal), 3.28–3.22 (2H, m, NCH2), 2.06–1.98 (2H, m, CH2), 1.29 (6H, t, J = 7.2 Hz, CH3). 13C NMR (101 MHz, DMSO) δ 159.44 (NCH), 152.03, 151.57, 149.84, 125.38, 124.88, 122.74, 117.37, 114.74, 114.56, 114.06, 112.40, 64.10 (OCH2), 57.08 (OCH3), 49.72 (NCH2), 47.66 (NCH2), 18.17 (CH2), 8.99 (CH3). Anal. calcd. for C24H31N5O2 × 2HCl × 2.5H2O (Mr = 539.49): C 53.43, H 7.10, N 12.98; found: C 53.65, H 6.98, N 13.07.
2-(4-(2-Morpholinoethoxy)phenyl)-5(6)-(3,4,5,6-tetrahydropyrimidin-2-yl)-1H-benzimidazole dihydrochloride (13a)
Compound 13a was prepared using the above described method from 4a (150 mg, 0.64 mmol) and o-phenylenediamine 11 (122 mg, 0.64 mmol) to obtain 13a as an orange solid (20.3 mg, 7%, m.p. > 240 °C). 1H NMR (400 MHz, DMSO-d6) δ 11.57 (1H, s, NH+), 10.20 (2H, s, CNH), 8.50 (2H, d, J = 8.8 Hz), 8.17 (1H, s), 7.94 (1H, d, J = 8.5 Hz), 7.77 (1H, d, J = 8.5 Hz), 7.34 (2H, d, J = 9.0 Hz), 4.62 (2H, t, J = 4.9 Hz, OCH2), 3.98 (4H, d, J = 12.1 Hz), 3.91–3.83 (4H, m), 3.54 (4H, s, NCH2), 3.24 (2H, dd, J = 19.3, 9.6 Hz, NCH2), 2.08–1.96 (2H, m, CH2). 13C NMR (75 MHz, DMSO) δ 161.87, 159.42 (CNH), 151.84, 136.37, 130.98, 125.41, 124.89, 117.17, 116.25, 114.78, 114.61, 63.58 (OCH2), 63.22 (OCH2), 55.03 (NCH2), 52.09 (NCH2), 39.33 (NCH2), 18.15 (CH2). Anal. calcd. for C23H27N5O2 × 2HCl × 3H2O (Mr = 532.46): C 51.88, H 6.63, N 13.15; found: C 51.73, H 6.51, N 13.27.
2-(3-Fluoro-4-(2-morpholinoethoxy)phenyl)-5(6)-(3,4,5,6-tetrahydropyrimidin-2-yl)-1H-benzimidazole dihydrochloride (13b)
Compound 13b was prepared using the above described method from 4b (150 mg, 0.59 mmol) and o-phenylenediamine 11 (113 mg, 0.59 mmol) to obtain 13b as a white solid (40.9 mg, 15%, m.p. > 240 °C). 1H NMR (400 MHz, DMSO-d6) δ 11.59 (1H, s, NH+), 10.09 (2H, s, CNH), 8.31 (1H, J = 12.2 Hz), 8.26 (1H, d, J = 8.8 Hz), 8.11 (1H, d, J = 1.7 Hz), 7.86 (1H, d, J = 8.5 Hz), 7.67 (1H, dd, J = 8.5, 1.7 Hz), 7.54 (1H, t, J = 8.7 Hz), 4.74–4.64 (2H, m, OCH2), 4.00 (4H, d, J = 11.5 Hz), 3.89–3.82 (4H, m), 3.54 (4H, d, J = 6.9 Hz, NCH2), 3.26 (2H, d, J = 8.6 Hz, NCH2), 2.08–1.94 (2H, m, CH2). 13C NMR (151 MHz, DMSO) δ 159.29 (CNH), 151.43 (d, JCF = 245.4 Hz), 151.77, 148.25 (d, JCF = 9.7 Hz), 124.89, 123.42, 122.83, 115.69, 115.36, 115.22, 63.92 (OCH2), 63.14 (OCH2), 54.57 (NCH2), 51.69 (NCH2), 38.83 (NCH2), 17.77 (CH2). Anal. calcd. for C23H26FN5O2 × 2HCl × 2.5H2O (Mr = 541.44): C 51.02, H 6.14, N 12.93; found: C 50.89, H 6.31, N 12.87.
2-(3-Methoxy-4-(2-morpholinoethoxy)phenyl)-5(6)-(3,4,5,6-tetrahydropyrimidin-2-yl)-1H-benzimidazole dihydrochloride (13c)
Compound 13c was prepared using the above described method from 4c (150 mg, 0.56 mmol) and o-phenylenediamine 11 (108 mg, 0.56 mmol) to obtain 13c as a yellow solid (22.8 mg, 8.6%, m.p. 205–209 °C).1H NMR (300 MHz, DMSO-d6) δ 11.88 (1H, s, NH+), 10.30 (2H, s, CNH), 8.37 (1H, s), 8.28–8.16 (2H, m), 7.95 (1H, d, J = 8.6 Hz), 7.81 (1H, dd, J = 8.6, 1.4 Hz), 7.38 (1H, d, J = 8.6 Hz), 4.64 (2H, t, J = 4.8 Hz, OCH2), 4.04–3.82 (10H, m, OCH3), 3.67–3.56 (4H, m, NCH2), 3.25 (2H, dd, J = 21.7, 11.5 Hz), 2.10–1.92 (2H, m, CH2). 13C NMR (75 MHz, DMSO) δ 159.91 (CNH), 149.87, 123.20, 121.45, 115.15, 114.57, 111.64, 64.16 (OCH2), 63.67 (OCH2), 56.70 (OCH3), 55.22 (NCH2), 52.38 (NCH2), 18.26 (CH2). Anal. calcd. for C24H29N5O3 × 2HCl × 1.75H2O (Mr = 539.97): C 53.39, H 6.44, N 12.97; found: C 53.51, H 6.30, N 13.04.
2-(4-(2-Morpholino-2-oxoethoxy)phenyl)-5(6)-(3,4,5,6-tetrahydropyrimidin-2-yl)-1H-benzimidazole hydrochloride (14a)
Compound 14a was prepared using the above described method from 5a (150 mg, 0.60 mmol) and o-phenylenediamine 11 (115 mg, 0.60 mmol) to obtain 14a as a yellow solid (87.1 mg, 31%, m.p. > 240 °C). 1H NMR (400 MHz, DMSO) δ 10.14 (2H, s, CNH), 8.39 (2H, d, J = 8.7 Hz), 8.13 (1H, d, J = 1.1 Hz), 7.92 (1H, d, J = 8.5 Hz), 7.73 (1H, dd, J = 8.5, 1.4 Hz), 7.23 (2H, d, J = 9.0 Hz), 5.05 (2H, s, OCH2), 3.62 (4H, m, OCH2), 3.53 (4H, s, NCH2), 3.48 (4H, s, NCH2), 2.09–1.94 (2H, m, CH2). 13C NMR (75 MHz, DMSO) δ 165.93 (C O), 162.31, 159.59 (CNH), 152.46, 130.45, 124.81, 124.27, 116.10, 114.79, 66.50 (OCH2), 66.19 (OCH2), 45.03 (NCH2), 42.05 (NCH2), 39.34 (CH2), 18.20 (CH2). Anal. calcd. for C23H25N5O3 × HCl × 2H2O (Mr = 491.97): C 56.15, H 6.15, N 14.24; found: C 56.29, H 6.01, N 14.12.
2-(3-Fluoro-4-(2-morpholino-2-oxoethoxy)phenyl)-5(6)-(3,4,5,6-tetrahydropyrimidin-2-yl)-1H-benzimidazole hydrochloride (14b)
Compound 14b was prepared using the above described method from 5b (150 mg, 0.56 mmol) and o-phenylenediamine 11 (107 mg, 0.56 mmol) to obtain 14b as a white solid (20.8 mg, 7%, m.p. > 240 °C). 1H NMR (400 MHz, DMSO) δ 10.13 (2H, s, CNH), 8.33 (1H, d, J = 12.2 Hz), 8.25 (1H, d, J = 8.8 Hz), 8.13 (1H, d, J = 1.2 Hz), 7.94–7.85 (1H, m), 7.71 (1H, dd, J = 8.5, 1.5 Hz), 7.38 (1H, t, J = 8.7 Hz), 5.15 (2H, s, OCH2), 3.63 (4H, s, OCH2), 3.56–3.44 (8H, m, NCH2, signal overlapping with H2O signal), 2.09–1.94 (2H, m, CH2). 13C NMR (151 MHz, DMSO) δ 165.16 (C O), 159.41 (CNH), 151.33 (d, JCF = 244.7 Hz), 148.85, 124.36, 123.15, 122.56, 122.02, 119.62, 115.64, 115.09, 114.95, 66.06 (OCH2), 63.17 (OCH2), 44.49 (NCH2), 41.58 (NCH2), 38.84 (NCH2), 17.78 (CH2). Anal. calcd. for C23H24FN5O3 × HCl × 1.5H2O (Mr = 500.95): C 55.15, H 5.63, N 13.98; found: C 55.01, H 5.78, N 14.07.
2-(4-(2-Oxo-2-phenylethoxy)phenyl)-5(6)-(3,4,5,6-tetrahydropyrimidin-2-yl)-1H-benzimidazole hydrochloride (15a)
Compound 15a was prepared using the above described method from 6a (150 mg, 0.62 mmol) and o-phenylenediamine 11 (119 mg, 0.62 mmol) to obtain 15a as a white solid (63.1 mg, 22%, m.p. > 240 °C). 1H NMR (400 MHz, DMSO-d6) δ 10.17 (2H, s, CNH), 8.41 (2H, d, J = 8.8 Hz), 8.15 (1H, d, J = 1.0 Hz), 8.11–8.01 (2H, m), 7.92 (1H, d, J = 8.5 Hz), 7.73–7.71 (2H, m), 7.61 (2H, t, J = 7.7 Hz), 7.31 (2H, d, J = 8.8 Hz), 5.81 (2H, s, OCH2), 3.53 (4H, s, CH2), 2.11–1.97 (2H, m, CH2). 13C NMR (101 MHz, DMSO) δ 194.36 (C O), 168.29, 162.06, 159.68 (CNH), 152.72, 134.71, 134.42, 130.37, 129.35, 128.40, 124.63, 124.04, 116.12, 114.84, 70.87 (OCH2), 18.22 (CH2). Anal. calcd. for C25H22N4O2 × HCl × 1.5H2O (Mr = 473.95): C 63.35, H 5.53, N 11.82; found: C 63.21, H 5.64, N 11.69.
2-(3-Fluoro-4-(2-oxo-2-phenylethoxy)phenyl)-5(6)-(3,4,5,6-tetrahydropyrimidin-2-yl)-1H-benzimidazole hydrochloride (15b)
Compound 15b was prepared using the above described method from 6b (150 mg, 0.58 mmol) and o-phenylenediamine 11 (111 mg, 0.58 mmol) to obtain 15b as a white solid (30.0 mg, 11%, m.p. 235–239 °C). 1H NMR (400 MHz, DMSO-d6) δ 10.07 (2H, s, CNH), 8.29 (1H, d, J = 12.3 Hz), 8.16 (1H, d, J = 8.8 Hz), 8.10 (1H, d, J = 1.0 Hz), 8.08–8.03 (1H, m), 7.87 (1H, d, J = 8.5 Hz), 7.77–7.71 (1H, m), 7.67 (1H, d, J = 8.5 Hz), 7.61 (2H, dd, J = 7.7 Hz), 7.45 (1H, t, J = 8.8 Hz), 5.90 (2H, s, OCH2), 3.53 (4H, s, CH2), 2.10–1.95 (2H, m, CH2). 13C NMR (75 MHz, DMSO) δ 194.13 (C O), 160.10 (CNH), 151.88 (d, JCF = 244.5 Hz), 134.59, 134.48, 129.36, 128.38, 124.42, 123.10, 122.45, 116.08, 115.42, 115.14, 71.30 (OCH2), 39.35 (NCH2), 18.32 (CH2). Anal. calcd. for C25H21FN4O2 × HCl × 2H2O (Mr = 500.95): C 59.94, H 5.23, N 11.18; found: C 59.78, H 5.37, N 11.03.
2-(3-Methoxy-4-(2-oxo-2-phenylethoxy)phenyl)-5(6)-(3,4,5,6-tetrahydropyrimidin-2-yl)-1H-benzimidazole hydrochloride (15c)
Compound 15c was prepared using the above described method from 6c (150 mg, 0.65 mmol) and o-phenylenediamine 11 (124 mg, 0.65 mmol) to obtain 15c as a beige solid (49.4 mg, 18%, m.p. > 240 °C). 1H NMR (400 MHz, DMSO-d6) δ 10.19 (2H, s, CNH), 8.26 (1H, s), 8.17 (1H, s), 8.13–8.00 (3H, m), 7.97 (1H, d, J = 8.5 Hz), 7.78 (1H, dd, J = 8.6, 1.5), 7.73 (1H, t, J = 7.4 Hz), 7.60 (2H, t, J = 7.7 Hz), 7.27 (1H, d, J = 8.7 Hz), 5.81 (2H, s, OCH2), 4.00 (3H, s, OCH3), 3.54 (4H, s, NCH2), 2.08–1.97 (2H, m, CH2). 13C NMR (75 MHz, DMSO) δ 194.19 (C O), 159.42 (CNH), 152.39, 151.95, 134.70, 134.39, 132.67, 129.32, 128.39, 125.68, 125.12, 122.76, 114.66, 114.40, 114.08, 112.51, 71.12 (OCH2), 56.93 (OCH3), 18.13 (CH2). Anal. calcd. for C26H24N4O3 × HCl × 1.75H2O (Mr = 508.48): C 61.41, H 5.65, N 11.02; found: C 61.23, H 5.58, N 11.12.
2-(4-(Pyridin-2-ylmethoxy)phenyl)-5(6)-(3,4,5,6-tetrahydropyrimidin-2-yl)-1H-benzimidazole hydrochloride (16a)
Compound 16a was prepared using the above described method from 7a (150 mg, 0.51 mmol) and o-phenylenediamine 11 (97.5 mg, 0.51 mmol) to obtain 16a as a white solid (46 mg, 21%, m.p. > 240 °C). 1H NMR (400 MHz, DMSO-d6) δ 10.24 (2H, s, CNH), 8.76 (1H, d, J = 4.8 Hz), 8.52 (2H, d, J = 8.8 Hz), 8.19 (1H, s), 7.96 (1H, d, J = 8.5 Hz), 7.86–7.76 (3H, m), 7.70–7.62 (1H, m), 7.41 (2H, d, J = 9.0 Hz), 5.51 (2H, s, OCH2), 3.53 (4H, s, NCH2), 2.06–1.98 (2H, m, CH2). 13C NMR (101 MHz, DMSO-d6) δ 162.06 (CNH), 159.52, 154.27, 152.11, 147.05, 141.25, 131.23, 130.88, 125.51, 125.20, 125.03, 124.63, 124.02, 116.96, 116.36, 114.70, 69.20 (OCH2), 18.18 (CH2). Anal. calcd. for C23H21N5O × HCl × 2.5H2O (Mr = 464.95): C 59.42, H 5.85, N 15.06; found: C 59.33, H 5.93, N 14.98.
2-(3-Fluoro-4-(pyridin-2-ylmethoxy)phenyl)-5(6)-(3,4,5,6-tetrahydropyrimidin-2-yl)-1H-benzimidazole hydrochloride (16b)
Compound 16b was prepared using the above described method from 7b (150 mg, 0.65 mmol) and o-phenylenediamine 11 (124 mg, 0.65 mmol) to obtain 16b as a white solid (76.6 mg, 26%, m.p. > 240 °C). 1H NMR (400 MHz, DMSO-d6) δ 10.19 (2H, s, CNH), 8.76 (1H, d, J = 4.4 Hz), 8.47 (1H, d, J = 12.2 Hz), 8.35 (1H, d, J = 8.7 Hz), 8.20–8.14 (2H, m), 7.93 (1H, d, J = 8.5 Hz), 7.81 (1H, d, J = 7.9 Hz), 7.75 (1H, dd, J = 8.6, 1.7 Hz), 7.63 (2H, m), 5.55 (2H, s, OCH2), 3.53 (4H, s, NCH2), 2.08–1.95 (2H, m, CH2). 13C NMR (151 MHz, DMSO-d6) δ 158.94 (CNH), 152.73, 151.39 (d, JCF = 245.8 Hz), 149.42 (d, JCF = 10.6 Hz), 146.02, 141.66, 125.87, 125.87, 125.00, 124.77, 124.23, 124.01, 117.92 (d, JCF = 10.3 Hz) 116.12 (d, JCF = 21.2 Hz), 116.00, 114.42 (d, JCF = 10.9 Hz), 68.99 (OCH2), 38.83 (NCH2), 17.67 (CH2). Anal. calcd. for C23H20FN5O × HCl × 1.5H2O (Mr = 464.92): C 59.42, H 5.20, N 15.06; found: C 59.32, H 5.03, N 15.23.
2-(3-Methoxy-4-(pyridin-2-ylmethoxy)phenyl)-5(6)-(3,4,5,6-tetrahydropyrimidin-2-yl)-1H-benzimidazole hydrochloride (16c)
Compound 16c was prepared using the above described method from 7c (150 mg, 0.46 mmol) and o-phenylenediamine 11 (88 mg, 0.46 mmol) to obtain 16c as a white solid (20.3 mg, 9%, m.p. 205–209 °C). 1H NMR (300 MHz, DMSO) δ 10.11 (2H, s, CNH), 8.69 (1H, d, J = 4.7 Hz), 8.14 (2H, d, J = 14.8 Hz), 8.04 (2H, t, J = 9.8 Hz), 7.91 (1H, d, J = 8.5 Hz), 7.76–7.66 (2H, m), 7.58–7.48 (1H, m), 7.37 (1H, d, J = 8.6 Hz), 5.39 (2H, s, OCH2), 3.97 (3H, s, OCH3), 3.53 (4H, s, NCH2), 2.02 (2H, s, CH2). 13C NMR (151 MHz, DMSO) δ 159.06 (CNH), 154.19, 151.85, 151.18, 149.41, 146.95, 140.20, 124.63, 124.32, 124.04, 123.30, 121.91, 114.32, 114.15, 113.84, 111.75, 69.43 (OCH2), 56.37 (OCH3), 38.85 (NCH2), 17.67 (CH2). Anal. calcd. for C24H23N5O2 × HCl × H2O (Mr = 467.95): C 61.60, H 5.60, N 14.97; found: C 61.73, H 5.48, N 14.81.
2-(4-((1H-1,2,3-Triazol-4-yl)methoxy)phenyl)-5(6)-(3,4,5,6-tetrahydropyrimidin-2-yl)-1H-benzimidazole hydrochloride (17a)
Compound 17a was prepared using the above described method from 8a (150 mg, 0.74 mmol) and o-phenylenediamine 11 (141 mg, 0.74 mmol) to obtain 17a as a white solid (63.8 mg, 21%, m.p. > 240 °C). 1H NMR (400 MHz, DMSO-d6) δ 10.16 (2H, s, CNH), 8.42 (2H, d, J = 8.7 Hz), 8.14 (1H, s), 8.06 (1H, s, H), 7.92 (1H, d, J = 8.5 Hz), 7.73 (1H, d, J = 8.6 Hz), 7.36 (2H, d, J = 8.9 Hz), 5.36 (2H, s, OCH2), 3.53 (4H, s, NCH2), 2.10–1.95 (2H, m, CH2). 13C NMR (101 MHz, DMSO) δ 161.90, 159.69 (CNH), 152.77, 130.40, 124.59, 123.96, 116.16, 114.89, 61.69 (OCH2), 18.23 (CH2). Anal. calcd. for C20H19N7O × HCl × H2O (Mr = 427.89): C 56.14, H 5.18, N 22.91; found: C 56.29, H 5.01, N 22.79.
2-(4-((1H-1,2,3-Triazol-4-yl)methoxy)-3-fluorophenyl)-5(6)-(3,4,5,6-tetrahydropyrimidin-2-yl)-1H-benzimidazole hydrochloride (17b)
Compound 17b was prepared using the above described method from 8b (150 mg, 0.68 mmol) and o-phenylenediamine 11 (129 mg, 0.68 mmol) to obtain 17b as a white solid (105.1 mg, 36%, m.p. > 240 °C). 1H NMR (400 MHz, DMSO-d6) δ 10.17 (2H, s, CNH), 8.38 (1H, d, J = 12.2 Hz), 8.32 (1H, d, J = 8.8 Hz), 8.15 (1H, d, J = 1.2 Hz), 8.09 (1H, s), 7.91 (1H, d, J = 8.5 Hz,), 7.76–7.66 (2H, m), 5.44 (2H, s, OCH2), 3.53 (4H, s, NCH2), 2.08–1.95 (2H, m, CH2). 13C NMR (151 MHz, DMSO) δ 159.00 (NCH), 151.41 (d, JCH = 245.5 Hz), 150.90, 149.50 (d, JCF = 10.9 Hz), 140.86, 125.62, 124.48, 123.96, 115.88, 115.72, 114.44, 62.02 (OCH2), 38.83 (NCH2), 17.69 (CH2). Anal. calcd. for C20H18FN7O × HCl × 0.75H2O (Mr = 441.38): C 54.42, H 4.68, N 22.21; found: C 54.57, H 4.60, N 22.30.
2-(4-((1H-1,2,3-Triazol-4-yl)methoxy)-3-methoxyophenyl)-5(6)-(3,4,5,6-tetrahydropyrimidin-2-yl)-1H-benzimidazole hydrochloride (17c)
Compound 17c was prepared using the above described method from 8c (150 mg, 0.64 mmol) and o-phenylenediamine 11 (123 mg, 0.64 mmol) to obtain 17c as a white solid (25.0 mg, 8%, m.p. > 240 °C). 1H NMR (300 MHz, DMSO-d6) δ 10.24 (2H, s, CNH), 8.29 (1H, s), 8.18 (2H, m), 8.05 (1H, s), 7.96 (1H, d, J = 8.5 Hz), 7.80 (1H, dd, J = 8.6, 1.5 Hz), 7.50 (1H, d, J = 8.7 Hz), 5.35 (2H, s, OCH2), 3.93 (3H, s, OCH3), 3.53 (4H, s, NCH2), 2.02 (2H, s, CH2). 13C NMR (75 MHz, DMSO) δ 159.50 (CNH), 152.05, 149.83, 141.64, 125.33, 124.75, 122.61, 114.75, 114.53, 114.04, 112.15, 61.96 (OCH2), 56.76 (OCH3), 39.35 (NCH2), 18.16 (CH2). Anal. calcd. for C21H21N7O2 × HCl × H2O (Mr = 457.91): C 55.08, H 5.28, N 21.41; found: C 55.19, H 5.11, N 21.34.
2-(4-((1-Benzyl-1H-1,2,3-triazol-4-yl)methoxy)phenyl)-5(6)-(3,4,5,6-tetrahydropyrimidin-2-yl)-1H-benzimidazole hydrochloride (18a)
Compound 18a was prepared using the above described method from 9a (150 mg, 0.51 mmol) and o-phenylenediamine 11 (98 mg, 0.51 mmol) to obtain 18a as a brown solid (41.6 mg, 16%, m.p. > 240 °C). 1H NMR (600 MHz, DMSO) δ 10.20 (2H, s, CNH), 8.46 (2H, d, J = 8.7 Hz, 2H), 8.37 (1H, s), 8.17 (1H, s), 7.96 (1H, d, J = 8.5 Hz), 7.78 (1H, d, J = 8.4 Hz), 7.39 (4H, m), 7.36–7.32 (3H, m), 5.64 (2H, s, OCH2), 5.32 (2H, s, OCH2), 3.53 (4H, s, NCH2), 2.07–1.96 (2H, m, CH2). 13C NMR (151 MHz, DMSO) δ 162.05, 158.94 (NCH), 151.40, 142.24, 135.92, 130.46, 128.76, 128.16, 127.97, 125.01, 124.99, 115.78, 114.25, 114.05, 61.50 (OCH2), 52.85 (NCH2), 38.85 (NCH2), 17.65 (CH2). Anal. calcd. for C27H25N7O × HCl × 0.5H2O (Mr = 509.01): C 63.71, H 5.35, N 19.26; found: C 63.62, H 5.41, N 19.13.
2-(4-((1-Benzyl-1H-1,2,3-triazol-4-yl)methoxy)-3-fluorophenyl)-5(6)-(3,4,5,6-tetrahydropyrimidin-2-yl)-1H-benzimidazole hydrochloride (18b)
Compound 18b was prepared using the above described method from 9b (150 mg, 0.48 mmol) and o-phenylenediamine 11 (92 mg, 0.48 mmol) to obtain 18b as a white solid (37.0 mg, 14%, m.p. > 240 °C). 1H NMR (600 MHz, DMSO-d6) δ 10.05 (2H, s, CNH), 8.38 (1H, s), 8.22 (2H, m), 8.09 (1H, s), 7.86 (1H, d, J = 8.5 Hz), 7.66 (2H, t, J = 8.2 Hz), 7.41–7.36 (2H, m), 7.33 (3H, m), 5.64 (2H, s, OCH2), 5.37 (2H, s, NCH2), 3.52 (4H, s, NCH2), 2.06–1.96 (2H, m, CH2). 13C NMR (151 MHz, DMSO) δ 159.48 (CNH), 152.27, 151.53 (d, JCF = 244.9 Hz), 148.46 (d, JCF = 10.4 Hz), 142.03, 135.87, 128.76, 128.18, 127.95, 125.17, 124.37, 123.06, 122.38, 120.69, 115.81, 114.91 (d, JCF = 20.7 Hz), 62.14 (OCH2), 52.86 (NCH2), 38.84 (NCH2), 17.78 (CH2). Anal. calcd. for C27H24FN7O × HCl × H2O (Mr = 536.00): C 60.50, H 5.08, N 18.29; found: C 60.41, H 5.17, N 18.17.
2-(4-((1-Benzyl-1H-1,2,3-triazol-4-yl)methoxy)-3-methoxyphenyl)-5(6)-(3,4,5,6-tetrahydropyrimidin-2-yl)-1H-benzimidazole hydrochloride (18c)
Compound 18c was prepared using the above described method from 9c (150 mg, 0.46 mmol) and o-phenylenediamine 11 (89 mg, 0.46 mmol) to obtain 18c as a white solid (65.4 mg, 26%, m.p. > 240 °C). 1H NMR (600 MHz, DMSO-d6) δ 10.07 (2H, s, CNH), 8.35 (1H, s), 8.08 (2H, m), 8.00 (1H, d, J = 7.9 Hz), 7.89 (1H, d, J = 8.5 Hz), 7.68 (1H, d, J = 8.4 Hz), 7.45 (1H, d, J = 8.6 Hz), 7.43–7.29 (5H, m), 5.64 (2H, s, OCH2), 5.27 (2H, s, NCH2), 3.90 (3H, s, OCH3), 3.53 (4H, s, CH2), 2.07–1.97 (2H, m, CH2). 13C NMR (151 MHz, DMSO) δ 159.36 (CNH), 152.85, 150.73, 149.26, 142.37, 135.91, 128.76, 128.17, 127.99, 125.05, 123.60, 122.97, 121.11, 113.53, 110.94, 61.69 (OCH2), 55.93 (OCH3), 52.84 (NCH2), 38.85 (NCH2), 17.74 (CH2). Anal. calcd. for C28H27N7O2 × HCl × 1.5H2O (Mr = 557.05): C 60.37, H 5.61, N 17.60; found: C 63.52, H 5.47, N 17.51.
2-(4-((1-(2-Morpholinoethyl)-1H-1,2,3-triazol-4-yl)methoxy)phenyl)-5(6)-(3,4,5,6-tetrahydropyrimidin-2-yl)-1H-benzimidazole dihydrochloride (19a)
Compound 19a was prepared using the above described method from 10a (150 mg, 0.47 mmol) and o-phenylenediamine 11 (91 mg, 0.47 mmol) to obtain 19a as a white solid (53.4 mg, 21%, m.p. > 240 °C). 1H NMR (300 MHz, DMSO) δ 11.39 (1H, s, NH+), 10.04 (2H, s, CNH), 8.42 (1H, s), 8.34 (2H, d, J = 9.0 Hz), 8.08 (1H, s), 7.86 (1H, d, J = 8.4 Hz), 7.65 (1H, d, J = 8.4 Hz), 7.33 (2H, d, J = 8.9 Hz), 5.33 (2H, s, OCH2), 4.94 (2H, t, J = 6.7 Hz, NCH2), 3.96 (s), 3.53 (4H, s, NCH2), 3.43 (2H, m, NCH2), 2.09–1.95 (2H, m, CH2). 13C NMR (101 MHz, DMSO) δ 159.95 (CNH), 153.55, 144.37, 143.03, 129.88, 125.97, 125.69, 115.98, 63.59 (OCH2), 61.79 (OCH2), 54.75 (NCH2), 51.77 (NCH2), 44.13 (NCH2), 18.30 (CH2). Anal. calcd. for C26H30N8O2 × 2HCl × 2.25H2O (Mr = 600.03): C 52.05, H 6.13, N 18.68; found: C 52.13, H 6.01, N 18.60.
2-(4-((1-(2-Morpholinoethyl)-1H-1,2,3-triazol-4-yl)methoxy)-3-methoxyphenyl)-5(6)-(3,4,5,6-tetrahydropyrimidin-2-yl)-1H-benzimidazole dihydrochloride (19c)
Compound 19c was prepared using the above described method from 10c (150 mg, 0.43 mmol) and o-phenylenediamine 19 (83 mg, 0.43 mmol) to obtain 19c as a white solid (52.3 mg, 21%, m.p. = 205–209 °C). 1H NMR (400 MHz, DMSO) δ 11.68 (1H, s, NH+), 10.24 (2H, s, CNH), 8.43 (1H, s), 8.31 (1H, s), 8.20 (2H, d, J = 10.5 Hz), 7.98 (1H, d, J = 8.5 Hz), 7.81 (1H, d, J = 8.5 Hz), 7.53 (1H, d, J = 8.7 Hz), 5.35 (2H, s, OCH2), 4.97 (2H, t, J = 6.9 Hz, CH2), 3.94 (s, OCH3), 3.74–3.65 (m, CH2), 3.54 (s, CH2), 2.03 (2H, s, CH2). 13C NMR (151 MHz, DMSO) δ 158.92 (CNH), 151.77, 151.37, 149.34, 142.34, 135.26, 132.17, 125.66, 125.18, 124.64, 122.43, 115.71, 114.16, 113.92, 113.59, 111.92, 63.03 (OCH2), 61.76 (OCH2), 56.39 (OCH3), 54.14 (NCH2), 51.21 (NCH2), 43.61 (NCH2), 38.88 (NCH2), 17.67 (CH2). Anal. calcd. for C26H30N8O2 × 2HCl × 1.7.5H2O (Mr = 621.04): C 52.22, H 6.09, N 18.04; found: C 52.31, H 5.98, N 18.10.
4.2.3. General procedure for the synthesis of 2,6-disubstituted benzothiazole derivatives 21a–21b, 22c, 23a–23c, 24a, and 24c
To a stirred solution of amidino-substituted 2-aminobenzenethiolate 20 (1 eq.) in glacial acetic acid (3 ml), a corresponding benzaldehyde (1 eq.) was added. The reaction mixture was stirred and heated under nitrogen for 3 h, then poured onto ice and made alkaline (pH 10–11) with 20% NaOH. The resulting free base was filtered, washed with water and dried. The free base was suspended in ethanol/HCl(g) (10 ml) and stirred at room temperature for 24 h. The addition of ether resulted in precipitation of products. The solid was collected by filtration, washed with anhydrous ether, and dried under vacuum.
2-(4-(2-Oxo-2-phenylethoxy)phenyl)-6-(1,4,5,6-tetrahydropyrimidin-2-yl)benzothiazole hydrochloride (21a)
Compound 21a was prepared using the above mentioned procedure from 20 (60.0 mg, 0.29 mmol) and 6a (69.7 mg, 0.29 mmol) to obtain 21a as beige powder (46.6 mg, 33%; m.p. > 240 °C). 1H NMR (600 MHz, DMSO) δ 10.08 (2H, s, CNH), 8.53 (1H, d, J = 1.8 Hz), 8.21 (1H, d, J = 8.6 Hz), 8.11–8.07 (2H, m), 8.05 (2H, dd, J = 8.3, 1.2 Hz), 7.82 (1H, dd, J = 8.5, 1.9 Hz), 7.74–7.70 (1H, m), 7.62–7.57 (2H, m), 7.22–7.16 (2H, m), 5.76 (2H, s, OCH2), 3.52 (4H, s, NCH2), 2.01 (2H, dd, J = 11.0, 5.3 Hz, CH2). 13C NMR (75 MHz, DMSO) δ 194.49 (C O), 161.68, 159.59 (CNH), 156.82, 134.68, 134.43, 129.76, 129.36, 128.37, 126.38, 125.68, 125.50, 123.14, 123.01, 116.10, 70.80 (OCH2), 18.20 (CH2). Anal. calcd. for C25H21N3O2S × HCl × H2O (Mr = 481.99): C 62.30, H 5.02, N 8.72; found: C 62.41, H 4.94, N 8.63.
2-(3-Fluoro-4-(2-oxo-2-phenylethoxy)phenyl)-6-(1,4,5,6-tetrahydropyrimidin-2-yl)benzothiazole hydrochloride (21b)
Compound 21b was prepared using the above mentioned procedure from 20 (60.0 mg, 0.29 mmol) and 6b (74.9 mg, 0 29 mmol) to obtain 21b as brown powder (32.0 mg, 21%; m.p. > 240 °C). 1H NMR (300 MHz, DMSO) δ 10.15 (2H, s, CNH), 8.58 (1H, d, J = 1.7 Hz), 8.24 (1H, d, J = 8.6 Hz), 8.09–7.99 (3H, m), 7.89 (1H, d, J = 9.6 Hz), 7.85 (1H, dd, J = 8.6, 1.9 Hz), 7.73 (1H, t, J = 7.4 Hz), 7.61 (2H, t, J = 7.5 Hz), 7.35 (1H, t, J = 8.7 Hz), 5.90 (2H, s, OCH2), 3.53 (4H, s, NCH2), 2.09–1.93 (2H, m, CH2). 13C NMR (75 MHz, CDCl3) δ 198.82 (C O), 164.28, 161.31, 158.38 (CNH), 156.75 (d, JCF = 246.1 Hz), 154.49, 154.36, 139.96, 139.28, 134.13, 133.13, 131.25, 130.72 (d, JCF = 7.0 Hz), 130.67, 129.82, 128.00 (d, JCF = 13.8 Hz), 120.92, 120.23, 119.97, 76.11 (OCH2), 22.93 (CH2). Anal. calcd. for C25H20N3FO2S × HCl × 1.5H2O (Mr = 508.99): C 58.99, H 4.75, N 8.26; found: C 59.07, H 4.66, N 8.31.
2-(3-Methoxy-4-(pyridin-2-ylmethoxy)phenyl)-6-(1,4,5,6-tetrahydropyrimidin-2-yl)benzothiazole hydrochloride (22c)
Compound 22c was prepared using the above mentioned procedure from 20 (60.0 mg, 0.29 mmol) and 7c (70.5 mg, 0.29 mmol) to obtain 22c as brown powder (45.2 mg, 30%; m.p. > 240 °C). 1H NMR (300 MHz, DMSO) δ 10.13 (2H, s, CNH), 8.67 (1H, d, J = 5.0 Hz), 8.55 (1H, s), 8.24 (1H, d, J = 8.6 Hz), 8.01 (1H, t, J = 7.2 Hz), 7.83 (1H, dd, J = 8.6, 1.8 Hz), 7.75–7.64 (3H, m), 7.51 (1H, dd, J = 6.9, 5.3 Hz), 7.26 (1H, d, J = 8.4 Hz), 5.36 (2H, s, OCH2), 3.95 (3H, s, OCH3), 3.53 (4H, s, NCH2), 2.07–1.95 (2H, m, CH2). 13C NMR (75 MHz, DMSO) δ 171.50, 159.62 (CNH), 156.71, 156.11, 151.41, 149.91, 149.09, 138.43, 135.10, 126.39, 125.97, 125.58, 123.96, 123.17, 122.98, 122.70, 121.94, 114.09, 110.40, 71.02 (OCH2), 56.29 (OCH3), 18.19 (CH2). Anal. calcd. for C24H22N4O2S × HCl × 2H2O (Mr = 503.01): C 57.31, H 5.41, N 11.14; found: C 57.46, H 5.29, N 11.02.
2-(4-((1H-1,2,3-Triazol-4-yl)methoxy)phenyl)-6-(1,4,5,6-tetrahydropyrimidin-2-yl)benzothiazole hydrochloride (23a)
Compound 23a was prepared using the above mentioned procedure from 20 (60.0 mg, 0.29 mmol) and 8a (58.9 mg, 0.29 mmol) to obtain 23a as beige powder (31.6 mg, 24%; m.p. > 240 °C). 1H NMR (300 MHz, DMSO) δ 10.09 (2H, s, CNH), 8.50 (1H, d, J = 1.6 Hz), 8.15 (1H, d, J = 8.6 Hz), 8.05 (2H, d, J = 8.8 Hz), 7.97 (1H, s), 7.77 (1H, dd, J = 8.6, 1.8 Hz), 7.20 (2H, d, J = 8.9 Hz), 5.26 (2H, s, OCH2), 3.46 (4H, s, NCH2), 1.94 (2H, s, CH2). 13C NMR (75 MHz, DMSO) δ 171.39, 161.61, 159.56 (NCH), 156.80, 135.01, 131.04, 129.85, 126.39, 125.69, 125.50, 123.14, 123.02, 116.18, 61.61 (OCH2), 39.36 (NCH2), 18.19 (CH2). Anal. calcd. for C20H18N6OS × HCl × 1.25H2O (Mr = 449.44): C 53.45, H 4.82, N 18.70; found: C 53.59, H 4.73, N 18.57.
2-(3-Fluoro-4-((1H-1,2,3-triazol-4-yl)methoxy)phenyl)-6-(1,4,5,6-tetrahydropyrimidin-2-yl)benzothiazole hydrochloride (23b)
Compound 23b was prepared using the above mentioned procedure from 20 (60.0 mg, 0.29 mmol) and 8b (64.1 mg, 0.29 mmol) to obtain 23b as brown powder (49.6 mg, 36%; m.p. 187–190 °C). 1H NMR (300 MHz, DMSO) δ 10.40 (2H, s, CNH), 8.67 (1H, d, J = 1.2 Hz), 8.21 (1H, d, J = 8.6 Hz), 8.08 (1H, s), 8.04–7.86 (3H, m), 7.60 (1H, t, J = 8.8 Hz), 5.42 (2H, s, OCH2), 3.52 (4H, s, NCH2), 2.00 (2H, s, CH2). 13C NMR (75 MHz, DMSO) δ 169.60, 158.66 (CNH), 156.02, 151.69 (d, JCF = 246.2 Hz), 149.07, 148.93, 134.60, 126.07, 125.56 (d, JCF = 7.1 Hz), 125.10, 124.80, 124.76, 122.78, 122.72, 115.84, 114.85, 114.59, 61.96 (OCH2), 38.79 (NCH2), 17.70 (CH2). Anal. calcd. for C20H17FN6OS × HCl × 1.25H2O (Mr = 467.43): C 51.39, H 4.42, N 17.98; found: C 51.45, H 4.35, N 17.86.
2-(3-Methoxy-4-((1H-1,2,3-triazol-4-yl)methoxy)phenyl)-6-(1,4,5,6-tetrahydropyrimidin-2-yl)benzothiazole hydrochloride (23c)
Compound 23c was prepared using the above mentioned procedure from 20 (60.0 mg, 0.29 mmol) and 8c (67.6 mg, 0.29 mmol) to obtain 23c as yellow powder (54.9 mg, 38%; m.p. 178–181 °C). 1H NMR (300 MHz, DMSO) δ 10.28 (2H, s, CNH), 8.56 (1H, d, J = 1.6 Hz), 8.14 (1H, d, J = 8.6 Hz), 7.96 (1H, s), 7.83 (1H, dd, J = 8.6, 1.8 Hz, 1H), 7.67–7.59 (2H, m), 7.30 (1H, d, J = 8.4 Hz), 5.23 (2H, s, OCH2), 3.82 (3H, s, OCH3), 3.46 (4H, s, NCH2), 1.93 (2H, s, CH2). 13C NMR (151 MHz, DMSO) δ 170.98, 158.80 (CNH), 156.21, 150.83, 149.37, 134.51, 125.94, 125.37, 124.90, 122.57, 122.55, 121.36, 113.56, 109.84, 61.39 (OCH2), 55.66 (OCH3), 38.81 (NCH2), 17.71 (CH2). Anal. calcd. for C21H20N6O2S × HCl × 1.75H2O (Mr = 488.48): C 51.64, H 5.06, N 17.20; found: C 51.52, H 5.18, N 17.26.
2-(4-((1-Benzyl-1H-1,2,3-triazol-4-yl)methoxy)phenyl)-6-(1,4,5,6-tetrahydropyrimidin-2-yl)benzothiazole hydrochloride (24a)
Compound 24a was prepared using the above mentioned procedure from 20 (60.0 mg, 0.29 mmol) and 9a (85.1 mg, 0.29 mmol) to obtain 24a as brown powder (24.5 mg, 15%; m.p. 220–223 °C). 1H NMR (300 MHz, DMSO) δ 10.12 (2H, s, CNH), 8.51 (1H, d, J = 1.6 Hz), 8.28 (1H, s), 8.14 (1H, d, J = 8.6 Hz), 8.04 (2H, d, J = 8.8 Hz), 7.78 (1H, dd, J = 8.6, 1.8 Hz), 7.36–7.23 (5H, m), 7.19 (2H, d, J = 8.9 Hz), 5.57 (2H, s, NCH2), 5.21 (2H, s, OCH2), 3.46 (4H, s, NCH2), 1.94 (2H, s, CH2). 13C NMR (75 MHz, DMSO) δ 171.35, 161.64, 159.43 (CNH), 156.81, 142.94, 136.44, 134.98, 129.83, 129.25, 128.65, 128.45, 126.41, 125.68, 125.43, 125.40, 123.08, 116.16, 61.88 (OCH2), 53.34 (NCH2), 39.33 (NCH2), 18.21 (CH2). Anal. calcd. for C21H24N6OS × HCl × 2H2O (Mr = 553.08): C 57.67, H 5.36, N 14.41; found: C 57.83, H 5.23, N 14.29.
2-(3-Methoxy-4-((1-benzyl-1H-1,2,3-triazol-4-yl)methoxy)phenyl)-6-(1,4,5,6-tetrahydropyrimidin-2-yl)benzothiazole hydrochloride (24c)
Compound 24c was prepared using the above mentioned procedure from 20 (60.0 mg, 0.29 mmol) and 9c (93.8 mg, 0.29 mmol) to obtain 24c as brown powder (13.2 mg, 7%; m.p. 183–186 °C). 1H NMR (300 MHz, DMSO) δ 10.13 (2H, s, CNH), 8.56 (1H, s), 8.34 (1H, s), 8.23 (1H, d, J = 8.6 Hz), 7.83 (1H, d, J = 8.9 Hz), 7.74–7.67 (2H, m), 7.41–7.29 (5H, m), 5.63 (2H, s, NCH2), 5.26 (2H, s, OCH2), 3.87 (3H, s, OCH3), 3.53 (4H, s, NCH2), 2.01 (2H, s, CH2). 13C NMR (75 MHz, DMSO) δ 171.01, 158.96 (CNH), 156.23, 153.52, 150.87, 142.39, 135.94, 134.55, 128.75, 127.98, 125.92, 125.34, 125.04, 124.97, 122.59, 121.36, 113.57, 112.16, 109.78, 61.71 (OCH2), 55.62 (OCH3), 52.82 (NCH2), 38.85 (NCH2), 17.71 (CH2). Anal. calcd. for C28H26N6O2S × HCl × 2.5H2O (Mr = 592.11): C 56.80, H 5.45, N 14.19; found: C 56.97, H 5.36, N 14.03.
4.3. Antibacterial screening
Antibacterial susceptibility was determined against standard bacterial strains obtained from ATCC: Staphylococcus aureus (ATCC 29213), Enterococcus faecalis (ATCC29212) and Moraxella catarrhalis (ATCC 23246). The hypersensitive strain of Escherichia coli due to efflux pump deficiency (AcrAB−) was used to enable compounds to reach the intercellular targets. Clinical strains of streptococci were used: S. pneumoniae B0652 (sensitive), S. pneumoniae B0326 (M (efflux) phenotype), S. pyogenes B0542 (sensitive), and S. pyogenes B0545 (M (efflux) phenotype).
Minimum inhibitory concentrations (MICs) were determined by the broth microdilution method according to the guidelines of the Clinical Laboratory Standards Institute.77 Double dilutions of tested compounds in 96-well microtitre plates were prepared in the 128–0.25 mg mL−1 concentration range, in cation adjusted Mueller Hinton broth (MHB). For testing of streptococci MHB was supplemented with 5% horse serum (Gibco, Life technologies Europe, Bleiswijk, Netherlands). E. coli and S. aureus were grown on Mueller–Hinton agar (MHA) plates (by Becton Dickinson, USA), while E. faecalis, M. catarrhalis and streptococci strains were grown on MHA with 5% sheep blood (Biognost, Zagreb, Croatia). Inocula were prepared by the direct colony suspension method and plates were inoculated with 5 × 104 CFU per well. Results were determined by visual inspection after 20–22 h of incubation at 37 °C in ambient air.
4.4. Cytotoxic assay
Cytotoxicity of compounds was determined on HepG2 human hepatocellular carcinoma cell line HB-8065 (ATCC, Manassas, VA, USA). Cells were maintained in complete DMEM/F12 medium supplemented with 10% fetal bovine serum (both Sigma-Aldrich Chemie GmbH, Taufkirchen, Germany), at 37 °C in 5% CO2 atmosphere. The assay was performed using the MTS CellTiter 96 AQueous One Solution Cell Proliferation Assay (Promega, G3580, Madison, USA).58 Double dilutions of compounds were prepared in the concentration range of 100–0.05 μM within microplate wells. In each well, 5 × 104 cells were added and the plates were incubated overnight at 37 °C in 5% CO2 atmosphere. Following this, 10 μL of MTS (3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfo-phenyl)-2H-tetrazolium) reagent was dispensed per well and the plates were incubated for 1–6 h at 37 °C in 5% CO2 atmosphere. Control wells consisted of media only (blank) or cells with 1% DMSO added (control). The absorbance was measured at 490 nm using a 96-well Spectramax i3x plate reader (Agilent, Santa Clara, USA). Results were analyzed in GraphPad Prism software (GraphPad Software, San Diego, USA).
4.5. DNA and RNA binding study
Compounds 15a–15c, 16a, 16c, 21a and 21b were dissolved (c = 1 × 10−3 mol dm−3) in water while compound 17a was dissolved (c = 1 × 10−3 mol dm−3) in DMSO. Those solutions were used for measurements in aqueous buffer (pH = 7.0, sodium cacodylate buffer, I = 0.05 mol dm−3). Polynucleotides were purchased as noted: calf thymus ctDNA, poly A–poly U and poly(dAdT)2 (Sigma-Aldrich, St. Louis, MI, USA). Polynucleotides were dissolved in Na-cacodylate buffer, I = 0.05 mol dm−3, pH = 7.0. The calf thymus ctDNA was additionally sonicated and filtered through a 0.45 mm filter.78 Polynucleotide concentration was determined spectroscopically79,80 as the concentration of phosphates.
4.5.1. UV/vis measurements
The UV/vis spectra were recorded on a Varian Cary 100 Bio spectrophotometer using 1 cm path quartz cuvettes. Calibration experiments were performed at 25 °C and pH = 7.0 (I = 0.05 mol dm−3, sodium cacodylate buffer). Absorption maxima and corresponding molar extinction coefficients (ε) of benzothiazole derivatives are given in Table S2 (ESI†). Thermal melting curves for DNA and their complexes with the studied compounds were determined as previously described by following the absorption change at 260 nm as a function of temperature. The absorbance of the ligands was subtracted from every curve and the absorbance scale was normalized. Tm values are the midpoints of the transition curves determined from the maximum of the first derivative and checked graphically by the tangent method. The ΔTm values were calculated by subtracting Tm of the free nucleic acid from Tm of the complex. Every ΔTm value here reported was the average of at least two measurements. The error in ΔTm is ±0.5 °C.
4.5.2. Fluorimetric measurements
Fluorescence spectra were recorded on a Varian Cary Eclipse spectrophotometer at 25 °C using appropriate 1 cm path quartz cuvettes. Fluorimetric experiments were performed at pH = 7.0 (I = 0.05 mol dm−3, sodium cacodylate buffer) by adding portions of polynucleotide solution into the solution of the studied compound. In fluorimetric experiments, an excitation wavelength of λexc ≥ 300 nm was used to avoid the inner filter effect caused by the increasing absorbance of the polynucleotide. Emission was collected in the range λem = 320–500 nm. Values for Ks obtained by processing titration data using the Scatchard equation63 all have satisfactory correlation coefficients (>0.99).
4.5.3. CD measurements
CD spectra were recorded on a JASCO J815 spectrophotometer in 1 cm path quartz cuvettes. CD parameters: range = 500–220 nm, data pitch = 2, standard sensitivity, scanning speed = 200 nm min−1, accumulation = 3. Titrations were performed at the temperature of 25 °C and pH = 7 (I = 0.05 mol dm−3, sodium cacodylate buffer). CD experiments were done by adding portions of the compound stock solution into the solution of polynucleotide.
4.6. Computational methods
Available X-ray structures of DNA complexes with small ligands were downloaded from the Protein data bank.81 The QM calculations and the ligand docking studies were carried out using the Glide docking protocol with extra precision (XP)82 within the Schrödinger suite of software.83 Binding poses were refined and binding energy was estimated using the MM–GBSA84–86 protocol and OPLS3 force-field with flexible residues with the distance being 5 Å.
Molecular dynamics simulations were carried out using Desmond software within the Schrödinger package.87,88 Simulations were carried out at room temperature for 20 ns. In silico ADME properties as well as structural parameters were calculated by ACD Percepta software.89
Conflicts of interest
There are no conflicts to declare.
Supplementary Material
Acknowledgments
We greatly appreciate the financial support of the Croatian Science Foundation (project No. IP-2018-01-4682, project No. IP-2018-01-4694).
Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2md00143h
Notes and references
- Zaman S. B. Hussain M. A. Nye R. Mehta V. Mamun K. T. Hossain N. A review on antibiotic resistance: Alarm bells are ringing. Cureus. 2017;9:e1403. doi: 10.7759/cureus.1403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aslam B. Wang W. Arshad M. I. Khurshid M. Muzammil S. Rasool M. H. Nisar M. A. Alvi R. F. Aslam M. A. Qamar M. U. Salamat M. K. F. Baloch Z. Antibiotic resistance: a rundown of a global crisis. Infect. Drug Resist. 2018;11:1645–1658. doi: 10.2147/IDR.S173867. doi: 10.2147/IDR.S173867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vivas R. Barbosa A. A. T. Dolabela S. S. Jain S. Multidrug-resistant bacteria and alternative methods to control them: An overview. Microb. Drug Resist. 2019;25:890–908. doi: 10.1089/mdr.2018.0319. doi: 10.1089/mdr.2018.0319. [DOI] [PubMed] [Google Scholar]
- van Duin D. Paterson D. L. Multidrug-resistant bacteria in the community: Trends and lessons learned. Infect. Dis. Clin. North Am. 2016;30:377–390. doi: 10.1016/j.idc.2016.02.004. doi: 10.1016/j.idc.2016.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hasan C. M. Dutta D. Nguyen A. N. T. Revisiting antibiotic resistance: Mechanistic foundations to evolutionary outlook. Antibiotics. 2022;11:40. doi: 10.3390/antibiotics11010040. doi: 10.3390/antibiotics11010040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jubeh B. Breijyeh Z. Karaman R. Resistance of Gram-positive bacteria to current antibacterial agents and overcoming approaches. Molecules. 2020;25:2888. doi: 10.3390/molecules25122888. doi: 10.3390/molecules25122888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- el Chakhtoura N. G. Saade E. Iovleva A. Yasmin M. Wilson B. Perez F. Bonomo R. A. Therapies for multidrug resistant and extensively drug-resistant non-fermenting Gram-negative bacteria causing nosocomial infections: a perilous journey toward “molecularly targeted” therapy. Expert Rev. Anti-infect. Ther. 2018;16:89–110. doi: 10.1080/14787210.2018.1425139. doi: 10.1080/14787210.2018.1425139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bassetti M. Peghin M. Vena A. Giacobbe D. R. Treatment of infections due to MDR Gram-negative bacteria. Front. Med. 2019;6:74. doi: 10.3389/fmed.2019.00074. doi: 10.3389/fmed.2019.00074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reygaert W. C. An overview of the antimicrobial resistance mechanisms of bacteria. AIMS Microbiol. 2019;4:482–501. doi: 10.3934/microbiol.2018.3.482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garousi M. Monazami Tabar S. Mirazi H. Asgari P. Sabeghi P. Salehi A. Khaledi A. Sanani M. G. P. Mirzahosseini H. K. A global systematic review and meta-analysis on correlation between biofilm producers and non-biofilm producers with antibiotic resistance in Uropathogenic Escherichia coli. Microb. Pathog. 2022;164:105412. doi: 10.1016/j.micpath.2022.105412. doi: 10.1016/j.micpath.2022.105412. [DOI] [PubMed] [Google Scholar]
- de Vries S. P. W. Eleveld M. J. Hermans P. W. M. Bootsma H. J. Characterization of the molecular interplay between Moraxella catarrhalis and human respiratory tract epithelial cells. PLoS One. 2013;8:e72193. doi: 10.1371/journal.pone.0072193. doi: 10.1371/journal.pone.0072193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maračić S. Kraljević T. G. Čipić Paljetak H. Perić M. Matijašić M. Verbanac D. Cetina M. Raić-Malić S. 1,2,3-Triazole pharmacophore-based benzofused nitrogen/sulfur heterocycles with potential anti-Moraxella catarrhalis activity. Bioorg. Med. Chem. 2015;23:7448–7463. doi: 10.1016/j.bmc.2015.10.042. doi: 10.1016/j.bmc.2015.10.042. [DOI] [PubMed] [Google Scholar]
- Perin N. Starčević K. Perić M. Čipić Paljetak H. Matijašić M. Stepanić V. Verbanac D. Karminski-Zamola G. Hranjec M. Synthesis and SAR study of novel amidino 2-substituted benzimidazoles as potential antibacterial agents. Croat. Chem. Acta. 2017;90:145–154. doi: 10.5562/cca3147. doi: 10.5562/cca3147. [DOI] [Google Scholar]
- de Vries S. P. W. van Hijum S. A. F. T. Schueler W. Riesbeck K. Hays J. P. Hermans P. W. M. Bootsma H. J. Genome analysis of Moraxella catarrhalis strain RH4, a human respiratory tract pathogen. J. Bacteriol. 2010;192:3574–3583. doi: 10.1128/JB.00121-10. doi: 10.1128/JB.00121-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larsson D. G. J. Flach C. F. Antibiotic resistance in the environment. Nat. Rev. Microbiol. 2022;20:257–269. doi: 10.1038/s41579-021-00649-x. doi: 10.1038/s41579-021-00649-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hiramatsu K. Cui L. Kuroda M. Ito T. The emergence and evolution of methicillin-resistant Staphylococcus aureus. Trends Microbiol. 2001;9:486–493. doi: 10.1016/S0966-842X(01)02175-8. doi: 10.1016/S0966-842X(01)02175-8. [DOI] [PubMed] [Google Scholar]
- Bistrović A. Krstulović L. Stolić I. Drenjančević D. Talapko J. Taylor M. C. Kelly J. M. Bajić M. Raić-Malić S. Synthesis, anti-bacterial and anti-protozoal activities of amidinobenzimidazole derivatives and their interactions with DNA and RNA. J. Enzyme Inhib. Med. Chem. 2018;33:1323–1334. doi: 10.1080/14756366.2018.1484733. doi: 10.1080/14756366.2018.1484733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang H. Z. Gan L. L. Wang H. Zhou C. H. New progress in azole compounds as antimicrobial agents. Mini-Rev. Med. Chem. 2017;17:122–166. doi: 10.2174/1389557516666160630120725. doi: 10.2174/1389557516666160630120725. [DOI] [PubMed] [Google Scholar]
- Peng X. M. Cai G. X. Zhou C.-H. Recent developments in azole compounds as antibacterial and antifungal agents. Curr. Top. Med. Chem. 2013;13:1963–2010. doi: 10.2174/15680266113139990125. doi: 10.2174/15680266113139990125. [DOI] [PubMed] [Google Scholar]
- Devasia J. Nizam A. Vasantha V. L. Azole-based antibacterial agents: A review on multistep synthesis strategies and biology. Polycyclic Aromat. Compd. 2021 doi: 10.1080/10406638.2021.1938615. [DOI] [Google Scholar]; , In Press, Corrected Proof
- Ahmad K. Khan M. K. A. Baig M. H. Imran M. Gupta G. K. Role of azoles in cancer prevention and treatment: Present and future perspectives. Anti-Cancer Agents Med. Chem. 2018;18:46–56. doi: 10.2174/1871520616666161221112042. doi: 10.2174/1871520616666161221112042. [DOI] [PubMed] [Google Scholar]
- Bistrović A. Krstulović L. Harej A. Grbčić P. Sedić M. Koštrun S. Kraljević Pavelić S. Bajić M. Raić-Malić S. Design, synthesis and biological evaluation of novel benzimidazole amidines as potent multi-target inhibitors for the treatment of non-small cell lung cancer. Eur. J. Med. Chem. 2018;143:1616–1634. doi: 10.1016/j.ejmech.2017.10.061. doi: 10.1016/j.ejmech.2017.10.061. [DOI] [PubMed] [Google Scholar]
- Kanwal A. Ahmad M. Aslam S. Naqvi S. A. R. Saif M. J. Recent advances in antiviral benzimidazole derivatives: A mini review. Pharm. Chem. J. 2019;53:179–187. doi: 10.1007/s11094-019-01976-3. doi: 10.1007/s11094-019-01976-3. [DOI] [Google Scholar]
- Biastrović Popov A. Stolić I. Krstulović L. Taylor M. C. Kelly J. M. Tomić S. Tumir L. Bajić M. Raić-Malić S. Novel symmetric bis-benzimidazoles: Synthesis, DNA/RNA binding and antitrypanosomal activity. Eur. J. Med. Chem. 2019;173:63–75. doi: 10.1016/j.ejmech.2019.04.007. doi: 10.1016/j.ejmech.2019.04.007. [DOI] [PubMed] [Google Scholar]
- Bistrović Popov A. Krstulović L. Koštrun S. Jelić D. Bokulić A. Radić Stojković M. Zonjić I. Taylor M. C. Kelly J. M. Bajić M. Raić-Malić S. Design, synthesis, antitrypanosomal activity, DNA/RNA binding and in vitro ADME profiling of novel imidazoline-substituted 2-arylbenzimidazoles. Eur. J. Med. Chem. 2020;207:112802. doi: 10.1016/j.ejmech.2020.112802. doi: 10.1016/j.ejmech.2020.112802. [DOI] [PubMed] [Google Scholar]
- Veerasamy R. Roy A. Karunakaran R. Rajak H. Structure–activity relationship analysis of benzimidazoles as emerging anti-inflammatory agents: An overview. Pharmaceuticals. 2021;14:663. doi: 10.3390/ph14070663. doi: 10.3390/ph14070663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prasher P. Sharma M. Zacconi F. Gupta G. Aljabali A. A. A. Mishra V. Tambuwala M. M. Kapoor D. N. Negi P. Pinto T. D. J. A. Singh I. Chellappan D. K. Dua K. Synthesis and anticancer properties of ‘azole’ based chemotherapeutics as emerging chemical moieties: A comprehensive review. Curr. Org. Chem. 2020;25:654–668. doi: 10.2174/1385272824999200820152501. doi: 10.2174/1385272824999200820152501. [DOI] [Google Scholar]
- Vasava M. S. Bhoi M. N. Rathwa S. K. Jethava D. J. Acharya P. T. Patel D. B. Patel H. D. Benzimidazole: A milestone in the field of medicinal chemistry. Mini-Rev. Med. Chem. 2019;20:532–565. doi: 10.2174/1389557519666191122125453. doi: 10.2174/1389557519666191122125453. [DOI] [PubMed] [Google Scholar]
- Yurttaş L. Çiftçi G. A. Aksoy M. O. Demirayak Ş. Novel benzimidazole derivatives: Cytotoxic and apoptotic properties on lung cancer cell line. Lett. Drug Des. Discovery. 2020;17:1227–1236. doi: 10.2174/1570180817999200513091613. doi: 10.2174/1570180817999200513091613. [DOI] [Google Scholar]
- Tahlan S. Kumar S. Kakkar S. Narasimhan B. Benzimidazole scaffolds as promising antiproliferative agents: A review. BMC Chem. 2019;13:66. doi: 10.1186/s13065-019-0579-6. doi: 10.1186/s13065-019-0579-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hashem H. E. el Bakri Y. An overview on novel synthetic approaches and medicinal applications of benzimidazole compounds. Arabian J. Chem. 2021;14:103418. doi: 10.1016/j.arabjc.2021.103418. doi: 10.1016/j.arabjc.2021.103418. [DOI] [Google Scholar]
- Bansal Y. Kaur M. Bansal G. Antimicrobial potential of benzimidazole derived molecules. Mini-Rev. Med. Chem. 2019;19:624–646. doi: 10.2174/1389557517666171101104024. doi: 10.2174/1389557517666171101104024. [DOI] [PubMed] [Google Scholar]
- Song D. Ma S. Recent Development of Benzimidazole-Containing Antibacterial Agents. ChemMedChem. 2016;11:646–659. doi: 10.1002/cmdc.201600041. doi: 10.1002/cmdc.201600041. [DOI] [PubMed] [Google Scholar]
- Zhang B. Pang L. Nautiyal M. de Graef S. Gadakh B. Lescrinier E. Rozenski J. Strelkov S. V. Weeks S. D. van Aerschot A. Synthesis and biological evaluation of 1,3-dideazapurine-like 7-amino-5-hydroxymethyl-benzimidazole ribonucleoside analogues as aminoacyl-tRNA synthetase inhibitors. Molecules. 2020;25:4751. doi: 10.3390/molecules25204751. doi: 10.3390/molecules25204751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y. N. Bheemanaboina R. R. Y. Cai G. X. Zhou C. H. Novel purine benzimidazoles as antimicrobial agents by regulating ROS generation and targeting clinically resistant Staphylococcus aureus DNA groove. Bioorg. Med. Chem. Lett. 2018;28:1621–1628. doi: 10.1016/j.bmcl.2018.03.046. doi: 10.1016/j.bmcl.2018.03.046. [DOI] [PubMed] [Google Scholar]
- Bansal S. Sur S. Tandon V. Benzimidazoles: Selective inhibitors of topoisomerase i with differential modes of action. Biochemistry. 2019;58:809–817. doi: 10.1021/acs.biochem.8b01102. doi: 10.1021/acs.biochem.8b01102. [DOI] [PubMed] [Google Scholar]
- Paul A. Guo P. Boykin D. W. Wilson W. D. A new generation of minor-groove-binding-heterocyclic diamidines that recognize G·C base pairs in an at sequence context. Molecules. 2019;24:946. doi: 10.3390/molecules24050946. doi: 10.3390/molecules24050946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paul A. Singh P. Kuznetsov M. L. Karmakar A. Guedes da Silva M. F. C. Koch B. Pombeiro A. J. L. Influence of anchoring moieties on new benzimidazole-based Schiff base copper(ii) complexes towards estrogen dependent breast cancer cells. Dalton Trans. 2021;50:3701–3716. doi: 10.1039/d0dt03873c. doi: 10.1039/d0dt03873c. [DOI] [PubMed] [Google Scholar]
- Singhal S. Khanna P. Khanna L. Synthesis, DFT studies, molecular docking, antimicrobial screening and UV fluorescence studies on ct-DNA for novel Schiff bases of 2-(1-aminobenzyl) benzimidazole. Heliyon. 2019;5:e02596. doi: 10.1016/j.heliyon.2019.e02596. doi: 10.1016/j.heliyon.2019.e02596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barančoková M. Kikelj D. Ilaš J. Recent progress in the discovery and development of DNA gyrase B inhibitors. Future Med. Chem. 2018;10:1207–1227. doi: 10.4155/fmc-2017-0257. doi: 10.4155/fmc-2017-0257. [DOI] [PubMed] [Google Scholar]
- Santos J. A. Lamers M. H. Novel antibiotics targeting bacterial replicative DNA polymerases. Antibiotics. 2020;9:1–14. doi: 10.3390/antibiotics9110776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Eijk E. Wittekoek B. Kuijper E. J. Smits W. K. DNA replication proteins as potential targets for antimicrobials in drug-resistant bacterial pathogens. J. Antimicrob. Chemother. 2017;72:1275–1284. doi: 10.1093/jac/dkw548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tse-Dinh Y. C. Targeting bacterial topoisomerase I to meet the challenge of finding new antibiotics. Future Med. Chem. 2015;7:459–471. doi: 10.4155/fmc.14.157. doi: 10.4155/fmc.14.157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barrett M. P. Gemmell C. G. Suckling C. J. Minor groove binders as anti-infective agents. Pharmacol. Ther. 2013;139:12–23. doi: 10.1016/j.pharmthera.2013.03.002. doi: 10.1016/j.pharmthera.2013.03.002. [DOI] [PubMed] [Google Scholar]
- Mann J. Taylor P. W. Dorgan C. R. Johnson P. D. Wilson F. X. Vickers R. Dale A. G. Neidle S. The discovery of a novel antibiotic for the treatment of Clostridium difficile infections: A story of an effective academic-industrial partnership. MedChemComm. 2015;6:1420–1426. doi: 10.1039/c5md00238a. doi: 10.1039/c5md00238a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sattar A. Thommes P. Payne L. Warn P. Vickers R. J. SMT19969 for Clostridium difficile infection (CDI): In vivo efficacy compared with fidaxomicin and vancomycin in the hamster model of CDI. J. Antimicrob. Chemother. 2014;70:1757–1762. doi: 10.1093/jac/dkv005. doi: 10.1093/jac/dkv005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qian X. Yanagi K. Kane A. V. Alden N. Lei M. Snydman D. R. Vickers R. J. Lee X. K. Thorpe C. M. Ridinilazole, a narrow spectrum antibiotic for treatment of Clostridioides difficile infection, enhances preservation of microbiota-dependent bile acids. Am. J. Physiol. 2020;319:227–237. doi: 10.1152/ajpgi.00046.2020.-Antibiotic. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu L. Kully M. L. Boykin D. W. Abood N. Synthesis and structure-activity relationship of dicationic diaryl ethers as novel potent anti-MRSA and anti-VRE agents. Bioorg. Med. Chem. Lett. 2009;19:4626–4629. doi: 10.1016/j.bmcl.2009.06.077. doi: 10.1016/j.bmcl.2009.06.077. [DOI] [PubMed] [Google Scholar]
- Hu L. Kully M. L. Boykin D. W. Abood N. Synthesis and in vitro activity of dicationic bis-benzimidazoles as a new class of anti-MRSA and anti-VRE agents. Bioorg. Med. Chem. Lett. 2009;19:1292–1295. doi: 10.1016/j.bmcl.2009.01.075. doi: 10.1016/j.bmcl.2009.01.075. [DOI] [PubMed] [Google Scholar]
- Pathania S. Narang R. K. Rawal R. K. Role of sulphur-heterocycles in medicinal chemistry: An update. Eur. J. Med. Chem. 2019;180:486–508. doi: 10.1016/j.ejmech.2019.07.043. doi: 10.1016/j.ejmech.2019.07.043. [DOI] [PubMed] [Google Scholar]
- Kamal A. Syed M. A. H. Mohammed S. M. Therapeutic potential of benzothiazoles: A patent review (2010-2014) Expert Opin. Ther. Pat. 2015;25:335–349. doi: 10.1517/13543776.2014.999764. doi: 10.1517/13543776.2014.999764. [DOI] [PubMed] [Google Scholar]
- Pathak N. Rathi E. Kumar N. Kini S. G. Rao C. M. A review on anticancer potentials of benzothiazole derivatives. Mini-Rev. Med. Chem. 2019;20:12–23. doi: 10.2174/1389557519666190617153213. doi: 10.2174/1389557519666190617153213. [DOI] [PubMed] [Google Scholar]
- Sumit A. Kumar A. K. Mishra, Advancement in pharmacological activities of benzothiazole and its derivatives: An up to date review. Mini-Rev. Med. Chem. 2020;21:314–335. doi: 10.2174/1389557520666200820133252. doi: 10.2174/1389557520666200820133252. [DOI] [PubMed] [Google Scholar]
- Racané L. Cindrić M. Zlatar I. Kezele T. Milić A. Brajša K. Hranjec M. Preclinical in vitro screening of newly synthesised amidino substituted benzimidazoles and benzothiazoles. J. Enzyme Inhib. Med. Chem. 2021;36:163–174. doi: 10.1080/14756366.2020.1850711. doi: 10.1080/14756366.2020.1850711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bohnert J. A. Karamian B. Nikaido H. Optimized nile red efflux assay of AcrAB-TolC multidrug efflux system shows competition between substrates. Antimicrob. Agents Chemother. 2010;54:3770–3775. doi: 10.1128/AAC.00620-10. doi: 10.1128/AAC.00620-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ansari K. F. Lal C. Synthesis, physicochemical properties and antimicrobial activity of some new benzimidazole derivatives. Eur. J. Med. Chem. 2009;44:4028–4033. doi: 10.1016/j.ejmech.2009.04.037. doi: 10.1016/j.ejmech.2009.04.037. [DOI] [PubMed] [Google Scholar]
- Wierzbowski A. K. Boyd D. Mulvey M. Hoban D. J. Zhanel G. G. Expression of the mef (E) gene encoding the macrolide efflux pump protein increases in Streptococcus pneumoniae with increasing resistance to macrolides. Antimicrob. Agents Chemother. 2005;49:4635–4640. doi: 10.1128/AAC.49.11.4635-4640.2005. doi: 10.1128/AAC.49.11.4635-4640.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mosmann T. Rapid colorimetric assay for cellular growth and survival: application to proliferation and cytotoxicity assays. J. Immunol. Methods. 1983;65:55–63. doi: 10.1016/0022-1759(83)90303-4. doi: 10.1016/0022-1759(83)90303-4. [DOI] [PubMed] [Google Scholar]
- Kaizerman J. A. Gross M. I. Ge Y. White S. Hu W. Duan J.-X. Baird E. E. Johnson K. W. Tanaka R. D. Moser H. E. Bürli R. W. DNA binding ligands targeting drug-resistant bacteria: Structure, activity, and pharmacology. J. Med. Chem. 2003;46:3914–3929. doi: 10.1021/jm030097a. doi: 10.1021/jm030097a. [DOI] [PubMed] [Google Scholar]
- Arif R. Nayab P. S. Akrema Abid M. Yadava U. Uddin R. Investigation of DNA binding and molecular docking propensity of phthalimide derivatives: in vitro antibacterial and antioxidant assay. J. Anal. Sci. Technol. 2019;10:19. doi: 10.1186/s40543-019-0177-1. doi: 10.1186/s40543-019-0177-1. [DOI] [Google Scholar]
- Hadjivassileva T. Thurston D. E. Taylor P. W. Pyrrolobenzodiazepine dimers: novel sequence-selective, DNA-interactive, cross-linking agents with activity against Gram-positive bacteria. J. Antimicrob. Chemother. 2005;56:513–518. doi: 10.1093/jac/dki256. doi: 10.1093/jac/dki256. [DOI] [PubMed] [Google Scholar]
- Cantor C. R. and Schimmel P. R., Biophysical chemistry, W. H. Freeman, San Francisco, 1980 [Google Scholar]
- Scatchard G. The attractions of proteins for small molecules and ions. Ann. N. Y. Acad. Sci. 1949;51:660–672. doi: 10.1111/j.1749-6632.1949.tb27297.x. doi: 10.1111/j.1749-6632.1949.tb27297.x. [DOI] [Google Scholar]
- McGhee J. D. von Hippel P. H. Theoretical aspects of DNA-protein interactions: Co-operative and non-co-operative binding of large ligands to a one-dimensional homogeneous lattice. J. Mol. Biol. 1974;86:469–489. doi: 10.1016/0022-2836(74)90031-X. doi: 10.1016/0022-2836(74)90031-X. [DOI] [PubMed] [Google Scholar]
- Mergny J.-L. Lacroix L. Analysis of Thermal Melting Curves. Oligonucleotides. 2003;13:515–537. doi: 10.1089/154545703322860825. doi: 10.1089/154545703322860825. [DOI] [PubMed] [Google Scholar]
- Radić Stojković M. Miljanić S. Mišković K. Glavaš-Obrovac L. Piantanida I. The phenanthridine biguanides efficiently differentiate between dGdC, dAdT and rArU sequences by two independent, sensitive spectroscopic methods. Mol. BioSyst. 2011;7:1753. doi: 10.1039/c1mb05030c. doi: 10.1039/c1mb05030c. [DOI] [PubMed] [Google Scholar]
- Demeunynck M., Bailly C. and Wilson D. W., Small Molecule DNA and RNA Binders, Wiley-VCH, Weinheim, 2002, 10.1002/3527601783 [DOI] [Google Scholar]
- Rodger A., Circular Dichroism and Linear Dichroism, in Encyclopedia of Analytical Chemistry, John Wiley & Sons, Ltd, Chichester, UK, 2014, pp. 1–34, 10.1002/9780470027318.a5402.pub2 [DOI] [Google Scholar]
- Berova N., Nakanishi K. and Woody R. W., Circular Dichroism: Principles and Applications, Wiley-VCH, New York, 2nd edn, 2000 [Google Scholar]
- Eriksson M. and Nordén B., Linear and circular dichroism of drug-nucleic acid complexes, in Methods in Enzymology. Vol. 340. Drug−Nucleic Acid Interactions, ed. J. B. Chaires and M. J. Waring, Academic Press, New York, 1st edn, 2001, pp. 68–98 [DOI] [PubMed] [Google Scholar]
- Tidwell R. R. and Boykin D. W., Dicationic DNA minor groove binders as antimicrobial agents, in Small Molecule DNA and RNA Binders: From Synthesis to Nucleic Acid Complexes, ed. M. Demeunynck, C. Bailly and W. D. Wilson, Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim, 2002, pp. 414–460, 10.1002/3527601783 [DOI] [Google Scholar]
- Tanious F. A. Veal J. M. Buczak H. Ratmeyer L. S. Wilson W. D. DAPI (4′,6-diamidino-2-phenylindole) binds differently to DNA and RNA: minor-groove binding at AT sites and intercalation at AU sites. Biochemistry. 1992;31:3103–3112. doi: 10.1021/bi00127a010. doi: 10.1021/bi00127a010. [DOI] [PubMed] [Google Scholar]
- Šmidlehner T. Piantanida I. Pescitelli G. Polarization spectroscopy methods in the determination of interactions of small molecules with nucleic acids – tutorial. Beilstein J. Org. Chem. 2018;14:84–105. doi: 10.3762/bjoc.14.5. doi: 10.3762/bjoc.14.5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olsen K. Bohr J. The generic geometry of helices and their close-packed structures. Theor. Chem. Acc. 2010;125:207–215. doi: 10.1007/s00214-009-0639-4. [DOI] [Google Scholar]
- Chandrika N. T. Shrestha S. K. Ngo H. X. Garneau-Tsodikova S. Synthesis and investigation of novel benzimidazole derivatives as antifungal agents. Bioorg. Med. Chem. 2016;24:3680–3686. doi: 10.1016/j.bmc.2016.06.010. doi: 10.1016/j.bmc.2016.06.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greenidge P. A. Kramer C. Mozziconacci J.-C. Wolf R. M. MM/GBSA Binding Energy Prediction on the PDBbind Data Set: Successes, Failures, and Directions for Further Improvement. J. Chem. Inf. Model. 2013;53:201–209. doi: 10.1021/ci300425v. doi: 10.1021/ci300425v. [DOI] [PubMed] [Google Scholar]
- Clinical Laboratory Standard Institute CLSI, Methods for dilution antimicrobial susceptibility tests for bacteria that grow aerobically, M07–A8, Wayne, PA, 2009 [Google Scholar]
- Fritzsche H. Triebel H. Chaires J. B. Dattagupta N. Crothers D. M. Studies on the interaction of anthracycline antibiotics and deoxyribonucleic acid: geometry of intercalation of iremycin and daunomycin. Biochemistry. 1982;21:3940–3946. doi: 10.1021/bi00260a006. doi: 10.1021/bi00260a006. [DOI] [PubMed] [Google Scholar]
- Bresloff J. L. Crothers D. M. Equilibrium studies of ethidium-polynucleotide interactions. Biochemistry. 1981;20:3547–3553. doi: 10.1021/bi00515a038. doi: 10.1021/bi00515a038. [DOI] [PubMed] [Google Scholar]
- Chalikian T. V. Volker J. Plum G. E. Breslauer K. J. A more unified picture for the thermodynamics of nucleic acid duplex melting: A characterization by calorimetric and volumetric techniques. Proc. Natl. Acad. Sci. U. S. A. 1999;96:7853–7858. doi: 10.1073/pnas.96.14.7853. doi: 10.1073/pnas.96.14.7853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berman H. M. The Protein Data Bank. Nucleic Acids Res. 2000;28:235–242. doi: 10.1093/nar/28.1.235. doi: 10.1093/nar/28.1.235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schrödinger Release 2019-3: Glide, Schrödinger, LLC, New York, NY, 2019 [Google Scholar]
- Schrödinger Release 2019-3, Schrödinger, LLC, New York, NY, 2017 [Google Scholar]
- Li J. Abel R. Zhu K. Cao Y. Zhao S. Friesner R. A. The VSGB 2.0 model: a next generation energy model for high resolution protein structure modeling. Proteins: Struct., Funct., Bioinf. 2011;79:2794–2812. doi: 10.1002/prot.23106. doi: 10.1002/prot.23106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jorgensen W. L. Maxwell D. S. Tirado-Rives J. Development and testing of the OPLS all-atom force field on conformational energetics and properties of organic liquids. J. Am. Chem. Soc. 1996;118:11225–11236. doi: 10.1021/ja9621760. doi: 10.1021/ja9621760. [DOI] [Google Scholar]
- Shivakumar D. Williams J. Wu Y. Damm W. Shelley J. Sherman W. Prediction of absolute solvation free energies using molecular dynamics free energy perturbation and the opls force field. J. Chem. Theory Comput. 2010;6:1509–1519. doi: 10.1021/ct900587b. doi: 10.1021/ct900587b. [DOI] [PubMed] [Google Scholar]
- Schrödinger Release 2019-3: Desmond Molecular Dynamics System, D. E. Shaw Research, New York, NY, 2019 [Google Scholar]
- Maestro-Desmond Interoperability Tools, Schrödinger, New York, NY, 2019 [Google Scholar]
- ACD/Percepta, version 2017.2.1, Advanced Chemistry Development, Inc., Toronto, On, Canada, 2019, https://www.acdlabs.com [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.