

Abstract
Degradation strategies have shown enormous promise after the inception of molecules like PROTACs (PRoteolysis TArgeting Chimeras) that induce the degradation of the substrate of choice rather than depending on blocking their catalytic activity like conventional inhibitory drugs. Over the past two decades, the application of PROTACs has made quite an impact, even reaching clinical translations. However, a major class of macromolecular targets, be that large proteins, aggregates, organelles or non-protein substrates, remain untouched when utilizing the ubiquitin–proteasomal pathway of degradation. In this review, we have attempted to cover modalities of targeted degradation that instead focus on recruiting the lysosomal pathway of degradation, which is gaining importance and being explored extensively as alternate and efficient approaches for treating disease-related milieus.
This review provides an overview of the recent development of targeted protein degradation strategies beyond PROTACs, which utilize the lysosomal pathway to clear up extracellular, membrane and/or cytosolic proteins.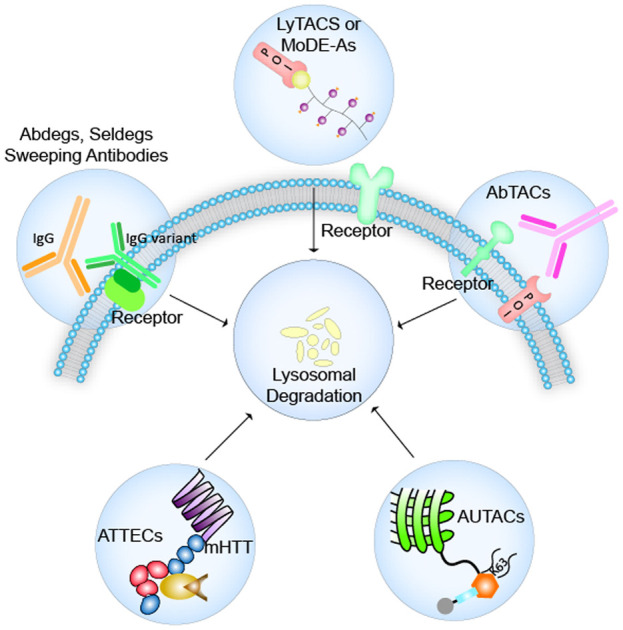
1. Introduction
Recent advancements in the development of therapeutic strategies have seen scientists focus more on the body's natural protein turnover system. Out of them, the ubiquitin–proteasome system (UPS) and autophagy–lysosome system are found to be the most significant degradation pathways.1,2 Targeting traditionally considered undruggable3 proteins, PROTACs are heterobifunctional molecules that have the ability to bind with the POI on one end and its substrate specific E3 ligase on the other end, such that the POI is brought in close proximity of the E3 ligase for poly-ubiquitination, and thus tagged for proteolysis.4 PROTACs show high selectivity for targets at sub-stoichiometric concentrations and can reduce compensatory downstream signaling pathways as they entirely degrade the target protein.5 However, the essence of the ubiquitin–proteasomal degradation system remains restricted to the intracellular targets only, whereby 40% of imminent targets are proteins found on the cytoplasmic membrane and in the extracellular cellular matrix.6,7 Several abnormal signal transduction pathways as a trigger for cancer and neurodegenerative diseases consist of potential targets such as large scaffold proteins, extracellular secreted proteins, and membrane bound receptors, which can be collected as cargo by exploiting the endocytosis–autophagy pathways leading to their lysosomal degradation.8 Here, we have explored the details of few such strategies that have been designed to extend their target beyond cytosolic proteins to membrane-bound receptors, dysfunctional mitochondria, and antibodies from the ECM (extracellular matrix).9,10 Intrigued by the action of PROTACs and specifically, their mode of exploitation of the UPS, Bertozzi and Tang in 2021 reported independently the design of two types of LYTACs (LYsosome TArgeting Chimeras) in an effort to clear extracellular and membrane-bound proteins via the lysosome.11–13 Following a similar mechanism of action, MoDE-As (molecular DEgraders of extracellular proteins through the asialoglycoprotein receptor) were developed by Spiegel.14 In 2019, Arimoto developed AUTACs (AUtophagy TArgeting Chimeras), and Lu developed ATTECs (autophagosome tethering compounds), with both strategies focusing on tethering the intended substrate to autophagosomal membrane proteins to be led to lysosomal degradation.15–17 The approach of exploiting mutant antibodies, especially IgG variants to enhance clearance of wild type IgGs in auto-immune diseases, was tackled in the development of Abdegs (antibodies targeting FcRn for IgG degradation), Seldegs (selective degradation) and sweeping antibodies.18–20 AbTACs (antibody targeting chimeras), reported by Cotton et al., were designed to pick up membrane-bound receptors for clearance in the lysosome, resembling the degradation principle similar to what is seen in LYTACs and MoDE-As.21
Over the last two decades, much attention has been given to the extensive study of the ubiquitin proteasomal system (UPS) with the report of PROTACs which target intracellular proteins. However, a major limitation of PROTACs and the UPS is that they can only be used to degrade single protein molecules at a time.22,23 This is where the endosomal–lysosomal and the autophagy–lysosomal system (ALS), if recruited, can be employed to remove protein aggregates, large non-protein components and even dysfunctional organelles and their fragments, extending the target scope from intracellular to extracellular cargo as well as cell membrane-bound proteins and receptors.
The endosomal–lysosomal pathway (Fig. 1) is a part of endocytosis employed by mammalian cells as a mechanism for clearing up extracellular and membrane-bound substrates by lysosomal degradation.24 Endocytic trafficking is initiated with the invagination of the plasma membrane leading to the formation of clathrin-coated bilayered vesicles.25 These can also be referred to as early endosomes (EEs) which can then sort the extracellular cargo or the membrane receptor-bound proteins to three locations: (1) late endosomes (Les) and finally to lysosome for acidified proteolytic degradation, (2) recycling endosomes (REs) for returning the proteins and receptors back to the plasma membrane for reuse, (3) transport to Golgi complexes.8 For the purposes of discussing the modalities exploited by LYTACs, MoDE-As and AbTACs, we shall keep the explanation simplified by only focussing on the sorting destinations (1) and (2) the above mentioned. A major portion of the internalized plasma membrane and contents such as receptors are usually transported back by the REs to the cell membrane, while other substrates remain in the EEs that then undergo maturation to form LEs and fuse with already formed lysosomal compartments or themselves mature further to form lysosomes.
Fig. 1. Endocytic–lysosomal pathway: endocytosis is initiated with plasma membrane invagination which leads to formation of bilayered vesicles called early endosomes (EEs) carrying cargo from the extracellular matrix (ECM) and/or cell membrane. From here the components of the EEs can either get recycled back to the membrane and ECM via recycling endosomes (REs) or can get sorted into matured late endosomes (LEs) that later fuse to the lysosome for degradation of the targets.

Autophagy regulated by the autophagy related gene (ATG) protein family is a catabolic process usually initiated in response to cell starvation, which essentially involves the generation of bilayer membrane vesicles, autophagosomes that merge with lysosomes where the contents are cleaved by hydrolytic enzymes. The metabolites released can be either recycled as an energy source or can undergo further modifications to be secreted out of the cell. Autophagy can be categorized into three forms, microautophagy, chaperone-mediated autophagy (CMA) and macroautophagy, which is the most extensively studied out of the three (Fig. 2).26,27 Microautophagy (Fig. 2) involves the direct delivery of organelles to be degraded in the lysosomal compartment. In CMA (Fig. 2), intracellular proteins with a KFERQ sequence are targeted with heat shock chaperone proteins HSC70 that in turn attach to lysosomal membrane proteins LAMP2A leading to the translocation of the cytosolic protein inside the lysosome to be hydrolyzed. Macroautophagy, hitherto being referred to as simply autophagy, (Fig. 2), first recruit ATG proteins to cup-shaped vesicles called isolation membranes (IM) that start elongating around the particle to be degraded, until they form closed circular double-membraned carriers called autophagosomes, containing the portion of cytosol with the substrate. These autophagosomes finally undergo fusion with the lysosome, forming autolysosomes to deliver the target molecules to be degraded.28
Fig. 2. Types of autophagy: in microautophagy, there is direct engulfment of a portion of cytoplasm containing components to be degrade via the lysosome. Heat shock chaperone proteins (HSC70) target proteins with the KFERQ sequence for degradation in chaperone-mediated autophagy (CMA). Macrophagy involves the recruitment of bilayered cup shaped vesicles called isolation membranes, which engulf and elongate around targets specific to binding to lipidated LC3 proteins present on the membrane surface leading to formation of autophagosomes carrying the cargo to lysosomes for degradation.

While autophagy in response to nutrient requirement in the cell is largely non selective, later studies have evolved to show that macroautophagy can be exploited to breakdown selective cargo via the lysosomal pathway. Cargo selectivity specifically involves interaction of LC3-interacting receptors (LIRs) present on the target molecule with LC3 proteins (light chain protein family associated with microtubules)28 that are recruited by ATG proteins during IM elongation (LC3 proteins get phospholipid conjugated to be bound to the membrane surface of the phagophores). Selective autophagy is initiated in response to the cargo bound autophagosomal receptors. Exploring ligand structures to recruit intended proteins and dysfunctional mitochondria to the LIRs on phagophores for selective autophagy is hereby one of the key concepts of the novel protein degradation techniques discussed in the next sections (AUTACs and ATTECs).
The mechanism of cargo selectivity in both the ubiquitin–proteasomal system and the autophagy–lysosomal system of degradation remains to be linked to the ubiquitination of the target substrate.22 In ubiquitin-dependent selective autophagy, intracellular polyubiquitinated protein molecule aggregates are recognized by autophagosomal receptors like p62, and this further prompts phagosomal vesicle formation around the aggregates. Well-established reports suggest proteins that carry K63 polyubiquitin chains have a higher affinity to be bound to the p62 autophagosomal receptor than proteins with K48 polyubiquitin chains, which are mostly targeted by the proteasomal complex of the UPS for degradation.
2. Endosomal–lysosomal pathway based degraders
2.1. LYTACs
LYTACs or the LYsosome TArgeting Chimeras are currently at the forefront of all non-cytosolic protein-degradation platforms. As the name suggests, the tool delivers proteins to the lysosome for degradation via the endosome–lysosome pathway with the help of lysosome-targeting receptors (LTRs) present on the cell surface (Fig. 3). The LTRs, after releasing their cargo in the early endosomes, shuttle back to the cell surface upon exposure to low pH, and thus are recycled.29 LYTACs are composed of a protein of interest (POI)-binding component (small molecule or antibody) attached to an LTR-binding tag whose structure depends on the LTR being employed. Banik et al.11 have synthesized two types of LYTACs: one utilizing the broadly expressed cation-independent mannose 6-phosphate receptor (CI-M6PR)11 and another using the liver-specific asialoglycoprotein receptor (ASGPR),13 which has also been designed and studied independently by the groups of Tang12 and Spiegel.14
Fig. 3. Mechanism of lysosomal delivery and subsequent degradation of target proteins by M6Pn-LYTAC, GalNAc-LYTAC, MoDE-A and AbTAC.

2.1.1. M6P/M6Pn LYTAC
The first LYTAC, developed by Bertozzi group, would transport non-cytosolic proteins into the cell through CI-M6PR.11 Proteins transported by this receptor have been observed to bear N-glycans capped with mannose-6-phosphate residues.30 Efforts were made to design the optimal CI-M6PR binding ligand by referring to earlier lysosomal enzyme replacement therapies that utilized the same receptor.31,32 The group finally arrived upon N-carboxyanhydride (NCA)-derived glycopeptides which bore multiple serine-O-mannose-6-phosphate (M6P) or serine-O-mannose-6-phosphonate (M6Pn) residues (Fig. 4A). Steps involved in the synthesis of the glycopeptides include: (1) 13-step process from mannose pentaacetate to M6P/M6Pn-NCA; (2) copolymerization of M6P/M6Pn-NCA with alanine-NCA to obtain poly(M6P)/poly(M6Pn) of different lengths (Fig. 4B).
Fig. 4. (A) and (B) Chemical structure of poly(M6P) and poly(M6Pn). (C) Synthesis of poly(M6Pn). (D) Chemical structure of poly(M6Pn)-biotin. (E) Conjugation of poly(M6Pn) on antibody (Ab-1, Ab-2, Ab-3), conjugation of poly(M6Pn) on Ctx-Fab (Fab-1).

Cellular uptake studies employing biotinylated glycopeptides (Fig. 4C) as the LYTACs and NeutrAvidin-647 (NA-647), an Alexa Fluor-647 (AF647)-labeled protein as the target protein, revealed that both poly(M6P) and poly(M6Pn) were internalized 5–6 times more than the controls (poly(GalNAc) and poly(mannose)), with poly(M6Pn) showing a slightly higher uptake compared to poly(M6P). Excess exogenous M6P hindered M6Pn LYTAC uptake, indicating that it is internalized through binding to CI-M6PR via the M6Pn ligand. Gene-knockdown studies using CRISPR interference (CRISPRi) demonstrated that IGF2R (the gene that codes for CI-M6PR) and genes that were required for the proper functioning of the endosome–lysosome pathway were essential for uptake of the POI, reflecting the fundamental role of CI-M6PR and the endosome–lysosome pathway for uptake and subsequent degradation of the target protein.
Various types of LYTACs with antibodies as the POI-binder were designed (Fig. 4D). (i) Ab-1: poly(M6Pn) and goat anti-mouse IgG. Ab-1 enabled targeted degradation of proteins bound to their primary IgG antibody, such as apolipoprotein (ApoE4): a protein involved in the pathogenesis of Alzheimer's disease.33 (ii) Ab-2: poly(M6Pn) and cetuximab (Ctx: an EGFR-blocking antibody). Ab-2 allowed for the targeted degradation of EGFR, a membrane-bound protein whose cancer proliferation-inducing properties cannot be eliminated solely by inhibition of its kinase activity.34 (iii) Fab-1: poly(M6Pn) and Ctx-Fab (Ctx digested with papain, binds in a monovalent manner to EGFR). Fab-1 was designed to confirm that cross-linking between EGFR and Ctx did not cause EGFR downregulation. (iv) Ab-3: poly(M6Pn) and anti-PD-L1. Ab-3 enabled the degradation of PD-L1, a protein that is usually protected against lysosomal degradation and aids cancer cells in evading the immune system.35,36 All LYTACs outperformed their antibody-counterparts. In addition, in vivo testing in mice was performed to examine the pharmacokinetic features of M6Pn LYTACs
2.1.2. GalNAc LYTAC
The second LYTAC was created to precisely target the liver tissues by exploiting ASGPR, an LTR found exclusively in liver cells. There are three groups of independently designed degraders of target proteins based on ASGPR.12–14 They are all composed of a POI-binder attached to triantennary N-acetylgalactosamine (tri-GalNAc), a ligand of ASGPR.37 GalNAc LYTACs have a few benefits over M6Pn LYTACs, including: (i) tissue-specific action; (ii) homogeneous nature of ligands, which allows for exact computation of the ligand-to-antibody ratio; (iii) proteins are specifically delivered to liver cells, where protein catabolism mostly occurs.
Banik et al. carried out several studies to demonstrate the feasibility of the GalNAc LYTACs (Fig. 5A). The internalization efficiency of the GalNAc LYTACs was measured in a hepatocellular carcinoma (HCC) cell line HEPG2. Cells were treated with IgG-647 and with one of the following: goat anti-rabbit (control), goat anti-rabbit M6Pn (M6Pn LYTAC), or goat anti-rabbit GalNAc (GalNAc LYTAC). Cells treated with GalNAc LYTAC showed a 16-fold increase in uptake relative to the control, while M6Pn-LYTAC treated cells only showed a 2-fold increase in uptake, presumably as a consequence of higher expression of ASGPR in hepatocytes.
Fig. 5. (A) Chemical structure of triGalNAc-DBCO. (B) Conjugation of tri-GalNAc on antibody (Ctx–GalNAc, Ptz–GalNAc). (C) Conjugation of tri-GalNAc on peptide (PIP-GalNAc). (D) Site-specific conjugation of tri-GalNAc on antibody.

GalNAc LYTACs targeting EGFR (Fig. 5B) were also designed as it is overexpressed in HCC.38–40 Ctx–GalNAc (10.5 GalNAc per Ctx) induced degradation of more than 70% of EGFR in HEP3B, HEPG2 and HUH7 HCC cell lines combined, which is comparable to Ctx-M6Pn levels. Ctx–GalNAc did not exhibit the hook effect up to a concentration of 100 nM. siRNA targeting ASGPR showcased the necessity of the receptor for the degradation of EGFR. Moreover, excess exogenous tri-GalNAc moieties reduced the degradation of EGFR, demonstrating the binding of GalNAc to ASGPR as a significant step in LYTAC-mediated degradation. HEP3B cells (ASGPR+, EGFR+, M6PR+) and HeLa-GFP cells (ASGPR−, EGFR+, M6PR+) were co-cultured and treated with either Ctx–GalNAc or Ctx-M6Pn. Ctx–GalNAc could deplete EGFR only in HEP3B cells while Ctx-M6Pn caused EGFR degradation in both cell lines, evidencing the tissue-specific activity of the GalNAc LYTACs. GalNAc LYTACs targeting HER2 (Fig. 5B), another membrane protein implicated in HCC,41 were developed using pertuzumab as the POI-binder, an approved antibody to treat HER2+ breast cancer. HEPG2 cells treated with Ptz–GalNAc showed a 75% reduction, while Ptz treated cells only showed a 30% reduction in HER2 levels. Another notable observation was that HER2 levels began to decline within 2 hours of incubation in Ptz–GalNAc treated cells, which was not observed in Ptz-M6Pn treated cells. The effect of GalNAc LYTACs on lysosomal health was investigated utilizing multiple methodologies, all of which concluded that GalNAc LYTACs have no significant impact on lysosomal health when compared to untreated cells.
Polyspecific integrin-binding peptide (PIP)42 was linked to a single tri-GalNAc ligand to form PIP-GalNAc (Fig. 5C), a peptide-based LYTAC that targets integrins (proteins involved in tumor progression)43 for degradation. PIP-GalNAc was able to decrease surface levels of integrins by 60% and inhibit the proliferation of HEPG2 cells. The success of PIP-GalNAc encouraged Banik et al. to create more simplified versions of LYTACs that employed antibodies as well. These site-specific conjugates would enable a more thorough evaluation for improving LYTAC design. Site-specific conjugates based on Ctx and Ptz were synthesized (Fig. 5D). 3 sites on the antibodies were chosen for conjugation: C terminus, hinge, and CH1 heavy chain, and studies were conducted for both Ctx–GalNAc and Ptz–GalNAc. Site-specific Ctx–GalNAc did not perform as well as non-specific Ctx–GalNAc, whereas site-specific Ptz–GalNAc performed similarly to non-specific Ptz–GalNAc. The heterogeneity in results could be due to the fact that the optimal binding site for GalNAc on Ctx has yet to be discovered. Furthermore, tests comparing the clearance rates of site-specific and non-specific Ctx–GalNAc conjugates in mice revealed that non-specific conjugates cleared quickly (within 6 hours), whereas site-specific conjugates remained even after 72 hours, implying the ability to regulate clearance rates by changing the mode of conjugation
Zhou et al.12 presented their findings on GalNAc LYTACs about the same time as Banik et al.13 They designed both small molecule and antibody-based LYTACs for their studies. They analyzed the uptake of neutravidin or NA-650 (POI) in HepG2 cells treated with tri-GalNAc-biotin (LYTAC) (Fig. 6A), and observed a time-dependent internalization. After 6 μM, uptake reduced as the concentration of LYTAC increased, demonstrating the typical hook effect reported in bifunctional species. The introduction of tri-GalNAc–CO2H as a competitive ASGPR binder reduced the tri-GalNAc uptake of NA-650, as evidenced by a drop in fluorescence. Incubation with cells having different expression levels of ASGPR showed a positive correlation of fluorescence to ASGPR expression levels. Knockdown of ASGPR by siRNA also decreased fluorescence. These results together suggest the LYTAC-mediated internalization of NA-650 via ASGPR.
Fig. 6. (A) Chemical structure of tri-GalNAc-biotin. (B) Conjugation of tri-GalNAc on antibody (Ab-GN, Ab-GN1, Ab-GN2) or Fab (Fab-GN).

Zhou et al. next sought to prove the lysosomal degradation of NA-650. Lysosomal delivery was observed by colocalization of NA-650 with Lysotracker. NA-650 levels decreased with time upon removal of medium containing NA-650 and tri-GalNAc-biotin after 1 hour of incubation. Additionally, the use of lysosomal inhibitors was seen to relatively increase NA-650 levels, confirming the depletion of POI via lysosomes.
The group went on to synthesize antibody-based LYTACs (Fig. 6B), by conjugating tri-GalNAc on goat-anti mouse IgG (Ab-GN). The first such antibody-LYTAC had 25 tri-GalNAc moieties per antibody. Surprisingly, the internalization of mouse anti-biotin IgG 647 was quite inefficient. Several efforts were undertaken to determine the root cause behind this inefficiency. To account for the size factor, 3 additional antibody LYTACs were developed: (i) Fab-GN composed of goat anti-mouse IgG Fab monomer (MW = 50 kDa) and an average of 3.2 tri-GalNAc residues per antibody; (ii) Ab-GN1 composed of full-size goat anti-mouse IgG (MW = 150 kDa) and an average of 5.7 tri-GalNAc residues per antibody; (iii) Ab-GN2 composed of full-size goat anti-mouse IgG (MW = 150 kDa) and an average of 4.7 tri-GalNAc residues per antibody. Even though Ab-GN1 showed a slightly higher uptake than Ab-GN2, Fab-GN showed the highest uptake, indicating the role of size in internalization by ASGPR. The importance of size was also investigated in the small molecule-based tri-GalNAc-biotin. Cells were treated with either mouse anti-biotin IgG-647, or complexes of the same protein with antibodies to increase its size and molecular weight. The decrease in fluorescence with increasing size established the dependence of internalization on the size of the target protein.
The antibody-LYTACs were further tested for their viability using earlier studies (ASGPR expression, lysosomal inhibition, etc.). A LYTAC targeting EGFR was also created in order to expand their range to include endogenous proteins. Ctx-GN, with an average of 6 tri-GalNAc residues per Ctx, significantly reduced EGFR levels, thus extending the scope of LYTACs to endogenous proteins.
2.2. MoDE-As
MoDE-As or the molecular degraders of extracellular proteins through the asialoglycoprotein receptor (ASGPR) are the most recent extracellular protein degradation technology.14 They are very similar to the GalNAc LYTACs in that they both possess tri-GalNAc ligands and POI binders to target extracellular proteins for lysosomal degradation (Fig. 3). They are also the first nonproteinogenic extracellular protein degrading tool to have been evaluated in vivo. MoDE-As are composed of 3 components: the ASGPR-binding tri-GalNAc ligand, a POI-binding element, and a polyethylene glycol (PEG) spacer that connects the first two (Fig. 7). Two MoDE-As have been designed and tested: (i) D-MoDE-A whose POI binding element is a dinitrophenyl group which binds α-DNP antibodies; (ii) M-MoDE-A whose POI binding element is a carboxylic acid terminated MIF tautomerase inhibitor which binds to cytokine macrophage migration inhibitory factor (MIF).
Fig. 7. Chemical structure of MoDE-As (D-MoDE-A and M-MoDE-A).

The formation of a ternary complex involving the target protein, MoDE-A and ASGPR, and internalization of the complex was analyzed by labeling α-DNP antibodies with Alexa Fluor 488 (AF488), and monitoring fluorescence with increasing concentrations of D-MoDE-A in HepG2 cells. Fluorescence increased with increasing D-MoDE-A concentration to a certain point, and then declined, displaying the characteristic hook effect seen in ternary complexes. DNP-OH3 (contains three hydroxyl groups in place of the GalNAc residues) which competitively binds to α-DNP antibodies, monomeric GalNAc sugar which competitively binds to ASGPR, and proteins binding to ASGPR all reduced fluorescence levels. Broad inhibitors of endocytosis, as well as specific inhibitors of clathrin-dependent endocytosis decreased fluorescence, whereas inhibitors of caveolae-mediated endocytosis, macropinocytosis and phagocytosis did not decrease fluorescence, implying a clathrin-dependent endocytosis pathway (the pathway used by ASGPR). All of these findings point towards the creation of a ternary complex involving the POI, D-MoDE-A, and ASGPR, as well as uptake via clathrin-dependent endocytosis. The same tests on M-MoDE-A yielded similar results, with the sole difference being that M-MoDE-A did not exhibit the hook effect, which the group attributes to M-MoDE-A's reduced affinity for MIF compared to D-MoDE-A's affinity for the α-DNP antibodies. Additionally, α-DNP antibody was found to be colocalized with lysosomal membrane protein LAMP2, but not with early endosome marker EEA1, indicating a rapid delivery of the antibodies to the lysosomes.
In order to explore the degrading properties of MoDE-A, western blotting was performed to identify the internalized α-DNP antibodies, with the help of an antibody that binds to AF488. The full-length protein (150 kDa) was detected in the cell lysate after 2 hours of incubation. After 6 hours of incubation, a fragment weighing between 37 and 50 kDa was recovered, as well as a fragment weighing more than 150 kDa, which could be intracellular aggregates of the protein. An additional fragment weighing between 25 and 37 kDa was discovered after 24 hours of incubation. None of these fragments were detected in the supernatant, clearly proving the targeted degradation of the α-DNP antibodies in the lysosome. Furthermore, treatment with lysosomal protease inhibitors reduced the amounts of low molecular weight bands, reaffirming the lysosomal degradation of the target protein.
As previously stated, MoDE-As are as of now the only fully synthetic extracellular protein degrading machinery that has been tested both in vitro and in vivo. Both D-MoDE-A and M-MoDE-A showcased promising results in mice injected with the target protein, followed by treatment with the respective MoDE-A. Optimization of MoDE-A structure to target other proteins of therapeutic relevance will aid in broadening the scope of MoDE-As and adopting it as a potential therapeutic method.
LYTACs and MoDE-As provide a promising means of addressing the existing shortage of therapies that target extracellular and membrane proteins. The three groups have individually demonstrated the internalization, LTR-mediated uptake, and lysosomal degradation of the target protein, and have also assessed the efficacy of LYTACs by their own unique methodologies. The spatially confined action of GalNAc LYTACs and MoDE-As suggests the prospect of developing more tissue-specific degrading tools. Identifying other tissue-selective LTRs and developing corresponding ligands will aid in the construction of such LYTACs. The site-specific conjugates of GalNAc LYTACs have demonstrated the capacity of simple structures, consequently facilitating a more comprehensive analysis for enhancing LYTAC efficiency. The tests by Zhou et al. on how the size of the target protein influences uptake have provided a different angle to the study. MoDE-As have proven the success of fully synthetic LYTACs both in vitro and in vivo, thus implying the potentiality of other synthetic species. Bertozzi and Spiegel's studies in mice have shown that LYTACs are also viable in vivo. Taken together, these results clearly establish the feasibility of LYTACs and pave the way for their continued development and optimization for future therapeutic applications.
3. Autophagy–lysosomal pathway based degraders
3.1. AUTACs
In the broad discussion above on small molecule inducers of autophagy, the disadvantage found in strategies invoking bulk autophagy is the non-selectivity of the substrates to be disposed of. Based on the concepts of targeted protein degradation as a high throughput strategy to target biomolecules using bifunctional chimeric adducts that can recruit the substrate as well as traffic them towards the cellular proteolysis machineries available to degrade them, autophagy-targeting-chimeras (AUTACs) developed by Takahashi et al.17 use selective autophagy to target protein aggregates and dysfunctional mitochondria and their fragments (Fig. 8). The discovery of AUTACs, as acknowledged by the Arimoto group themselves, was a consequence of a serendipitous observation where cytoplasm invading group A streptococci are cleared bacterial selective autophagy or xenophagy,17,44–46 orchestrated by the accumulation of 8-nitroguanosine 3′,5′-cyclic monophosphate (8-nitro-cGMP) that has been guanine modified. The attachment of an S-guanylation tag at the cysteine residues of the cGMP proteins was noted to be correlated to the recognition of pathogens in the cytoplasm of the host cell and their subsequent selective clearance by the autophagic machinery.
Fig. 8. (A) Degradation of EGFP-HT proteins via cGMP unit with a HaloTag linker by S-guanylation and subsequently tethering the EGFP-HT to LC3 proteins. (B) Structure of S-guanylated cGMP unit with a chloro based HaloTag binder.

They use specific recruiting ligands for attachment of an S-guanylation degradation tag for the adduct to be identified as a target for lysosomal elimination by the isolation membrane of the phagosomal cup shaped vesicles. From previous studies of xenophagy,45 the group's earlier work on S-guanylation of endogenous nucleotide 8-nitro-cGMP leading to K63 linked polyubiquitination46 of GAS for sequestration by autophagosome vacuoles,44 a prior knowledge of p62/SQSTM1 receptors47 that interact with the LC3 (ubiquitin like human Atg homolog) regions (LIRs)48 and selectively link cargo to such lysosomal autophagosomes, they reported the design of cGMP-HTL (Fig. 8B), a cGMP containing ligand which has the ability to covalently bind to HaloTags fused on EGFP proteins and destine them for autophagosomal uptake (Fig. 8A). The HaloTag technology is required for S-guanylation of the model substrate to tag it for polyubiquitination and to be carried to sequestered by autophagophores. Even with high selectivity shown by cGMP-HTLs for EGFP as cargo, the ligand showed slow reactivity towards the EGFP-HT fusion proteins and the c-GMP substructures could also activate cGMP related protein kinase G causing undesired outcomes. To reform the restricted clearance rate of EGFP shown by cGMP-HTLs they modified the guanine tag, introducing a new ligand devoid of a cyclic phosphate moiety, p-fluorobenzylguanine, FBnG-HTL that showed much faster reactivity to bind to the substrate (Fig. 9).
Fig. 9. Chemical structure of the AUTACs reported by Takashi et al.

Whether S-guanylation could serve as an inducer of selective autophagy alone was justified by designing HeLa cells expressing EGFP-HT proteins that were treated with cGMP-HT ligands. Over time it showed reduction in the levels of cGMP modified EGFP-HT seen on conducting a series of Western blotting experiments and the fact that they were getting degraded could be seen in colocalization experiments where EGFP dots colocalised with LC3B, K63-linked polyubiquitin suggesting S-guanylation based recruitment of EGFP into autophagosomes. This importance of the autophagosomal degradation mechanism involved was corroborated by showing a lack of EGFP degradation in autophagy deficient mouse embryonic fibroblasts (Atg−/− MEF), even in the presence of cGMP-HTL.
However, due to some limited physiochemical properties of cGMP-HTL such as slow reactivity with HT fusion proteins, unwanted side effects that could arise due to the activation of cGMP-dependent protein kinase G, the group was prompted to find a new ligand lacking a cyclic phosphate moiety, p-fluorobenzylguanine (FBnG) ligand. As expected, FBnG-HTL showed rapid reaction with HT-fusion proteins, as well as kept the rest of the proof-of-concept results similar to cGMP-HTL, such as colocalization with LC3B, p62/SQSTM-1, K63-linked polyubiquitin chains. The use of ubiquitin 1 inhibitor PYR41 halted ubiquitination and subsequently suspended autophagic degradation of the proteins. Lack in reduction of protein levels of FbnG modified EGFP-HT were seen in Atg−/− MEF cell lines. Cargo selectivity of FBnG over TMR-modified protein was also demonstrated.
Subsequently, for the proof-of-concept study, the first AUTAC (AUTAC1, Fig. 9) was built with a hydrolysed fumagillin moiety (fumagillol) warhead to recruit Met-AP2 protein (methionine-aminopeptidase 2) linked via a polyethylene glycol unit to the FBnG unit. 1–100 μm of AUTAC1 was seen to silence endogenous Met-AP2 protein levels in HeLa cells, which was stopped by using lysosomal inhibitor bafilomycin A1 proving the involvement of autophagy. This was followed by AUTAC2 (Fig. 9) which was designed to degrade FK506 binding protein (FKBP12) using the warhead as a non-covalently targeting synthetic ligand of FKBP12 (SLF). AUTAC3 (Fig. 9) was built with the epigenetic anticancer agent JQ1 acid as a specific recruiting ligand for the BET family of proteins. Downregulation of Brd4, a transcriptional factor belonging to the BET family, is essential for treatment of melanoma tumour suppression. However, a major hindrance for targeting these BET proteins is their nuclear localization thus making the process heavily cell-cycle dependent. Since canonical autophagy is fundamentally a cytoplasmic process, the AUTAC3s can attack these Brd4 proteins only during mitosis when the nuclear envelope is disintegrated and the nuclear contents are poured into the cytosol. Thus, colocalization of Brd4 with LC3B (family of LC3 proteins), a marker of autophagy, during mitosis when the nuclear contents are poured into the cytoplasm, and reduction of BrD4 levels during G2–G1 transition, showed partial efficacy in lysosomal proteolysis of the substrate with the help of AUTAC3.
The group also conducted an extensive study on whether these AUTACs can potentiate mitophagy to eliminate fragmented mitochondria, a phenotype observed in Down's syndrome.49 A fusion protein of outer mitochondrial membrane (OMM) 25 protein and EGFP-HT (mito-EGFP-HT) was linked to FBnG-HTL for S-guanylation of the targeted mitochondria (Fig. 10A). Expected colocalization of LC3B with mitochondria in FBnG-HTL treated cells was seen without FBnG-HTL causing any cytotoxicity at the concentrations used (Fig. 10A). cGMP-HTL, like FBnG-HTL, also showed K63-linked polyubiquitination. However, guanylation alone was found to be ineffective for mitophagy under such conditions of targeting healthy mitochondria, and these mito-EGFP-HT expressing cells had to be treated with FBnG-HTLs under OPA1-knockdown conditions, (OPA1 is a 120 kDa dynamin like protein which causes fragmentation of the mitochondria by suppressing the fusion of the inner mitochondrial membrane) to observe significant reduction in the mitochondrial protein levels during 24 h to 48 h after treatment. S-Guanylation of mitochondria was proposed to trigger mitophagy under OPA1-KD conditions seen from the downregulation of OMM-localising Tom20 and matrix-localising complex III core 1 proteins. Under the OPA1-KD conditions the mitochondria would be fragmented into small sized dysfunctional particles. This size difference accelerated their engulfment by isolation membranes for mitophagy. To test this concept, they inhibited dynamin-1-like protein (Drp1) by Mdivi-1 which forms elongated fused structures of mitochondria that showed reduced levels of mitochondrial degradation.
Fig. 10. (A) Mitoautophagy observed for mitochondria whose outer mitochondrial membrane (OMM) 25 protein was fused with EGFP-HT which was found to be S-guanylated by FBnG-HTL and degraded under OPA1-knockdown conditions (where the inner mitochondrial membrane fails to fuse with the outer mitochondrial membrane leading to fragmentation of the organelle). (B) AUTAC4s observed targeting fragmented mitochondria via recruitment of a translocator protein on OMM to LC3 proteins for lysosomal degradation.

Another action of these S-guanylation agents was seen in the upregulation of mitophagy of dysfunctional mitochondria to prevent cell death. Treatment with CCCP (carbonyl cyanide m-chlorophenylhydrazone) destructs mitochondrial membrane potential being a depolarising agent. Incubating with FBnG-HTL before CCCP treatment destined the impaired mitochondria to be LC3B recruited and returned partial membrane potential 6 h after treatment inside the cellular environment. This restoration was studied using TMRE (tetramethylrhodamine ethyl ester), which was inhibited on the use of bafilomycin A1. Damaged mitochondria release cytochrome c into the cytosol leading to caspase activation dependent cell death. Reducing levels of dysfunctional mitochondrial fragments impeded cyt c levels. These experiments were also conducted in Atg MEF cells as the control showing the importance of mitophagy for inducing these cytoprotective effects when treated with FBnG-HTL. Only FBnG-HTL could not reduce protein levels without treatment with CCCP. The rapid fall in the levels of mitochondrial proteins partially recovered after 18 h suggesting interestingly, that the mitochondrial biogenesis through adenosine monophosphate-activated receptor gamma coactivator 1alpha (AMPK-PGC1a) can be a result of mitochondrial degradation inside the cell in an attempt to prevent apoptosis. These effects on cytoprotection, improving mitochondrial membrane potential and fragmented mitochondrial degradation, were all inhibited on using bafilomycin A1. FBnG-HTL also increased S-guanylation induced mitophagy in phosphatase and tensin homolog (PTEN)-induced kinase 1 (PINK1)-knockout HeLa cells, suggesting the non-requirement of Parkin-mediated mitophagy protein PINK1.
To conclusively characterise the vitality of K63-polyubiquitination of the selective autophagy targets, Takahashi et al.17 prepared a thalidomide bound HTL moiety (Thal-HTL). This moiety prompted the mito-EGFP-HT targets to be K48 polyubiquitinated. However, as expected K48 polyubiquitin chains did not cause LC3B colocalization of the targets and subsequently neither did they improve the mitochondrial membrane protection after CCCP treatment nor show any other cytoprotective effects under the OPA1-KD conditions. As such, AUTAC4 (Fig. 9 and 10B) was prepared to deliver S-guanine tags on mitochondria, using a 2-phenylindole derivative as a mitochondrial membrane binder (2-phenylindole 3-glyoxyamide is the ligand for the translocator protein on OMM) linked to the FBnG unit via a PEG linker on the other side. Mitophagy detection assays determined the formation of lysosomal mitochondria. Using the MitoTracker Red (MTR) dye they demonstrated that the fluorescence intensity of MTR decreased upon treatment of AUTAC4 in CCCP incubated cells suggesting the removal of damaged mitochondria along with fluorescence signals emitted by newly generated mito-EGFP proteins. AUTAC4 activity (Fig. 5B) supported the improvement in mitochondrial quality control which can be concluded from the maintenance of mitochondria levels by upregulating biogenesis for proper functioning of the cell, reduction in release of cyt c in cytosol, and inhibiting caspase activation and potential cell death. ATP generation in the cells also improved after AUTAC4 treatment of the CCCP incubated cell. To implement the therapeutic effect of these chimeric molecules, fibroblast cells from a patient suffering with Down syndrome were taken and subjected to AUTAC4 treatment. Major mitochondrial respiratory complex components ATP5A1 and MTCO1 are downregulated in DS where their membrane potential is almost destroyed. AUTAC4 restored 50% increased energy production in these cells compared to control cells, which can be concluded as a direct effect of allowing healthy mitochondria to regenerate in these cells by removal of dysfunctional mitochondrial fragments by autophagosome-mediated lysosomal degradation.
3.2. ATTECs
Groups of Lu and Fei15,16 together reported in 2019 the use of proximity-based ligands that would allele selectively target mutant HTT proteins to lipidated LC3 autophagosome membrane bound proteins for their subsequent lysosomal degradation (Fig. 11). Huntington's disease is a monogenetic neurodegenerative disorder caused by an expansion of a poly-glutamine (polyQ) chain with 36 glutamine residues.15,50–55 As such these aggregates of proteins with excess glutamine stretches cannot be targeted with the PROTAC technology as these heterobifunctional molecules are incapable of degrading more than one single protein molecule at a time (Fig. 11). The group screened 3375 compounds using microarray-based screening with the help of a pathogenic mHTT exon1 fragment with an expanded 72 glutamine residue containing polyQ stretch (mHTTexon1(Q72)) and a wtHTT protein with 25 glutamine residue containing polyQ stretch (HTTexon1(Q25)) as the control. They identified the compounds that selectively interacted with the mHTT proteins and LC3B phagophore membrane proteins using a scanning oblique-incidence reflectivity difference (OI-RD) microscope56,57 from these single molecule/small molecule microarrays (SMMs). Excluding non-specific ligands using HTTexon1(Q25) alone, they elucidated two compounds 10O5 (GW5074, 3-3-((3,5-dibromo-4-hydroxyphenyl)methylidene)-5-iodo-1H-indol-2-one) and 8F20 (ispinesib, N-(3-aminopropyl)-N-((1R)-(7-chloro-4-oxo-3-(phenylmethyl)-2-quinqzolinyl)-2-methylpropyl)-4-methylbenzamide) (Fig. 7), that interact with both LC3B and the full length mHTT (flHTT(Q73)), at a Kd of 100 nM, but not with HTTexon1(Q25) or the flHTT(Q23) fragment or any irrelevant proteins.
Fig. 11. ATTECs allele selectively tethering only mutant HTT proteins with poly-glutamine expansion (72 glutamine residues) to isolation membrane proteins to be carried for lysosomal degradation.

Keeping the hydroxyl group in 10O5 and the amino group in 8F20 inaccessible to the mHTT and LC3B, thus halting compound–protein interactions during screening, they tested for several other compounds. They found that the exposed structure of these two hit compounds shares a similarity in bearing an aryl ring connected to a lactam-based bicyclic structure with a halogen-substituted aryl group. Based on these structural data, they identified two more compounds that selectively tether to mHTT and LC3B proteins, AN1 (3-5-bromo-3-((3-bromo-4,5-dihydroxyphenyl)methylidene)-1H-indol-2-one) and AN2 (5,7-dihydroxy-4-phenylcoumarin) (Fig. 12).
Fig. 12. Chemical structures two compounds 10O5 and 8F20 that bind allele selectively to mHTT proteins and LC3 membrane proteins, and ATTECs, AN1 and AN2.

All four compounds showed an optimal dose range of 100 nM for 10O5, 8F20 and AN1 and 50 nM for AN2, for maximum effective mHTT and LC3B tethering as expectantly they also showed the hook effect. That is, at excessively higher concentrations they form dimers separately with the mHTT and LC3B proteins without any tethering and subsequent autophagosomal clearance of the mHTT proteins. Autophagy inhibitors used in control experiments confirmed degradation via autophagy. Linker induced mHTT lowering effects were also increased by using rapamycin that promotes autophagosome formation. The Lu group then confirmed whether degradation was due to mHTT antibodies and found that the protein levels were not lowered due to interaction with any specific antibodies that have the ability to detect the poly expansion neither did mHTT clearance occur as a result of site-specific cleavage of the flHTT(Q73) in smaller fragments which would then be picked by autophagosomes for degradation.
ATG5 gene, which plays a key role in autophagosome formation and knockdown in fibroblasts of HD patients, resulted in lowering of LC3 proteins and directly affected the efficiency of the mHTT–LC3 linker compounds. 10O5 and 8F20 also show c-Raf and KSP activity inhibition. While 10O5 reduced c-Raf levels of c-Raf significantly, the other three were ineffective. In similar experiments, KSP activity was also not seen to get affected by the four compounds thus suggesting that mHTT lowering via autophagy did not affect c-Raf and KSP activity. Two known inhibitors of c-Raf bore structural similarity to 10O5 and 8F20 but had no effects on the mHTT levels, further confirming that these two processes were not affected by each other. Drosophila and HD-knock-in mouse models were treated with the four compounds to confirm their in vivo efficacy. 10O5 and AN2 at 0.5 mg kg−1 crossed the blood brain barrier in mice and significantly lowered mHTT levels without any aggregates being formed in the cortical tissues of these mice. AN1 also showed similar effects in these HD mice but not 8F20. In vitro pull-down experiments were conducted showing 10O5 and AN2 enhancing mHTT–LC3 interactions allele selectively without wtHTT tethering with LC3 proteins. In both, HeLa cells transiently expressing GFP-LC3B and His tagged mHTT fragments and mouse strial cells endogenously expressing LC3 and flHTT proteins, on treatment with the linker compounds, subsequent engulfment by autophagosomes was seen.
These ATTECs did not function by changing the number or size of autophagosomal compartments formed nor did they influence the fusion of autophagosomes with lysosomes or any other autophagosomal processes, as the LC3 levels remained unaffected in the absence or presence of lysosome inhibitor bafilomycin A. Important proteins involved in the autophagy pathway such as the SQSTM1 as well as control proteins like tubulin and GAPDH along with other wild type polyQ protein levels were also not affected on treatment with ATTECs. In an order to explain the action of these compounds selectively in only tethering to the expanded polyQ chain of the mutant HTT proteins, the group predicted that these linker compounds possibly recognize the unique conformation of the polyQ stretch that it adapts unlike in the short glutamine chains in wild type proteins. To confirm the concept, they found that 10O5, AN1, and AN2 reduced the levels of the mutant type ATXN3 in fibroblasts over wilt type ATXN3 proteins taken from patients with spinocerebellar ataxia type 3 diseasealong with targeting exogenously expressed 72Q-GFP, 46Q-GFP, and 38Q-GFP but not 25Q-GFP in HEK293T cells. This suggested that the region where the ATTECs bound to the polyexpansion was within a range of 25Q to 38Q which they confirmed with polyQ–GFP conjugation experiments. Lastly, they studied whether HD phenotypes were rescued in HD-knock-in-mouse models on treatment with 10O5 and AN2 ATTECs. Further proof of concept studies for elucidating the therapeutic applications of ATTECs against polyQ expansion-based diseases remain to be reported.
3.3. LD-ATTECs
Lipid droplets are small, nascent vesicular bodies that contain neutral lipids in their core and are closed off by a monolayer phospholipid membrane, embedding various classes of membrane proteins that the lipid droplets utilize for membrane-based contact with many cellular organelles (Fig. 13).58 Accumulation of lipid droplets can give rise to abnormalities in lipid homeostasis leading to obesity, cardiovascular diseases, and cell signaling.59 So far, targeted therapeutic technologies have mostly shown promise in picking up protein based substrates. The Lu group had earlier reported the use of ATTECs in a ubiquitination-independent manner to target protein substrates for autophagosomal degradation. Here, they realized that small lipid droplet aggregations show affinity for being engulfed as cargo by LC3 protein bound isolation membranes to subsequently target them for lipophagy. This is essentially dependent on the lipidation of LC3 proteins into their LC3-II forms, which behave as biomarkers for autophagosomal compartments, to which ATTECs can bind to directly thus attaching the POI cargo to the LC3-II positive membranes (Fig. 8).
Fig. 13. Lipid droplets (LDs) containing neutral core lipids like triacylglycerol (TAG) and sterol esters (cholesterol), targeted by LD-ATTECs to be carried as cargo after being tethers to autophagosomes for lysosomal degradation.

For the design they chose two compounds GW5074 (GW) and 5,7-dihydroxy-4-phenylcoumarin (DP) as the LC3B binding protein warheads, and for the LD-detection probe head of the LD-ATTECs they used Sudan IV (SIV, 1-2-methyl-4-[(2-methylphenylazo)phenylazo]-2-naphthalenol) or Sudan III (SIII, 1-[4-(phenylazo)phenylazo]-2-naphthalenol) connected via a linear alkyl chain linker (Fig. 14). They induced LDs in wild-type (WT) mouse embryonic fibroblasts by extracellular oleic acid (OA) treatment and subsequently exposed them to LD-ATTEC1 (C1) and LD-ATTEC2 (C2) (consisting of SIV as the LD recruiting warhead) (Fig. 14). Using the LD-detecting probe BIODIPY (BIODIPY 493/503, 4,4-difluoro-1,3,5,7,8-pentamethyl-4-bora-3a,4a-diaza-s-indacene), they found near removal of LDs after 24 h of incubation at a concentration of 5–15 μm. The involvement of autophagy was confirmed by showing a lack of reduction in the levels of OA induced LDs in Atg5-knockout MEF cells. Similar results were also observed in the neuroblastoma cell line, SH-SY5Y, along with which they confirmed that enhancing or inhibiting global autophagy using starvation or NH4Cl treatment respectively did not affect LD levels. An alternate mechanism which could be proposed for the lowering in BIODIPY levels was the binding of LD-ATTECs in a competitive manner to BIODIPY rather than the targeted LDs. This idea was rejected when they found that in the presence of SIV (LD binding ligand) even at the highest concentration, SIV showed no affinity for BIODIPY versus the DMSO control as BIODIPY levels in both remained unchanged. Following similar experiments, they confirmed the activity of two more LD-ATTECs C3 and C4 (which use SIII as the LD recruiting warhead, Fig. 14). Endogenous LDs found in 3T3-L1 differentiated adipocytes were also found to be lowered by the compounds, which could be blocked by autophagy inhibition or Atg5 knockdown. Microscale thermophoresis (MST) was used to measure the affinity of LD-ATTECs to the recombinant purified LC3B protein to confirm that the LC3B binding moiety is required for tethering LD-ATTECs to LC3 proteins. However, LD-ATTECs only selectively targeted the LDs carrying neutral lipids unlike the polar lipids which are found in plasma and intracellular membranes thus maintaining their integrity and keeping their functions unaffected after treatment.
Fig. 14. Chemical structures of lipid-droplet (LD) recruiters, GW and DP, LC3 protein binders, Sudan (III) and Sudan (IV). LD-ATTECs C1 and C2 carry Sudan (IV) as the LC3 binders. while C3 and C4 utilise Sudan (III) to bind to LC3 proteins.

They also confirmed the formation of a TAG–LD-ATTEC–LC3 ternary complex. TAG is a core lipid component of LDs that recruit the SIII or SIV moiety of the LD-ATTECs and attach them to the LC3 on the autophagosomes. Colocalisation of exogenously expressed mCherry-LC3B in MEFs and BIODIPY stained LDs in the presence of LD-ATTECs after 2 h of treatment along with lysosomes (LAMP1-mCherry) was observed confirming enhanced engulfment of LDs by autophagosomes. LC3B knockout largely inhibited LD clearance by LD-ATTECs but due to unavailability of confirmed studies on LC3B–LD interaction it was difficult to conclude whether LD clearance is wholly dependent on LD-ATTEC–LC3B interaction or whether LC3B knockout affects macroautophagy as a whole. There was no change in global autophagosomal numbers or sizes. Since plasma, nuclear and mitochondrial membranes also carry lipids, it was confirmed whether these LD-ATTECs affected their composition or not.
Lastly, the in vivo efficacy of LD-ATTECs was confirmed in a genetic model of db/db mice with obesity and the NASH mouse model with a choline deficient, l-amino acid-defined high fat diet fed for 10 weeks. They intraperitoneally injected 30 mg kg−1 of C3 and C4 (due to their lower molecular weights and higher LC3 B binding affinities) and observed that the whole body weight in both models was reduced in two weeks of continuous treatment. They also observed a decrease in levels of the fat/lean ratio, liver weight, TAG and TC levels, FFA levels in serum and liver, and LDs, which eventually became comparable to WT levels. The decrease in FFA levels was later on shown from studies that LD lowering possibly led to mitochondria consuming FFA for energy production. The total lipid concentration was confirmed by lipidomics analysis and found to be lowered by 62%. The prediction that LD-ATTECs tethered selectively to neutral lipid also found evidence in studies that showed selective lowering of TAG, cholesterol ester (ChE), and diglyceride (DG) compared to phosphatidylethanolamine (PE) and phosphatidylinositol (PI) carrying polar lipids. There was a slight increase in the levels of some low abundance neutral lipids on being treated with LD-ATTECs suggesting secondary metabolite effects. They also found that LD lowering had an effect on the levels of Plin2 protein (LD maker protein). However, any lipases or related cofactors involved in lipid synthesis were unaffected suggesting that lipid synthesis pathways were not hijacked by the LD-ATTECs. On the whole, LD-ATTECS may not affect protein levels directly but some cascading effects on protein levels discussed above were observed. The SIII or S1V analogue Sudan red dye I produces cytotoxicity and so prolonged treatment with higher dosage of these compounds was not suggested. However, the group is trying to find other LD binding moieties like small molecule lipid based steroids and cholesterols to give a proof-of-concept model of these LD-ATTECs.
4. Antibody based targeted protein degradation
Antibodies are the natural defence mechanism of the body and are an integral part of the humoral immunity. They consist of mainly two functional regions, the antigen binding fragment (Fab, light chain, recognizes different antigens with high affinity and specificity) and the constant fragment crystallizable region (Fc, heavy chain, involved in receptor recognition linked to different immune effector pathways).60 Out of the five classes of antibodies (Abs), IgA, IgD, IgE, IgG and IgM, IgGs are the most prevalent class of antibodies in human serum and have the capability to recognize a wide range of pathogens and toxins. Engineering these heavy chains on IgGs to vary the antibody effector functions, their tissue localizations and receptor recognition is what forms a basis for target POI degradation using therapeutic antibodies via Fc receptors.61 To shed more light on the antibody based approaches discussed later in the review, we'll particularly focus on the interaction between IgG and FcRn receptors. FcRn receptors are known to regulate the levels and transport of Abs throughout the body and their inhibition hold potential therapeutic possibilities to control IgG mediated autoimmune diseases.62 Certain autoimmune based diseases enhance IgG–FcRn interaction and lead to the implementation of inflammatory pathways which may cause extensive tissue damage such as in rheumatoid arthritis and systemic lupus erythematosus63 and other diseases like myasthenia gravis and idiopathic thrombocytopenic purpura where the concentration of pathogenic antibodies is too high.64
The neonatal crystallizable fragment receptor (FcRn) is an MHC class I protein like receptor which binds to the Fc region of the IgGs and is widely present on the cell surface of endothelial cells as a heterodimer of heavy (alpha) chain and beta-2 microglobulin (Fig. 15A).64 His residues located on the outer side of the CH2–CH3 region of IgGs are responsible for their pH dependent reversible binding to the FcRn receptors. The protonated imidazole side chains of the His residues interact at around an acidic pH of 6.0 with the acidic residues on FcRn with high affinity and show nearly negligible binding to the same residues on FcRn receptors at a near neutral pH of 7.3–7.4. Hijacking the internalization of these IgGs and designing the antibodies with antigen specific recruiters can lead to dissemination of cargo using lysosomal degradation. The most common strategy of maintaining IgG homeostasis is through fluid state pinocytosis61 (Fig. 15B), where at the extracellular near neutral pH, IgG–FcRn binding is low but once they enter early endosomes, where an acidic pH allows receptor–antibody complexation, the excess, free floating IgGs, antibodies with naturally less tendency to receptor bind, as well as cross-linked species, are sorted into lysosomes for degradation while the rest of the bound IgGs are recycled back to be released into the blood with the FcRn re-expressed on the cell surface. The application of these antibodies could be seen in sweeping up soluble as well as membrane bound targets.64
Fig. 15. (A) Interaction of wild type IgGs with a heterodimer of FcRn receptor at their CH2–CH3 region located on their Fc (heavy chain fragment). This interaction is responsible for the pH dependent reversible binding of IgGs to the FcRn receptors. (B) Mechanism of action of Abdegs, Seldegs and sweeping antibodies: the IgG variants in Abdegs have the ability to show higher affinity binding to the FcRn receptors at neutral pH (7.3–7.4) as compared to wild type IgGs, thus remaining bound tightly to the FcRn receptors at acidic pH (<6.0) inside the endosomes allowing themselves to be rescued via exocytosis but causing lysosomal degradation of the intended wild type IgGs. Similarly, Seldegs lead to the formation of the antigen-IgG variant–FcRn complex to internalise the antigen related membrane proteins to lead to lysosomal degradation. In sweeping antibodies, the IgG variants utilise the endocytic receptor properties of asialoglycoproteins and high mannose receptors to take up membrane bound antigen like tumor surface marker proteins. In all three cases, the targets get sorted into matured endosomes for being carried to lysosome for degradation while the IgG variants and the endocytic receptors are recycled back to the plasma membrane for reuse.

4.1. Abdegs
One of the approaches used in Abdegs (antibody degraders) is to reduce the serum half-lives of endogenous pathogenic antibodies by using recombinant IgGs that can outcompete their binding to the FcRn. This would prompt non-receptor-bound proteins in the serum internalized by endocytosis to be degraded while both the Fc receptor and the IgGs with engineered Fc regions would be recycled by IgG salvation pathways61 to maintain beneficial levels in the human serum (Fig. 15B).
Vaccaro et al.20 in 2005 reported the use of MST-HN Abdeg which carries a recombinant IgG, IG1 variant which showed enhanced affinity to bind to FcRn at acidic and near-neutral pH compared to the parent wild type (Fig. 15B). This reduced pH dependency essential at neutral pH. FcRn blocking is important in treating diseases due to deleterious auto-antibodies (effects seen in myasthenia gravis, rheumatoid arthritis, immune thrombocytopenic purpura and Guillain Barre syndrome,65 systemic lupus erythematosus63) and due to transcytosis,61 these can also get transferred from the mother to the offspring across the placenta. Patel et al.62 in 2011 used a murine model where arthritis develops in normal mice followed by transfer of anti-glucose-6-phosphate isomerase (anti-GPI) Abs that conjugates with endogenous glucose-6-phosphate isomerases. They used MST-HN (a wild type IgG1 variant, with the following amino acid modifications, Met252 to Try, Ser254 to Thr, Thr256 to Glu, His433 to Lys, Asn434 to Phe) like the previous group, to perform inhibition of FcRn functions in antibody-mediated arthritis in mice.62
4.2. Sweeping antibodies
Similar to Abdegs, sweeping antibodies also implement the pH-dependent binding to Fc-receptors and the clearance of a specific antigen from the blood plasma via receptor-mediated degradation pathways. Igawa et al.66 reported the utilization of these sweeping antibodies as advantageous to conventional IgGs which would remain bound to the antigen in plasma increasing the antibody mediated antigen accumulation/concentration since conventional IgGs have long serum half-life and take time to clear these antigens. Even with infinite affinity of antigen binding, the concentration of conventional antibodies in the blood plasma should be higher than the antigen concentration and they can bind to the antigen only once which would also in turn increase the total antigen concentration in the plasma since free antigen gets cleared up from plasma comparatively much faster than antibody bound antigens as the serum half-life of IgGs is much longer.
FDA-approved targets of monoclonal antibodies for membrane bound antigens include tumor surface markers (HER2, CD20, CD19), tyrosine kinase receptors (EGFR and VEGFR), and a cytokine receptor (IL-6R), while soluble antigens include cytokines (TNFα, IL-6, IL-12, IL-17), growth factor (VEGF), and other soluble disease mediators (IgE and C5).18 They employed the concept of endocytic-receptor like properties shown by asialoglycoprotein receptors, low density lipoprotein (LDL) receptors, and high mannose receptors to engineer monoclonal antibodies that would mimic binding in neutral pH (plasma) shown in endosomal cellular uptake and show pH-dependent ligand binding affinity so as to release the antigens in acidic pH of lysosomal compartments, thus rapidly eliminating soluble targets from circulation (Fig. 15B).18 These sweeping antibodies require modification in both their variable region so as to bind or dissociate from the antigens in plasma and endosome respectively, as well as in the constant region to enable efficient cellular uptake during endocytosis. pH dependent binding antibody against human soluble IL-6 receptor (hsIL-6R) with neutralizing activity (PH-IgG1), PHX-IgG1 (non-neutralizing activity) was generated from non pH dependent antibody (NPH-IgG1) using tocilizumab (TCZ),67 a humanized IgG antibody found effective in treatment against rheumatoid arthritis, and engineering its unmodified form to dissociate from IL-6R in endosomes (PH1 and PH2 variants with several histidine mutations that confer pH dependency), decreasing its binding affinity at pH < 6.0 without affecting its binding affinity in plasma.
4.3. Seldegs
Devanaboyina et al.19 in their report on the novel design of Seldegs (selectively degrading antigen-specific antibodies) backed their work on the disadvantages of the previously reported FcRn blocking technology, i.e., the inhibitors that block the interaction between the Fc region of the IgGs and the receptor led to clearance of all the specificities of IgGs, including protective antibodies. A few challenges posed in the development of Seldegs are firstly the concentration of antigen specific antibodies to the non-specific ones is comparatively low and secondly, since their design would be based on the bivalent antibodies, error could arise due to cross-linking leading to inflammatory immune complexes. These Seldegs display a recombinant antigen, binding as a monomer to a dimeric human IgG1 derived Fc fragment similar to monomeric erythropoietin (EPO)–Fc fusions,68 with the mutations allowing interactions with FcγRs and enhanced binding with FcRn at pH 6 (in endosomes)–7.4 (neutral).64 This would enable an antibody specific to the recruiting antigen–Fc fusion to be bound to the latter followed by internalization of the antibody–antigen–Fc–FcRn complex (Fig. 15B) and entry into the lysosomal degradation pathway, selectively co-opting the antibodies for destruction.19 Their work demonstrated the development of two antigen specific Seldegs, MOG-Seldeg, targeting the extracellular domain of myelin oligodendrocyte glycoprotein, and HER2-Seldeg, targeting the ECD of the tumor target, HER2. They were successful in showcasing the selectivity of these Seldegs in a time and concentration dependent manner at being cleared with higher efficiency compared to their wild type parents.
4.4. AbTACs
Cotton et al. devised AbTACs or antibody-based PROTACs as a strategy to target membrane-bound proteins for lysosomal degradation (Fig. 3).21 AbTACs are bispecific IgGs or BsIgGs that recruit both a membrane protein and a transmembrane E3 ligase, obviating the necessity for a ligand to bind the POI. The POI is then ubiquitinated by the E3 ligase, thereby marking it for lysosomal degradation. For this study, Cotton et al. chose to degrade the membrane protein PD-L1 using BsIgGs that bind to PD-L1 (ref. 35 and 36) and the transmembrane E3 ligase RNF43.69 These BsIgGs are fully recombinant and developed using phage display,70,71 hence making them renewable and highly specific binders.
The group initially investigated the ability of RNF43 to cause degradation of membrane proteins using GFP as the POI. An anti-GFP single chain Fab or scFab was fused to RNF43, while GFP was fused to a NanoLuc domain via a transmembrane domain (Fig. 16A). Fluorescence was observed in the lysosomes, and a NanoLuciferase assay reported a decrease by 20%, proving the RNF43 mediated lysosomal degradation of GFP.
Fig. 16. (A) Schematic representation of the cellular uptake study using engineered RNF43 constructs and GFP–Nanoluciferase reporter. (B) Schematic representation of synthesis of AbTACs that bind to RNF43 and PD-L1.

Cotton et al. proceeded to synthesize the BsIgGs using a recombinant antibody (called R3) that binds to the ectodomain of RNF43 (generated using phage display), and atezolizumab that binds to PD-L1. Employing the knobs-into-holes Fc construct,72 they were expressed as half IgGs and combined to give a BsIgG called AC-1 (Fig. 16B). Bio-layer interferometry studies proved that AC-1 can bind both to PD-L1 as well as RNF43. In fact, AC-1 and R3 Fab bind with similar affinity to RNF43.
MDA-MB-231 cells, a triple negative breast cancer cell line with high expression levels of PD-L1, were treated with AC-1 to validate the degrading properties of AbTAC. PD-L1 levels reduced drastically, while the same was not observed for cells treated with the half IgGs either alone or jointly. Knockdown of RNF43 using CRISPRi established the essential role of RNF43 in the degradation of PD-L1, while treatment with MG-132 (a proteasome inhibitor) or bafilomycin (a lysosome acidification inhibitor) indicated the lysosomal degradation of PD-L1. AC-1 also showcased its capability in other cell lines, revealing its wide cellular action.
The mode of action of AbTACs is still largely unknown. It would be useful to learn whether AbTACs, like PROTACs, are catalytic in nature, and look deeper into other aspects such as the recycling rate of RNF43 and the utilization of other transmembrane E3 ligases. Such research will aid in the degradation of more POIs linked to disease and will be useful for future clinical use.
5. Conclusions
While PROTACs have grabbed a lot of attention with the advent of orally bioavailable PROTACs (ARV-110, against prostate cancer and ARV-471, against breast cancer) entering clinical trials, strategies using the lysosomal pathway of degradation can be made to target a much-extended scope of proteins. Lysosomes are present in nearly all animal cell types making them an excellent basis for orchestrating targeted degradation of a huge class of biomolecules. As an attempt to modulate the function of macromolecules through these chimeric compounds, the emerging concepts recruiting the autophagy/lysosomal pathway, the above strategies can provide a bank of available tools that can be co-opted for targeting a wide range of biomolecules whether inside or outside the cell. These remain fairly a new concept and further mechanistic studies are required to probe into a deeper understanding of the technologies. Lysosome targeting approaches can be explored further to have the ability to remove deleterious proteins with promising translational potential.
A vast majority of extracellular and membrane bound proteins include abnormal signaling factors like receptor tyrosine kinases (RTKs), ion channels, immune effectors, scaffolding proteins which are polyfunctional and proteins that lack an epitope specific site. Hijacking the endosome–lysosomal pathway in such cases would help in rounding up these proteins for degradation seen in LYTACs and MoDE-As, utilising the concept of POI specific antibodies, either conjugated to M6P glycans (M6Pn LYTACs) or asialoglycoprotein receptor harnessing lysosomal targeting chimeric degraders (GalNAc-LYTACs and MoDE-As). A few limitations of LYTACs remain in their design due to their large sizes unlike small molecule-based drugs and their ease in clinical translation, and the use of antibodies which may cause an unwanted immune response.
A direct approach of recruiting the autophagosome–lysosomal system is observed in AUTACs and ATTECs which can adeptly pick up cytosolic targets such as single proteins, macromolecules like protein aggregates, non-protein cargoes such as lipid droplets, and even organellar components such as dysfunctional mitochondria. These open up the avenue for targeting other non-protein targets such as DNA/RNA too. However, a major limitation of the AUTAC system is the lack of clear knowledge of all the proteins involved in the mechanistic pathway of the AUTAC function, which makes discovering potent new sites where to attach the cargo other than the LC3 proteins difficult. Extensive studies are required to answer issues such as whether the global autophagy is affected due to the mechanism of action of AUTACs and ATTECs. In the clearance of mHTT proteins related to Alzheimer's disease, ATTECs have shown the promise of being able to cross the blood brain barrier15 and in such scenarios, along with their typically smaller sizes than other technologies like LYTACs and AUTACs, they promise showing better translational properties.
Abdegs, Seldegs and sweeping antibodies have shown clearance of soluble proteins outside the cell and even deleterious membrane bound receptor targets. While engineering the Fc region of these antibodies can ideally lead to an increase of the target scope, limitations remain in optimising their binding affinity which is heavily pH dependent, the prior knowledge of deleterious epitopes or markers on target antigen for selective degradation with the help of antibodies, and setting of consequential unwanted immune responses in the body. AbTACs have been developed as a promising strategy in protein degradation where they ubiquitinate their substrates to be tagged for proteasomal degradation, quite similar to PROTACs. The mechanism of AbTACs is not yet fully understood and whether they function catalytically like PROTACs.
Future studies on the deeper understanding of these strategies focussing mainly on targeting proteins by degrading them via the lysosome have the potential to increase the target scope beyond intracellular proteins, to extracellular soluble proteins, antigens, antibodies, membrane bound deleterious receptors and even non protein, macromolecular substrates, aggregates, etc.
Conflicts of interest
There are no conflicts to declare.
Supplementary Material
Acknowledgments
DM thanks IISER-Bhopal and SERB (CRG/2019/001667) for funding. SG and BR thank DST for INSPIRE fellowship.
Biographies
Biography
Samrajni Ghosh.

Samrajni Ghosh received her BS-MS Dual Degree in 2022 with a Major in Chemistry from the Indian Institute of Science Education & Research Bhopal, India. She was born in Kolkata, India. She completed her master's thesis in the lab of Dr. Debasish Manna, where she worked on the synthesis and characterization of optically controlled proteolysis targeting chimeras. She has recently joined, as a PhD student, the lab of Prof. Dr. Philippe Bastiaens, in the Systemic Cell Biology department of the Max Planck Institute of Molecular Physiology, Dortmund.
Biography
Bhavana Ramadas.

Bhavana Ramadas was born in Thrissur, Kerala, India. She received her BS-MS dual degree in Chemistry in 2021 from the Indian Institute of Science Education & Research Bhopal, India. She completed her master's thesis project in the lab of Dr. Debasish Manna, where she worked on achieving optical control of CRISPR/Cas9. She is currently a PhD student in the lab of Dr. Sidney Becker in the Chemical Biology department of the Max Planck Institute of Molecular Physiology, Dortmund.
Biography
Debasish Manna.

Dr. Debasish Manna completed his MS and PhD at the Indian Institute of Science, Bangalore, India. Then he moved to the Weizmann Institute of Science, Israel for his 1st post-doctoral research. After that, he joined as a research fellow the Broad Institute and Harvard Medical School, USA. In May 2019, he joined IISER Bhopal as an assistant professor. His research interest includes optochemical control of various biological functions, targeted protein degradation and development of small molecule mimics of various metalloenzymes to understand their mechanism.
Notes and references
- Bonam S. R. Wang F. Muller S. Nat. Rev. Drug Discovery. 2019;18:923–948. doi: 10.1038/s41573-019-0036-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y. Li S. Wu H. Cells. 2022;11:851. doi: 10.3390/cells11050851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hopkins A. L. Groom C. R. Nat. Rev. Drug Discovery. 2002;1:727–730. doi: 10.1038/nrd892. [DOI] [PubMed] [Google Scholar]
- Burslem G. M. Crews C. M. Chem. Rev. 2017;117:11269–11301. doi: 10.1021/acs.chemrev.7b00077. [DOI] [PubMed] [Google Scholar]
- An S. Fu L. EBioMedicine. 2018;36:553–562. doi: 10.1016/j.ebiom.2018.09.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong E. Cuervo A. M. Cold Spring Harbor Perspect. Biol. 2010;2:a006734. doi: 10.1101/cshperspect.a006734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uhlen M. Fagerberg L. Hallstrom B. M. Lindskog C. Oksvold P. Mardinoglu A. Sivertsson A. Kampf C. Sjostedt E. Asplund A. Olsson I. Edlund K. Lundberg E. Navani S. Szigyarto C. A. Odeberg J. Djureinovic D. Takanen J. O. Hober S. Alm T. Edqvist P. H. Berling H. Tegel H. Mulder J. Rockberg J. Nilsson P. Schwenk J. M. Hamsten M. von Feilitzen K. Forsberg M. Persson L. Johansson F. Zwahlen M. von Heijne G. Nielsen J. Ponten F. Science. 2015;347:1260419. doi: 10.1126/science.1260419. [DOI] [PubMed] [Google Scholar]
- Tooze S. A. Abada A. Elazar Z. Cold Spring Harbor Perspect. Biol. 2014;6:a018358. doi: 10.1101/cshperspect.a018358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bagdanoff J. T., Allan M., Barnes D. W., Clairmont K., Smith T. and Wang S., Patent Appl, WO/2021/156792, 2022
- Wells Z. B., Wells J. and Martinko A., Patent Appl, WO/2018/213848, 2018
- Banik S. M. Pedram K. Wisnovsky S. Ahn G. Riley N. M. Bertozzi C. R. Nat. Chem. Biol. 2020;584:291–297. doi: 10.1038/s41586-020-2545-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Y. Teng P. Montgomery N. T. Li X. Tang W. ACS Cent. Sci. 2021;7:499–506. doi: 10.1021/acscentsci.1c00146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ahn G. Banik S. M. Miller C. L. Riley N. M. Cochran J. R. Bertozzi C. R. Nat. Chem. Biol. 2021;17:937–946. doi: 10.1038/s41589-021-00770-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caianiello D. F. Zhang M. Ray J. D. Howell R. A. Swartzel J. C. Branham E. M. J. Chirkin E. Sabbasani V. R. Gong A. Z. McDonald D. M. Muthusamy V. Spiegel D. A. Nat. Chem. Biol. 2021;17:947–953. doi: 10.1038/s41589-021-00851-1. [DOI] [PubMed] [Google Scholar]
- Li Z. Wang C. Wang Z. Zhu C. Li J. Sha T. Ma L. Gao C. Yang Y. Sun Y. Wang J. Sun X. Lu C. Difiglia M. Mei Y. Ding C. Luo S. Dang Y. Ding Y. Fei Y. Lu B. Nat. Biotechnol. 2019;575:203–209. doi: 10.1038/s41586-019-1722-1. [DOI] [PubMed] [Google Scholar]
- Li Z. Zhu C. Ding Y. Fei Y. Lu B. Autophagy. 2020;16:185–187. doi: 10.1080/15548627.2019.1688556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahashi D. Moriyama J. Nakamura T. Miki E. Takahashi E. Sato A. Akaike T. Itto-Nakama K. Arimoto H. Mol. Cell. 2019;76:797–810. doi: 10.1016/j.molcel.2019.09.009. [DOI] [PubMed] [Google Scholar]; , e710
- Igawa T. Haraya K. Hattori K. Immunol. Rev. 2016;270:132–151. doi: 10.1111/imr.12392. [DOI] [PubMed] [Google Scholar]
- Devanaboyina S. C. Khare P. Challa D. K. Ober R. J. Ward E. S. Nat. Commun. 2017;8:15314. doi: 10.1038/ncomms15314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vaccaro C. Zhou J. Ober R. J. Ward E. S. Nat. Biotechnol. 2005;23:1283–1288. doi: 10.1038/nbt1143. [DOI] [PubMed] [Google Scholar]
- Cotton A. D. Nguyen D. P. Gramespacher J. A. Seiple I. B. Wells J. A. J. Am. Chem. Soc. 2021;143:593–598. doi: 10.1021/jacs.0c10008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kudriaeva A. A. Sokolov A. V. Belogurov A. A. J. Acta Naturae. 2020;12:18–32. doi: 10.32607/actanaturae.10936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eskelinen E. L. Saftig P. Biochim. Biophys. Acta, Mol. Basis Dis. 2009;1793:664–673. doi: 10.1016/j.bbamcr.2008.07.014. [DOI] [PubMed] [Google Scholar]
- Lai S. S. M. Ng K. Y. Koh R. Y. Chok K. C. Chye S. M. Metab. Brain Dis. 2021;36:1087–1100. doi: 10.1007/s11011-021-00737-0. [DOI] [PubMed] [Google Scholar]
- Lamb C. A. Dooley H. C. Tooze S. A. BioEssays. 2013;35:34–45. doi: 10.1002/bies.201200130. [DOI] [PubMed] [Google Scholar]
- Mizushima N. Komatsu M. Cell. 2011;147:728–741. doi: 10.1016/j.cell.2011.10.026. [DOI] [PubMed] [Google Scholar]
- Shacka J. J. Roth K. A. Zhang J. Front. Biosci. 2008;13:718–736. doi: 10.2741/2714. [DOI] [PubMed] [Google Scholar]
- Alabi S. B. Crews C. M. J. Biol. Chem. 2021;296:100647. doi: 10.1016/j.jbc.2021.100647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mellman I. Fuchs R. Helenius A. Annu. Rev. Biochem. 1986;55:663–700. doi: 10.1146/annurev.bi.55.070186.003311. [DOI] [PubMed] [Google Scholar]
- Ghosh P. Dahms N. M. Kornfeld S. Nat. Rev. Mol. Cell Biol. 2003;4:202–212. doi: 10.1038/nrm1050. [DOI] [PubMed] [Google Scholar]
- Zhu Y. Li X. Kyazike J. Zhou Q. Thurberg B. L. Raben N. Mattaliano R. J. Cheng S. H. J. Biol. Chem. 2004;279:50336–50341. doi: 10.1074/jbc.M409676200. [DOI] [PubMed] [Google Scholar]
- Liu L. Lee W. S. Doray B. Kornfeld S. Mol. Ther.--Methods Clin. Dev. 2017;5:59–65. doi: 10.1016/j.omtm.2017.03.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamazaki Y. Painter M. M. Bu G. Kanekiyo T. CNS Drugs. 2016;30:773–789. doi: 10.1007/s40263-016-0361-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sigismund S. Avanzato D. Lanzetti L. Mol. Oncol. 2018;12:3–20. doi: 10.1002/1878-0261.12155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burr M. L. Sparbier C. E. Chan Y. C. Williamson J. C. Woods K. Beavis P. A. Lam E. Y. N. Henderson M. A. Bell C. C. Stolzenburg S. Gilan O. Bloor S. Noori T. Morgens D. W. Bassik M. C. Neeson P. J. Behren A. Darcy P. K. Dawson S. J. Voskoboinik I. Trapani J. A. Cebon J. Lehner P. J. Dawson M. A. Nat. Biotechnol. 2017;549:101–105. doi: 10.1038/nature23643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freeman G. J. Long A. J. Iwai Y. Bourque K. Chernova T. Nishimura H. Fitz L. J. Malenkovich N. Okazaki T. Byrne M. C. Horton H. F. Fouser L. Carter L. Ling V. Bowman M. R. Carreno B. M. Collins M. Wood C. R. Honjo T. J. Exp. Med. 2000;192:1027–1034. doi: 10.1084/jem.192.7.1027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spiess M. Biochemistry. 1990;29:10009–10018. doi: 10.1021/bi00495a001. [DOI] [PubMed] [Google Scholar]
- Ito Y. Takeda T. Sakon M. Tsujimoto M. Higashiyama S. Noda K. Miyoshi E. Monden M. Matsuura N. Br. J. Cancer. 2001;84:1377–1383. doi: 10.1054/bjoc.2000.1580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blivet-Van Eggelpoël M.-J. Chettouh H. Fartoux L. Aoudjehane L. Barbu V. Rey C. Priam S. Housset C. Rosmorduc O. Desbois-Mouthon C. J. Hepatol. 2012;57:108–115. doi: 10.1016/j.jhep.2012.02.019. [DOI] [PubMed] [Google Scholar]
- Ezzoukhry Z. Louandre C. Trécherel E. Godin C. Chauffert B. Dupont S. Diouf M. Barbare J.-C. Mazière J.-C. Galmiche A. Int. J. Cancer Res. 2012;131:2961–2969. doi: 10.1002/ijc.27604. [DOI] [PubMed] [Google Scholar]
- Shi J. H. Guo W. Z. Jin Y. Zhang H. P. Pang C. Li J. Line P. D. Zhang S. J. Cancer Med. 2019;8:1269–1278. doi: 10.1002/cam4.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kimura R. H. Levin A. M. Cochran F. V. Cochran J. R. Proteins. 2009;77:359–369. doi: 10.1002/prot.22441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Desgrosellier J. S. Cheresh D. A. Nat. Rev. Cancer. 2010;10:9–22. doi: 10.1038/nrc2748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakagawa I. Amano A. Mizushima N. Yamamoto A. Yamaguchi H. Kamimoto T. Nara A. Funao J. Nakata M. Tsuda K. Hamada S. Yoshimori T. Science. 2004;306:1037–1040. doi: 10.1126/science.1103966. [DOI] [PubMed] [Google Scholar]
- Bauckman K. A. Owusu-Boaitey N. Mysorekar I. U. Methods. 2015;75:120–127. doi: 10.1016/j.ymeth.2014.12.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sawa T. Zaki M. H. Okamoto T. Akuta T. Tokutomi Y. Kim-Mitsuyama S. Ihara H. Kobayashi A. Yamamoto M. Fujii S. Arimoto H. Akaike T. Nat. Chem. Biol. 2007;3:727–735. doi: 10.1038/nchembio.2007.33. [DOI] [PubMed] [Google Scholar]
- Narendra D. Kane L. A. Hauser D. N. Fearnley I. M. Youle R. J. Autophagy. 2010;6:1090–1106. doi: 10.4161/auto.6.8.13426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hartmann M. Huber J. Kramer J. S. Heering J. Pietsch L. Stark H. Odadzic D. Bischoff I. Furst R. Schroder M. Akutsu M. Chaikuad A. Dotsch V. Knapp S. Biondi R. M. Rogov V. V. Proschak E. J. Med. Chem. 2021;64:3720–3746. doi: 10.1021/acs.jmedchem.0c01564. [DOI] [PubMed] [Google Scholar]
- Whitmarsh-Everiss T. Laraia L. Nat. Chem. Biol. 2021;17:653–664. doi: 10.1038/s41589-021-00768-9. [DOI] [PubMed] [Google Scholar]
- Abou-Sleymane G. Chalmel F. Helmlinger D. Lardenois A. Thibault C. Weber C. Merienne K. Mandel J. L. Poch O. Devys D. Trottier Y. Hum. Mol. Genet. 2006;15:691–703. doi: 10.1093/hmg/ddi483. [DOI] [PubMed] [Google Scholar]
- Cornett J. Cao F. Wang C. E. Ross C. A. Bates G. P. Li S. H. Li X. J. Nat. Genet. 2005;37:198–204. doi: 10.1038/ng1503. [DOI] [PubMed] [Google Scholar]
- Jones A. L. Wood J. D. Harper P. S. J. Inherited Metab. Dis. 1997;20:125–138. doi: 10.1023/A:1005340302695. [DOI] [PubMed] [Google Scholar]
- Nukina N. Rinsho Shinkeigaku. 1997;37:1139–1140. [PubMed] [Google Scholar]
- Ross C. A. Margolis R. L. Rosenblatt A. Ranen N. G. Becher M. W. Aylward E. Medicine. 1997;76:305–338. doi: 10.1097/00005792-199709000-00001. [DOI] [PubMed] [Google Scholar]
- Sugaya K. Matsubara S. Kagamihara Y. Kawata A. Hayashi H. PLoS One. 2007;2:e635. doi: 10.1371/journal.pone.0000635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Landry J. P. Zhu X. D. Gregg J. P. Opt. Lett. 2004;29:581–583. doi: 10.1364/OL.29.000581. [DOI] [PubMed] [Google Scholar]
- Zhu X. Landry J. P. Sun Y. S. Gregg J. P. Lam K. S. Guo X. Appl. Opt. 2007;46:1890–1895. doi: 10.1364/AO.46.001890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olzmann J. A. Carvalho P. Nat. Rev. Mol. Cell Biol. 2019;20:137–155. doi: 10.1038/s41580-018-0085-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu S. Zhang X. Liu P. Biochim. Biophys. Acta, Mol. Basis Dis. 2018;1864:1968–1983. doi: 10.1016/j.bbadis.2017.07.019. [DOI] [PubMed] [Google Scholar]
- Hanson Q. M. Barb A. W. Biochemistry. 2015;54:2931–2942. doi: 10.1021/acs.biochem.5b00299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roopenian D. C. Akilesh S. Nat. Rev. Immunol. 2007;7:715–725. doi: 10.1038/nri2155. [DOI] [PubMed] [Google Scholar]
- Patel D. A. Puig-Canto A. Challa D. K. Perez Montoyo H. Ober R. J. Ward E. S. J. Immunol. 2011;187:1015–1022. doi: 10.4049/jimmunol.1003780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hogarth P. M. Pietersz G. A. Nat. Rev. Drug Discovery. 2012;11:311–331. doi: 10.1038/nrd2909. [DOI] [PubMed] [Google Scholar]
- Ward E. S. Ober R. J. Trends Pharmacol. Sci. 2018;39:892–904. doi: 10.1016/j.tips.2018.07.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blumberg R. S. Lencer W. I. Nat. Biotechnol. 2005;23:1232–1234. doi: 10.1038/nbt1005-1232. [DOI] [PubMed] [Google Scholar]
- Igawa T. Maeda A. Haraya K. Tachibana T. Iwayanagi Y. Mimoto F. Higuchi Y. Ishii S. Tamba S. Hironiwa N. Nagano K. Wakabayashi T. Tsunoda H. Hattori K. PLoS One. 2013;8:e63236. doi: 10.1371/journal.pone.0063236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Igawa T. Ishii S. Tachibana T. Maeda A. Higuchi Y. Shimaoka S. Moriyama C. Watanabe T. Takubo R. Doi Y. Wakabayashi T. Hayasaka A. Kadono S. Miyazaki T. Haraya K. Sekimori Y. Kojima T. Nabuchi Y. Aso Y. Kawabe Y. Hattori K. Nat. Biotechnol. 2010;28:1203–1207. doi: 10.1038/nbt.1691. [DOI] [PubMed] [Google Scholar]
- Bitonti A. J. Dumont J. A. Low S. C. Peters R. T. Kropp K. E. Palombella V. J. Stattel J. M. Lu Y. Tan C. A. Song J. J. Garcia A. M. Simister N. E. Spiekermann G. M. Lencer W. I. Blumberg R. S. Proc. Natl. Acad. Sci. U. S. A. 2004;101:9763–9768. doi: 10.1073/pnas.0403235101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zebisch M. Xu Y. Krastev C. MacDonald B. T. Chen M. Gilbert R. J. He X. Jones E. Y. Nat. Commun. 2013;4:2787. doi: 10.1038/ncomms3787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hornsby M. Paduch M. Miersch S. Saaf A. Matsuguchi T. Lee B. Wypisniak K. Doak A. King D. Usatyuk S. Perry K. Lu V. Thomas W. Luke J. Goodman J. Hoey R. J. Lai D. Griffin C. Li Z. Vizeacoumar F. J. Dong D. Campbell E. Anderson S. Zhong N. Graslund S. Koide S. Moffat J. Sidhu S. Kossiakoff A. Wells J. Mol. Cell. Proteomics. 2015;14:2833–2847. doi: 10.1074/mcp.O115.052209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lowman H. B. Bass S. H. Simpson N. Wells J. A. Biochemistry. 1991;30:10832–10838. doi: 10.1021/bi00109a004. [DOI] [PubMed] [Google Scholar]
- Ridgway J. B. Presta Lg Fau-Carter P. Carter P. Protein Eng., Des. Sel. 1996;9:617–621. doi: 10.1093/protein/9.7.617. [DOI] [PubMed] [Google Scholar]