

Abstract
Background and Objectives
To determine the diagnostic efficacy of clinical exome-targeted sequencing (CES) and spinocerebellar ataxia 36 (SCA36) screening in a real-life cohort of patients with cerebellar ataxia (CA) from Eastern Spain.
Methods
A total of 130 unrelated patients with CA, negative for common trinucleotide repeat expansions (SCA1, SCA2, SCA3, SCA6, SCA7, SCA8, SCA12, SCA17, dentatorubral pallidoluysian atrophy [DRPLA], and Friedreich ataxia), were studied with CES. Bioinformatic and genotype-phenotype analyses were performed to assess the pathogenicity of the variants encountered. Copy number variants were analyzed when appropriate. In undiagnosed dominant and sporadic cases, repeat primed PCR was used to screen for the presence of a repeat expansion in the NOP56 gene.
Results
CES identified pathogenic or likely pathogenic variants in 50 families (39%), including 23 novel variants. Overall, there was a high genetic heterogeneity, and the most frequent genetic diagnosis was SPG7 (n = 15), followed by SETX (n = 6), CACNA1A (n = 5), POLR3A (n = 4), and SYNE1 (n = 3). In addition, 17 families displayed likely pathogenic/pathogenic variants in 14 different genes: KCND3 (n = 2), KIF1C (n = 2), CYP27A1A (n = 2), AFG3L2 (n = 1), ANO10 (n = 1), CAPN1 (n = 1), CWF19L1 (n = 1), ITPR1 (n = 1), KCNA1 (n = 1), OPA1 (n = 1), PNPLA6 (n = 1), SPG11 (n = 1), SPTBN2 (n = 1), and TPP1 (n = 1). Twenty-two novel variants were characterized. SCA36 was diagnosed in 11 families, all with autosomal dominant (AD) presentation. SCA36 screening increased the total diagnostic rate to 47% (n = 61/130). Ultimately, undiagnosed patients showed delayed age at onset (p < 0.05) and were more frequently sporadic.
Discussion
Our study provides insight into the genetic landscape of CA in Eastern Spain. Although CES was an effective approach to capture genetic heterogeneity, most patients remained undiagnosed. SCA36 was found to be a relatively frequent form and, therefore, should be tested prior to CES in familial AD presentations in particular geographical regions.
Hereditary cerebellar ataxias (CAs) are highly heterogeneous disorders frequently associated with additional neurologic and extraneurologic manifestations.1,2 The genetic diagnosis in the clinical practice is challenging. Nearly 200 causal genes have been associated with ataxia,3-5 and different technologies are needed to cover the full mutational spectrum: dynamic expansions, point mutations, and small and large duplications, insertions, or deletions. Sanger sequencing of candidate genes has been superseded in recent years by next-generation sequencing (NGS) techniques.6,7 However, conventional NGS is not technically capable of detecting tandem repeat expansions, the most common sequence variant type in adult-onset CA.1 Furthermore, ethnic and geographical background should be considered in diagnostic algorithms because some SCA subtypes are more common in certain populations.8,9 Accordingly, spinocerebellar ataxia 36 (SCA36) was reported as the most frequent hereditary CA in the northwest regions of Spain.10
In this study, the authors aimed to analyze the diagnostic yield of clinical exome-targeted sequencing (CES) in our cohort of patients with CA. In addition, the authors screened for SCA36 in undiagnosed dominant and sporadic cases. This study may help clarify the most appropriate diagnostic algorithm in our population.
Methods
This is a retrospective descriptive study which includes clinical and genetic data collected from January 2012 to October 2021 at Hospital Universitari i Politècnic La Fe, Valencia (Spain), a national referral center for Hereditary Cerebellar Ataxia and Spastic Paraplegia. This work describes the experience of a Spanish Ataxia Unit and is influenced by local standards of care.
Standard Protocol Approvals, Registrations, and Patient Consents
This study was approved by the institutional ethics committee at Health Research Institute Hospital La Fe (PI18/00147; PI2022-388-1). Informed consent was obtained before the genetic analysis. Genetic counseling was offered prior to and after the genetic results.
Patient Recruitment
Between January 2012 and October 2021, a total of 234 index cases with familial or sporadic noncongenital CA were recruited from the Neurology and Genetics Department at Hospital Universitari I Politècnic La Fe. All included patients had familial or sporadic noncongenital CA,11 with onset after 2 years. In all cases, acquired etiologies were fully excluded.
Common trinucleotide dynamic expansions in ATXN1 (SCA1), ATXN2 (SCA2), ATXN3 (SCA3), CACNA1A (SCA6), ATXN7 (SCA7), ATXN8 (SCA8), PPP2R2B (SCA12), TBP (SCA17), ATN1 (DRPLA), and FXN (Friedreich ataxia) were previously ruled out in all patients by PCR, followed by capillary electrophoresis. In addition, mutations in specific genes (FMR1 and RFC1) were studied in patients with a highly suggestive phenotype. A total of 31 SCA3, 23 Friedreich ataxia, 14 SCA2, 14 SCA8, 7 SCA6, 3 SCA7, 2 SCA17, 1 DRPLA, 5 cerebellar ataxia neuropathy and vestibular areflexia syndrome, and 4 fragile X–associated tremor/ataxia syndrome families were identified and excluded from further analysis. A total of 172 patients from 130 unrelated families were ultimately selected to determine the diagnostic efficacy of CES and SCA36 screening in a real-life CA cohort.
Clinical Assessments
All patients were examined by consultant neurologists (J.J.V., L.B., N.M., and R.B.-M.) in clinical settings during their follow-up visits. Demographic, familial, age at onset, and phenotypic information was systematically collected in a prospective database. Dominant transmission was considered if vertical inheritance.12 Recessive inheritance was assumed if consanguinity or family history of at least 2 affected siblings, with no cases in other generations. In all, a biological workup including albumin, immunoglobulin, cholesterol, triglycerides, α-fetoprotein, and vitamin E was routinely carried out. Additional metabolic testing (lactate, cholestanol, very long chain fatty acids, enzymatic activity assays, plasma amino acids, or urine organic acids) were performed based on clinical presumption. Brain MRI, nerve conduction studies, EMG, vestibular function testing, and muscle biopsy were performed and reviewed whenever needed.
Genetic Procedures
Genomic DNA was extracted from peripheral blood samples following standard procedures. A total of 130 probands from unrelated families were studied with a singleton Targeted-Exome Sequencing Panel (Agilent Technologies, Santa Clara, CA) for Illumina (San Diego, CA), in which 184 genes involved in CA and related forms were studied (additional data are provided in eAppendix 1, links.lww.com/NXG/A554). Family ATX-1 and ATX-27 were studied with the MovDisord-498 panel previously reported.13 Genes related to episodic ataxia were initially studied in cases with prominent and recurrent spells of ataxia and mild CA at interictal examination. If negative, all genes involved in CA were analyzed.
The library preparation was carried out according to the Bravo NGS SureSelectQXT Automated Target Enrichment protocol (Agilent Technologies) for Illumina Multiplexed Sequencing. The captured libraries were sequenced on NextSeq500 (Illumina) in a paired-end mode to generate a minimum median raw target coverage of 100×. The obtained sequences were aligned against the genome reference sequence (GRCh37/hg19) to perform the calling of variants with the Alissa Clinical Informatics Platform (Agilent Technologies). The annotated variants were initially filtered according to a minor allele frequency value ≤ 0.02, but only variants with a MAF ≤0.01 were considered. The frequency of the variants was explored in the Exome Aggregation Consortium database/gnomAD (gnomad.broadinstitute.org/) and 1,000 genomes (internationalgenome.org/). Filtered DNA variants were classified according to the American College of Medical Genetics guidelines.14 To classify the variants, the authors consulted their annotation in the single nucleotide polymorphism database (dbSNP, ncbi.nlm.nih.gov/SNP/) and their description in ClinVar (ncbi.nlm.nih.gov/clinvar/), varsome (varsome.com/), Human Gene Mutation Database (hgmd.cf.ac.uk), and Leiden Open Variation Database (lovd.nl/). In addition, base conservation was assessed with Genomic Evolutionary Rate Profiling,15 and in silico analysis were performed with the predictive tools: Protein Variation Effect Analyzer,16 Sorting Intolerant from Tolerant,17 Polyphen,18 and MutationTaster.19 All the likely pathogenic/pathogenic point mutations detected were confirmed by Sanger sequencing, and segregation of the variant in available family members was done when possible. Matching of phenotypic presentation to molecular diagnoses was evaluated in all cases. The presence of copy number variants (CNVs) was investigated by multiple ligation–dependent probe amplification analysis (MLPA; MRC Holland, Amsterdam, the Netherlands) and arrayCGH (aCGH) when appropriate. In all episodic ataxia cases undiagnosed after CES, CACNA1A CNVs were analyzed using SALSA P279. Furthermore, in the context of a compatible phenotype, CNVs in autosomal-recessive (AR) genes were analyzed if a single heterozygous pathogenic variant was detected by CES. Whenever MLPA commercial kit was not available for a gene, cytogenomic microarray (CytoScan XON array, Thermofisher, Waltham, MA) was performed. Coffalyser software and Chromosome Analysis Suite were used to analyze MLPA and aCGH results, respectively.
SCA36 Screening
NOP56 GGCCTG hexanucleotide repeat expansion was firstly studied by conventional PCR following standard procedures [sequence forward primer: TTTCGGCCTGCGTTCGGG (fluorescently labeled) and reverse: AGCCGACCGCGTGCTCAA; annealing temperature: 60°C]. Then, repeat-primed PCR (RP-PCR) was carried out as previously described.20 PCR products were separated on an ABI Prism 3,130 Analyzer (Applied Biosystems, Vernon Hills, IL), and data were examined using GeneMapper software (Applied Biosystems, Vernon Hills, IL). A positive result was defined by the combination of a single peak in the conventional PCR electropherogram plus the typical sawtooth pattern in the RP-PCR.
Statistical Methods
Descriptive analysis of clinical and paraclinical data was performed. In addition, differences in clinical features between diagnosed and undiagnosed patients were studied. Statistical analysis was performed using IBM SPSS Statistics for Mac version 27.0.1. Distribution of continuous variables was assessed with the Kolmogorov-Smirnov test (*p < 0.05), and subsequently, nonparametric tests were used for comparisons between groups. In all analysis, p < 0.05 was considered statistically significant.
Data Availability
Anonymized data not published within this article will be made available by request from any qualified investigator.
Results
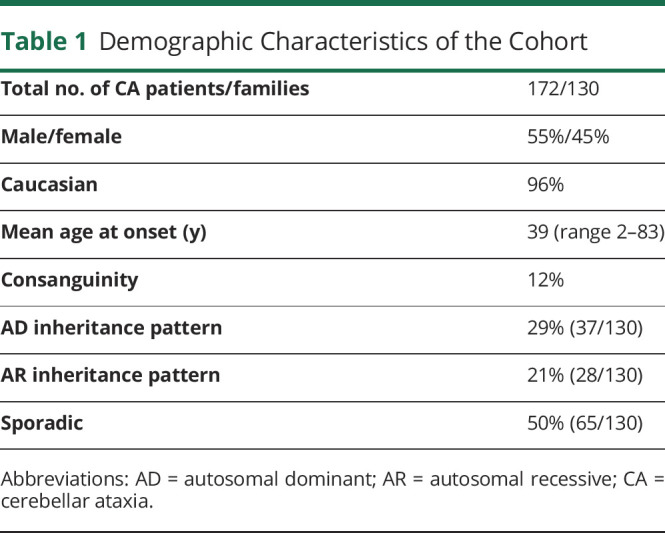
Overall, 172 individuals from 130 unrelated families were studied by CES. Screening of NOP56 expansion was done in all undiagnosed families with a possible dominant disease and sporadic cases with a suggestive phenotype of hereditary CA (Figure 1 and eFigure 1, links.lww.com/NXG/A555). The mean age at onset was 39 years (range 2–83, SD ± 19.5). Ninety-four patients (55%) were male. Ethnicity was predominantly Caucasian (n = 125, 96%). Four families had Romani ancestry and one African. The pattern of disease presentation was autosomal dominant (AD) in 37 (29%), AR in 28 (21%), and sporadic (S) in 65 (50%). First-degree consanguinity was present in 16 families (12%), Table 1.
Figure 1. Diagnostic Flowchart and Genetic Results of the Studied Patients.
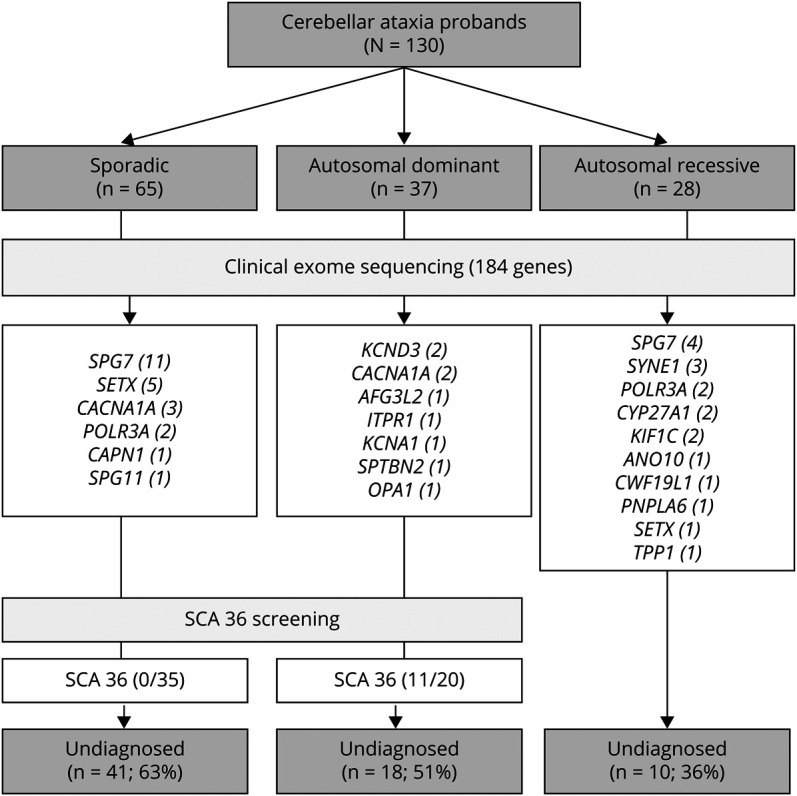
Table 1.
Demographic Characteristics of the Cohort
CES reached very probable or definite diagnoses in 50 families (39%). Clinical and genetic features of these patients are displayed in Table 2. Diagnosis yield was higher for AR presentation (18/28, 64%) than sporadic (23/65, 35%) or AD (9/37, 24%). Overall, the most frequently mutated gene was SPG7 in 15 of 130 families (12%), followed by SETX in 6 (5%), CACNA1A in 5 (4%), POLR3A in 4 (3%), and SYNE1 in 3 (2%). Sporadic cases harbored more frequently AR-associated genes (11 SPG7, 5 SETX, 2 POLR3A, 1 CAPN, and 1 SPG11) than AD genes (3 CACNA1A). Our population showed a high degree of genetic heterogeneity. Only 2 recurrent variants were identified: the SPG7 change c.1529C > T; p.(Ala510Val)21 was found in 19 alleles and the POLR3A variant c.1909 + 22G > A22 in 4 alleles. Seventeen families displayed likely pathogenic/pathogenic variants in 14 different genes: KCND3 (n = 2), KIF1C (n = 2), CYP27A1 (n = 2), AFG3L2 (n = 1), ANO10 (n = 1), CAPN1 (n = 1), CWF19L1 (n = 1), ITPR1 (n = 1), KCNA1 (n = 1), OPA1 (n = 1), PNPLA6 (n = 1), SPG11 (n = 1), SPTBN2 (n = 1), and TPP1 (n = 1). Twenty-two novel likely pathogenic/pathogenic variants were detected. Pathogenicity assessments are provided in Supplementary material, eTable 1 (links.lww.com/NXG/A556). Any gene with X-linked dominant or recessive transmission was identified. Copy number variations (CNVs) were studied by MLPA in CACNA1A, SALSA P279 (n = 7); SPG7, SALSA P213 (n = 5); ATM, SALSAs P041, and P042 (n = 1); SACS, SALSA P441 (n = 1); and SETX, SALSA P316 (n = 1), but any CNVs were identified. aCGH was performed only in family ATX-5, which carried a heterozygous pathogenic variant in POLR3A.
Table 2.
Clinical and Genetic Characteristics of the Cases With a Genetically Confirmed Diagnosis

Clinical features were generally highly concordant with the genetic diagnosis. Spastic ataxia was the most frequent SPG7 phenotype, but rarer clinical features such as progressive external ophthalmoplegia or dystonia were also identified.23 Alpha-fetoprotein was elevated in 8 patients, and molecular analysis later confirmed that 7 of them harbored SETX (ataxia with oculomotor apraxia type 2, AOA2) biallelic pathogenic variants. AOA2 was a frequent cause of early-onset ataxia (mean age at onset 12.6 years, range 3–17). Patients displayed a phenotype consistent of ataxia, oculomotor apraxia, sensorimotor neuropathy, and elevated alpha-fetoprotein. All patients with POLR3A-related ataxia were compound heterozygotes carriers of the intronic variant c.1909 + 22G > A.24 Three novel pathogenic/likely pathogenic POLR3A variants were identified: c.685C > T; p.(Arg229Ter), c.1628A > C; p.(Gln542Pro) and c.3688G > A; p.(Asp1230Asn). Phenotypes were rather homogeneous across different families. All 6 cases displayed short stature and central sensory tracts impairment with normal nerve conduction studies. Dystonia and dystonic tremor were prominent features in 4/6. Abnormal dentition (5/6) and mild MRI white matter hyperintensities (5/6) were also characteristic hallmarks. In 2 monozygotic twin siblings (ATX-5) with a highly suggestive phenotype, Figure 2, a cytogenic microarray (CytoScan XON array, Thermofisher, Waltham, MA) detected a formerly reported deletion (arr[GRCh37] 10q22.3(79781064_79782608) x 1) including exons 6, 7, and 8.25 Recessive ataxia due to SYNE1 was relatively common in our population (2%); the authors detected 2 novel pathogenic variants: c.368T > C; p.(Leu123Pro) and c.11253 + 2_11253+4dupTAG in 2 families a pure CA. Two siblings who carried a known pathogenic variant c.21148C > T; p.(Arg7050Ter)26 presented with early-onset ataxia and upper motor neuron involvement. The most severely affected brother had a history of congenital cataracts.
Figure 2. Family ATX-5, 2 Female Monozygotic Twins With Spastic Ataxia Had Compound Heterozygous Variants in POLR3A c.1909 + 22G > A and arr[GRCh37] 10q22.3(79781064_79782608)x1.
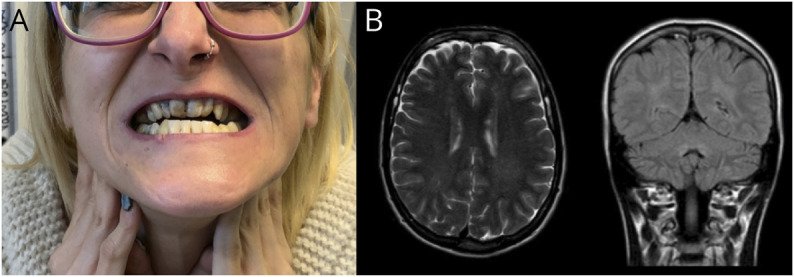
(A) Both patients had typically abnormal dentition; (B) T2-weighted brain MRI (left) and fluid attenuated recovery (FLAIR) brain MRI (right) showing characteristic mild supratentorial and infratentorial white matter hyperintensities.
Channelopathies were the most frequent AD ataxia detected by NGS (9/13). CACNA1A was the channel gene most commonly mutated. Patients with episodic ataxia, belonging to the families ATX-39, ATX-41, ATX-42, and ATX-43, had loss of function variants while the family ATX-40's patients with the missense change c.1748G > A; p.(Arg583Gln)27 displayed a complex phenotype. Both siblings in family ATX-40 showed marked axial weakness on examination, but EMG, including repetitive nerve stimulation and single fiber jitter, studies showed no abnormalities. Two novel changes in KCNA1 c.904A > C; p.(Ile302Leu) and KCND3 c.117A > G; p.(Met373Val) were identified. Patient 60, with the novel pathogenic variant KCND3 c.117A > G; p.(Met373Val), had frequent paroxysmal ataxic exacerbations responsive to acetazolamide. Patient 50, with the previously reported KCND3 c.680_682delTCT; p.(Phe227del),28 displayed occasional ataxic exacerbations triggered by emotional stress.
Very rare genetic etiologies were identified by CES. A novel homozygous pathogenic splicing variant in CWF19L1 (c.24-1G > C) was identified in 3 affected siblings with late-onset pure CA. In addition, a homozygous likely pathogenic variants in PNPLA6 c.3373G > A; p.(Asp1125Asn) was detected in a Romaní proband with adult-onset pure CA.
Spinocerebellar Ataxia 36 Screening
NOP56 hexanucleotide repeat expansion (GGCCTG) was screened in all undiagnosed families with an AD presentation (n = 20) and sporadic cases with a phenotype of late-onset (older than 20 years) progressive CA (n = 35). SCA36 was diagnosed in 31 patients of 11 families, all with AD presentation, and in none sporadic. Patients displayed a phenotype of pure or spastic CA. Sensorineural hearing loss was frequent (15/31, 48%), but facial and lingual myokymia were identified only in 3/31. Progressive external ophthalmoplegia and mild parkinsonism were present in 13/31 and 4/31 cases, respectively. NOP56 hexanucleotide expansion represented the most common dominant variants in this study, which accounted for 30% (11/37) of cases with AD, increasing the overall diagnostic rate to 47% (61/130). Of interest most of the SCA36 apparently unrelated families lived in the geographical areas of La Costera and La Vall d’Albaida, pointing to a founder effect, as reported in other SCA36 series.7
Sixty-nine families (53%) were not genetically diagnosed despite CES and SCA36 screening. Undiagnosed patients showed a significantly delayed age at onset (n = 81, 47, SD ± 17.2 years) compared with diagnosed cases (n = 86, 32, SD ± 18.5 years), p < 0.05, but without differences in sex distribution. Negative cases were more frequently sporadic (n = 39/69, 49%), followed by AD (n = 18/69, 30%) and AR (n = 10/69, 21%).
Discussion
In this study, CES was performed in a series of 130 probands with CA, leading to an overall diagnostic yield of 39%. Although common classic repeat expansions were excluded, the NOP56 hexanucleotide repeat expansion was further analyzed, which allowed us to diagnose 11 additional AD cases with SCA36. Several practical conclusions can be established from our study: (1) Diagnostic efficacy of CES applied to hereditary CA remains unsatisfactory because it missed the genetic diagnosis in more than 60% of cases in our series. This is concordant with other adult-onset CA cohorts, which reported a yield between 12.1% and 52%.6,29-32 (2) There is an enormous genetic heterogeneity underlying CA phenotypes, and the routine clinical use of CES allows the diagnosis of very rare genetic etiologies. Most genes mutated were detected in individual families, and diverse novel pathogenic/likely pathogenic variants were identified. (3) Because SCA36 is a common form of AD CA in Eastern Spain,10 the diagnostic workup of HCA in our geographical region should consider SCA36 testing in advance to NGS, especially if AD presentation. (4) The diagnostic efficacy was slightly higher in patients with a positive family history of dominant or, more frequently, recessive disease. The lowest diagnostic efficacy was found in sporadic and elderly cases. In this particular group, care should be taken to exclude neurodegenerative nonmendelian forms of CA such as multiple system atrophy. Future studies are also needed to ascertain if the so-called “idiopathic late-onset cerebellar ataxia” represents a distinct neurodegenerative disease entity.33-36
The correct interpretation of the countless number of detected DNA variants is the main challenge in NGS-based studies. Despite this, this study's CES allowed the identification of unexpected etiologies and expanded the clinical spectrum of known genes. A pathogenic variant in CWF19L1 (c.24-1G > C) was detected in 3 siblings with adult-onset pure CA. CWF19L1 is a very rare gene associated with congenital or early-onset CA and mental retardation. Only 3 families have been described to date.37-39 Thus, the clinical findings reported here contribute to broaden CWF19L1 phenotypical spectrum. Similarly, PNPLA6, a gene traditionally related with complex congenital or childhood disorders, was recently associated with adult-onset CA presentations.40 The novel PNPLA6 c.3373G > A; p.(Asp1125Asn), substitution described here, reinforces previously reported observations.
The availability of updated genome variant databases (e.g., ExAc/gnomAD or ClinVar) is critical in the diagnostic workup of diseases with variable expressivity or incomplete penetrance and, also, in late-onset diseases where segregation studies are often difficult. Familial trio (proband and parents) whole-exome or genome sequencing approaches can further improve diagnostic efficacy by identifying new ataxia-related genes and mutations.
NGS studies across different populations have contributed to identify underdiagnosed mutations in commonly mutated genes. In keeping with other CA cohorts, few genes harbored most pathogenic variants in our series. SPG7 (12%) and SETX (5%) were the most common AR genes detected,6 and channelopathies were the most frequent AD ataxias.41 Originally involved in 4H-leukodystrophy (hypomyelination and hypodontia), POLR3A is increasingly found in adult CA/hereditary spastic paraplegia cohorts (3.1%).22,24 Concordantly, in this study, POLR3A represented 3% of the studied population. All cases carried the recurrent intronic variant c.1909 + 22G > A in compound heterozygosity and displayed a uniform phenotype of ataxia, spasticity, central sensory tract impairment, and dystonia. SYNE1 mutations, initially described in Quebec, have also been reported as a frequent cause of ataxia outside French-Canadian population, with an estimated frequency of 5.3%.26 Herein, homozygous SYNE1 mutations were identified in 2% of cases. The authors reported 2 novel deleterious variants and described a family with ataxia, spasticity, and congenital cataracts.
The main limitations of this work are the retrospective nature of this study and the influence of local standards of care. Extensive metabolic testing was not systematically performed. Specific biochemical tests were carried out solely based on clinical presumption. Although CES is increasingly available and shall ultimately identify mutations in inherited metabolic disorders genes, metabolic screening should be done prior to molecular testing.42 Many adult-onset forms of inherited metabolic disorders can present either with complex or isolated ataxia phenotypes43,44 and some benefit from specific treatments that improve prognosis or prevent early death.45 Therefore, our approach could have missed the diagnosis of some patients or led to a delay in the identification of treatable conditions.
Because CES does not detect dynamic expansions and SCA36 was a common cause of AD ataxia in Spain, the authors systematically studied NOP56 hexanucleotide repeat expansion in AD and sporadic cases. SCA36 was a common cause of AD ataxia in this study, increasing the diagnostic yield from 39 to 47%. Studies in other populations showed that SCA36 has a worldwide distribution with reduced prevalence (France 1.9%, Japan 1.5%, and the United States 0.7%).46,47 These findings support that SCA36 should be tested prior to CES in Spanish families with AD presentations. New AD repeat expansion disorders are continuously being discovered. For instance, SCA37, caused by the intronic ATTTC repeat expansion in DAB1, was recently identified in families from Portugal and Spain.48-50 Screening of newly recognized repeat disorders should be carefully incorporated to genetic diagnostic algorithms.
In recent decades, advances in high-throughput sequencing technologies have enabled impressive achievements in the molecular diagnosis of mendelian diseases. However, an unsatisfactorily high proportion of patients remain undiagnosed among different CA series. Implementation of advanced sequencing strategies such as whole-genome sequencing together with collaborative international networks focused on ataxia are necessary steps toward reducing this diagnostic gap.
Glossary
- aCGH
arrayCGH
- AD
autosomal dominant
- AOA2
ataxia with oculomotor apraxia type 2
- AR
autosomal recessive
- CA
cerebellar ataxia
- CES
clinical exome-targeted sequencing
- CNV
copy number variant
- MLPA
multiple ligation-dependent probe amplification analysis
- NGS
next-generation sequencing
- RP-PCR
repeat primed PCR
- SCA
spinocerebellar ataxia
Appendix. Authors
Study Funding
This work was supported by the Instituto de Salud Carlos III (ISCIII)-Subdirección General de Evaluación y Fomento de la Investigación [PI18/00147; PI21/00103] and by the Generalitat Valenciana [PROMETEO/2018/135; PROMETEO/2019/075], within the framework of the National R + D + I Plan co-funded with ERDF funds. JFVC is funded by the ISCIII (JR19/00030). Part of the equipment employed in this work has been funded by Generalitat Valenciana and co-financed with ERDF funds (OP ERDF of Comunitat Valenciana 2014–2020).
Disclosure
The authors report no conflict of interest. Go to Neurology.org/NG for full disclosure.
References
- 1.Klockgether T, Mariotti C, Paulson HL. Spinocerebellar ataxia. Nat Rev Dis Primers. 2019;5(1):24-21. [DOI] [PubMed] [Google Scholar]
- 2.Durr A. Autosomal dominant cerebellar ataxias: polyglutamine expansions and beyond. Lancet Neurol. 2010;9(9):885-894. [DOI] [PubMed] [Google Scholar]
- 3.Synofzik M, Schüle R. Overcoming the divide between ataxias and spastic paraplegias: shared phenotypes, genes, and pathways. Mov Disord. 2017;32(3):332-345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Beaudin M, Klein CJ, Rouleau GA, Dupré N. Systematic review of autosomal recessive ataxias and proposal for a classification. Cerebellum Ataxias. 2017;4(1):3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Coutelier M, Stevanin G, Brice A. Genetic landscape remodelling in spinocerebellar ataxias: the influence of next-generation sequencing. J Neurol. 2015;262(10):2382-2395. [DOI] [PubMed] [Google Scholar]
- 6.Coutelier M, Hammer MB, Stevanin G, et al. Efficacy of exome-targeted capture sequencing to detect mutations in known cerebellar ataxia genes. JAMA Neurol. 2018;75(5):591-599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Pyle A, Smertenko T, Bargiela D, et al. Exome sequencing in undiagnosed inherited and sporadic ataxias. Brain. 2015;138(2):276-283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ruano L, Melo C, Silva MC, Coutinho P. The global epidemiology of hereditary ataxia and spastic paraplegia: a systematic review of prevalence studies. Neuroepidemiology. 2014;42(3):174-183. [DOI] [PubMed] [Google Scholar]
- 9.Obayashi M, Stevanin G, Synofzik M, et al. Spinocerebellar ataxia type 36 exists in diverse populations and can be caused by a short hexanucleotide GGCCTG repeat expansion. J Neurol Neurosurg Psychiatry. 2015;86(9):986-995. [DOI] [PubMed] [Google Scholar]
- 10.García-Murias M, Quintáns B, Arias M, et al. “Costa da Morte” ataxia is spinocerebellar ataxia 36: clinical and genetic characterization. Brain. 2012;135(5):1423-1435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Valence S, Cochet E, Rougeot C, et al. Exome sequencing in congenital ataxia identifies two new candidate genes and highlights a pathophysiological link between some congenital ataxias and early infantile epileptic encephalopathies. Genet Med. 2019;21(3):553-563. [DOI] [PubMed] [Google Scholar]
- 12.Hilton-Jones D, Turner MR. Oxford Textbook of Neuromuscular Disorders. Vol 1. First. In: Hilton-Jones D, Turner MR, eds. Oxford University Press; 2014. [Google Scholar]
- 13.Correa-Vela M, Lupo V, Montpeyó M, et al. Impaired proteasome activity and neurodegeneration with brain iron accumulation in FBXO7 defect. Ann Clin Translational Neurol. 2020;7(8):1436-1442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Richards S, Aziz N, Bale S, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of medical genetics and genomics and the association for molecular pathology. Genet Med. 2015;17(5):405-424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Davydov Ev, Goode DL, Sirota M, Cooper GM, Sidow A, Batzoglou S. Identifying a high fraction of the human genome to be under selective constraint using GERP++. Plos Comput Biol. 2010;6(12):e1001025, [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Choi Y, Sims GE, Murphy S, Miller JR, Chan AP. Predicting the functional effect of amino acid substitutions and indels. PLoS One. 2012;7(10):e46688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Vaser R, Adusumalli S, Leng SN, Sikic M, Ng PC. SIFT missense predictions for genomes. Nat Protoc. 2016;11(1):1-9. [DOI] [PubMed] [Google Scholar]
- 18.Adzhubei IA, Schmidt S, Peshkin L, et al. A method and server for predicting damaging missense mutations. Nat Methods. 2010;7(4):248-249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Schwarz JM, Cooper DN, Schuelke M, Seelow D. MutationTaster2: mutation prediction for the deep-sequencing age. Nat Methods. 2014;11(4):361-362. [DOI] [PubMed] [Google Scholar]
- 20.Kobayashi H, Abe K, Matsuura T, et al. Expansion of intronic GGCCTG hexanucleotide repeat in NOP56 causes SCA36, a type of spinocerebellar ataxia accompanied by motor neuron involvement. Am J Hum Genet. 2011;89(1):121-130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Brugman F, Scheffer H, Wokke JHJ, et al. Paraplegin mutations in sporadic adult-onset upper motor neuron syndromes. Neurology. 2008;71(19):1500-1505. [DOI] [PubMed] [Google Scholar]
- 22.la Piana R, Cayami FK, Tran LT, et al. Diffuse hypomyelination is not obligate for POLR3-related disorders. Neurology. 2016;86(17):1622-1626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Baviera-Muñoz R, Campins-Romeu M, Carretero-Vilarroig L, et al. Clinical and genetic characteristics of 21 Spanish patients with biallelic pathogenic SPG7 mutations. J Neurol Sci. 2021;429(August):118062. [DOI] [PubMed] [Google Scholar]
- 24.Minnerop M, Kurzwelly D, Wagner H, et al. Hypomorphic mutations in POLR3A are a frequent cause of sporadic and recessive spastic ataxia. Brain. 2017;140(6):1561-1578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Infante J, Serrano-Cárdenas KM, Corral‐Juan M, et al. POLR3A-related spastic ataxia: new mutations and a look into the phenotype. J Neurol. 2020;267(2):324-330. [DOI] [PubMed] [Google Scholar]
- 26.Synofzik M, Smets K, Mallaret M, et al. SYNE1 ataxia is a common recessive ataxia with major non-cerebellar features: a large multi-centre study. Brain. 2016;139(5):1378-1393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Battistini S, Stenirri S, Piatti M, et al. A new CACNA1A gene mutation in acetazolamide-responsive familial hemiplegic migraine and ataxia. Neurology. 1999;53(1):38-43. [DOI] [PubMed] [Google Scholar]
- 28.Lee YC, Durr A, Majczenko K, et al. Mutations in KCND3 cause spinocerebellar ataxia type 22. Ann Neurol. 2012;72(6):859-869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Pyle A, Smertenko T, Bargiela D, et al. Exome sequencing in undiagnosed inherited and sporadic ataxias. Brain. 2015;138(2):276-283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Fogel BL, Lee H, Deignan JL, et al. Exome sequencing in the clinical diagnosis of sporadic or familial cerebellar ataxia. JAMA Neurol. 2014;71(10):1237-1246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ngo KJ, Rexach JE, Lee H, et al. A diagnostic ceiling for exome sequencing in cerebellar ataxia and related neurological disorders. Hum Mutat. 2020;41(2):487-501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Sun M, Johnson AK, Nelakuditi V, et al. Targeted exome analysis identifies the genetic basis of disease in over 50% of patients with a wide range of ataxia-related phenotypes. Genet Med. 2019;21(1):195-206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Giordano I, Harmuth F, Jacobi H, et al. Clinical and genetic characteristics of sporadic adult-onset degenerative ataxia. Neurology. 2017;89(10):1043-1049. [DOI] [PubMed] [Google Scholar]
- 34.Abele M, Bürk K, Schöls L, et al. The aetiology of sporadic adult-onset ataxia. Brain. 2002;125(5):961-968. [DOI] [PubMed] [Google Scholar]
- 35.Lin DJ, Hermann KL, Schmahmann JD. Multiple system atrophy of the cerebellar type: clinical state of the art. Mov Disord. 2014;29(3):294-304. [DOI] [PubMed] [Google Scholar]
- 36.Harding AE “‘Idiopathic’ late onset cerebellar ataxia. J Neurol Sci. 1981;51(2):259-271. [DOI] [PubMed] [Google Scholar]
- 37.Burns R, Majczenko K, Xu J, et al. Homozygous splice mutation in CWF19L1 in a Turkish family with recessive ataxia syndrome. Neurology. 2014;83(23):2175-2182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Evers C, Kaufmann L, Seitz A, et al. Exome sequencing reveals a novel CWF19L1 mutation associated with intellectual disability and cerebellar atrophy. Am J Med Genet A. 2016;170(6):1502-1509. [DOI] [PubMed] [Google Scholar]
- 39.Nguyen M, Boesten I, Hellebrekers DMEI, et al. Pathogenic CWF19L1 variants as a novel cause of autosomal recessive cerebellar ataxia and atrophy. Eur J Hum Genet. 2016;24(4):619-622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Wiethoff S, Bettencourt C, Paudel R, et al. Pure cerebellar ataxia with homozygous mutations in the PNPLA6 gene. Cerebellum. 2017;16(1):262-267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Coutelier M, Coarelli G, Monin ML, et al. A panel study on patients with dominant cerebellar ataxia highlights the frequency of channelopathies. Brain. 2017;140(6):1579-1594. [DOI] [PubMed] [Google Scholar]
- 42.de Silva R, Greenfield J, Cook A, et al. Guidelines on the diagnosis and management of the progressive ataxias. Orphanet J Rare Dis. 2019;14(1):51, [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Masingue M, Dufour L, Lenglet T, et al. Natural history of adult patients with GM2 gangliosidosis. Ann Neurol. 2020;87(4):609-617. [DOI] [PubMed] [Google Scholar]
- 44.Sévin M, Lesca G, Baumann N, et al. The adult form of Niemann-Pick disease type C. Brain. 2006;130(1):120-133. [DOI] [PubMed] [Google Scholar]
- 45.Silver G, Mercimek-Andrews S. Inherited metabolic disorders presenting with ataxia. Int J Mol Sci. 2020;21(15):5519-5618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Valera JM, Diaz T, Petty LE, et al. Prevalence of spinocerebellar ataxia 36 in a US population. Neurol Genet. 2017;3(4):e174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Obayashi M, Stevanin G, Synofzik M, et al. Spinocerebellar ataxia type 36 exists in diverse populations and can be caused by a short hexanucleotide GGCCTG repeat expansion. J Neurol Neurosurg Psychiatry. 2015;86(9):986-995. [DOI] [PubMed] [Google Scholar]
- 48.Corral-Juan M, Serrano-Munuera C, Rábano A, et al. Clinical, genetic and neuropathological characterization of spinocerebellar ataxia type 37. Brain. 2018;141(7):1981-1997. [DOI] [PubMed] [Google Scholar]
- 49.Loureiro JR, Oliveira CL, Mota C, et al. Mutational mechanism for DAB1 (ATTTC) n insertion in SCA37: ATTTT repeat lengthening and nucleotide substitution. Hum Mutat. 2019;40(4):404-412. [DOI] [PubMed] [Google Scholar]
- 50.Seixas AI, Loureiro JR, Costa C, et al. A pentanucleotide ATTTC repeat insertion in the non-coding region of DAB1, mapping to SCA37, causes spinocerebellar ataxia. Am J Hum Genet. 2017;101(1):87-103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Bessa C, Teixeira CA, Dias A, et al. CLN2/TPP1 deficiency: the novel mutation IVS7-10A>G causes intron retention and is associated with a mild disease phenotype. Mol Genet Metab. 2008;93(1):66-73. [DOI] [PubMed] [Google Scholar]
- 52.Renaud M, Anheim M, Kamsteeg EJ, et al. Autosomal recessive cerebellar ataxia type 3 due to ANO10 mutations: delineation and genotype-phenotype correlation study. JAMA Neurol. 2014;71(10):1305-1310. [DOI] [PubMed] [Google Scholar]
- 53.Arnoldi A, Tonelli A, Crippa F, et al. A clinical, genetic, and biochemical characterization of SPG7 mutations in a large cohort of patients with hereditary spastic paraplegia. Hum Mutat. 2008;29(4):522-531. [DOI] [PubMed] [Google Scholar]
- 54.Moreira MC, Klur S, Watanabe M, et al. Senataxin, the ortholog of a yeast RNA helicase, is mutant in ataxia-ocular apraxia 2. Nat Genet. 2004;36(3):225-227. [DOI] [PubMed] [Google Scholar]
- 55.Anheim M, Monga B, Fleury M, et al. Ataxia with oculomotor apraxia type 2: clinical, biological and genotype/phenotype correlation study of a cohort of 90 patients. Brain. 2009;132(10):2688-2698. [DOI] [PubMed] [Google Scholar]
- 56.Pera J, Lechner S, Biskup S, Strach M, Grodzicki T, Slowik A. Two novel mutations of the SETX gene and ataxia with oculomotor apraxia type 2. Clin Neurol Neurosurg. 2015;128:44-46. [DOI] [PubMed] [Google Scholar]
- 57.Stevanin G, Santorelli FM, Azzedine H, et al. Mutations in SPG11, encoding spatacsin, are a major cause of spastic paraplegia with thin corpus callosum. Nat Genet. 2007;39(3):366-372. [DOI] [PubMed] [Google Scholar]
- 58.Guyant-Maréchal L, Verrips A, Girard C, et al. Unusual cerebrotendinous xanthomatosis with fronto-temporal dementia phenotype. Am J Med Genet A. 2005;139A(2):114-117. [DOI] [PubMed] [Google Scholar]
- 59.Melo US, Freua F, Lynch DS, et al. Clinical aspects of hereditary spastic paraplegia 76 and novel CAPN1 mutations. Clin Genet. 2018;94(5):482-483. [DOI] [PubMed] [Google Scholar]
- 60.Gan-Or Z, Bouslam N, Birouk N, et al. Mutations in CAPN1 cause autosomal-recessive hereditary spastic paraplegia. Am J Hum Genet. 2016;98(5):1038-1046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Denier C, Ducros A, Vahedi K, et al. High prevalence of CACNA1A truncations and broader clinical spectrum in episodic ataxia type 2. Neurology. 1999;52(9):1816-1821. [DOI] [PubMed] [Google Scholar]
- 62.Yugrakh MS, Levy OA. Clinical reasoning: a middle-aged man with episodes of gait imbalance and a newly found genetic mutation. Neurology. 2012;79(16):e135, e139, [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Eunson LH, Graves TD, Hanna MG. New calcium channel mutations predict aberrant RNA splicing in episodic ataxia. Neurology. 2005;65(2):308-310. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Anonymized data not published within this article will be made available by request from any qualified investigator.