

SUMMARY
G protein-coupled receptors (GPCRs) interact with intracellular transducers to control both signal initiation and desensitization, but the distinct mechanisms that control the regulation of different GPCR subtypes are unclear. Here we use fluorescence imaging and electrophysiology to examine the metabotropic glutamate receptor (mGluR) family. We find distinct properties across subtypes in both rapid desensitization and internalization, with striking differences between the group II mGluRs. mGluR3, but not mGluR2, undergoes glutamate-dependent rapid desensitization, internalization, trafficking, and recycling. We map differences between mGluRs to variable Ser/Thr-rich sequences in the C-terminal domain (CTD) that control interaction with both GPCR kinases and β-arrestins. Finally, we identify a cancer-associated mutation, G848E, within the mGluR3 CTD that enhances β-arrestin coupling and internalization, enabling an analysis of mGluR3 β-arrestin-coupling properties and revealing biased variants. Together, this work provides a framework for understanding the distinct regulation and functional roles of mGluR subtypes.
Graphical abstract
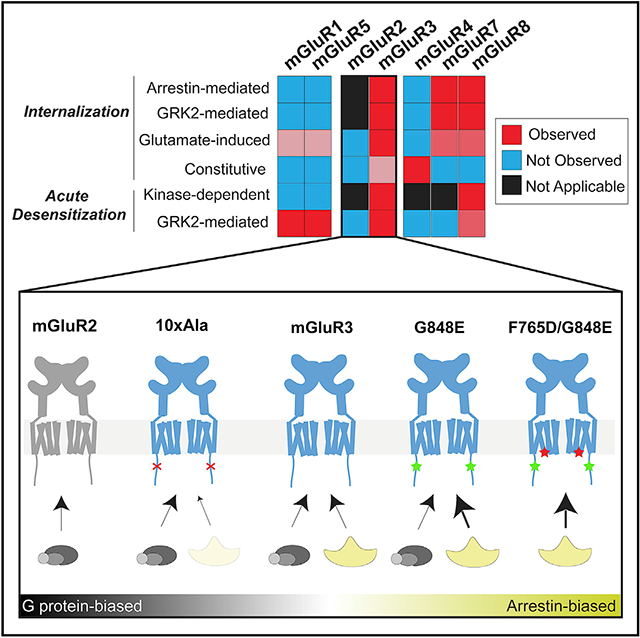
In brief
Abreu et al. use high-resolution imaging and electrophysiology to reveal distinct desensitization properties across subtypes of the metabotropic glutamate receptor (mGluR) family. Comparative analysis of group II mGluR subtypes identifies a Ser/Thr-rich region in the C-terminal domain of mGluR3 that mediates both rapid GRK-mediated desensitization and arrestin-mediated internalization.
INTRODUCTION
G protein-coupled receptors (GPCRs) sense diverse extracellular signals to initiate intracellular responses. The temporal properties of GPCR signaling are regulated by desensitization, a process that involves both uncoupling of the receptor from G proteins via steric blockage and receptor internalization. Generally, GPCR kinases (GRKs) and arrestins regulate GPCR desensitization, although GRK- and arrestin-independent mechanisms also exist (Ferguson et al., 1996; Gainetdinov et al., 2004; Ménard et al., 1997; Rajagopal and Shenoy, 2018). Canonically, GRKs are recruited to activated GPCRs and either compete with G proteins for receptor access or directly bind G proteins to contribute to rapid desensitization (Lodowski et al., 2003; Tesmer et al., 2005). GRKs can also phosphorylate the cytosolic C-terminal domain (CTD) of the receptor to enable the recruitment of β-arrestin (β-arr) 1 or β-arr2. β-arr serves as a scaffold for the recruitment of either endocytic proteins (e.g., clathrin) or signaling proteins (e.g., ERK) (Hilger et al., 2018; Peterson and Luttrell, 2017; Shenoy and Lefkowitz, 2011). This desensitization framework has been established through the study of prototypical GPCRs, but different GPCR subtypes use distinct sets of mechanisms that are only partially deciphered.
Although a structural understanding of GPCR/GRK interaction is limited (Komolov and Benovic, 2018), structural and biophysical studies have revealed complexity in the interactions between β-arr and receptors. This includes modes of interactions in which β-arr binds the CTD, the transmembrane core, or both domains of the receptor (Cahill et al., 2017; Kang et al., 2015; Kumari et al., 2016; Shukla et al., 2014; Staus et al., 2020) and associated conformational dynamics within both β-arr subtypes (Latorraca et al., 2018; Lee et al., 2016; Nuber et al., 2016). Many GPCRs have been shown to couple to β-arr, but the nature of this interaction depends on the receptor subtype, the β-arr subtype, and the receptor ligand used. Key determinants of β-arr-coupling propensity are the degree and the pattern of receptor phosphorylation (Miess et al., 2018; Nobles et al., 2011; Sente et al., 2018), leading to the proposal of phosphorylation codes (phospho-codes) that are recognized by β-arr (Zhou et al., 2017). GPCRs are broadly characterized into class A and class B based on the nature of their β-arr coupling (Kohout et al., 2001; Oakley et al., 2000). In the strong interactions of class B GPCRs, such as the V2 vasopressin receptor, β-arr internalizes with the receptor, which can lead to slow recycling and degradation (DeWire et al., 2007). In the weaker interactions of class A GPCRs, such as the beta-2 adrenergic receptor (β2AR), receptor/β-arr complexes do not typically persist into the endosome and receptors are recycled relatively quickly compared with class B GPCRs. Recently, it was shown that some GPCRs can engage in transient interactions with β-arr to produce non-stoichiometric catalytic activation and accumulation of β-arr in clathrin-coated structures (Eichel et al., 2016, 2018). The complexity of β-arr coupling suggests that GRK coupling may be similarly complex, motivating a thorough analysis of transducer coupling across receptor subtypes.
In contrast to family A and B GPCRs, which form the basis of existing desensitization models, the desensitization of family C GPCRs is less understood (Ellaithy et al., 2020). In particular, there is a lack of consensus regarding desensitization for metabotropic glutamate receptors (mGluRs) (Iacovelli et al., 2013; Suh et al., 2018), and comparative analysis across subtypes is lacking. Among the well-studied group I mGluRs (mGluR1 and mGluR5), evidence exists for constitutive clathrin-independent internalization (Fourgeaud et al., 2003; Francesconi et al., 2009), and control of receptor surface levels via kinases and accessory proteins have been observed (Suh et al., 2018). Evidence for interactions between mGluRs and GRKs or β-arr is limited and indirect (Emery et al., 2010; Mundell et al., 2001; Stoppel et al., 2017), leaving mechanistic details unknown.
The group II mGluRs (mGluR2 and mGluR3) are ubiquitous throughout the nervous system, where they couple to Gi/o-family G proteins to play roles in neuromodulation and are drug targets for neurological and psychiatric disorders (Nicoletti et al., 2011; Reiner and Levitz, 2018). mGluR3 is also expressed in the skin, where it has been linked to melanoma (Neto and Ceol, 2018; O’Hayre et al., 2013; Prickett et al., 2011). Like most family C GPCRs, mGluR2 and mGluR3 have large extracellular ligand binding domains (LBDs), 7-helix transmembrane domains (TMDs), and relatively short (~50 aa) intracellular CTDs (Ellaithy et al., 2020). Although the highly homologous (~70% sequence identity) mGluR2 and mGluR3 have been treated as a single entity, biophysical studies have shown that they have distinct activation properties, with mGluR3 showing ~10-fold higher apparent glutamate affinity and more basal activation (Tora et al., 2018; Vafabakhsh et al., 2015). This suggests that these receptors have unique physiological profiles, raising the question of their desensitization and signaling dynamics. A previous study used an endpoint cyclic AMP assay to suggest that mGluR2, but not mGluR3, is resistant to GRK- and β-arr-mediated desensitization (Iacovelli et al., 2009), but otherwise little is known about group II mGluR desensitization.
In this study, we elucidate the molecular mechanisms of mGluR desensitization using fluorescence imaging and functional assays. Among the group II mGluRs, we find that mGluR3, but not mGluR2, is subject to extensive glutamate-dependent internalization via GRK and β-arr. Compared with mGluR3, group I and group III mGluRs show distinct properties, including β-arr-independent glutamate-induced (mGluR1 and mGluR5) and constitutive (mGluR4) internalization. A subset of mGluRs (mGluR1, mGluR3, mGluR5, and mGluR8) shows GRK-mediated functional desensitization on the timescale of seconds. After characterizing GRK and β-arr sensitivity across subtypes, we focus on deciphering the β-arr-coupling dynamics of mGluR3 and probing the mechanistic basis of the differences between group II mGluRs. Altogether, our results indicate that different mGluRs have fundamentally different signaling properties and provide insight into the mechanisms of GRK and β-arr coupling of family C GPCRs.
RESULTS
Glutamate-dependent internalization and trafficking of mGluRs
We first asked whether mGluRs undergo internalization following activation by labeling N-terminal SNAP-tagged mGluRs with the membrane impermeable BG-Alexa 546 fluorophore in HEK293T cells and measuring the loss of surface receptor fluorescence following agonist incubation. Saturating glutamate led to a ~40% reduction in surface fluorescence for mGluR3 (Figure 1A; Figure S1A). A similar agonist-induced decrease in surface levels was observed for β2AR (Figure 1A; Figure S1A), a prototypical family A GPCR known to undergo agonist-induced internalization (von Zastrow and Kobilka, 1992). The group III mGluRs, mGluR7 and mGluR8, also showed a clear but smaller (~25%) reduction in surface levels, and the group I mGluRs, mGluR1 and mGluR5, showed a small (~10%) decrease (Figure 1A). Strikingly, surface levels of both mGluR2 (group II) and mGluR4 (group III) showed no sensitivity to glutamate treatment. Altogether, these results motivated a comparative analysis of the closely related group II mGluRs, mGluR2 and mGluR3, which showed different extents of internalization.
Figure 1. Glutamate-dependent internalization of mGluRs.
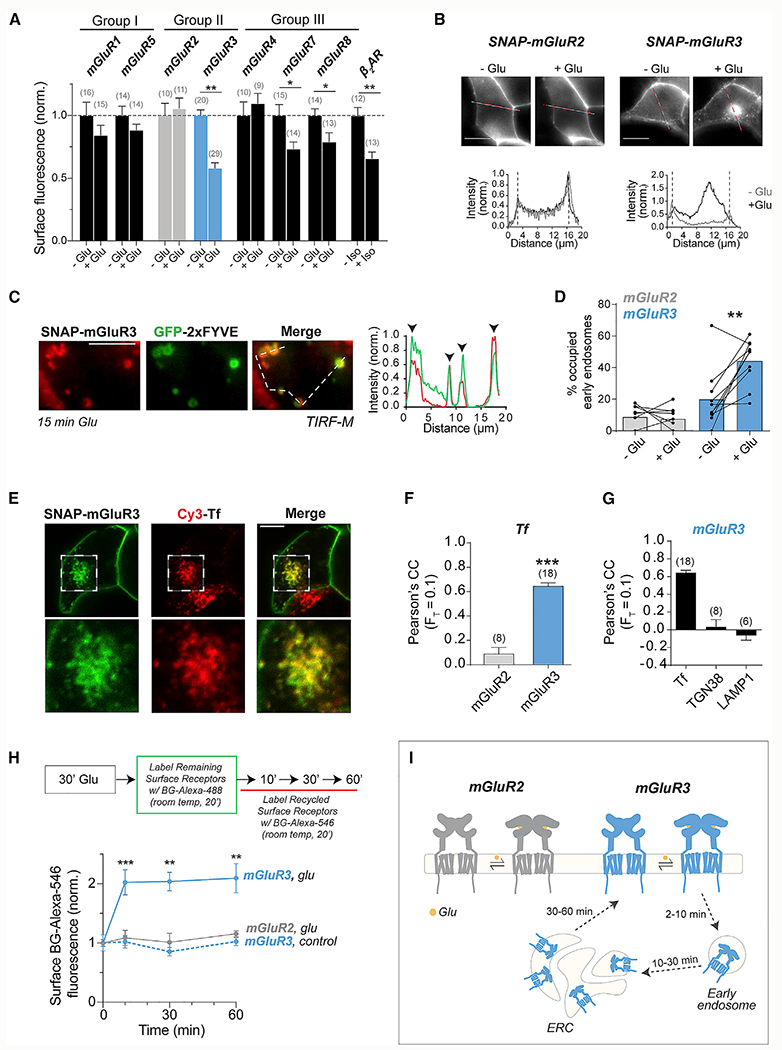
(A) Surface fluorescence from HEK293T cells expressing either SNAP-tagged mGluRs or β2AR with and without 60 min agonist stimulation (1–10 mM Glu and 10 μM Iso) before BG-Alexa 546 labeling. Values were normalized to the fluorescence of a given receptor without an agonist. Unpaired t tests, ***p < 0.0001 for mGluR3, *p = 0.02 for mGluR7, *p = 0.03 for mGluR8, and ***p = 0.0004 for β2AR.
(B) Images of cells expressing SNAP-mGluR2 (left) or SNAP-mGluR3 (right) before and after 30 min 1 mM Glu treatment with intensity line scans (bottom). Dotted lines denote the plasma membrane location.
(C) TIRF image (left) and line scan (right) showing colocalization of SNAP-mGluR3 (red) and GFP-2xFYVE (green) following Glu treatment.
(D) Quantification of the percentage of GFP-2xFYVE puncta that show colocalization with SNAP-mGluR2 or SNAP-mGluR3 before and after 15 min of Glu application. Lines connect values for individual cells, and bars show average values. Paired t test, **p = 0.004.
(E) Confocal images showing colocalization of SNAP-mGluR3 and Cy3-transferrin (Cy3-Tf) following 30 min treatment with 1 mM Glu.
(F) Pearson’s correlation coefficient (PCC) comparing the top 10% of pixels between mGluR2 or mGluR3 with Cy3-Tf following incubation in 1 nM Glu for 30 min. Unpaired t test, ***p < 0.0001.
(G) Quantification of PCC comparing colocalization of mGluR3 and Cy3-Tf, mCh-TGN38, or LAMP1-YFP following incubation in 1 mM Glu for 30 min.
(H) Top, schematic describing a recycling experiment in which receptors are internalized with Glu, remaining surface receptors are labeled with BG-Alexa 488, and subsequent labeling with BG-Alexa 546 is indicative of recycled receptors. For the control (blue dotted line), no Glu treatment was given. Unpaired t tests, ***p = 0.0002 for 10 min, ***p = 0.0002 for 30 min, and ***p = 0.0003 for 60 min.
(I) Schematic summarizing major results. mGluR2 remains on the surface following activation, whereas mGluR3 internalizes into early endosomes, traffics to the ERC, and recycles back to the surface.
Error bars show SEM. Scale bar, 10 μm (B and E) or 5 μm (C). Number of cells tested (F and G) or fields of cells analyzed (A) is in parentheses.
We next imaged cells expressing receptors labeled with BG-Alexa 546 before glutamate treatment. Although mGluR2 remained on the surface, mGluR3 showed an increase in intracellular fluorescence (Figure 1B; Figure S1B), similar to β2AR (Figure S1C). Some accumulation of mGluR3 was also seen in the absence of glutamate addition (Figure S1D), likely because of its basal activity (Vafabakhsh et al., 2015) and the release of glutamate from HEK293T cells (Tora et al., 2018). About 60% of glutamate-treated cells showed mGluR3 internalization versus <10% of cells showing negligible levels of mGluR2 internalization (Figure S1E). Treating cells with an mGluR3-negative allosteric modulator (ML289) or competitive antagonist (LY341495) decreased mGluR3 internalization (Figure S1E), confirming the requirement of receptor activation. Importantly, an EC50 level (1 μM) of glutamate (Figure S1F) or 5 min of glutamate stimulation was sufficient to internalize mGluR3 (Figure S1G). Similar results, in which mGluR3, but not mGluR2, showed internalization, were observed in HEK293 cells (Figure S1H).
We also imaged HEK293T cells to test internalization of group I and III mGluRs, and results agreed with the surface labeling assay (Figure S1I). mGluR7 and mGluR8 showed modest, glutamate-induced intracellular fluorescence, whereas limited intracellular puncta were observed for mGluR1 and mGluR5. Unexpectedly, mGluR4 showed intracellular puncta in the presence and absence of glutamate. Treatment with a saturating concentration of LY341495 did not alter mGluR4 internalization (Figure S1J), confirming that it is constitutive and glutamate independent. This result shows that despite both mGluR2 and mGluR4 showing no net glutamate-induced internalization (Figure 1A), they have distinct behavior.
We next characterized the trafficking of mGluR3 following internalization. Using total internal reflection fluorescence (TIRF) microscopy, we observed an increase in cytosolic fluorescent puncta for mGluR3, but not mGluR2, within 5 min of glutamate treatment (Figure S2B). A sparse population of fluorescent puncta was observed for all other mGluRs (Figure S2A). mGluR3 puncta showed glutamate-dependent colocalization with GFP-2xFYVE (Gillooly et al., 2000), indicating that puncta represent internalized receptors within early endosomes (Figures 1C and 1D; Figure S2C).
Following internalization into endosomes, GPCRs can traffic to lysosomes for degradation (Marchese and Benovic, 2001; Shenoy et al., 2001), the trans-Golgi network (TGN) for sorting (Abdullah et al., 2016), or the endocytic recycling compartment (ERC) (von Zastrow and Kobilka, 1992). We tested whether mGluR3 accumulates in the ERC using transferrin (Tf) as a marker (Maxfield and McGraw, 2004). Confocal microscopy revealed that mGluR3, but not mGluR2, colocalizes with internalized Cy3-Tf after glutamate treatment (Figures 1E and 1F; Figure S2D). In contrast, minimal colocalization of mGluR3 was observed with TGN and lysosome markers (Figure 1G; Figures S2E and S2F). Trafficking to the ERC suggests that mGluR3 recycles to the surface following internalization, which we confirmed by measuring the return of surface mGluR3 following glutamate incubation using a modified version of the surface labeling assay (Figure 1H; Figure S2G). We conclude that mGluR2 remains on the surface following activation, whereas mGluR3 internalizes, traffics to the ERC, and traffics back to the surface within 1 h (Figure 1I).
GRKs and β-arrestins mediate the internalization of a subset of mGluRs
The canonical pathway of GPCR endocytosis requires β-arr binding followed by targeting to clathrin-coated pits (CCPs). However, there is evidence that family C GPCRs, including group I mGluRs, internalize via β-arr and clathrin-independent mechanisms (Fourgeaud et al., 2003; Francesconi et al., 2009). Therefore, we decided to test which transducers mediate mGluR internalization.
To test whether G protein coupling is required for mGluR3 internalization, we transfected cells with pertussis toxin (PTX) (Mangmool and Kurose, 2011), which abolished the ability to activate G protein-coupled inward rectifier potassium (GIRK) channels in a patch-clamp assay (Figure S3A). Glutamate-induced mGluR3 internalization was slightly enhanced with PTX expression (Figures 2A and 2B; Figure S3B), indicating that G proteins do not drive mGluR3 internalization and may compete with other transducers. In contrast, treatment with the GRK2/3 inhibitor, compound 101 (cmpd101), or coexpression of a dominant-negative (DN) β-arr1 (Lin et al., 1997), greatly reduced mGluR3 internalization (Figures 2A and 2B; Figure S3B). Moreover, GRK2 or β-arr expression increased internalization of mGluR3, but not mGluR2 (Figures 2C and 2D; Figure S3B). mGluR3 internalization depended on the GRK2 kinase domain, because overexpressing a kinase-dead mutant (K220R) (Kong et al., 1994) exhibited a dominant-negative effect (Figures S3C and S3D). In contrast, a GRK2 mutant that is unable to bind Gβγ (GRK2-R587Q) (Carman et al., 2000) could still enhance internalization (Figures S3C and S3D).
Figure 2. GRK and β-arrestin dependence of mGluR internalization.
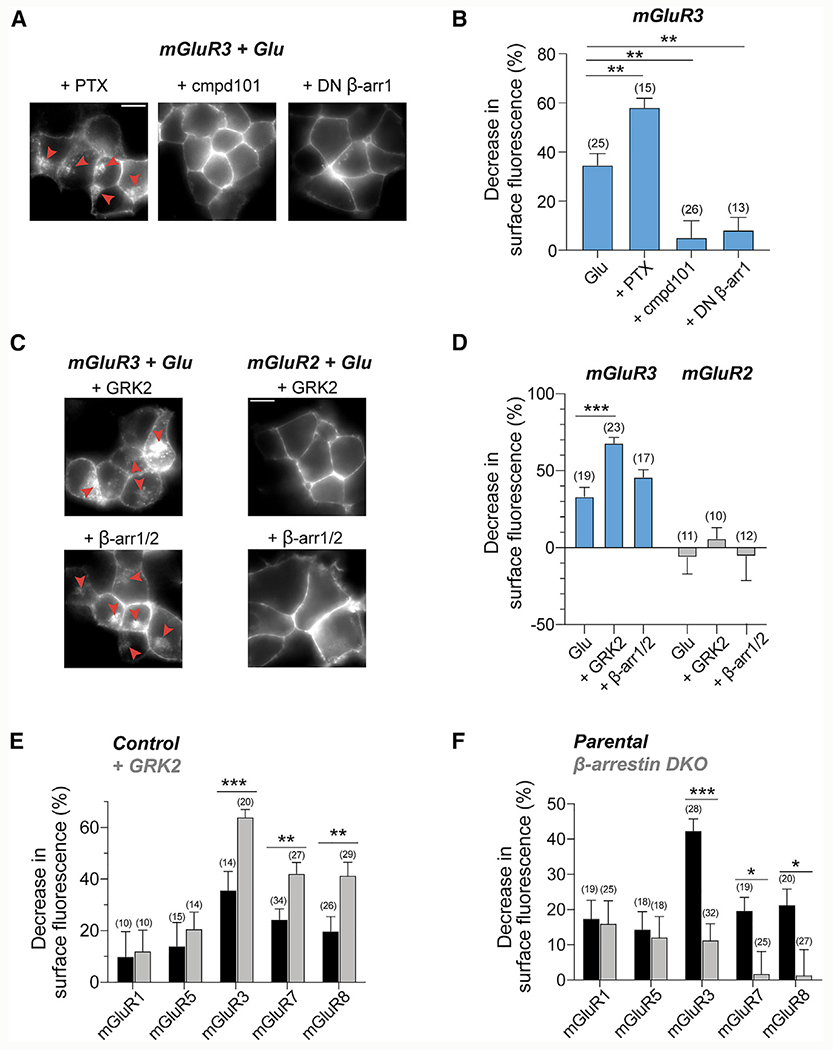
(A and B) DN β-arr1 (S412D) and cmpd101 reduce internalization of mGluR3, whereas PTX enhances internalization, as seen in representative images
(A) and quantification of surface fluorescence reduction following Glu application (B). Red arrows denote internalized receptors. Unpaired t tests, **p = 0.001 for PTX, **p = 0.001 for cmpd101, and **p = 0.001 for DN β-arr1.
(C and D) Overexpression of GRK2 or β-arr enhances internalization of mGluR3, but not mGluR2. Unpaired t test, ***p < 0.0001 for GRK2.
(E) Overexpression of GRK2 enhances internalization of mGluR7 and mGluR8, but not mGluR1 or mGluR5. Unpaired t tests, ***p = 0.0003 for mGluR3, **p = 0.004 for mGluR7, and **p = 0.006 for mGluR8.
(F) SNAP-mGluRs were expressed in a CRISPR β-arr DKO HEK293 cell line. Internalization for mGluRs in β-arr DKO cells compared with the parental cell line using quantification of surface fluorescence reduction following Glu application. Unpaired t tests, ***p < 0.0001 for mGluR3, *p = 0.02 for mGluR7, and *p = 0.04 for mGluR8.
Number of fields of cells analyzed is in parentheses. Error bars show SEM. Scale bar, 10 μm.
We next asked whether internalization of other mGluR subtypes depends on GRK and β-arr. GRK2 expression enhanced internalization of mGluR7 and mGluR8 (Figure 2E) but did not affect internalization of mGluR1 and mGluR5 (Figure 2E) or mGluR4 (Figure S3E). Next, we tested arrestin dependence by using β-arr double-knockout (DKO) cells (Luttrell et al., 2018), which showed dramatically reduced mGluR3 internalization (Figure 2F; Figure S3F). Similarly, mGluR7 and mGluR8 showed reduced internalization in DKO cells, whereas the internalization of mGluR1 and mGluR5 was not altered (Figure 2F; Figure S3G). Basal internalization of mGluR4 was maintained in β-arr DKO cells, suggesting that its constitutive internalization proceeds through a β-arr-independent mechanism (Figure S3G). These data show that internalization of mGluR3, mGluR7, and mGluR8 depend on β-arr and GRK.
GRKs mediate rapid desensitization of mGluRs
Given that internalization of a subset of mGluRs depends on GRKs and β-arr (Figure 2), we asked whether these transducers also produce rapid functional desensitization using GIRK currents as a readout. GIRK channels are activated by binding of Gβγ proteins (Logothetis et al., 1987; Whorton and MacKinnon, 2013) and are major native effectors of Gi/o-coupled GPCRs, including group II/III mGluRs (Dutar et al., 1999; Saugstad et al., 1996). mGluR2-mediated GIRK currents showed minimal desensitization, but mGluR3-mediated currents showed pronounced desensitization (Figures 3A–3C). Consistent with this, following 3 consecutive applications of glutamate, the peak amplitudes of mGluR2-mediated currents were maintained, whereas mGluR3-mediated currents progressively decreased (Figures S4A–S4C). Despite higher surface expression levels of mGluR2 under our conditions (Figure S4D), mGluR3 activation led to substantially smaller current amplitudes (Figure S4E). This suggests that a population of desensitized surface mGluR3 is unable to signal.
Figure 3. GRK2-mediated rapid desensitization of mGluRs.
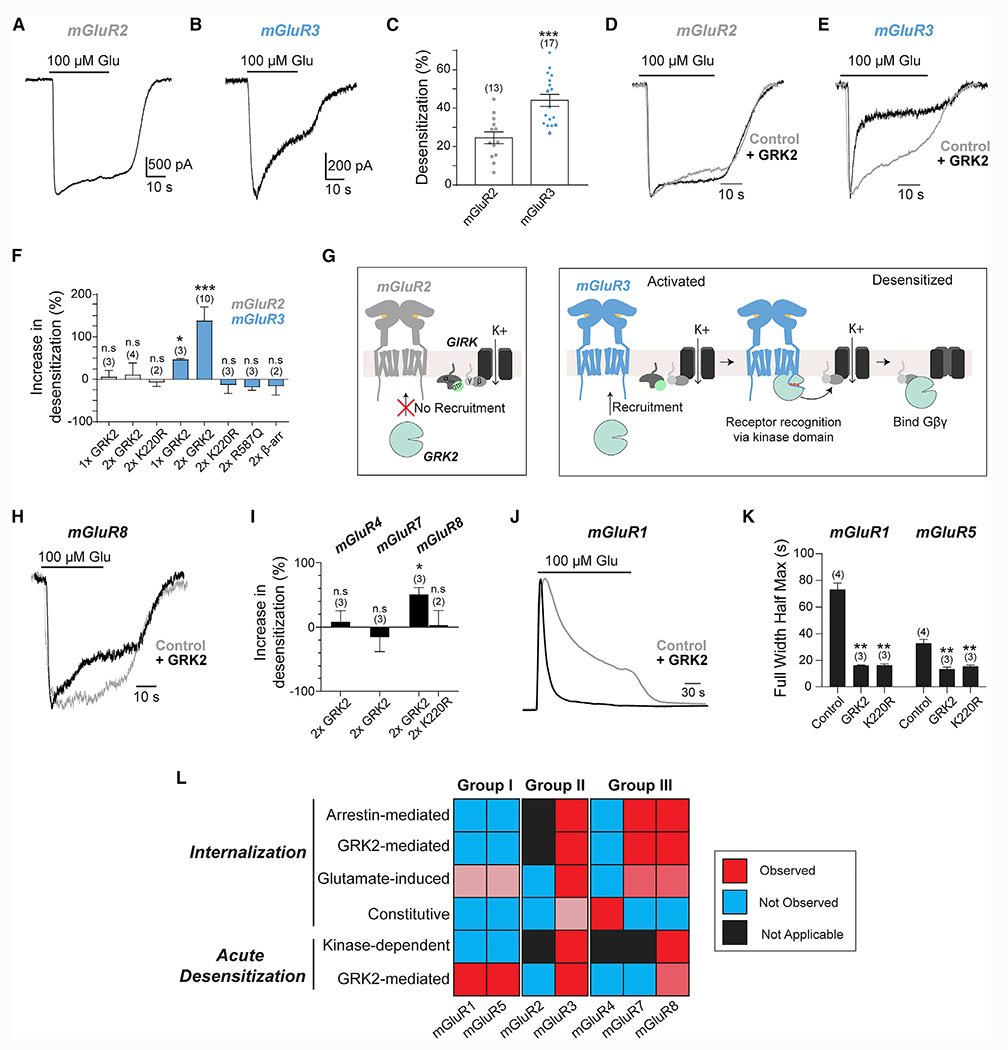
(A and B) Representative whole-cell patch-clamp recordings from HEK293T cell expressing SNAP-mGluR2 (A) or SNAP-mGluR3 (B) showing an inward GIRK current induced by Glu application.
(C) Quantification of the percentage of desensitization of GIRK currents showing larger desensitization of mGluR3 responses. Unpaired t test, ***p < 0.0001.
(D and E) Recordings showing sensitivity of GIRK currents induced by mGluR2 (D) or mGluR3 (E) with and without GRK2 overexpression.
(F) Summary bar graph showing the percentage increase in desensitization on mGluR2- and mGluR3-mediated GIRK currents with overexpression of GRK2, GRK2 mutants, or β-arr. Unpaired t test versus mGluR3 control, *p = 0.04 for 1xGRK2 and ***p < 0.0001 for 2xGRK2.
(G) Schematic showing differential GRK recruitment and rapid desensitization for mGluR2 (left) and mGluR3 (right).
(H) Recordings showing modest desensitization of mGluR8-mediated GIRK currents with and without GRK2 overexpression over 30 s Glu application.
(I) Summary bar graph showing the percentage increase in desensitization of mGluR4-, mGluR7-, and mGluR8-mediated GIRK currents with overexpression of WT GRK2 or K220R. Unpaired t test of mGluR8 control versus mGluR8 + GRK, *p = 0.03.
(J) Average trace representing calcium transients produced by ~50 cells expressing only mGluR1 or with GRK2 overexpression.
(K) Summary bar graph showing the full duration at half-maximum of calcium transients mediated by mGluR1 and the first calcium transient mediated by mGluR5 activation, without and with overexpression of WT GRK2 or K220R. ** indicates statistical significance. Unpaired t tests, p = 0.002 for mGluR1 control versus GRK2, p = 0.001 for mGluR1 control versus K220R, p = 0.005 for mGluR5 control versus GRK2, and p = 0.009 for mGluR5 control versus K220R. Number of cells analyzed are as follows: n = 142 for mGluR1 control, 134 for GRK2, and 149 for K220R; n = 287 for mGluR5 control, 126 for GRK2, and 88 for K220R.
(L) Summary heatmap showing the relative propensities for acute desensitization and internalization across mGluRs, and their dependence on interactions with β-arr and GRK2.
Error bars show SEM. Number of cells recorded from (A) or independent experiments (F, I, and K) is in parentheses.
We next asked how different transducers contribute to mGluR3 desensitization. Raveh et al. (2010) identified a mechanism whereby GRKs bind Gβγ proteins to desensitize GIRK currents following activation of the adenosine-1 receptor (A1R). mGluR2 and the M4 muscarinic acetylcholine receptor were not sensitive to GRKs, although no mechanism for this subtype specificity was identified. Here we focused on the GRK2 subtype, which is expressed throughout the nervous system (Erdtmann-Vourliotis et al., 2001) and in HEK293T cells (Violin et al., 2006). As previously reported (Raveh et al., 2010), GRK2 overexpression had no effect on the desensitization of mGluR2-mediated GIRK currents (Figure 3D). However, for mGluR3, GRK2 dose-dependently increased the magnitude and speed of desensitization (Figures 3E and 3F; Figures S4F and S4L). GRK2 expression also led to a large reduction in the amplitude of mGluR3-induced currents (Figure S4G), whereas a small (~25%) decrease of mGluR2-induced current amplitude was seen (Figure S4G). Importantly, no relationship between current amplitude and desensitization kinetics was observed, indicating that the enhanced desensitization produced by GRK2 is not a consequence of reduced mGluR3 surface expression (Figure S4H).
Next, we probed the mechanism of GRK2-mediated desensitization by first testing the role of GRK2-Gβγ binding. We coexpressed GRK2-R587Q (Carman et al., 2000) and found that it was unable to increase mGluR3 desensitization (Figure 3F; Figures S4I–S4K). We also tested kinase-dead GRK2-K220R (Kong et al., 1994), and in contrast to what was reported for the A1R (Raveh et al., 2010), it did not produce an effect on the desensitization of mGluR3-mediated currents, despite similar expression to wild-type (WT) GRK2 (Figure 3F; Figures S4I–S4K). The need for a catalytically active GRK2 raised the possibility that β-arr recruitment contributes to rapid desensitization. However, overexpression of β-arr1 and β-arr2 did not affect desensitization (Figure 3F; Figure S4K). Altogether, these results demonstrate that both functional kinase and Gβγ binding domains of GRK2 mediate rapid and specific mGluR3 desensitization (Figure 3G). The ability of mGluR3, but not mGluR2, to be desensitized by GRK2 despite both receptors producing free Gβγ argues that GRK2 is recruited directly to the receptor rather than to free G proteins (Figure 3G; Figure S3D).
Group III mGluRs also showed variable propensities for internalization (Figure 2F). Consistent with its sensitivity to GRK2 in internalization assays (Figure 2E), mGluR8 showed modest but clear enhancement of desensitization following GRK2 coexpression (Figures 3H and 3I; Figure S4L). Similar to mGluR3, the K220R mutation prevented GRK2-mediated desensitization of GIRK currents by mGluR8 (Figure 3I). Surprisingly, mGluR7-mediated currents did not exhibit enhanced desensitization with GRK2 overexpression (Figure 3I; Figures S4M and S4O). Slower ON-kinetics of mGluR7 currents (~5–10 versus ~1–3 s) may prevent the resolution of fast desensitization components. Consistent with its resistance to GRK- and β-arr-mediated internalization (Figure 2), mGluR4 did not show GRK2-mediated functional desensitization (Figure 3I; Figures S4N and S4O). However, current amplitudes were decreased for all three group III mGluRs tested (Figure S4P), which may indicate sensitivity over longer timescales.
We also examined the GRK2 sensitivity of the Gq-coupled group I mGluRs using the fluorescent calcium sensor R-GECO (Zhao et al., 2011). The duration and rate of decay, but not the amplitude, of glutamate-induced calcium transients were dramatically reduced with expression of GRK2 for both mGluR1 and mGluR5 (Figures 3J and 3K; Figures S4Q–S4S). Furthermore, extended mGluR5 activation produces oscillatory calcium signals (Kawabata et al., 1996). GRK2 expression reduced the percentage of cells producing oscillatory responses, leading to many cells that showed a single calcium transient (Figures S4T and S4U). Unlike group II and III mGluRs, GRK2-mediated desensitization of group I mGluRs did not depend on a functional kinase domain based on the strong effect of GRK2-K220R (Figure 3K; Figures S4S and S4U).
Altogether, these results show that variable propensities for and modes of desensitization and internalization are seen throughout the mGluR family (Figure 3L), which can shape the signaling properties of each subtype.
Mechanistic analysis of group II mGluRs: Coupling of β-arrestins to mGluR3
We next focused on deciphering the mechanisms of differential transducer coupling of the group II subfamily. To measure surface translocation of cytosolic β-arr, we coexpressed either β-arr1-YFP or β-arr2-YFP with mGluR3 or mGluR2 and observed β-arr surface recruitment only with mGluR3 activation (Figure 4A). As expected, β-arr recruitment was insensitive to PTX but was blocked by mGluR3 antagonists (Figures S5A and S5B). Similar levels of recruitment of β-arr1 or β-arr2 indicates that mGluR3 has no major β-arr subtype preference, unlike the preference for β-arr2 seen with the β2AR (Figures S5C and S5D) (Kohout et al., 2001; Oakley et al., 2000).
Figure 4. mGluR3 recruits β-arrestins via scaffold and catalytic coupling.
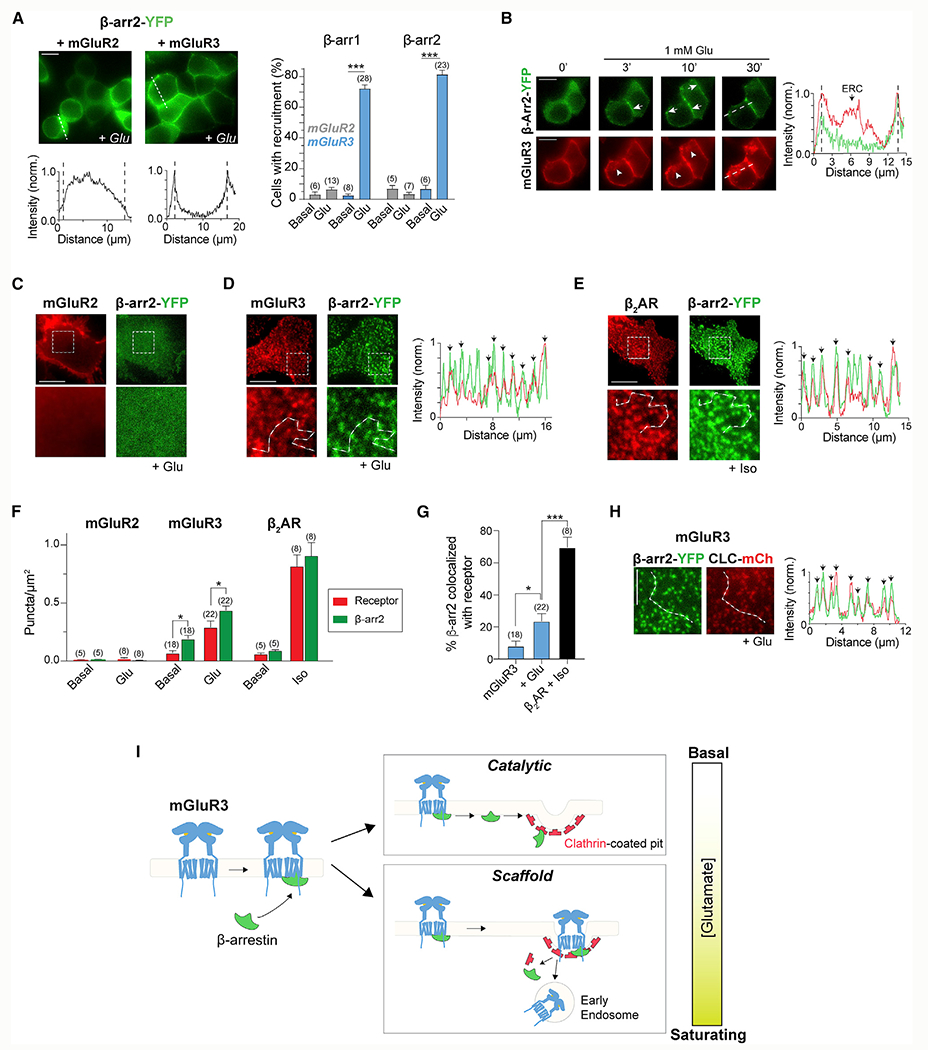
(A) Left, representative images and intensity line scans showing Glu-dependent surface recruitment of β-arr2 by mGluR3. Right, quantification ofthe percentage of cells that exhibit β-arr surface recruitment. Unpaired t tests for mGluR3, ***p < 0.0001 for β-arr1 and ***p < 0.0001 for β-arr2.
(B) Time-lapse images showing Glu-dependent surface recruitment of β-arr2 and subsequent internalization of only mGluR3. Arrows point at β-arr2 accumulation on the cell surface. Arrowheads denote internalized receptors.
(C–E) Representative TIRF images showing a lackof β-arr2 puncta for mGluR2 (C) but a high density of β-arr2 puncta generated by activation of mGluR3 (D) and β2AR (E). Line scans reveal colocalization of receptor (red) and β-arr2 (green) puncta. Black arrows denote overlapping peaks.
(F) Summary bar graph of receptor and β-arr2 puncta densities under basal conditions and agonist treatment. Paired t tests, *p = 0.03 for mGluR3 basal and *p = 0.01 for mGluR3 + Glu.
(G) Quantification of the percentage of β-arr2 puncta that are colocalized with either mGluR3 (under basal or +Glu conditions) or β2AR (+Iso) puncta. Unpaired t tests, *p = 0.03 for mGluR3 basal versus +Glu and ***p = 0.0005 for mGluR3 + Glu versus β2AR + Iso.
(H) Left, representative TIRF images showing that mGluR3 activation leads to β-arr2 puncta that colocalize extensively with the CCP marker, CLC. Right, intensity line scan through the white dotted line on the images that shows overlapping β-arr2 (green) and CLC (red) peaks, denoted by the black arrows.
(I) Schematic of the working model for β-arr coupling with mGluR3.
Error bars show SEM. Scale bar, 10 μm (B–E) or 5 μm (H). Number of fields of cells (A) or cells (F and G) analyzed is in parentheses.
We characterized the timing and stability of mGluR3/β-arr complexes by simultaneously imaging mGluR3 and β-arr-YFP. We observed surface recruitment of β-arr1 or β-arr2 by mGluR3 within 3 min, with further accumulation until ~10 min after glutamate application (Figure 4B; Figure S5E). After 10 min, β-arr remained on the surface as mGluR3 internalized, indicating that β-arr does not cotraffic into the ERC (Figure 4B; Figure S5E). β-arr translocation was not detectable following activation of mGluR2 at any time point (Figure S5F), but the β2AR produced a fluorescence distribution similar to that of mGluR3 (Figure S5G). Consistent with this, minimal colocalization of β-arr2-RFP and GFP-2xFYVE was observed following mGluR3 activation (Figures S5H and S5I), although the level of colocalization was slightly higher with β2AR activation compared with mGluR3 (Figure S5I).
To further characterize the coupling of group II mGluRs and β-arr at the cell surface, we simultaneously imaged receptors and β-arr using TIRF microscopy. Following 10–15 min of glutamate application, no features were observed in mGluR2-expressing cells (Figure 4C; Figure S5J), but we observed bright puncta for both β-arr and mGluR3 (Figure 4D; Figure S5K). Although β-arr puncta were largely immobile, receptor puncta showed a mix of immobile and mobile spots (Videos S1 and S2). Similar puncta formed with β2AR and β-arr2, consistent with previous reports (Eichel et al., 2016, 2018) (Figure 4E). Quantification of puncta density revealed higher levels of both receptor and β-arr2 puncta for β2AR than mGluR3, with few puncta in mGluR2-expressing cells (Figure 4F; Figure S5L). In mGluR3-expressing cells, ~40% fewer receptor puncta were observed compared with β-arr2 puncta (Figure 4F; Figure S5L). In contrast, similar densities of receptor and β-arr2 puncta were observed for β2AR (Figure 4F). This discrepancy in puncta densities suggests that one mGluR3 can activate multiple β-arr molecules, as was reported for the beta-1 adrenergic receptor (β-1AR) (Eichel et al., 2018). In line with this, some puncta were detected under basal conditions (Figure S5M) but with a nearly 3:1 ratio of β-arr to receptor puncta (Figure 4F; Figure S5L). This supports the notion of catalytic coupling and suggests that a low level of receptor activation leads to a transient form of coupling, whereas stronger agonism may enhance stability. Line scans revealed colocalization between receptor and β-arr, although the degree of colocalization was lower for mGluR3 than for β2AR (Figures 4D, 4E, and 4G).
We characterized the process of mGluR3/β-arr coupling by imaging CCPs and observed strong colocalization between β-arr and CLC-mCh puncta in mGluR3- or β2AR-expressing cells (Figure 4H; Figures S5M–S5O). Furthermore, in contrast to the mobility of GFP-2xFYVE-positive early endosomes (Video S3) most CLC-mCh-positive CCPs appear to be immobile (Video S4). This suggests that the immobile population of receptors occurs in CCPs with immobile β-arr clusters and the mobile receptors are likely in early endosomes.
Altogether, our data show interaction between mGluR3 and both β-arr and indicate that formation of this complex is critical for recruitment of mGluR3 to CCPs and receptor internalization.
In addition, our data suggest that mGluR3 shows a mix of transient coupling that leads to independent clusters of activated β-arr (i.e., catalytic coupling) and colocalized mGluR3/β-arr complexes that persist into CCPs, but not endosomes (i.e., scaffold coupling) (Figure 4I).
A Ser/Thr-rich subregion of the mGluR3 CTD controls internalization and rapid desensitization
Having observed GRK- and β-arr-dependent desensitization of mGluR3, but not mGluR2, we aimed to identify the molecular determinants of the differences between receptors with a focus on the CTD. Removing the CTD of mGluR3 (mGluR3-ΔCTD) substantially reduced internalization (Figures 5A and 5B), although the degree of internalization for mGluR3-ΔCTD was still above the background levels seen with mGluR2. We next swapped the CTDs of mGluR2 and mGluR3 and observed a clear effect on each subtype. mGluR3-mGluR2CTD showed no internalization, whereas mGluR2-mGluR3CTD exhibited internalization (Figures 5A and 5B), demonstrating that the CTD is the major determinant of group II mGluR desensitization. Surprisingly, mGluR3-mGluR2CTD exhibited even less internalization than mGluR3-ΔCTD, suggesting that the mGluR2 CTD may serve an inhibitory role (Figures 5A and 5B). Consistent with this, removing the CTD from mGluR2 (mGluR2-ΔCTD) resulted in a small increase in internalization (Figures 5A and 5B). We tested the aforementioned CTD perturbations in the surface labeling assay and found that only WT mGluR3 and mGluR2-mGluR3CTD showed reductions of surface receptor levels following glutamate treatment (Figure 5C). This is in line with cell imaging, because the weak internalization of mGluR3-ΔCTD and mGluR2-ΔCTD (Figures 5A and 5B) is unlikely to produce a measurable change in the total level of surface receptors. For each CTD chimera, we also tested the ability to recruit β-arr following glutamate application under expression-matched conditions (Figure S6A) and found consistent results in which swapping mGluR CTDs swapped the ability to recruit β-arr (Figures S6B–S6D).
Figure 5. The C-terminal domains mediate differential internalization and rapid desensitization of mGluRs.
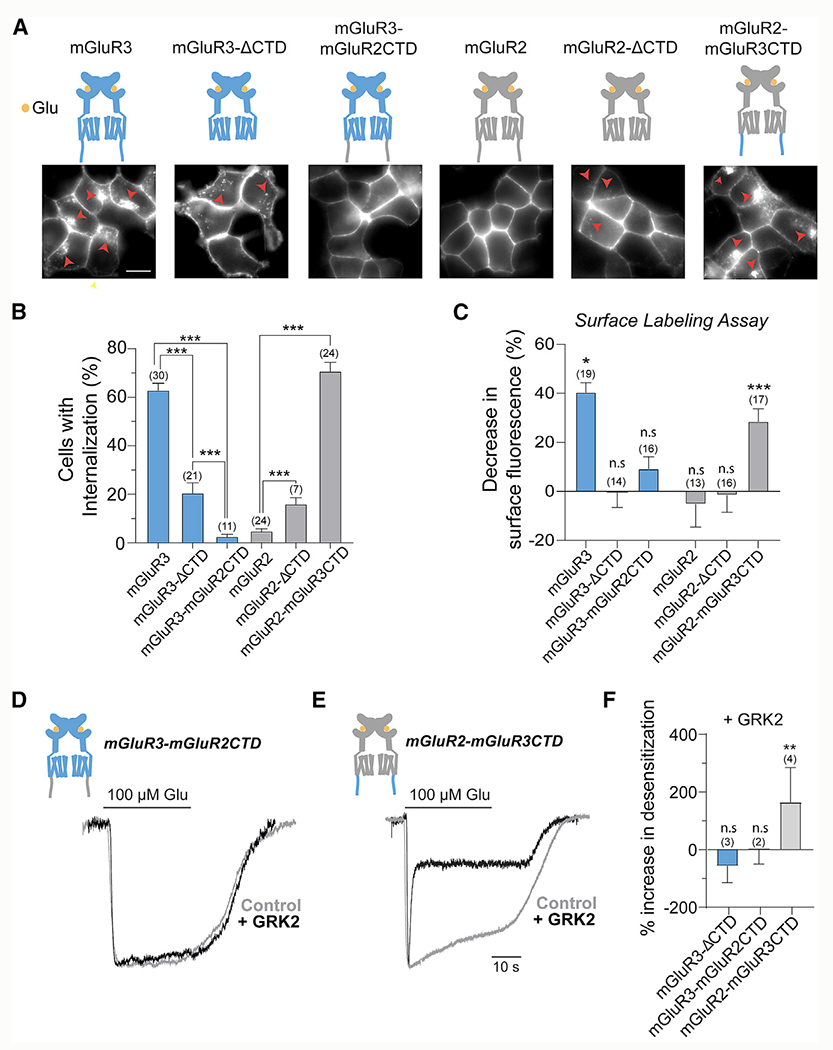
(A) Top, illustrations of CTD deletions and chimeras for mGluR2 and mGluR3. Bottom, representative images of HEK293T cells expressing SNAP-tagged receptors and incubated for 30 min in Glu. Red arrowheads show internalized receptors. Scale bar, 10 μm.
(B) Quantification of the percentage of cells that exhibit internalization of the various CTD variants. *** indicates statistical significance. Unpaired t test, p < 0.0001 for mGluR3 versus mGluR3-ΔCTD, p < 0.0001 for mGluR3 versus mGluR3-mGluR2CTD, p < 0.0001 for mGluR2 versus mGluR2-ΔCTD, p < 0.0001 for mGluR2 versus mGluR2-mGluR3CTD, and p = 0.0005 for mGluR3-ΔCTD versus mGluR2.
(C) Quantification of surface fluorescence reduction following Glu application, where the control was no Glu. Unpaired t tests, *p = 0.03 mGluR3 and ***p < 0.0001 for mGluR2-mGluR3CTD.
(D and E) Representative GIRK current traces for mGluR3-mGluR2CTD (D) and mGluR2-mGluR3CTD (E) with and without GRK2 overexpression.
(F) Summary plot showing the percentage increase in desensitization of GIRK currents in the presence of GRK2 overexpression. Unpaired t test versus no GRK2 overexpression condition, **p = 0.005.
Error bars show SEM. Number of fields of cells analyzed (B and C) or independent experiments (F) is in parentheses.
We also asked whether the CTDs control the differences in rapid desensitization between mGluR2 and mGluR3. The traditional view is that the CTD is not involved in the GRK-mediated rapid uncoupling of GPCRs from G proteins. However, GIRK currents induced by mGluR3-mGluR2CTD showed no sensitivity to GRK2 expression, whereas currents induced by mGluR2-mGluR3CTD showed enhanced desensitization (Figures 5D–5F; Figures S6F and S6G). To confirm the importance of the CTD to rapid GRK-mediated desensitization, we tested mGluR3-ΔCTD, which showed no change in desensitization with GRK2 (Figures S6E and S6F). GRK2 decreased amplitudes mediated by both mGluR3-ΔCTD and mGluR2-mGluR3CTD, but not mGluR3-mGluR2CTD (Figure S6H).
Having identified the CTD as the major mediator of differential mGluR desensitization, we sought to identify critical CTD subregions. Sequence alignment revealed a stretch of residues in mGluR2 and mGluR3 that contains a high density of Ser and Thr, which we termed the ST-rich region (Figure 6A), and exhibits low-sequence identity between mGluR2 and mGluR3. The mGluR3 ST-rich region contains 6 overlapping Px(x)PxxP/E/D full phospho-codes, as defined based on an arrestin-bound rhodopsin crystal structure (Zhou et al., 2017), whereas the mGluR2 CTD contains 0 phospho-codes (Figure 6A). Consistent with our results (Figures 1 and 2), mGluR7 and mGluR8 have full phospho-codes within the membrane proximal region of their CTDs, whereas mGluR4 has only 1 phosphorylation code in its distal CTD (Figure S7A). However, both mGluR1 and mGluR5 contain many phospho-codes along their extended CTDs, indicating that the mere presence of phospho-codes is not sufficient to ensure β-arr coupling (Figure S7A).
Figure 6. A Ser/Thr-rich stretch of the mGluR3CTD controls GRK- and β-arrestin-dependent internalization and rapid desensitization.
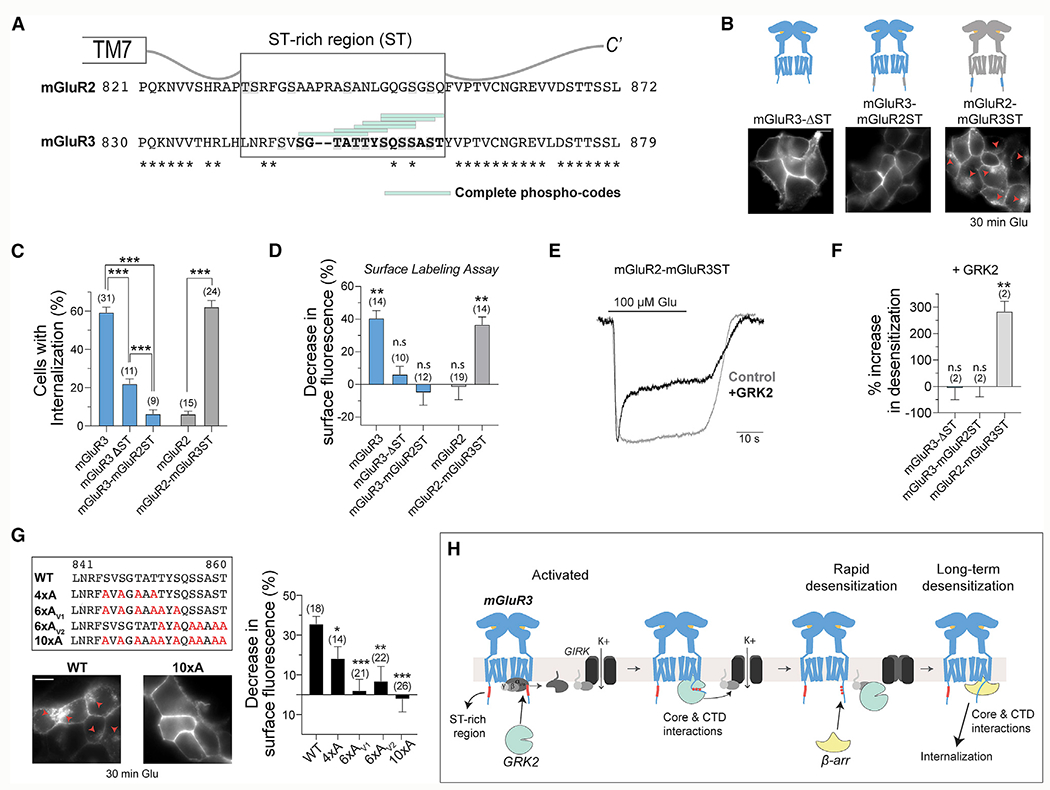
(A) Sequence alignment of the CTDs of mGluR2 and mGluR3 displaying the Ser/Thr (ST)-rich sequence and phospho-codes.
(B) Top, illustrations of ST-rich region deletion and chimeric constructs. Bottom, representative images of HEK293T cells expressing SNAP-tagged receptor constructs and incubated in 1 mM Glu for 30 min.
(C) Quantification of the percentage of cells that exhibit internalization of the various ST sequence variants. Unpaired t tests, ***p < 0.0001 for all comparisons made.
(D) Quantification ofthe percentage of the internalized surface receptor population as determined bythe surface labeling assay, in which the control was no Glu. Unpaired t tests, **p = 0.003 for mGluR3 and **p = 0.001 for mGluR2-mGluR3ST.
(E) Recordings showing the GRK2 sensitivity of mGluR2-mGluR3ST-mediated GIRK currents.
(F) Summary plot showing the percentage increase in desensitization of GIRK currents with GRK2 overexpression. Unpaired t test versus no GRK2 overexpression, **p = 0.006.
(G) Top left, sequence alignments of the ST-rich region of mGluR3, showing the residues that were mutated to Ala. Bottom left, images of cells expressing either WT mGluR3 or mGluR3-10xA following 30 min of 1 mM Glu treatment. Red arrows point to internalized receptors. Right, summary bar graph showing the percentage decrease in surface fluorescence following treatment with 1 mM Glu across mutants. Unpaired t tests, *p = 0.02 for WT versus 4xA, ***p < 0.0001 for WT versus 6xAv1, **p = 0.003 for WT versus 6xAv2, and ***p < 0.0001 for WT versus 10xA.
(H) Working model of GRK2- and β-arr-mediated rapid and long-term desensitization of mGluR3.
Error bars show SEM. Scale bars, 10 μm. Number of fields of cells analyzed (B, D, and G) or independent experiments (F) is shown in parentheses.
We first deleted the ST-rich region from mGluR3 (mGluR3-ΔST) and observed a large decrease in receptor internalization (Figures 6B–6D) and β-arr recruitment (Figures S7C–S7E), despite normal surface expression (Figure S7B). Introducing the ST-rich region of mGluR2 into mGluR3 (mGluR3-mGluR2ST) also attenuated internalization (Figures 6B–6D) and β-arr recruitment (Figures S7C–S7E). Interestingly, mGluR3-ΔST exhibited higher levels of internalization than mGluR3-mGluR2ST (Figure 6C), providing further evidence that the mGluR2 CTD has inhibitory properties. In contrast, mGluR2-mGluR3ST showed robust internalization (Figures 6B–6D) and β-arr recruitment (Figures S7C–S7E). Altogether, these results indicate that the ST-rich region of mGluR3 contains residues that are important for its desensitization.
Consistent with the requirement of a functional GRK2 kinase domain (Figure 4F), the ST-rich region plays a role in the rapid desensitization of mGluR3. GRK2 expression did not enhance the rapid desensitization of GIRK currents mediated by mGluR3-ΔST or mGluR3-mGluR2ST but did enhance desensitization of currents mediated by mGluR2-mGluR3ST (Figures 6E and 6F; Figures S7F and S7G). Moreover, GRK2 expression decreased the amplitudes of mGluR2-mGluR3ST-mediated currents (Figure S7H).
To identify key residues, we mutated Ser and Thr residues in the ST-rich region to Ala and found that mutating all ten (10xA) abolished mGluR3 internalization and β-arr recruitment (Figure 6G; Figures S7I–S7K). mGluR3-10xA also showed a lack of sensitivity to GRK2 in GIRK measurements (Figures S7L and S7M). Mutating either the first six (6xAv1) or last six (6xAv2) Ser/Thr residues also abolished internalization and β-arr recruitment (Figure 6G; Figures S7I–S7K), while mutating the first four (4xA) Ser/Thr residues produced partial effects (Figure 6G; Figures S7I–S7K). This shows that efficient β-arr coupling and subsequent internalization of mGluR3 requires multiple Ser and Thr residues.
Based on our findings, we propose a model for mGluR3 desensitization (Figure 6H) in which GRK2 is specifically recruited to active mGluR3 by binding to the transmembrane core and recognizing the ST-rich region of the CTD via its kinase domain. This recruitment allows GRK2 to phosphorylate CTD residues and to bind Gβγ to desensitize GIRK channels and other effectors. Phosphorylated mGluR3 can then recruit β-arr to initiate internalization of the receptor. Critically, receptor subtype specificity between group II mGluRs is mediated primarily by a differential ability to recruit GRK2 directly to the receptor, and this ability is primarily governed by the identity of the CTD.
A cancer-associated CTD mutation enhances β-arrestin coupling and internalization of mGluR3
A large-scale mutational analysis of GPCRs identified mutations in mGluR3 in 17% of melanoma tumor samples Prickett et al., 2011). One mutation, G848E, is located within the ST-rich region (Figure 7A), raising the possibility that this may modify β-arr coupling. Consistent with this hypothesis, we found that cells express mGluR3-G848E to similar levels as WT (Figure S8A) but exhibit enhanced internalization, even in the absence of glutamate stimulation (Figures 7A and 7B; Figure S8B). The basal internalization of mGluR3-G848E was blocked only following 3 h of incubation in ML289, indicating that it likely depends on some accumulation of phosphorylation (Figure S8C). The glutamate-dependent internalization of G848E was only partially sensitive to cmpd101 treatment (Figures S8D and S8E), and full block required prolonged incubation with cmpd101 and ML289 (Figure S8E). These data suggest that the threshold for sufficient CTD phosphorylation to initiate internalization is lowered in this mutant.
Figure 7. Cancer-associated G848E mutation in the mGluR3CTD enables the identification of biased mGluR3 variants.
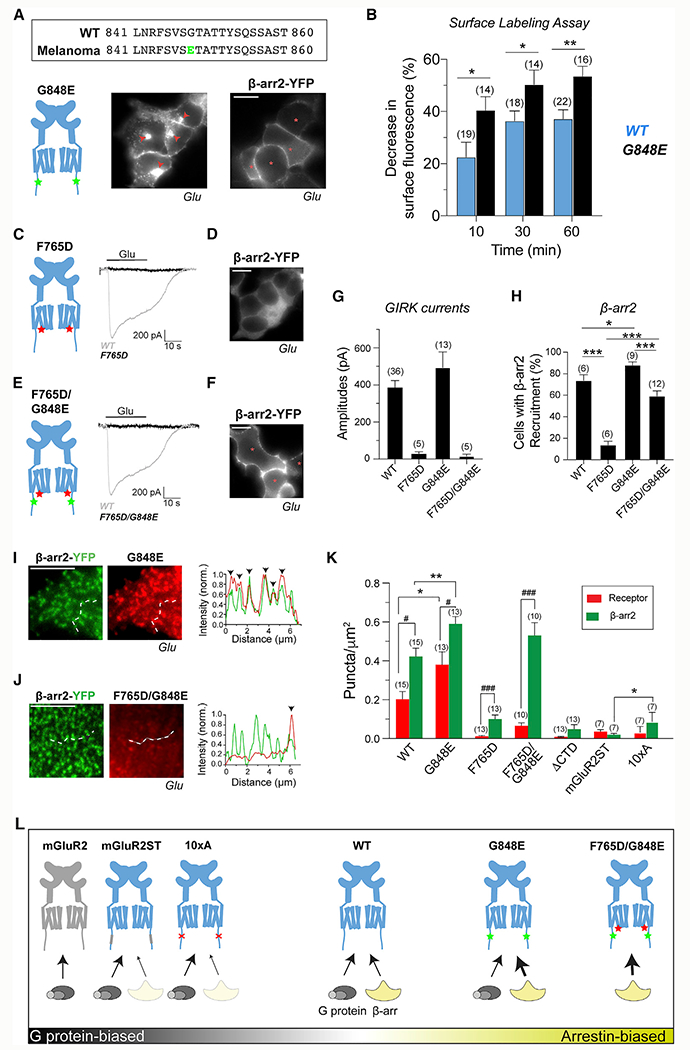
(A) Top, mutation G848E (green) within the ST-rich region ofthe mGluR3CTD is associated with melanoma. Bottom, mGluR3-G848E exhibits internalization and surface recruitment of β-arr2.
(B) Surface labeling assay summary graph showing enhanced internalization for G848E compared with WT. Unpaired t tests, *p = 0.03 for 10 min, *p = 0.04 for 30 min, and **p = 0.003 for 60 min.
(C and D) mGluR3-F765D is unable to produce Glu-induced GIRK channel activation (C) or β-arr2-YFP recruitment (D).
(E and F) mGluR3-F765D/G848E is unable to produce Glu-induced GIRK channel activation (E) but exhibits β-arr2 recruitment (F).
(G and H) Summary bar graphs showing the efficiency of G protein activation as detected via GIRK currents (G) and the efficiency of β-arr recruitment as measured by percentage cell analysis (H). Unpaired t tests, ***p < 0.0001 for WT versus F765D, *p = 0.02 for WT versus G848E, ***p = 0.0001 for G848E versus F765D/G848E, and ***p < 0.0001 for F765D versus F765D/G848E.
(I and J) Differential receptor and β-arr2 puncta formation for G848E (I) and F765D/G848E (J) mutants.
(K) Summary of mGluR3 and β-arr2 puncta density produced across receptor variants. # indicates statistical significance. Paired t tests, ###p = 0.0008 for WT, ##p = 0.008 for G848E, and ###p = 0.0008 for F765D. * indicates statistical significance. Unpaired t tests, *p = 0.04 for WT versus G848E receptor bars, **p = 0.002 for WT versus G848E β-arr2 bars, and **p = 0.008 for mGluR2ST versus 10xA β-arr2 bars. n = 15 cells for WT, 13 for G848E, 13 for F765D, 10 for F765D/G848E, 13 for ΔCTD, 7 for mGluR2ST, 7 for 10xA).
(L) Schematic showing the relative G protein- and β-arr-coupling propensities of group II mGluR variants.
Scale bar, 10 μm (A, C, and E) or 5 μm (I and J). Number of fields of cells analyzed (B and H), cells recorded from (G), or cells analyzed (K) is in parentheses.
Consistent with internalization data, mGluR3-G848E was able to recruit β-arr2 to the surface in the absence of glutamate application and to higher levels with glutamate (Figure 7A; Figures S8F–S8H). Despite the apparent enhanced interaction, β-arr remained on the surface following internalization of mGluR3-G848E (Figure S8I), indicating that this mutation does not alter the trafficking of mGluR3/β-arr complexes. In contrast to the effects on β-arr coupling, mGluR3-G848E showed slightly decreased basal desensitization and similar sensitivity to GRK2 (Figures S8J–S8L). mGluR3-G848E produced larger GIRK currents than WT (Figure S8L), suggesting that effects on G protein coupling may also exist.
Tuning β-arrestin coupling of mGluR3 via mutations to the TMD and CTD
We investigated the interactions of β-arr and mGluR3 by asking how modifications to the receptor alter coupling. We first asked whether β-arr interacts with the G protein binding site of the TMD core of mGluR3, because such an interaction has been shown for GPCRs (Huang et al., 2020; Staus et al., 2020; Hilger et al., 2018). We first mutated a conserved phenylalanine (F765D) in intracellular loop 3 that is known to abolish G protein coupling across mGluRs (Figure S8M) (Francesconi and Duvoisin, 1998; Kniazeff et al., 2004; Levitz et al., 2016) and observed undetectable GIRK currents (Figure 7C), despite normal expression (Figure S8N). F765D also nearly abolished the ability of mGluR3 to recruit β-arr to the surface following activation (Figure 7D) and exhibited negligible internalization (Figures S8O and S8P), indicating that β-arr interacts with the TMD core of mGluR3 in a similar way to G proteins. Given the enhanced coupling to β-arr by mGluR3-G848E, we asked whether introduction of the F765D mutation into this mutant background would prevent β-arr coupling. The double mutant (G848E/F765D) did not produce GIRK currents (Figure 7E) but, surprisingly, showed substantial surface recruitment of β-arr to nearly WT levels (Figure 7F). This indicates that the enhanced CTD coupling of G848E can overcome the weak TMD coupling of F765D to produce a β-arr-biased mutant (Figures 7G and 7H).
To further understand the effects of TMD and CTD perturbations, we turned to TIRF imaging, with which we observed various levels of glutamate-induced receptor and β-arr2 puncta for WT mGluR3, G848E, F765D, F765D/G848E, mGluR3-mGluR2ST, mGluR3-10xA, and mGluR3-ΔCTD (Figures 7I–7K; Figure S8Q). Consistent with receptor internalization and β-arr recruitment data (Figure 7), G848E produced higher levels of receptor and β-arr2 puncta compared with WT (Figures 7I and 7K). However, β-arr2 showed a slightly higher degree of colocalization with G848E (Figure S8R), suggesting a more stable form of coupling. In addition, under basal conditions, G848E showed substantially more β-arr2 puncta than receptor puncta (Figure S8S), consistent with what was seen with WT (Figure 3F). As expected, F765D/G848E also produced a high density of β-arr2 puncta but, interestingly, a lower level of receptor puncta (Figures 7J and 7K). Correspondingly, a low level of β-arr puncta colocalized with the double mutant (Figure S8R). This lack of apparent tight association with β-arr may explain why F765D/G848E internalization is weaker than WT (Figure S7K).
The density of receptor and β-arr2 puncta was low but higher than that seen with mGluR2 for both mGluR3-F765D and mGluR3-ΔCTD (mGluR2 = 0.006 ± 0.002 puncta/μm2, mGluR3-F765D = 0.10 ± 0.02 puncta/μm2, and mGluR3-ΔCTD = 0.04 ± 0.01 puncta/μm2) (Figure 7K; Figure S8Q). For all constructs that weakly recruit β-arr, a higher density was observed for β-arr2 compared with the receptor (Figure 7K) and the degree of colocalization between β-arr2 and mGluR3 was very low (Figure S8R), suggesting that weak β-arr coupling typically proceeds through a catalytic mechanism (Eichel et al., 2016, 2018).
Having developed a β-arr-biased mGluR3, we sought to define a G protein-biased variant. We observed minimal receptor and β-arr puncta formation and colocalization with glutamate for both mGluR3-mGluR2ST and mGluR3-10xA (Figure 7K; Figures S8P and S8R), making mGluR3-mGluR2ST and mGluR3-10xA good candidates. As summarized in Figure 7L, this analysis led us to identify a range of mGluR3 variants that show different degrees of bias toward G protein and β-arr coupling, as well as variability in the mode of β-arr coupling (i.e., scaffold versus catalytic).
DISCUSSION
Functional diversity of mGluRs
We report a comparative analysis of the diverse desensitization and trafficking properties of mGluRs, in which only a subset of subtypes undergoes β-arr-mediated internalization, with mGluR3 showing the most pronounced effects. In a recent high-throughput screen of GPCR/transducer coupling, all mGluR subtypes tested (mGluR2, mGluR4, mGluR5, mGluR6, and mGluR8) showed little to no β-arr recruitment, although mGluR3 was not included (Avet et al., 2020). However, another study (Lee et al., 2019) used biochemical assays to show coupling between mGluR7 and β-arr. Our data were consistent with β-arr-driven internalization of a subset of group III mGluRs, with the major exception of mGluR4, which undergoes β-arr-independent constitutive internalization. We found no evidence for β-arr-dependent internalization of group I mGluRs, in contrast to some previous reports (Dale et al., 2001; Mundell et al., 2001) but in line with the aforementioned screen (Avet et al., 2020). Two studies used knockout mice to report that mGluR5-mediated hippocampal long-term depression partially depends on β-arr (Eng et al., 2016; Stoppel et al., 2017), suggesting that a direct or indirect functional interaction between group I mGluRs and β-arr can occur. GPCRs can be internalized via other mechanisms, which may involve various kinases and protein scaffolding complexes, as shown for mGluR5 and the scaffold Shank (Scheefhals et al., 2019). Thus, future work is needed to analyze mGluR internalization and β-arr-mediated signaling in biological contexts (Suh et al., 2018; Reiner and Levitz, 2018).
We also show that many mGluRs are sensitive to GRK2-mediated rapid functional desensitization. This includes mGluR1 and mGluR5, which are resistant to GRK- and β-arr-mediated internalization under the conditions tested here. Previous studies showed mixed results for functional GRK sensitivity of group I mGluRs, with some reporting kinase-dependent effects (Dhami et al., 2002, 2004; Sorensen and Conn, 2003) versus kinase-independent effects (Dale et al., 2000; Dhami et al., 2002; Ribeiro et al., 2009) using endpoint assays. Using time-resolved measurements of calcium responses, we show that group I mGluR signaling is rapidly desensitized on the ~10 s timescale by GRK2 in a kinase-independent manner. In contrast to group I mGluRs, although mGluR7 shows GRK- and β-arr-dependent internalization, it eluded rapid GRK2-mediated functional effects in our patch-clamp assay. These data suggest that distinct modes of GRK coupling are required for rapid versus slower desensitization effects. An important caveat of our analysis is that although we have focused on GRK2, other GRK subtypes that have distinct domain architectures (Komolov and Benovic, 2018) may show unique effects.
Overall, with the exception of mGluR2, which we found is uniquely resistant to all forms of desensitization tested, all mGluRs show varying degrees and forms of regulation (Figure 3L). This suggests that distinct regulatory properties are a major aspect of the functional diversity of mGluRs. Recent work has revealed that mGluRs heterodimerize (Doumazane et al., 2011; Lee et al., 2020), likely providing further desensitization diversity.
Differential GRK- and arrestin-coupling mechanisms
Our mechanistic analysis focused on group II mGluRs, which have typically been treated monolithically because of their high degree of sequence identity and difficulty distinguishing them with pharmacological compounds or antibodies. We find that mGluR3, but not mGluR2, is subject to rapid functional desensitization and internalization. These differences are underscored by different abilities to couple to GRKs and β-arr and are primarily encoded within the mGluR CTDs.
Classically, GRKs are thought to be recruited to the membrane via free Gβγ proteins released following receptor activation, which then enables coupling to the GPCR (Pitcher et al., 1992). Our data, in which only mGluR3 and mGluR8 are desensitized despite mGluR2, mGluR4, and mGluR7 also producing free Gβγ, argue for a mechanism, in which cytosolic GRK is recruited directly to the active receptor instead of to Gβγ. This is consistent with GRK2-dependent mGluR3 internalization in the presence of PTX, which prevents the release of Gβγ. Recruitment of GRK2 directly to the activated receptor could explain the GPCR subtype specificity of GRK2-mediated GIRK desensitization reported here and previously (Raveh et al., 2010), as well as the PTX-insensitive surface recruitment of GRK2 reported in the same study. A recent study (Stoeber et al., 2020) found that GRK2 recruitment by the kappa-opioid receptor was abolished by PTX for etorphine, but not dynorphin A, showing that the mechanism of GRK recruitment can be both agonist and receptor specific. Our data also argue that critical interactions take place between the mGluR3 CTD and the kinase domain of GRK2 to mediate rapid desensitization. Swapping the ST-rich region of the CTDs swapped the ability of mGluR2 and mGluR3 to undergo GRK2-mediated desensitization, and kinase-dead GRK2 was unable to desensitize mGluR3 signaling. This is in contrast to what was reported previously (Raveh et al., 2010) for A1R, in which the kinase-dead mutant produced desensitization, suggesting that different GPCRs differ in their dependence on interactions with the GRK kinase domain. mGluR8 also showed GRK2-mediated functional desensitization that was abolished in the kinase-dead mutant, suggesting a mode of interaction similar to that of mGluR3. Our data argue for a bitopic interaction between GRKs and both the transmembrane domain and the ST-rich region of the CTD of mGluR3 (or mGluR8) in a manner that depends on an intact GRK kinase domain. This is consistent with a biochemical study that revealed extensive interactions between GRK5 and both intracellular loops and the CTD of the β2AR (Komolov et al., 2017). Studies have also shown that GRK2 recruitment and rapid desensitization of the muopioid receptor require intact CTD phospho-sites (Arttamangkul et al., 2019; Leff et al., 2020; Miess et al., 2018).
Our data show that mGluR3 falls roughly into the class A distinction of GPCRs based on its transient β-arr coupling. However, unlike many class A GPCRs, such as β2AR, mGluR3 showed no preference between β-arr subtypes. Furthermore, our TIRF imaging supports recent studies (Eichel et al., 2016, 2018) showing that catalytic coupling can occur, in which a single GPCR briefly interacts with a β-arr that subsequently remains bound to the membrane and clusters at CCPs. We find that the degree of activation of mGluR3, whether it is via the low ambient levels of glutamate that produce basal activity or saturating 1 mM glutamate, determines the degree of scaffold versus catalytic coupling. Basal activity led to more catalytic coupling, whereas saturating glutamate produced more scaffold coupling with a higher proportion of colocalized mGluR3 and β-arr. Both the extent of occupancy of the active TMD conformation of the receptor and the degree of CTD phosphorylation likely determine the lifetime of the receptor-transducer complex. Thus, the level and dynamics of extracellular glutamate likely shape the mode of β-arr coupling for mGluR3 in physiological settings.
Our finding that mGluR3, but not mGluR2, undergoes agonist-dependent internalization into endosomes is consistent with a mobility study that found mGluR3 accumulation at CCPs (Yanagawa et al., 2018) but did not investigate receptor internalization. Following internalization, we find that mGluR3 primarily recycles back to the surface with minimal occupancy of lysosomes, suggesting that degradation is not a major downstream consequence of internalization. Future work will be needed to identify the proteins that mediate mGluR3 trafficking to different cellular compartments, the roles of different phosphorylation sites in controlling the trafficking itinerary, and whether endosomal mGluR3 can signal.
Physiological relevance of differential regulation of group II mGluRs
Our results suggest that major differences in downstream signaling properties likely exist between group II mGluRs. The ability of mGluR3 to couple to β-arr opens the possibility of signaling to various effectors, including ERK, AKT, and JNKs (Peterson and Luttrell, 2017). Importantly, many of these same effectors are also targeted via G protein-dependent pathways, and debate exists over the relative contributions of G protein and β-arr pathways to ERK activation (Luttrell et al., 2018; O’Hayre et al., 2017).
Our results also have implications for the roles of group II mGluRs in their classical location of excitatory synapses, where we anticipate distinct surface lifetimes and signaling dynamics for mGluR2 and mGluR3. One possibility is that the relative desensitization of group II mGluRs can determine their effects following activation over synaptic plasticity-relevant timescales of minutes to hours. For example, studies of the effects of acute stress on group II mGluRs in the cortex have found downregulation of mGluR3, but not mGluR2 (Joffe et al., 2019). Further work is needed to address a potential role for GRK and β-arr coupling and dynamic trafficking of mGluR3 in intact physiological settings.
Finally, our findings highlight the need for techniques to dissect β-arr versus G protein signaling of mGluR3, especially in physiologically relevant settings. The ability of mGluR3 to couple to β-arr and G proteins opens the possibility that biased ligands may be developed. Recent work (Ellaithy et al., 2020; Gutzeit et al., 2019) has revealed heterogeneity in the kinetics, efficacy, and chemical scaffolds of allosteric modulators, suggesting that sufficient chemical and conformational complexity exists to harness pharmacology for these purposes. Alternatively, the G protein- and β-arr-biased mGluR3 variants reported here (Figure 7) may be useful for probing the relative roles of each transducer in mGluR3 biology. In the long term, the ability to combine biased mutants with genetically targeted photopharmacology, as we reported for mGluR3 (Acosta-Ruiz et al., 2020), offers a potentially powerful approach for dissecting G protein versus β-arr signaling in vivo.
STAR★METHODS
RESOURCE AVAILABILITY
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Joshua Levitz (jtl2003@med.cornell.edu).
Materials availability
Requests for resources and reagents will be fulfilled by the Lead Contact with a completed Materials Transfer Agreement.
Data and code availability
This study did not generate large datasets or code. The datasets analyzed during the study are available from the corresponding author on reasonable request.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
HEK293, HEK293T, parental and HAR CRISPR β-Arr1/2 KO cells (Luttrell et al., 2018) were cultured in DMEM (Fisher Scientific) supplemented with 5%–10% FBS and maintained at 37°C in a 5% CO2 humidified incubator. HEK293 and 293T were obtained from ATCC and tested negative for mycoplasma contamination using a kit (Molecular Probes).
METHOD DETAILS
Molecular biology
Point mutations were made using site-directed mutagenesis. CTD or ST-rich region chimeras were made using a Gibson Assembly Cloning Kit (New England BioLabs). mGluR2-ΔCTD (Δaa821-872) and mGluR3-ΔCTD (Δaa830-879) were generated by introducing a premature stop codon at P821 and P830, respectively. ST chimeras and mGluR3-ΔST (Δaa841-860) were made utilizing a PCR-based DNA ligation method using primers with 5′ phosphates and the enzyme T4 DNA Ligase (Invitrogen).
Cell culture and transfection
Cells were seeded on poly-L-lysine-coated 18 mm coverslips and transfected ~18 hr later with either Lipofectamine 2000 or 3000 (Invitrogen). Cells were maintained in 5-10 μM LY341495 post-transfection to maintain cell health. All experiments were done at least 24 hours post-transfection unless otherwise stated.
Surface labeling assay
For the surface labeling assay, HEK293T cells transfected with 0.35 μg SNAP-tagged receptors (with or without 0.7 μg of the PTX-S1 subunit, YFP-tagged β-arr, or GFP-tagged GRK) were incubated in either no glutamate (media or 20 μM LY34) or 1 mM Glu (10 mM for mGluR7; 10 μM isoproterenol for β2AR) in media for 60 min. To label remaining surface receptors, the cells were incubated in 1 μM BG-Alexa-546 (New England BioLabs) for 20 min at room temperature in an extracellular (EX) solution composed of (in mM) 135 NaCl, 5.4 KCl, 10 HEPES, 5 L-Glucose, 2 CaCl2, and 1 MgCl2 (pH 7.4). To block GRK2/3 kinase activity, cells were pre-incubated in 30 μM cmpd101 for 20 min. Following fluorophore labeling, live cells were washed and imaged on an inverted microscopy (Olympus IX83) with a 60x 1.49 NA objective. Alexa-546 was excited using a 560 nm laser and snapshots were acquired to measure the mean fluorescence intensity of labeled surface receptors using ImageJ. Fluorescence was normalized to the average fluorescence of the control. The raw fluorescence values were normalized to 20 uM LY34 rather than zero glutamate as a control in Figures 2 and 7. The decrease in surface fluorescence was calculated as 100 * (1-normalized surface fluorescence).
Internalization and arrestin recruitment assays
To observe the internalization of SNAP-tagged surface receptors, cells were incubated in 1 μM BG-Alexa-546 for 30-45 min at 37°C in extracellular solution. To induce internalization of SNAP-tagged receptors, cells were incubated with agonist in EX solution for 30 min at 37°C and imaged. mGluR7 was stimulated with 10 mM glutamate due to its low affinity, while other mGluRs were stimulated with 1 mM glutamate, Alexa-546 was excited with a 560 nm laser and snapshots of randomized fields were acquired. To block internalization of SNAP-mGluR3, cells were preincubated in 20 μM LY34 or 100 μM ML289 for 10-15 min, and the drugs were maintained during the glutamate incubation that followed. The basal condition was always run side-by-side with other conditions with the same allotted time but in EX solution that did not contain drugs.
For the β-arr recruitment experiments, HEK293T cells were co-transfected with 0.5-0.7 μg SNAP-mGluR2 or SNAP-mGluR3 and either 0.1-0.3 μg β-arr1-YFP or β-arr2-YFP. Since the expression levels of mGluR2 and mGluR3 differ at 24 hr, we expression-matched conditions by measuring arrestin-recruitment of mGluR2-CTD and -ST variants at 48 hr expression. For time lapse experiments, internalization of Alexa 546-labeled SNAP-mGluR and β-arr surface recruitment was followed on the microscope following bath application of 1 mM glutamate. Cells were maintained at 37°C using a temperature control device (TC-326C, Warner Instruments). For simple surface recruitment of β-arr experiments to quantify the percentage of cells with recruitment, cells were incubated in 1 mM glutamate for 15 min at room temperature and snapshots of the 488 channel were taken of randomized fields of cells.
The percentage of cells with internalization or β-arr recruitment was manually counted following blinding to the conditions. Internalization was defined by cells that exhibit clear intracellular accumulation of fluorescence. β-arr recruitment was defined by cells that exhibited higher YFP intensity on the cell surface compared with the inside. Cell-to-cell variability is also present and can likely be attributed to transient transfection of variable amounts of receptor and varying expression patterns of endogenous regulatory proteins. Values for % cells with internalization or β-arr recruitment are compared on the same days within the same bar graph, with at least two separate days for each condition, as there is modest day-to-day variability that can confound results. This day-to-day variability explains the ~10%–15% fluctuation of some values for common conditions between figures.
Recycling assay
After 48 hr expression, HEK293T cells seeded on 12 mm coverslips transfected with ~0.2 μg SNAP-mGluR2 or SNAP-mGluR3 were incubated in 1 mM glutamate in complete media for 30 min at 37°C to internalize a population of receptors. Cells were then washed and labeled with 5 μM BG-Alexa 488 for 20 minutes at room temperature to label and occupy the SNAP-tag on all remaining surface receptors. Cells were then washed and incubated in media for either 30, 60, or 90 min to allow internalized receptors to recycle back to the surface. Following incubation, cells were then washed with media and labeled in 5 μM Alexa 546 for 20 min at room temperature to label receptors that trafficked back to the surface. Control coverslips did not receive a 30 min glutamate treatment. For all conditions, cells were maintained in 10 μM LY341495 when not treated with glutamate to avoid any confounding effects from ambient glutamate. Cells were washed in PBS and fixed in a 4% PFA/4% sucrose solution and coverslips were mounted on slides for imaging. Fixed cells were imaged with the 60x 1.49 NA objective. Alexa 488 and Alexa-546 were excited with 488 nm and 560 nm lasers respectively. For surface fluorescence analysis, mean intensity of ROIs draw around receptor-positive cells (based on the 488 channel) were measured on ImageJ. All raw fluorescence values were normalized to the BG-Alexa-546 fluorescence observed prior to post-glutamate incubation.
Confocal microscopy
To measure colocalization with the ERC, HEK293T cells expressing SNAP-mGluR3 or SNAP-mGluR2 were incubated in 1 μM BG-Alexa 488 in EX solution for ~45 min at 37°C followed by 5 μg/ml Cy3-transferrin + 1 mM glutamate in EX solution for 30 min at 37°C and fixed in a solution of 4% PFA/4% Sucrose. To measure colocalization of mGluR3 with lysosomes and TGN, cells expressing either SNAP-mGluR3 and LAMP1-YFP or mCh-TGN38, respectively, were incubated in 1 mM glutamate in EX solution for 30 min at 37°C and fixed. Cells were imaged using a 63x objective with 1.2x zoom on a Zeiss LSM880 scanning confocal microscope and the ZEN Black software. Fluorophores were excited using 488 nm and/or 561 nm lasers. The plane of imaging was chosen based on the organelle marker channel. Images were captured using one directional scanning and a pinhole of 1 Airy unit. For the image analysis, region of interests (ROIs) were drawn within the cells to exclude the surface receptors. The pixels with the top 10% highest intensity (FT) in each channel was used to measure the Pearson’s correlation coefficient (PCC) via the plugin EzColocalization (Stauffer et al., 2018) on ImageJ.
Total internal reflection fluorescence microscopy
HEK293T cells were seeded sparsely and transfected with 0.5-0.7 μg SNAP-tagged receptors with or without 0.1-0.3 μg GFP-2xFYVE, β-arr1-YFP, β-arr2-YFP, β-arr2-RFP, or CLC-mCh. SNAP-tags were labeled with BG-Alexa 546 or 488 and then maintained at 37°C on the microscope. Agonist was applied by bath application and images were captured with a 100x 1.49 NA Apo N objective in TIRF mode after ~15 min. Receptor and β-arr puncta were manually counted using the multi-point tool on ImageJ. To determine colocalization and overlap of puncta, the puncta of one channel were marked in the center with the multi-point tool. The markers denoting the location of puncta from one channel was transferred to the next channel to determine the number of puncta that contained a marker in its center. These puncta were counted as overlapping.
Patch clamp electrophysiology
HEK293T cells were transfected with 0.35-0.7 μg SNAP-tagged mGluRs, GIRK1-F137S homotetramerization mutant (Vivaudou et al., 1997), tdTomato (as a transfection marker), with or without WT or mutant 0.35 μg (1x) or 0.7 μg (2x) GRK2-GFP, or 0.7 μg each of β-arr1-YFP and β-arr2-YFP. Whole-cell patch clamp experiments were performed 24-36 hr after transfection in a high potassium extracellular solution composed of (in mM) 120 KCl, 25 NaCl, 10 HEPES, 2 CaCl2, and 1 MgCl2 (pH 7.4). Solutions were delivered to a recording chamber using a gravity-driven perfusion system with exchange times of ~1 s. Cells were voltage-clamped at −60 mV using an Axopatch 200B amplifier (Molecular Devices). Patch pipettes with resistances of 4-10 MΩ were filled with an intracellular solution composed of (in mM): 140 KCl, 10 HEPES, 3 Na2ATP, 0.2 Na2GTP, 5 EGTA, 3 MgCl2 (pH 7.4). Inward GIRK currents were induced with the perfusion of glutamate. Desensitization of currents were measured over 30-60 s glutamate application (40 s for mGluR7, 60 s for mGluR3-ΔCTD, 30 s for all other receptors). For the repeated glutamate application experiment, 100 μM glutamate was applied for ~1 min and washed out for ~1 min before reapplying glutamate. Currents were sampled at 10 kHz and filtered using a low-pass Bessel (8-pole) at 2 kHz.
Recordings were analyzed using Clampfit (Molecular Devices) and Prism (GraphPad) software. Calculation of glutamate-dependent current amplitudes were quantified relative to the average baseline amplitude. Desensitization of the glutamate-induced responses was calculated over 30 s or 60 s as follows: 100* (1 − (amplitude prior to glutamate washout) / (peak amplitude following glutamate application)). Comparisons were done on the same and across multiple days to account for variability. Percent increase in desensitization (Figures 3F, 3I, 5F, and 6F) was calculated as: 100* ((mean desensitization of currents) − (mean desensitization of control currents)) / (mean desensitization of control currents). The mean desensitization of currents was averaged from the same experimental day. The standard error of the mean (s.e.m.) was calculated from the average of multiple experimental days. The tau of desensitization was calculated from the peak amplitude to glutamate washout, fit to a single exponential curve in Clampfit.
Calcium imaging
HEK293T cells were transfected with the calcium indicator, 0.2 μg R-GECO (Zhao et al., 2011), and 0.4-0.7 μg SNAP-mGluR1 or SNAP-mGluR5, with or without 0.4 μg WT or mutant GRK2-GFP, were imaged on an inverted microscope (Olympus IX83) with a 20x objective at room temperature in EX solution with continuous perfusion following 24 hr expression. R-GECO was excited using a 561 nm laser at 0.5 Hz with a 100 ms exposure time. Receptors were activated with a perfusion of 100 μM glutamate. Time-lapse movies were recorded with an scMOS camera (Hamamatsu ORCA-Flash4v3.0). Image analysis was performed using ImageJ (Fiji) (Schindelin et al., 2012). Intensities were normalized to baseline prior to glutamate application. Full width at half maximum (FWHM) was calculated as the width (duration of time, measured in seconds) of an average trace (40-60 cells) representing the calcium transient achieved after glutamate application measured at half of its maximum amplitude. Peak amplitude was determined as the highest value of arbitrary unit fluorescence reached after glutamate application.
QUANTIFICATION AND STATISTICAL ANALYSIS
Statistics were calculated using GraphPad Prism. Only two-tailed t tests were performed unless otherwise stated, and pairing is specified in the figure legends. Data is represented as mean ± s.e.m. and averaged across multiple independent experiments.
Supplementary Material
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Chemicals, peptides, and recombinant proteins | ||
| Dulbecco’s Modified Eagle Medium (DMEM) | Thermo Fisher Scientific | Cat#11995073 |
| Fetal Bovine Serum (FBS) | Thermo Fisher Scientific | Cat#10437028 |
| SNAP-Surface Alexa Fluor 546 (BG-Alexa 546) | New England Biolabs | Cat#S9132S |
| SNAP-Surface Alexa Fluor 488 (BG-Alexa 488) | New England Biolabs | Cat#S9232S |
| Lipofectamine 2000 Transfection Reagent | Thermo Fisher Scientific | Cat#11668019 |
| Lipofectamine 3000 Transfection Reagent | Thermo Fisher Scientific | Ca#L300015 |
| Poly-L-lysine (PLL) hydrobromide | Sigma Aldrich | Cat#P2636 |
| Cy3-Transferrin (CY3-Tf) | Subtil et al., 2000 | N/A |
| ML289 | Tocris | Cat#4976 |
| CMPD101 | Tocris | Cat#5642 |
| LY341495 | Tocris | Cat#1209 |
| Experimental models: Cell lines | ||
| HEK293T | ATCC | Cat#CRL-11268; RRID:CVCL_1926 |
| HEK293 | ATCC | Cat#CRL-1573; RRID:CVCL_0045 |
| HAR-HEK293 β-Arrestin1/2 KO Cell Lines | Luttrell et al., 2018 | N/A |
| Recombinant DNA | ||
| SNAP-tagged mGluRs | Doumazane et al., 2011 | N/A |
| SNAP-β2AR | Addgene | Cat#101123 |
| GIRK1-F137S | Vivaudou et al., 1997 | N/A |
| R-GECO | Zhao et al., 2011 | N/A |
| β-arrestin1-YFP | Addgene | Cat#36916 |
| β-arrestin2-YFP | Addgene | Cat#36917 |
| β-arrestin2-RFP | Ahn et al., 2004 | N/A |
| GRK2-GFP | Raveh et al., 2010 | N/A |
| GFP-2xFYVE | Gillooly et al., 2000 | N/A |
| mCherry-TGN38 (mCh-TGN38) | Addgene | Cat#55145 |
| LAMP1-YFP | Addgene | Cat#1816 |
| PTX-S1 | Raveh et al., 2010 | N/A |
| CLC-mCherry (CLC-mCh) | Addgene | Cat#27680 |
| Software and algorithms | ||
| ImageJ (Fiji) | Schneider et al., 2012 | RRID:SCR_003070 |
| Microsoft Excel | Microsoft Office | RRID:SCR_016137 |
| Adobe Illustrator | Adobe | RRID:SCR_010279 |
| Prism | GraphPad | RRID:SCR_002798 |
| pCLAMP (Clampfit) | Molecular Devices | RRID:SCR_011323 |
Highlights.
GRKs and arrestins mediate the fast and slow desensitization of a subset of mGluRs
TIRF imaging reveals scaffold and catalytic coupling between mGluR3 and β-arrestins
Different patterns of Ser and Thr control the desensitization of group II mGluRs
Mutational analysis reveals G protein- and β-arrestin-biased mGluR3 variants
ACKNOWLEDGMENTS
We thank Drs. Frederick R. Maxfield, Timothy E. McGraw, Jeremy S. Dittman, David Eliezer, Robert J. Lefkowitz, and Seungkirl Ahn for discussion and critical reagents. N.A. is supported by a National Science Foundation (NSF) Graduate Research Fellowship. J.L. is supported by an R35 grant (R35 GM124731) from NIGMS and the Rohr Family Research Scholar Award.
Footnotes
SUPPLEMENTAL INFORMATION
Supplemental information can be found online at https://doi.org/10.1016/j.celrep.2021.109050.
DECLARATION OF INTERESTS
The authors declare no competing interests.
REFERENCES
- Abdullah N, Beg M, Soares D, Dittman JS, and McGraw TE (2016). Downregulation of a GPCR by β-Arrestin2-Mediated Switch from an Endosomal to a TGN Recycling Pathway. Cell Rep. 17, 2966–2978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Acosta-Ruiz A, Gutzeit VA, Skelly MJ, Meadows S, Lee J, Parekh P, Orr AG, Liston C, Pleil KE, Broichhagen J, and Levitz J (2020). Branched Photoswitchable Tethered Ligands Enable Ultra-efficient Optical Control and Detection of G Protein-Coupled Receptors In Vivo. Neuron 105, 446–463.e13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ahn S, Shenoy SK, Wei H, and Lefkowitz RJ (2004). Differential kinetic and spatial patterns of beta-arrestin and G protein-mediated ERK activation by the angiotensin II receptor. J. Biol. Chem 279, 35518–35525. [DOI] [PubMed] [Google Scholar]
- Arttamangkul S, Leff ER, Koita O, Birdsong WT, and Williams JT (2019). Separation of Acute Desensitization and Long-Term Tolerance of μ-Opioid Receptors Is Determined by the Degree of C-Terminal Phosphorylation. Mol. Pharmacol 96, 505–514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Avet C, Mancini A, Breton B, Le Gouill C, Hauser AS, Normand C, Kobayashi H, Gross F, Hogue M, Lukasheva V, et al. (2020). Selectivity Landscape of 100 Therapeutically Relevant GPCR Profiled by an Effector Translocation-Based BRET Platform. bioRxiv. 10.1101/2020.04.20.052027. [DOI] [Google Scholar]
- Cahill TJ 3rd, Thomsen AR, Tarrasch JT, Plouffe B, Nguyen AH, Yang F, Huang LY, Kahsai AW, Bassoni DL, Gavino BJ, et al. (2017). Distinct conformations of GPCR-β-arrestin complexes mediate desensitization, signaling, and endocytosis. Proc. Natl. Acad. Sci. USA 114, 2562–2567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carman CV, Barak LS, Chen C, Liu-Chen LY, Onorato JJ, Kennedy SP, Caron MG, and Benovic JL (2000). Mutational analysis of Gbetagamma and phospholipid interaction with G protein-coupled receptor kinase 2. J. Biol. Chem 275, 10443–10452. [DOI] [PubMed] [Google Scholar]
- Dale LB, Bhattacharya M, Anborgh PH, Murdoch B, Bhatia M, Nakanishi S, and Ferguson SS (2000). G protein-coupled receptor kinase-mediated desensitization of metabotropic glutamate receptor 1A protects against cell death. J. Biol. Chem 275, 38213–38220. [DOI] [PubMed] [Google Scholar]
- Dale LB, Bhattacharya M, Seachrist JL, Anborgh PH, and Ferguson SS (2001). Agonist-stimulated and tonic internalization of metabotropic glutamate receptor 1a in human embryonic kidney 293 cells: agonist-stimulated endocytosis is beta-arrestin1 isoform-specific. Mol. Pharmacol 60, 1243–1253. [DOI] [PubMed] [Google Scholar]
- DeWire SM, Ahn S, Lefkowitz RJ, and Shenoy SK (2007). Beta-arrestins and cell signaling. Annu. Rev. Physiol 69, 483–510. [DOI] [PubMed] [Google Scholar]
- Dhami GK, Anborgh PH, Dale LB, Sterne-Marr R, and Ferguson SS (2002). Phosphorylation-independent regulation of metabotropic glutamate receptor signaling by G protein-coupled receptor kinase 2. J. Biol. Chem 277, 25266–25272. [DOI] [PubMed] [Google Scholar]
- Dhami GK, Dale LB, Anborgh PH, O’Connor-Halligan KE, Sterne-Marr R, and Ferguson SS (2004). G Protein-coupled receptor kinase 2 regulator of G protein signaling homology domain binds to both metabotropic glutamate receptor 1a and Galphaq to attenuate signaling. J. Biol. Chem 279, 16614–16620. [DOI] [PubMed] [Google Scholar]
- Doumazane E, Scholler P, Zwier JM, Trinquet E, Rondard P, and Pin JP (2011). A new approach to analyze cell surface protein complexes reveals specific heterodimeric metabotropic glutamate receptors. FASEB J. 25, 66–77. [DOI] [PubMed] [Google Scholar]
- Dutar P, Vu HM, and Perkel DJ (1999). Pharmacological characterization of an unusual mGluR-evoked neuronal hyperpolarization mediated by activation of GIRK channels. Neuropharmacology 38, 467–475. [DOI] [PubMed] [Google Scholar]
- Eichel K, Jullié D, and von Zastrow M (2016). β-Arrestin drives MAP kinase signalling from clathrin-coated structures after GPCR dissociation. Nat. Cell Biol 18, 303–310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eichel K, Jullié D, Barsi-Rhyne B, Latorraca NR, Masureel M, Sibarita JB, Dror RO, and von Zastrow M (2018). Catalytic activation of β-arrestin by GPCRs. Nature 557, 381–386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ellaithy A, Gonzalez-Maeso J, Logothetis DA, and Levitz J (2020). Structural and Biophysical Mechanisms of Class C G Protein-Coupled Receptor Function. Trends Biochem. Sci 45, 1049–1064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emery AC, Pshenichkin S, Takoudjou GR, Grajkowska E, Wolfe BB, and Wroblewski JT (2010). The protective signaling of metabotropic glutamate receptor 1 Is mediated by sustained, beta-arrestin-1-dependent ERK phosphorylation. J. Biol. Chem 285, 26041–26048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eng AG, Kelver DA, Hedrick TP, and Swanson GT (2016).Transduction of group I mGluR-mediated synaptic plasticity by β-arrestin2 signalling. Nat. Commun 7, 13571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Erdtmann-Vourliotis M, Mayer P, Ammon S, Riechert U, and Höllt V (2001). Distribution of G-protein-coupled receptor kinase (GRK) isoforms 2, 3, 5 and 6 mRNA in the rat brain. Brain Res. Mol. Brain Res 95, 129–137. [DOI] [PubMed] [Google Scholar]
- Ferguson SS, Downey WE 3rd, Colapietro AM, Barak LS, Ménard L, and Caron MG (1996). Role of beta-arrestin in mediating agonist-promoted G protein-coupled receptor internalization. Science 271, 363–366. [DOI] [PubMed] [Google Scholar]
- Fourgeaud L, Bessis AS, Rossignol F, Pin JP, Olivo-Marin JC, and Hémar A (2003). The metabotropic glutamate receptor mGluR5 is endocytosed by a clathrin-independent pathway. J. Biol. Chem 278, 12222–12230. [DOI] [PubMed] [Google Scholar]
- Francesconi A, and Duvoisin RM (1998). Role of the second and third intracellular loops of metabotropic glutamate receptors in mediating dual signal transduction activation. J. Biol. Chem 273, 5615–5624. [DOI] [PubMed] [Google Scholar]
- Francesconi A, Kumari R, and Zukin RS (2009). Regulation of group I metabotropic glutamate receptor trafficking and signaling by the caveolar/lipid raft pathway. J. Neurosci 29, 3590–3602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gainetdinov RR, Premont RT, Bohn LM, Lefkowitz RJ, and Caron MG (2004). Desensitization of G protein-coupled receptors and neuronal functions. Annu. Rev. Neurosci 27, 107–144. [DOI] [PubMed] [Google Scholar]
- Gillooly DJ, Morrow IC, Lindsay M, Gould R, Bryant NJ, Gaullier JM, Parton RG, and Stenmark H (2000). Localization of phosphatidylinositol 3-phosphate in yeast and mammalian cells. EMBO J. 19, 4577–4588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gutzeit VA, Thibado J, Stor DS, Zhou Z, Blanchard SC, Andersen OS, and Levitz J (2019). Conformational dynamics between transmembrane domains and allosteric modulation of a metabotropic glutamate receptor. eLife 8, e45116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hilger D, Masureel M, and Kobilka BK (2018). Structure and dynamics of GPCR signaling complexes. Nat. Struct. Mol. Biol 25, 4–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang W, Masureel M, Qu Q, Janetzko J, Inoue A, Kato HE, Robertson MJ, Nguyen KC, Glenn JS, Skiniotis G, and Kobilka BK (2020). Structure of the neurotensin receptor 1 in complex with β-arrestin 1. Nature 579, 303–308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iacovelli L, Molinaro G, Battaglia G, Motolese M, Di Menna L, Alfiero M, Blahos J, Matrisciano F, Corsi M, Corti C, et al. (2009). Regulation of group II metabotropic glutamate receptors by G protein-coupled receptor kinases: mGlu2 receptors are resistant to homologous desensitization. Mol. Pharmacol 75, 991–1003. [DOI] [PubMed] [Google Scholar]
- Iacovelli L, Nicoletti F, and De Blasi A (2013). Molecular mechanisms that desensitize metabotropic glutamate receptor signaling: an overview. Neuropharmacology 66, 24–30. [DOI] [PubMed] [Google Scholar]
- Joffe ME, Santiago CI, Engers JL, Lindsley CW, and Conn PJ (2019). Metabotropic glutamate receptor subtype 3 gates acute stress-induced dysregulation of amygdalo-cortical function. Mol. Psychiatry 24, 916–927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang Y, Zhou XE, Gao X, He Y, Liu W, Ishchenko A, Barty A, White TA, Yefanov O, Han GW, et al. (2015). Crystal structure of rhodopsin bound to arrestin by femtosecond X-ray laser. Nature 523, 561–567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawabata S, Tsutsumi R, Kohara A, Yamaguchi T, Nakanishi S, and Okada M (1996). Control of calcium oscillations by phosphorylation of metabotropic glutamate receptors. Nature 383, 89–92. [DOI] [PubMed] [Google Scholar]
- Kniazeff J, Bessis AS, Maurel D, Ansanay H, Prézeau L, and Pin JP (2004). Closed state of both binding domains of homodimeric mGlu receptors is required for full activity. Nat. Struct. Mol. Biol 11, 706–713. [DOI] [PubMed] [Google Scholar]
- Kohout TA, Lin FS, Perry SJ, Conner DA, and Lefkowitz RJ (2001). beta-Arrestin 1 and 2 differentially regulate heptahelical receptor signaling and trafficking. Proc. Natl. Acad. Sci. USA 98, 1601–1606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Komolov KE, and Benovic JL (2018). G protein-coupled receptor kinases: Past, present and future. Cell. Signal 41, 17–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Komolov KE, Du Y, Duc NM, Betz RM, Rodrigues JPGLM, Leib RD, Patra D, Skiniotis G, Adams CM, Dror RO, et al. (2017). Structural and Functional Analysis of a β2-Adrenergic Receptor Complex with GRK5. Cell 169, 407–421.e16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kong G, Penn R, and Benovic JL (1994). A beta-adrenergic receptor kinase dominant negative mutant attenuates desensitization of the beta 2-adrenergic receptor. J. Biol. Chem 269, 13084–13087. [PubMed] [Google Scholar]
- Kumari P, Srivastava A, Banerjee R, Ghosh E, Gupta P, Ranjan R, Chen X, Gupta B, Gupta C, Jaiman D, and Shukla AK (2016). Functional competence of a partially engaged GPCR-β-arrestin complex. Nat. Commun 7, 13416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Latorraca NR, Wang JK, Bauer B, Townshend RJL, Hollingsworth SA, Olivieri JE, Xu HE, Sommer ME, and Dror RO (2018). Molecular mechanism of GPCR-mediated arrestin activation. Nature 557, 452–456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee MH, Appleton KM, Strungs EG, Kwon JY, Morinelli TA, Peterson YK, Laporte SA, and Luttrell LM (2016). The conformational signature of β-arrestin2 predicts its trafficking and signalling functions. Nature 531, 665–668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee S, Park S, Lee H, Han S, Song JM, Han D, and Suh YH (2019). Nedd4 E3 ligase and beta-arrestins regulate ubiquitination, trafficking, and stability of the mGlu7 receptor. eLife 8, e44502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee J, Munguba H, Gutzeit VA, Singh DR, Kristt M, Dittman JS, and Levitz J (2020). Defining the Homo- and Heterodimerization Propensities of Metabotropic Glutamate Receptors. Cell Rep. 31, 107605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leff ER, Arttamangkul S, and Williams JT (2020). Chronic Treatment with Morphine Disrupts Acute Kinase-Dependent Desensitization of GPCRs. Mol. Pharmacol 98, 497–507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levitz J, Habrian C, Bharill S, Fu Z, Vafabakhsh R, and Isacoff EY (2016). Mechanism of Assembly and Cooperativity of Homomeric and Heteromeric Metabotropic Glutamate Receptors. Neuron 92, 143–159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin FT, Krueger KM, Kendall HE, Daaka Y, Fredericks ZL, Pitcher JA, and Lefkowitz RJ (1997). Clathrin-mediated endocytosis of the beta-adrenergic receptor is regulated by phosphorylation/dephosphorylation of beta-arrestin1. J. Biol. Chem 272, 31051–31057. [DOI] [PubMed] [Google Scholar]
- Lodowski DT, Pitcher JA, Capel WD, Lefkowitz RJ, and Tesmer JJ (2003). Keeping G proteins at bay: a complex between G protein-coupled receptor kinase 2 and Gbetagamma. Science 300, 1256–1262. [DOI] [PubMed] [Google Scholar]
- Logothetis DE, Kurachi Y, Galper J, Neer EJ, and Clapham DE (1987). The beta gamma subunits of GTP-binding proteins activate the muscarinic K+ channel in heart. Nature 325, 321–326. [DOI] [PubMed] [Google Scholar]
- Luttrell LM, Wang J, Plouffe B, Smith JS, Yamani L, Kaur S, Jean-Charles PY, Gauthier C, Lee MH, Pani B, et al. (2018). Manifold roles of β-arrestins in GPCR signaling elucidated with siRNA and CRISPR/Cas9. Sci. Signal 11, eaat7650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mangmool S, and Kurose H (2011). G(i/o) protein-dependent and -independent actions of Pertussis Toxin (PTX). Toxins (Basel) 3, 884–899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marchese A, and Benovic JL (2001). Agonist-promoted ubiquitination of the G protein-coupled receptor CXCR4 mediates lysosomal sorting. J. Biol. Chem 276, 45509–45512. [DOI] [PubMed] [Google Scholar]
- Maxfield FR, and McGraw TE (2004). Endocytic recycling. Nat. Rev. Mol. Cell Biol 5, 121–132. [DOI] [PubMed] [Google Scholar]
- Ménard L, Ferguson SS, Zhang J, Lin FT, Lefkowitz RJ, Caron MG, and Barak LS (1997). Synergistic regulation of beta2-adrenergic receptor sequestration: intracellular complement of beta-adrenergic receptor kinase and beta-arrestin determine kinetics of internalization. Mol. Pharmacol 51, 800–808. [PubMed] [Google Scholar]
- Miess E, Gondin AB, Yousuf A, Steinborn R, Mösslein N, Yang Y, Göldner M, Ruland JG, Bünemann M, Krasel C, et al. (2018). Multisite phosphorylation is required for sustained interaction with GRKs and arrestins during rapid μ-opioid receptor desensitization. Sci. Signal 11, eaas9609. [DOI] [PubMed] [Google Scholar]
- Mundell SJ, Matharu AL, Pula G, Roberts PJ, and Kelly E (2001). Agonist-induced internalization of the metabotropic glutamate receptor 1a is arrestin- and dynamin-dependent. J. Neurochem 78, 546–551. [DOI] [PubMed] [Google Scholar]
- Neto A, and Ceol CJ (2018). Melanoma-associated GRM3 variants dysregulate melanosome trafficking and cAMP signaling. Pigment Cell Melanoma Res. 31, 115–119. [DOI] [PubMed] [Google Scholar]
- Nicoletti F, Bockaert J, Collingridge GL, Conn PJ, Ferraguti F, Schoepp DD, Wroblewski JT, and Pin JP (2011). Metabotropic glutamate receptors: from the workbench to the bedside. Neuropharmacology 60, 1017–1041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nobles KN, Xiao K, Ahn S, Shukla AK, Lam CM, Rajagopal S, Strachan RT, Huang TY, Bressler EA, Hara MR, et al. (2011). Distinct phosphorylation sites on the β(2)-adrenergic receptor establish a barcode that encodes differential functions of β-arrestin. Sci. Signal 4, ra51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nuber S, Zabel U, Lorenz K, Nuber A, Milligan G, Tobin AB, Lohse MJ, and Hoffmann C (2016). β-Arrestin biosensors reveal a rapid, receptor-dependent activation/deactivation cycle. Nature 531, 661–664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Hayre M, Vázquez-Prado J, Kufareva I, Stawiski EW, Handel TM, Seshagiri S, and Gutkind JS (2013). The emerging mutational landscape of G proteins and G-protein-coupled receptors in cancer. Nat. Rev. Cancer 13, 412–424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Hayre M, Eichel K, Avino S, Zhao X, Steffen DJ, Feng X, Kawakami K, Aoki J, Messer K, Sunahara R, et al. (2017). Genetic evidence that β-arrestins are dispensable for the initiation of β2-adrenergic receptor signaling to ERK. Sci. Signal 10, eaal3395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oakley RH, Laporte SA, Holt JA, Caron MG, and Barak LS (2000). Differential affinities of visual arrestin, beta arrestin1, and beta arrestin2 for G protein-coupled receptors delineate two major classes of receptors. J. Biol. Chem 275, 17201–17210. [DOI] [PubMed] [Google Scholar]
- Peterson YK, and Luttrell LM (2017). The Diverse Roles of Arrestin Scaffolds in G Protein-Coupled Receptor Signaling. Pharmacol. Rev 69, 256–297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pitcher JA, Inglese J, Higgins JB, Arriza JL, Casey PJ, Kim C, Benovic JL, Kwatra MM, Caron MG, and Lefkowitz RJ (1992). Role of beta gamma subunits of G proteins in targeting the beta-adrenergic receptor kinase to membrane-bound receptors. Science 257, 1264–1267. [DOI] [PubMed] [Google Scholar]
- Prickett TD, Wei X, Cardenas-Navia I, Teer JK, Lin JC, Walia V, Gartner J, Jiang J, Cherukuri PF, Molinolo A, et al. (2011). Exon capture analysis of G protein-coupled receptors identifies activating mutations in GRM3 in melanoma. Nat. Genet 43, 1119–1126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rajagopal S, and Shenoy SK (2018). GPCR desensitization: Acute and prolonged phases. Cell. Signal 41, 9–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raveh A, Cooper A, Guy-David L, and Reuveny E (2010). Nonenzymatic rapid control of GIRK channel function by a G protein-coupled receptor kinase. Cell 143, 750–760. [DOI] [PubMed] [Google Scholar]
- Reiner A, and Levitz J (2018). Glutamatergic Signaling in the Central Nervous System: Ionotropic and Metabotropic Receptors in Concert. Neuron 98, 1080–1098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ribeiro FM, Ferreira LT, Paquet M, Cregan T, Ding Q, Gros R, and Ferguson SS (2009). Phosphorylation-independent regulation of metabotropic glutamate receptor 5 desensitization and internalization by G protein-coupled receptor kinase 2 in neurons. J. Biol. Chem 284, 23444–23453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saugstad JA, Segerson TP, and Westbrook GL (1996). Metabotropic glutamate receptors activate G-protein-coupled inwardly rectifying potassium channels in Xenopus oocytes. J. Neurosci 16, 5979–5985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scheefhals N, Catsburg LAE, Westerveld ML, Blanpied TA, Hoogenraad CC, and MacGillavry HD (2019). Shank Proteins Couplethe Endocytic Zone to the Postsynaptic Density to Control Trafficking and Signaling of Metabotropic Glutamate Receptor 5. Cell Rep. 29, 258–269.e8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schindelin J, Arganda-Carreras I, Frise E, Kaynig V, Longair M, Pietzsch T, Preibisch S, Rueden C, Saalfeld S, Schmid B, et al. (2012). Fiji: an open-source platform for biological-image analysis. Nat. Methods 9, 676–682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneider CA, Rasband WS, and Eliceiri KW (2012). NIH Image to ImageJ: 25 years of image analysis. Nat. Methods 9, 671–675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sente A, Peer R, Srivastava A, Baidya M, Lesk AM, Balaji S, Shukla AK, Babu MM, and Flock T (2018). Molecular mechanism of modulating arrestin conformation by GPCR phosphorylation. Nat. Struct. Mol. Biol 25, 538–545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shenoy SK, and Lefkowitz RJ (2011). β-Arrestin-mediated receptor trafficking and signal transduction. Trends Pharmacol. Sci 32, 521–533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shenoy SK, McDonald PH, Kohout TA, and Lefkowitz RJ (2001). Regulation of receptor fate by ubiquitination of activated beta 2-adrenergic receptor and beta-arrestin. Science 294, 1307–1313. [DOI] [PubMed] [Google Scholar]
- Shukla AK, Westfield GH, Xiao K, Reis RI, Huang LY, Tripathi-Shukla P, Qian J, Li S, Blanc A, Oleskie AN, et al. (2014). Visualization of arrestin recruitment by a G-protein-coupled receptor. Nature 512, 218–222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sorensen SD, and Conn PJ (2003). G protein-coupled receptor kinases regulate metabotropic glutamate receptor 5 function and expression. Neuropharmacology 44, 699–706. [DOI] [PubMed] [Google Scholar]
- Stauffer W, Sheng H, and Lim HN (2018). EzColocalization:An ImageJ plugin for visualizing and measuring colocalization in cells and organisms. Sci. Rep 8, 15764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Staus DP, Hu H, Robertson MJ, Kleinhenz ALW, Wingler LM, Capel WD, Latorraca NR, Lefkowitz RJ, and Skiniotis G (2020). Structure of the M2 muscarinic receptor-β-arrestin complex in a lipid nanodisc. Nature 579, 297–302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stoeber M, Jullié D, Li J, Chakraborty S, Majumdar S, Lambert NA, Manglik A, and von Zastrow M (2020). Agonist-selective recruitment of engineered protein probes and of GRK2 by opioid receptors in living cells. eLife 9, e54208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stoppel LJ, Auerbach BD, Senter RK, Preza AR, Lefkowitz RJ, and Bear MF (2017). β-Arrestin2 Couples Metabotropic Glutamate Receptor 5 to Neuronal Protein Synthesis and Is a Potential Target to Treat Fragile X. Cell Rep 18, 2807–2814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Subtil A, Lampson MA, Keller SR, and McGraw TE (2000). Characterization of the insulin-regulated endocytic recycling mechanism in 3T3-L1 adipocytes using a novel reporter molecule. J. Biol. Chem 275, 4787–4795. [DOI] [PubMed] [Google Scholar]
- Suh YH, Chang K, and Roche KW (2018). Metabotropic glutamate receptor trafficking. Mol. Cell. Neurosci 91, 10–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tesmer VM, Kawano T, Shankaranarayanan A, Kozasa T, and Tesmer JJ (2005). Snapshot of activated G proteins at the membrane: the Galphaq-GRK2-Gbetagamma complex. Science 310, 1686–1690. [DOI] [PubMed] [Google Scholar]
- Tora AS, Rovira X, Cao AM, Cabayé A, Olofsson L, Malhaire F, Scholler P, Baik H, Van Eeckhaut A, Smolders I, et al. (2018). Chloride ions stabilize the glutamate-induced active state of the metabotropic glutamate receptor 3. Neuropharmacology 140, 275–286. [DOI] [PubMed] [Google Scholar]
- Vafabakhsh R, Levitz J, and Isacoff EY (2015). Conformational dynamics of a class C G-protein-coupled receptor. Nature 524, 497–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Violin JD, Dewire SM, Barnes WG, and Lefkowitz RJ (2006). G protein-coupled receptor kinase and beta-arrestin-mediated desensitization of the angiotensin II type 1A receptor elucidated by diacylglycerol dynamics. J. Biol. Chem 281, 36411–36419. [DOI] [PubMed] [Google Scholar]
- Vivaudou M, Chan KW, Sui JL, Jan LY, Reuveny E, and Logothetis DE (1997). Probing the G-protein regulation of GIRK1 and GIRK4, the two subunits of the KACh channel, using functional homomeric mutants. J. Biol. Chem 272, 31553–31560. [DOI] [PubMed] [Google Scholar]
- von Zastrow M, and Kobilka BK (1992). Ligand-regulated internalization and recycling of human beta 2-adrenergic receptors between the plasma membrane and endosomes containing transferrin receptors. J. Biol. Chem 267, 3530–3538. [PubMed] [Google Scholar]
- Whorton MR, and MacKinnon R (2013). X-ray structure of the mammalian GIRK2-βγ G-protein complex. Nature 498, 190–197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yanagawa M, Hiroshima M, Togashi Y, Abe M, Yamashita T, Shichida Y, Murata M, Ueda M, and Sako Y (2018). Single-molecule diffusion-based estimation of ligand effects on G protein-coupled receptors. Sci. Signal 11, eaao1917. [DOI] [PubMed] [Google Scholar]
- Zhao Y, Araki S, Wu J, Teramoto T, Chang YF, Nakano M, Abdelfattah AS, Fujiwara M, Ishihara T, Nagai T, and Campbell RE (2011). An expanded palette of genetically encoded Ca2+ indicators. Science 333, 1888–1891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou XE, He Y, de Waal PW, Gao X, Kang Y, Van Eps N, Yin Y, Pal K, Goswami D, White TA, et al. (2017). Identification of Phosphorylation Codes for Arrestin Recruitment by G Protein-Coupled Receptors. Cell 170, 457–469.e13. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
This study did not generate large datasets or code. The datasets analyzed during the study are available from the corresponding author on reasonable request.