

Abstract
Acute kidney injury occurs frequently in COVID-19 patients infected by the coronavirus SARS-CoV-2, and infection of kidney cells by this virus has been reported. However, little is known about the direct impact of the SARS-CoV-2 infection upon the renal tubular cells. We report that SARS-CoV-2 activated signal transducer and activator of transcription 3 (STAT3) signaling and caused cellular injury in the human renal tubular cell line. Mechanistically, the viral protein ORF3A of SARS-CoV-2 augmented both NF-κB and STAT3 signaling and increased the expression of kidney injury molecule 1. SARS-CoV-2 infection or expression of ORF3A alone elevated the protein level of tripartite motif-containing protein 59 (TRIM59), an E3 ubiquitin ligase, which interacts with both ORF3A and STAT3. The excessive TRIM59 in turn dissociated the phosphatase TCPTP from binding to STAT3 and hence inhibited the dephosphorylation of STAT3, leading to persistent STAT3 activation. Consistently, ORF3A induced renal injury in zebrafish and mice. In addition, expression of TRIM59 was elevated in the kidney autopsies of COVID-19 patients with acute kidney injury. Thus, the aberrant activation of STAT3 signaling by TRIM59 plays a significant role in the renal tubular cell injury caused by SARS-CoV-2, which suggests a potential targeted therapy for the renal complications of COVID-19.
Keywords: COVID-19, SARS-CoV-2, ORF3A, zebrafish, acute kidney injury, NF-κb, STAT, inflammation, phosphorylation, renal tubule
Graphical abstract
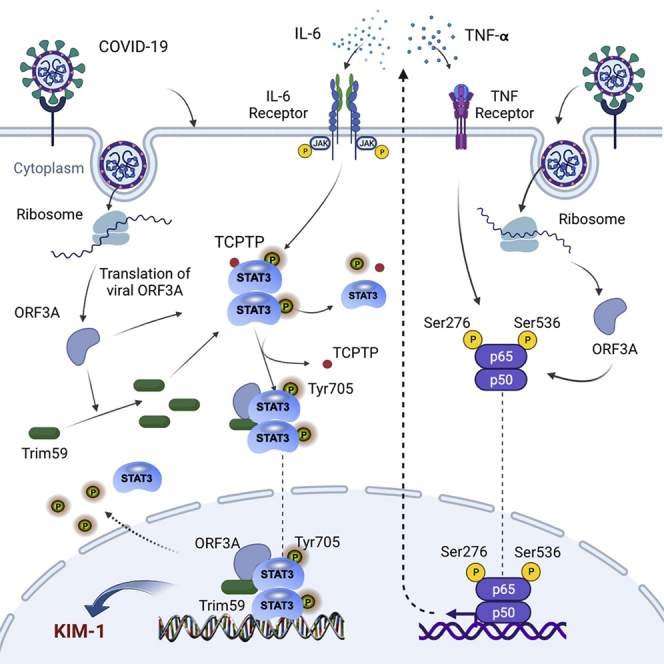
SARS-CoV-2 infection of renal tubular cells increases TRIM59 proteins via the viral protein ORF3A, resulting in the inhibition of STAT3 dephosphorylation and excessive activation of STAT3 that leads to renal tubular injury. This reveals a novel mechanism underlying the acute kidney injury occurring in severe COVID-19 patients.
Introduction
The COVID-19 pandemic has had an unprecedented impact on public health on a global scale. Although the causal virus SARS-CoV-2 primarily attacks the respiratory system and the major symptom of COVID-19 is acute respiratory disorder, acute kidney injury (AKI) is a frequent complication associated with the SARS-CoV-2 infection.1,2 Multiple mechanisms may underlie the pathogenesis of AKI in COVID-19. These include excessive immune response, abnormal metabolic condition, organ crosstalk, and direct viral infection of renal cells, etc.3 Accumulating evidence of direct infection of renal cells in COVID-19 patients has been documented recently.4,5,6 In particular, a recent study showed that the cellular receptor for SARS-CoV-2 was elevated in renal tubular cells under diabetic conditions, which increased the susceptibility of SARS-Cov-2 infection in human kidney cells.7 Therefore, cytotoxicity conferred by the viral proteins to renal tubular epithelial cells may be one of the potential pathogenic mechanisms contributing to the AKI in COVID-19 patients.
SARS-CoV-2 carries a single-strand RNA genome of about 30 kb.8 Two large open reading frames ORF1a and 1b can be translated and processed into 27 non-structural proteins. Additional open reading frames encode the spike protein (S), envelop protein (E), membrane protein (M), nucleocapsid protein (N), and several accessory proteins (3A, 3B, 6, 7A, 7B, 8, and 10). Following the viral infection, these proteins are synthesized in large quantities to establish a cellular environment that favors viral replication and generation of new viral particles. Several of the viral proteins encoded by the SARS-CoV-2 genome have been reported to interact with host cell proteins and perturb a series of cell signaling pathways that lead to cell injury.9 These include aberrant activation of NF-κB pathways,9 NLRP3 inflammasome pathway,10,11 interferon signaling pathways,12,13 TGF-β/SMAD pathways,14 and STAT pathways.15,16
The Janus kinase/signal transducers and activators of transcription (JAK/STAT) pathway regulates a wide range of cellular processes and is implicated in a variety of kidney diseases,17,18 such as AKI, diabetic nephropathy, and glomerular diseases. The selective and combinatorial actions of four JAK members and seven STAT members are involved in the JAK/STAT pathway in response to various extracellular signals, ranging from cytokines to growth factors. Among them, STAT3 plays an essential role in normal development, and its dysregulation is critical to many disease conditions, including kidney diseases.18,19 Canonical STAT3 activation is induced by interleukin-6 (IL-6), leading to the dimerization of IL-6 receptors and signal transducer gp130 and subsequent phosphorylation of STAT3. Phosphorylation of STAT3 at Tyr705 causes its dimerization and nuclear translocation that in turn results in the transcription of STAT3 target genes. Massive cytokine release (known as a cytokine storm) occurs in COVID-19 patients, in which IL-6 and TNF-α are two key cytokines,20 so aberrant STAT signaling has been proposed to play a central role in the pathogenesis of COVID-19 complications.19,21 Recently, we reported excessive phosphorylation of STAT3 in the kidney autopsy of COVID-19 patients with AKI.22,23 However, it remains unclear whether direct viral infection modulates the STAT3 activation in response to systematic cytokine exposure in kidney cells.
In this study, we hypothesized that SARS-CoV-2 viral protein(s) may interfere with host cell signaling in renal tubular cells and cause kidney cell injury. We used transgenic zebrafish to screen for viral proteins that caused tubular epithelial cell injury and observed in transgenic zebrafish that ORF3A expression in pronephric tubular epithelial cells resulted in the induction of kidney injury molecule 1 (KIM-1), a hallmark of renal tubular injury. Using in vitro cultured renal tubular cell line (HK-2), we found that the viral protein ORF3A interacted with the E3 ubiquitin ligase TRIM59, which modulated STAT3 phosphorylation and downstream activities. Furthermore, we showed that expression of ORF3A was sufficient to induce STAT3 phosphorylation, KIM-1 expression, and renal injury in mouse kidneys and could exacerbate ischemia/reperfusion-induced kidney injury. Moreover, the inhibition of STAT3 activities could ameliorate the renal injury induced by ORF3A expression in vivo. Consistently, we found that in HK-2 cells, SARS-CoV-2 infection also elevated the expression of TRIM59 and activated STAT3, which was confirmed by immunohistochemistry on kidney autopsies of human COVID-19 patients. Taken together, our findings have provided a previously unrecognized mechanism of the renal cell injury caused by SARS-CoV-2 infection via the action of viral protein ORF3A that dysregulates STAT3 signaling downstream to the cytokine-induced STAT3 phosphorylation. It is informative for the development of a potential therapeutic strategy for the renal complications associated with COVID-19.
Results
Functional screening identified the cytotoxicity of ORF3A in pronephric tubular cells in transgenic zebrafish
We used transgenic zebrafish as an in vivo functional screening platform to test the cytotoxicity of individual viral proteins of SARS-CoV-2 that may injure pronephric tubular epithelial cells. The binary Gal4-UAS system was employed to drive the expression of viral proteins in a renal tubular cell-specific manner (Figure 1A). A transgenic zebrafish line Tg(enpep: Gal4) we published previously24 was used to drive the renal tubular expression of the Gal4 transcription activator, which can drive the expression of transgene under the control of the UAS promoter sequence. We have generated seven transgenic zebrafish lines for viral proteins (ORF3A, ORF7A, ORF6, ORF8, M, N, E) using the UAS promoter and crossed these lines with Tg(enpep:Gal4) respectively (data not shown). Among them, the transgenic fish expressing ORF3A in the tubular epithelial cells displayed a significant peri-cardiac effusion phenotype at 48 hours postfertilization (hpf) (Figure 1B, B’) that resembled the morphology of other zebrafish models of AKI.25,26 At 48 hpf, 33.3% ±8.3% (n = 151 embryos in three individual clutches) of each clutch of double transgenic embryos developed peri-cardiac edema, while almost none of the non-transgenic siblings (0.15% ±0.05%, n = 105 embryos in three individual clutches) had this phenotype. The variable penetrance of the edematous phenotype may be due to the variable expression level of the transgene among different animals. The edema deteriorated and eventually led to whole-body edema and lethality around 7 days post fertilization. This phenotype is consistent with previously reported zebrafish models of renal injury.25,27 kim-1 expression in the renal epithelia in pronephros was detected by in situ hybridization in the transgenic fish expressing ORF3A but not in the control fish (Figure 1D, D’), ascertaining that renal injury was induced by ORF3A.
Figure 1.

SARS-CoV-2 ORF3A causes renal injury in pronephros of transgenic zebrafish
(A) Schematic graph of the transgene structure of Tg(enpep:Gal4) and Tg (UAS:ORF3A,myl7:Venus). (B) Tg(enpep:Gal4; UAS:NTR-mCherry) zebrafish embryo shows normal morphology at 48 hpf. (B’) zebrafish embryos show peri-cardiac edema at 48 hpf. (C) Red fluorescence illustrates the expression of NTR-mCherry transgene in pronephric tubules in Tg(enpep:Gal4; UAS:NTR-mCherry). (C’) Yellow fluorescence in the myocardium indicates the transgene of UAS:ORF3A,myl7:Venus. (D) Side view of a normal control zebrafish embryo at 72 hpf shows no detectable kim-1 expression in the pronephros. (D’) Side view of a transgenic zebrafish embryo expressing ORF3A in pronephric tubules shows kim-1 expression detected in the pronephros by in situ hybridization. (E) Quantitative real-time PCR for target genes in 72 hpf Tg(enpep:Gal4;UAS:NTR-mCherry; UAS:ORF3A,myl7:Venus) zebrafish embryos. The mRNA transcript levels are normalized to ef1a mRNA, and folds of change compared with age-matched Tg(enpep:Gal4; UAS:NTR-mCherry) embryos are plotted. Bar graphs indicate the means of triplicates and the standard deviations. ∗p < 0.05; ∗∗p < 0.01.
Quantitative PCR revealed the expression of kim-1 and a number of cytokines and interferon gamma target genes were elevated by ORF3A expression (Figure 1E). The other viral proteins that we tested in zebrafish did not produce any obvious morphological changes in the embryos (data not shown), so we concluded that ORF3A overexpression in pronephric tubular cells in zebrafish is sufficient to cause renal injury and thus carried out further functional studies of ORF3A.
ORF3A of SARS-CoV-2 modulates NF-kB and STAT3 signaling in HK-2 cells
To perform in vitro functional studies, we used HK-2, a human renal proximal tubular cell line that has been shown to be susceptible to SARS-CoV-2 infection.28 In these renal tubule-derived cells, expression of ORF3A effectively activated the NF-κB pathway by increasing the phosphorylation of p65 (at both S536 and S276 position) (Figure 2A). In particular, it also induced the expression of KIM-1 (Figures 2A and 2B), indicating renal cell injury. The level of cleaved caspase-3 was also elevated by ORF3A (Figure 2A), suggesting a role of ORF3A in apoptosis induction that has also been reported in other cell lines. TNF-α is a cytokine that can activate NF-κB pathway and cause renal tubular cell injury, and we found that ORF3A expression could potentiate the action of TNF-α in HK-2 cells (Figure 2A). The mRNA levels of TNFα, IL-6, and CXCL11, which are downstream targets of NF-kB, were elevated by ORF3A in HK-2 cells (Figures 2A and 2B), confirming that the NF-κB signaling pathway was modulated by ORF3A.
Figure 2.

SARS-CoV-2 ORF3A enhances NF-kB and STAT3 signaling and injures HK-2 cells
(A) Western blots for protein lysates from HK-2 cells transfected with pCMV-GFP (GFP) as controls and pCMV-ORF3A (ORF3A) following TNF-α treatment. Cells were treated with TNF-α for the given periods of time. (B) Quantitative real-time PCR for the expression of inflammation and renal injury markers genes in ORF3A-expressing cells. All data were normalized to 18S ribosomal RNA, and the ratios are shown for relative mRNA levels. Each bar represents the mean ± SD (n = 3). ∗p < 0.05; ∗∗p < 0.01. (C) Western blots for protein lysates from HK-2 cells transfected with pCMV-GFP (GFP) as controls and pCMV-ORF3A (ORF3A) following TNF-α treatment of given periods of time. Western blot signals were quantified by densitometry. (D) Western blots for protein lysates from HK-2 cells transfected with pCMV-GFP (GFP) as controls and pCMV-ORF3A (ORF3A) following IL-6 treatment of given periods of time. Western blot signals were quantified by densitometry.
Since IL-6 is a major activator of STAT3 signaling pathway,18 we examined the phosphorylation of STAT3 at Y705 position, a modification promoting the nuclear translocation of this transcription factor. We found ORF3A expression was sufficient to elevate the phosphorylation of STAT3 in HK-2 cells (Figures 2C and 2D). It also enhanced TNF-α or IL-6-induced phosphorylation of STAT3 (Figures 2C and 2D). Since KIM-1 is a downstream target gene of STAT3 signaling,29 ORF3A also significantly enhanced the induction of KIM-1 expression by IL-6, through the phosphorylation of STAT3 (Figure 2D). Thus, ORF3A expression could modulate the STAT3 signaling pathway and injure renal tubular cells. Our data suggest that ORF3A expression activates the NF-kB signaling pathway to increase the downstream target gene IL-6, which in turn activates STAT3 signaling pathway and enhances the injury of renal tubular cells.
ORF3A injures HK-2 cells via TRIM59-mediated STAT3 activation
Although ORF3A can possibly activate STAT3 signaling by induction of IL-6 via the modulation of NF-κB pathway, we sought to explore whether ORF3A can directly regulate STAT3 phosphorylation. A large-scale proteomic study revealed that ORF3A interacts with TRIM59, an E3 ubiquitin ligase that physically binds to STAT3.30 We thus tested these protein interactions in HK-2 cells expressing Strep-tagged ORF3A. Using co-immunoprecipitation or Strep-tag-based pull-down assays, we found that both TRIM59 and STAT3 could be reciprocally precipitated with ORF3A-Strep from HK-2 cells (Figure 3A). Interestingly, we noticed that ORF3A-Strep not only increased TRIM59 expression, but it also enhanced the interaction between STAT3 and TRIM59 in HK-2 cells (Figure 3A).
Figure 3.

SARS-CoV-2 ORF3A interacts with TRIM59 and STAT3 and interferes with STAT3 binding to TCPTP in HK-2 cells
(A) Co-immunoprecipitation was carried out for ORF3A, STAT3, and TRIM59 with lysates of HK-2 cells expressing ORF3A-Strep. (B) Co-immunoprecipitation was carried out for STAT3 and TCPTP with lysates of HK-2 cells that were transduced with lentiviruses encoding ORF3A-Strep or an shRNA targeting TRIM59. (C) Cytosol and nucleus fractions were isolated from HK-2 cells transduced with lentiviruses encoding ORF3A-Strep or GFP-Strep. (D) Confocal images of HK-2 cells transduced with lentiviruses encoding ORF3A-Strep showing nuclear localization of STAT3 (red) and ORF3A-strep (green). The nuclei were counter-stained with DAPI.
In glioma cells, TRIM59 can interfere with the dephosphorylation of STAT3 by the phosphatase TCPTP through blocking of the interaction between STAT3 and TCPTP.31,32 In HK-2 cells, we found that reducing TRIM59 expression by shRNA-mediated knockdown increased the amount of STAT3 that was co-immunoprecipitated with TCPTP, while expression of ORF3A decreased the interaction between STAT3 and TCPTP likely through increased TRIM59 expression (Figure 3B). This suggests that ORF3A protein of SARS-CoV-2 can not only increase TRIM59 expression, but it can also interact with TRIM59 in order to competitively inhibit the interaction between TCPTP and STAT3, leading to more phosphorylated STAT3. The phosphorylation of STAT3 at Y705 position facilitates the nuclear translocation of STAT3. In HK-2 cells expressing ORF3A, we found more phosphorylated STAT3 in the nuclear fraction compared to the GFP-expressing controls, confirming that ORF3A elevated the phosphorylated STAT3 translocated to the nuclei (Figure 3C). In addition, we found a small amount of ORF3A was also present in the nuclear fraction by western blot (Figure 3C), and this was confirmed by immunofluorescence staining (Figure 3D). These results indicate that a fraction of ORF3A protein could be translocated to the nuclei together with STAT3, although the majority of the ORF3A appears to be located in the cytoplasm (Figure 3D).
To further confirm that TRIM59 mediates the activation of STAT3 and the induction of kidney injury by ORF3A, we overexpressed TRIM59 or knocked down TRIM59 in HK-2 cells expressing V5-tagged ORF3A. In these cells, overexpression of TRIM59 significantly elevated the level of phosphorylated STAT3 and KIM-1, while knockdown of TRIM59 reduced the level of phosphorylated STAT3 and KIM-1 (Figures 4A–4C). We also confirmed that STAT3 activation plays a major role in ORF3A-induced renal cell injury by showing that within ORF3A-expressing HK-2 cells, the co-expression of a dominant-negative form of STAT3, which cannot be phosphorylated at Tyr705, reduced the KIM-1 expression, even though TRIM59 expression remained elevated by ORF3A (Figures 4D–4F).
Figure 4.

HK-2 cellular injury induced by ORF3A is mediated by upregulation of TRIM59 and STAT3 signaling activities
(A) Western blots for protein lysates from HK-2 cells. Expression of GFP, TRIM59, or ORF3A-v5 was achieved by Viafect transfection. TRIM59 was knocked down by shRNA. (B) Densitometric quantitation of western blot signals shown in (A). (C) Quantitative real-time PCR for HK-2 cells shown in (A). (D) Western blots for protein lysates from HK-2 cells. Expression of GFP, TRIM59, or dominant-negative STAT3 (DN-STAT3) was achieved by lentiviral transduction. (E) Densitometric quantitation of western blot signals shown in (D). (F) Quantitative real-time PCR for HK-2 cells shown in (D). ∗p < 0.05; ∗∗p < 0.01.
Since there is a lack of appropriate animal model for SARS-CoV-2 infection in kidneys, we had to utilize the in vitro cultured HK-2 cells to determine whether SARS-CoV-2 infection of kidney cells could induce similar activation of STAT3 signaling pathway and kidney cell injury. We found that SARS-CoV-2 infection of HK-2 cells induced KIM-1 expression and STAT3 phosphorylation as well as elevation of the TRIM59 expression (Figure 5A). Previously, we have reported that STAT3 is activated in the kidney autopsies of some COVID-19 patients with AKI.23 With immunohistochemical staining, we found TRIM59 was significantly highly expressed in the nuclei of renal tubular cells in these COVID-19 kidney autopsy specimens (Figures 5B–5D). These data are consistent with our in vitro findings and suggest that ORF3A is one of the viral proteins responsible for SARS-CoV-2-induced kidney cell injury by activating STAT3 via TRIM59.
Figure 5.

TRIM59 expression is increased in HK-2 cells by SARS-CoV-2 and in kidney autopsies of COVID-19 patients with AKI
(A) Western blots for protein lysates from HK-2 cells infected with SARS-CoV-2 (MOI = 1). Immunohistochemical staining of TRIM59 in a control kidney biopsy (B) and two COVID-19 kidney autopsies with AKI (C, D). Scale bar: 100 μm.
Taken together, our data suggest that SARS-CoV-2 ORF3A protein upregulates TRIM59 expression, forms a complex with TRIM59 and STAT3, and subsequently enhances the STAT3 phosphorylation by competing with TCPTP to cause renal cell injury.
ORF3A exacerbates kidney injury induced by ischemia-reperfusion (I/R) in mice
To investigate the in vivo effects of ORF3A expression on kidney cells, we injected lentiviruses encoding ORF3A-Strep into the mouse kidney, induced I/R-kidney injury, and analyzed the kidneys 3 weeks later (Figure 6A). We found that either overexpression of ORF3A or I/R alone induced KIM-1 expression, along with increased expression of TRIM59 and phosphorylated STAT3 (Figures 6B and 6C). BUN and serum creatinine levels were also increased (Figure 6D), and a number of downstream genes of the STAT3 pathway were induced (Figure 6E), indicating the impairment of renal function and activation of STAT3 signaling by ORF3A or I/R. Moreover, I/R in ORF3A-expressing kidney induced a significantly higher expression of TRIM59 and KIM-1 and phosphorylation of STAT3 than either I/R or ORF3A overexpression alone, as demonstrated by both western blots and immunohistochemical staining (Figures 6B and 6F). BUN and serum creatinine levels and the expression of downstream genes of the STAT3 pathway were further increase in ORF3A-expressing kidney with I/R (Figures 6D and 6E). These data suggest that ORF3A could activate STAT3 and exacerbate I/R-induced kidney injury in mice. This may explain a synergistic effect between SARS-CoV-2 infection and sepsis/ischemia on the development of AKI in sick COVID-19 patients.
Figure 6.

SARS-CoV-2 ORF3A enhances STAT3 signaling and exacerbates kidney injury in mice with ischemia-reperfusion
(A) Schematic graph showing the timeline of the experimental procedures. (B) Western blots for protein lysates from mouse kidneys following ORF3A expression (3A) and ischemia-reperfusion (I/R). (C) Densitometric quantitation of western blot results shown in (B) (normalized to Vaculin) and the mRNA level of KIM-1 measured by quantitative real-time PCR (normalized to 18S rRNA). (D) Measurements of BUN and serum creatinine in mice. (E) Quantitative PCR measurements of mRNA levels of STAT3 target genes, normalized to 18S rRNA. (F) Immunohistochemical staining of mouse kidney sections. (G) Quantification of IHC staining results shown in (F). ∗p < 0.05; ∗∗p < 0.01.
To examine the role of STAT3 in mediating the kidney injury caused by ORF3A expression, we tested whether inhibition of STAT3 activation could ameliorate the injury in mouse kidneys expressing ORF3A. We administered STAT3 inhibitor S3I-201 to mice injected with lentiviruses encoding ORF3A-Strep for 3 weeks and induced injury by I/R (Figure 7A). The STAT3 inhibitor S3I-201 effectively reduced the levels of KIM-1 and phosphorylation of STAT3 in mice with both ORF3A expression and I/R (Figures 7B and 7C). Consistently, it also reduced the BUN and serum creatinine levels as well as the expression of STAT3 downstream target gene (Figures 7D and 7E), but it did not significantly affect the expression of Trim59 (Figures 7B, 7C, 7F, and 7G). This suggests that STAT3 acted downstream of Trim59, and targeting STAT3 could be a potential approach to attenuate renal tubular cell injury associated with COVID-19.
Figure 7.

Inhibition of STAT3 activities in mice ameliorates kidney injury caused by ORF3A expression and I/R
(A) Schematic graph showing the timeline of the experimental procedures. (B) Western blots for protein lysates from mouse kidneys following the administration of S3I-201, ORF3A expression (3A), and ischemia-reperfusion (I/R). DMSO was used as a negative control for S3I-201 inhibitor. (C) Densitometric quantitation of western blot results shown in (B) (normalized to GAPDH) and the mRNA level of KIM-1 measured by quantitative real-time PCR (normalized to 18S rRNA). (D) Measurements of BUN and serum creatinine in mice. (E) Quantitative PCR measurements of mRNA levels of STAT3 target genes, normalized to 18S rRNA. (F) Immunohistochemical staining of mouse kidney sections. (G) Quantification of IHC staining results shown in (F). ∗p < 0.05; ∗∗p < 0.01.
Discussion
In this study, we employed both in vitro and in vivo model systems to uncover a previously unrecognized pathogenic mechanism of kidney injury associated with COVID-19, in which dysregulation of STAT3 signaling plays a central role. Our recent reports about increased STAT3 phosphorylation in COVID-19 patient kidney biopsy suggests that aberrant STAT3 signaling due to the SARS-CoV-2 infection could be one of the pathomechanisms underlying the renal injury in COVID-19 patients,22,23 given that STAT3 activation in kidney cells has been implicated in various kidney diseases.18 However, how the STAT3 signaling pathway in kidney cells is perturbed by SARS-CoV-2 infection remains unclear.
A massive secretion of cytokines is elicited by SARS-CoV-2 infection in COVID-19 patients, including IL-6, a potent activator of STAT3 signaling, so the systematic cytokine response to the viral infection is one of the triggers to activate the STAT3 signaling. However, recent reports about the detection of viral mRNA and proteins in kidney biopsies of COVID-19 patients33,34 and infection of human kidney organoids by SARS-CoV-2 viruses in vitro6 suggest that kidney cells could be infected directly by the virus. This opens a new question of whether direct viral infection by SARS-CoV-2 can alter STAT3 signaling and modulate the severity of kidney injury due to the exposure to massive cytokines. To address this question, we found that in vitro cultured HK-2 cells infected by SARS-CoV-2 had an elevated level of phosphorylated STAT3 and KIM-1 expression, demonstrating that direct infection of SARS-COV-2 can disturb the STAT3 signaling and cause renal tubular epithelial cell injury. This is consistent with a recent report showing that the JAK-STAT pathway was solely activated in the proximal tubule cells infected by the virus.6
In order to understand the mechanism for the renal cell injury caused by direct infection of SARS-CoV-2, we generated a number of transgenic zebrafish to express individual viral proteins of SARS-CoV-2 in the embryonic pronephric tubular epithelial cells and found that the viral protein ORF3A could induce renal tubular injury in zebrafish. ORF3A, one of the largest accessory proteins of SARS-CoV-2, encodes a viroporin with ion channel activities that is localized to plasma membrane and lysosomes and is involved in viral release.35,36,37,38,39 Like SARS-CoV ORF3A, SARS-CoV-2 ORF3A has been shown to induce apoptosis, necrosis, and pyroptosis in HEK293T and Vero E6 cells,40,41 and it can activate NF-κB and NLRP3 inflammatory pathways in HEK293T and A549 cells.42 However, the pathogenic role of ORF3A has not been characterized in the context of kidney cells or STAT3 signaling. Here, we presented both in vitro evidence with HK-2 cells and in vivo evidence with transgenic zebrafish that ORF3A in kidney cells is indeed able to modulate the NF-kB pathway and increase cytokine production in kidney cells, including TNF-α and IL-6, which are NF-kB target genes and strongly associated with the severity of COVID-19 patient outcomes.20,43 Therefore, our data support that, upon viral infection, certain viral proteins such as ORF3A affect the inflammatory pathways triggering cytokine release to activate the STAT3 pathway and induce renal cell injury.
Multiple molecular mechanisms have been proposed to mediate the dysregulation of STAT3 signaling by SARS-CoV-2, such as the imbalance between STAT1 and STAT3 activation and the crosstalk between plasminogen activator-inhibitor 1 and STAT3 signaling.21 We showed in HK-2 cells, both SARS-CoV-2 infection and expression of SARS-CoV-2 ORF3A alone could upregulate TRIM59, which competitively block the interaction between STAT3 and the phosphatase TCPTP and reduce the dephosphorylation of STAT3, leading to persistent activation of STAT3. This represents a previously unrecognized mechanism of the regulation of STAT3 signaling by a SARS-CoV-2 viral protein downstream to the JAK-mediated STAT3 phosphorylation. Furthermore, since ORF3A could simultaneously activate NF-kB and STAT3 signaling, our data suggest that direct infection by SARS-CoV-2 may create a positive feedback loop to enhance the cellular response to cytokines such as IL-6 in kidney cells and aggravate the injury. Such simultaneous activation of NF-kB and STAT3 has been observed in clinical samples from patients with other inflammatory diseases as an evidence of IL-6 signaling amplifier,44,45 so it is worthwhile to explore whether this mechanism is applicable to other cell types besides kidney cells, and further investigation is needed to test it in other cell types susceptible to SARS-CoV-2 infection, such as pulmonary alveolar cells and endothelial cells.
We discovered that ORF3A of SARS-CoV-2 could interact with TRIM59 and STAT3, but it is unclear whether these interactions are involved in the regulation of STAT3 activation. Previously ORF3A was found to be localized to the lysosome-endosome membranes and inhibiting autophagy in cultured cells.37,39 We found that ORF3A is also present in the nuclei of HK-2 cells, which is consistent with its interaction with STAT3 that translocates to the nuclei when it is phosphorylated. Whether nuclear ORF3A also affects activity of other transcriptional factors requires further studies.
To corroborate our in vitro findings, we also examined the expression TRIM59 in the kidney autopsies of COVID-19 patients with AKI and found that its expression was significantly increased in the renal tubular cells. However, it is still unclear how TRIM59 is regulated in the COVID-19-associated nephropathy. As ORF3A has been shown to inhibit autophagosome/lysosome function, it is possible that TRIM59 degradation is affected by ORF3A, which deserves further investigation.
To explore the potential role of ORF3A in COVID-19-associated AKI, we established a murine model with transient ORF3A expression in kidney and showed that ORF3A exacerbated kidney injury induced by ischemia/reperfusion, and inhibition of STAT3 activities could ameliorate this effect. These results demonstrate the cytotoxicity of ORF3A in kidney cells in a mammalian animal model and suggest that STAT3 is a critical mediator of renal injury in COVID-19-associated kidney injury. Recently, the N protein of SARS-CoV-2 has been reported to cause renal tubular necrosis in mice by dysregulating the TGF-β-Smad3 pathway.14 Thus, multiple pathogenic pathways may be involved in the COVID-19-related renal injury. An appropriate animal model for kidney injury induced by SARS-CoV-2 infection is needed to further confirm these findings.
Zebrafish is a small vertebrate model organism that has been applied to AKI research.25,26,46,47,48,49 Because of the ease of genetic manipulation in zebrafish and its organ similarity to human, we used transgenic zebrafish to perform the functional screening of several viral proteins of SARS-CoV-2 in relation to kidney injury and identified ORF3A as a cytotoxic gene for tubular epithelial cells. Although we did not identify the N gene of SARS-CoV-2 in our screening, presumably because zebrafish kidney is minimally susceptible to fibrosis and may have a different response to TGF-β signaling, the zebrafish may still be useful to test other viral proteins of SARS-CoV-2 to identify a virulent gene affecting different organs or cell types. In addition, the zebrafish models could be used to screen drugs for treatment of COVID-19-induced kidney injury.
In summary, SARS-CoV-2 ORF3A protein can augment both NF-kB and STAT3 signaling and exacerbate the injury of renal tubular cells in the presence of cytokines induced by SARS-CoV-2 infection. In addition, ORF3A modulates the STAT3 signaling via TRIM59-mediated inhibition of dephosphorylation. Therefore, targeting the aberrant STAT3 activation may serve as a potential therapy for AKI in COVID-19 patients.
Materials and methods
Cell culture, transfections, stimulations, and nuclear-cytoplasmic fractionation
HK-2 cells were grown at 37°C and 5% CO2 in supplemented RPMI 1640 medium. pCMV-ORF3A, TRIM59 KD, and STAT3 DN were transfected into HK-2 cells with ViaFect Transfection Reagent (Promega, USA). After transfecting 24 h, we changed the media without serum for 24 hours; then we treated with TNF-α (10 ng/mL) or IL-6 (10 ng/mL) for 15, 30, and 60 min, respectively. Cells were lysed for protein analysis. For the immunoprecipitation experiment, HK-2 cells were transduced with lentiviruses expressing ORF3A-Strep, TRIM59, or GFP-Strep respectively for 24 h, and total protein was harvested 48 h later. Nuclear-cytoplasmic fractionation was conducted using the NE-PER Nuclear and Cytoplasmic Extraction Reagents kit (Thermo Fisher Scientific, cat. no. 78835) according to the manufacture’s protocol. For the nuclear-cytoplasmic fractionation experiments, HK-2 cells were transduced with lentiviruses for ORF3A-Strep or GFP (control) expression.
Zebrafish maintenance
Wild-type (AB strain) zebrafish, transgenic zebrafish, and larvae were maintained in the zebrafish facility according to the Institutional Animal Care and Use Committee (IACUC) standards. All procedures were approved by IACUC at the Icahn School of Medicine at Mount Sinai (IACUC-2019-0081). Eggs were collected in Petri dishes and raised in an incubator with constant temperature at 28.5oC.
Transgenic zebrafish
The pDONR207-SARS-CoV-2 ORF3A were obtained from Addgene (ID: 141271).50 The Tol2-Gateway system was used to generate the transgenic construct Tg(UAS:ORF3A,myl7:Venus) following the published multisite gateway cloning method.51 The transgenic construct was co-injected with Tol2 transposase RNA into one-cell-stage embryos for genomic integration. The stable transgenic lines were screened by presence of Venus (yellow fluorescent protein) expression in the myocardium. Tg(UAS:Eco.NfsB-mCherry) (also termed Tg(UAS:NTR-mCherry)) transgenic fish were obtained from Zebrafish International Resource Center (ZFIN ID: ZDB-TGCONSTRCT-110215-5). Tg(enpep:Gal4) was previously reported.24 Triple transgenic zebrafish Tg(enpep:Gal4;UAS:NTR-mCherry; UAS:ORF3A,myl7:Venus) were obtained by multiple rounds of crossing the transgenic zebrafish. Imaging of transgenic zebrafish was captured under an Olympus SZX16 stereoscope.
Quantitative real-time PCR
Total RNA was extracted from zebrafish embryos (72 hpf) or HK-2 cells using Direct-zol RNA MiniPrep (Zymo Research). Radiant cDNA Synthesis kit (Radiant Molecular tools) was used for reverse transcription of 1 μg of total RNA. Quantitative real-time PCR was performed using SYBR Green Master Mix (Applied Biosystems) and the 7500 Real-Time PCR System (Applied Biosystems). Intron-spanning primer sets selectively targeting mRNA and not genomic DNA sequence were designed using Primer-BLAST (NCBI). Transcript levels of genes of interest were normalized to the housekeeping gene EF1a or 18S ribosomal RNA, and folds of change in transcript levels relative to the control group were calculated using the 2−ΔΔCT method. The primers for quantitative real-time PCR are listed in Table S1.
Western blot
Cells were lysed with M-PER mammalian protein extraction reagent containing protease and phosphatase inhibitor cocktail. The antibody against KIM-1 was from R&D (AF1817). Antibodies for phosphor-STAT3(Ser727) (9134), phosphor-STAT3(Tyr705) (9145), phosphor-p65(Ser536) (3033), phosphor- p65(Ser276) (3037), and cleaved-caspase3 (9664) were from Cell Signaling Technology. TRIM59 (ab69639), STAT3 (ab68153), and p65(ab52175) were from Abcam. Blots were developed with the enhanced chemiluminescence system and imaged with Odyssey FC Imager (LI-COR Biosciences, NE).
Whole-mount in situ hybridization
The complete coding sequence of zebrafish kim-1 was PCR amplified and cloned into pCR-blunt, and the riboprobe was synthesized using DIG RNA Labeling Kit (Roche). Zebrafish embryos were collected and fixed in 4% paraformaldehyde in PBS. In situ hybridization was carried out following a standard protocol.52 Following coloration, zebrafish embryos were cleared with 50% glycerol/50% PBS and deyolked and imaged under an Olympus SZX16 stereoscope.
Mouse models
B57BL/6 mice were generated by the Model Animal Center at Renji Hostal, School of Medicine, Shanghai JiaoTong University. The mice were housed in pathogen-free conditions. All animal experiments were performed according to the Animal Protocol Committee of Shanghai JiaoTong University and were approved by the animal care committee of Renji Hospital, School of Medicine, Shanghai JiaoTong University (Protocol: m20170324).
The I/R-AKI mice were constructed as described in previous studies.53,54 Six adult male mice were included in each group. The animal was anesthetized with 50–60 mg/kg of pentobarbital sodium by intraperitoneal injection. The skin in the surgical area was then wiped clean with 70% alcohol swab. Then the mice were placed on the homeothermic blanket of a homeothermic monitor system and covered by sterile gauze. The body temperature was monitored through a rectal probe and controlled at 36.5°C. The skin and muscle on the left side were cut open along the back to expose the left kidney. A micro-aneurysm was used to clamp the pedicle to block the blood flow to the kidney for 30 min to induce renal ischemia. Complete ischemia was indicated by color change of the kidney from red to dark purple in a few seconds. After the ischemia, the micro-aneurysm clips were released. Then the kidney would start reperfusion, which is indicated by the change of kidney color to red. At the same time, we removed the right kidney. 0.5 mL warm sterile saline was given intraperitoneally to each mouse. The mice were kept on a heating pad until they gained full consciousness before being returned to their housing cage.
A lentivirus expressing ORF-3A was prepared and directly injected into the left kidney of mice according to previously described methods.55,56,57 Briefly, we anesthetized the mice and occluded the left renal pedicle. Then, we inserted a 31G needle on a syringe from the lower pole to the left kidney to the upper pole along the long axis of the kidney. The needle was slowly withdrawn, while 100 μL lentivirus (∼1 x 105 IU/μL) expressing GFP (control) or ORF3A was continuously released. The occlusion of the renal pedicle was reversed after the injection.
For the S3I-201 treatment, mice were randomized to receive either S3I-201 10 mg/kg (Selleck) or DMSO intraperitoneally three times per week for 3 weeks.
Immunoprecipitation
For immunoprecipitation of cell lysates of cells expressing Strep-tagged ORF-3A, GFP was incubated with magstrep “type3” XT Beads (iba, cat. no. 2-4090-002) overnight at 4°C. After washing, immunoprecipitated protein was eluted in sample buffer according to the manufacture’s protocol and was immunoblotted using p-STAT3(Y705), STAT3, TRIM59, and Strep tag antibodies as indicated.
Lentiviral preparation and infection
HEK293T cells were transfected with either lentiviral plasmids expressing ORF3A-Strep, Trim59 shRNA, or GFP-Strep, plus psPAX2 packaging plasmid and pMD2.G envelop plasmid using Polyjet transfection reagent according to the manufacturer’s protocol (SignaGen Laboratories). The lentiviral particles were harvested by centrifugation from the HEK293T cell culture medium collected 48–96 h post transfection. Concentrated lentiviral particles were used to infect HK-2 cells.
Immunohistochemistry
Immunohistochemistry staining was performed on formalin-fixed, paraffin-embedded kidney sections. Sections were deparaffinized and heated in citrate for antigen retrieval. Then, sections were added with H2O2 to ablate endogenous peroxidase and blocked with diluted horse serum for 1 h at room temperature, followed by incubation with primary antibody overnight at 4°C. The next day, sections were washed three times with phosphate-buffered saline and then incubated with secondary antibody for 60 min. For quantification, 10 or more nonoverlapping fields were randomly selected and imaged per mouse. Positive area was quantified using the ImageJ software (National Institutes of Health) and shown as an average percentage of the positive area relative to the total area.
Cells immunofluorescence
Co-immunostaining of ORF3A was performed on HK-2 cells using anti-Strep and anti-STAT3 following the standard protocol. After washing, sections were incubated with fluorophore-conjugated secondary antibodies and mounted with aqueous mounting media containing DAPI. The samples were imaged with a Leica SP5 confocal microscope.
Immunohistochemistry for human autopsies
Autopsies were performed at the Mount Sinai Hospital in a negative pressure room, utilizing appropriate personal protective equipment and techniques as recommended by the Centers for Disease Control and Prevention. Entire kidneys were procured as part of a full autopsy. Immunofluorescence studies were performed on 4-μm-thick formalin-fixed paraffin-embedded tissue slides following antigen retrieval in boiling citrate buffer (pH 6) (Vector labs), blocking for 1 h at room temperature (10% normal donkey serum/0.5% triton-X), and primary antibody incubation overnight at 4°C with anti-TRIM59 antibody (Abcam ab69639, 1:250). On the next day, sections were incubated with secondary antibody for 1 h after being washed three times with PBS. Vectastain Elite ABC kit (Vector Laboratories, PK-6100) was used for detection of the antibody. Normal tissues from nephrectomy were used as negative controls.
Statistical analysis
Data were shown as mean ± SD. For comparisons of means between two groups, unpaired two-tailed t tests were performed. Two-way ANOVA with Tukey’s post hoc multiple comparison test was applied for analyses between three or more groups. Statistical significance was achieved when p < 0.05. GraphPad Prism (v.9) was used for statistical analysis.
Acknowledgments
The authors appreciate Dr. Zhengzhe Li for technical assistance and discussion. We also thank Dr. Randy Albrecht for support with the BSL3 facility and procedures at the ISMMS as well as Richard Cadagan and Daniel Flores for excellent technical assistance. W.Z. is supported by the startup fund from the Department of Medicine, Icahn School of Medicine at Mount Sinai. This work was supported by the following NIH grants: R01DK078897, R01DK088541, and P01-DK-56492 to J.C.H.; R01DK117913 to K.L.; R01DK121978 and R01DK129735 to L.K.; R01DK131047 to G.L.G. This work was also partly supported by CRIPT (Center for Research on Influenza Pathogenesis and Transmission), an NIAID-funded Center of Excellence for Influenza Research and Response (CEIRR, contract # 75N93021C00014), NIAID grant U19AI135972, NCI SeroNet grant U54CA260560, DARPA grant HR0011-19-2-002, and by anonymous donors to A.G.-S.
Author contributions
C.J.H. and W.Z. conceived and supervised the study. H.C., Y.C., Y.F., M.A., and W.Z. performed the studies. H.C., C.J.H., and W.Z. analyzed data. L.K., K.L., L.M., A.G.-S., G.L.G., L.G., Z.H., and S.M. shared research materials/resources and assisted the study. H.C., C.J.H., and W.Z. wrote the manuscript. All the authors reviewed and edited the manuscript.
Declaration of interests
The A.G.-S. laboratory has received research support from Pfizer, Senhwa Biosciences, Kenall Manufacturing, Avimex, Johnson & Johnson, Dynavax, 7Hills Pharma, Pharmamar, ImmunityBio, Accurius, Nanocomposix, Hexamer, N-fold LLC, Model Medicines, Atea Pharma, Applied Biological Laboratories, and Merck, outside of the reported work. A.G.-S. has consulting agreements for the following companies involving cash and/or stock: Vivaldi Biosciences, Contrafect, 7Hills Pharma, Avimex, Vaxalto, Pagoda, Accurius, Esperovax, Farmak, Applied Biological Laboratories, Pharmamar, Paratus, CureLab Oncology, CureLab Veterinary, Synairgen, and Pfizer, outside of the reported work. A.G.-S. has been an invited speaker in meeting events organized by Seqirus, Janssen, and Astrazeneca. A.G.-S. is inventor on patents and patent applications on the use of antivirals and vaccines for the treatment and prevention of virus infections and cancer, owned by the Icahn School of Medicine at Mount Sinai, New York, outside of the reported work.
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.ymthe.2022.12.008.
Contributor Information
Shan Mou, Email: shan_mou@shsmu.edu.cn.
John Cijiang He, Email: cijiang.he@mssm.edu.
Weibin Zhou, Email: weibin.zhou@mssm.edu.
Supplemental information
Data availability
All the research materials and data reported in this article are available upon request to the authors.
References
- 1.Chen N., Zhou M., Dong X., Qu J., Gong F., Han Y., Qiu Y., Wang J., Liu Y., Wei Y., et al. Epidemiological and clinical characteristics of 99 cases of 2019 novel coronavirus pneumonia in Wuhan, China: a descriptive study. Lancet. 2020;395:507–513. doi: 10.1016/S0140-6736(20)30211-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Chen A., Yin L., Lee K., He J.C. Similarities and differences between COVID-19-associated nephropathy and HIV-associated nephropathy. Kidney Dis. 2022;8:1–12. doi: 10.1159/000520235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Qian J.Y., Wang B., Lv L.L., Liu B.C. Pathogenesis of acute kidney injury in coronavirus disease 2019. Front. Physiol. 2021;12 doi: 10.3389/fphys.2021.586589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Diao B., Wang C., Wang R., Feng Z., Zhang J., Yang H., Tan Y., Wang H., Wang C., Liu L., et al. Human kidney is a target for novel severe acute respiratory syndrome coronavirus 2 infection. Nat. Commun. 2021;12:2506. doi: 10.1038/s41467-021-22781-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ondruschka B., Heinrich F., Lindenmeyer M., Edler C., Möbius D., Czogalla J., Heinemann A., Braun F., Aepfelbacher M., Lütgehetmann M., Huber T.B. Multiorgan tropism of SARS-CoV-2 lineage B.1.1.7. Int. J. Leg. Med. 2021;135:2347–2349. doi: 10.1007/s00414-021-02691-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Jansen J., Reimer K.C., Nagai J.S., Varghese F.S., Overheul G.J., de Beer M., Roverts R., Daviran D., Fermin L.A.S., Willemsen B., et al. SARS-CoV-2 infects the human kidney and drives fibrosis in kidney organoids. Cell Stem Cell. 2022;29:217–231.e8. doi: 10.1016/j.stem.2021.12.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Garreta E., Prado P., Stanifer M.L., Monteil V., Marco A., Ullate-Agote A., Moya-Rull D., Vilas-Zornoza A., Tarantino C., Romero J.P., et al. A diabetic milieu increases ACE2 expression and cellular susceptibility to SARS-CoV-2 infections in human kidney organoids and patient cells. Cell Metab. 2022;34:857–873.e9. doi: 10.1016/j.cmet.2022.04.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kim D., Lee J.Y., Yang J.S., Kim J.W., Kim V.N., Chang H. The architecture of SARS-CoV-2 transcriptome. Cell. 2020;181:914–921.e10. doi: 10.1016/j.cell.2020.04.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Su C.M., Wang L., Yoo D. Activation of NF-kappaB and induction of proinflammatory cytokine expressions mediated by ORF7a protein of SARS-CoV-2. Sci. Rep. 2021;11 doi: 10.1038/s41598-021-92941-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.van den Berg D.F., Te Velde A.A. Severe COVID-19: NLRP3 inflammasome dysregulated. Front. Immunol. 2020;11:1580. doi: 10.3389/fimmu.2020.01580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Freeman T.L., Swartz T.H. Targeting the NLRP3 inflammasome in severe COVID-19. Front. Immunol. 2020;11:1518. doi: 10.3389/fimmu.2020.01518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chen D.Y., Khan N., Close B.J., Goel R.K., Blum B., Tavares A.H., Kenney D., Conway H.L., Ewoldt J.K., Chitalia V.C., et al. SARS-CoV-2 disrupts proximal elements in the JAK-STAT pathway. J. Virol. 2021;95 doi: 10.1128/JVI.00862-21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Miorin L., Kehrer T., Sanchez-Aparicio M.T., Zhang K., Cohen P., Patel R.S., Cupic A., Makio T., Mei M., Moreno E., et al. SARS-CoV-2 Orf6 hijacks Nup98 to block STAT nuclear import and antagonize interferon signaling. Proc. Natl. Acad. Sci. USA. 2020;117:28344–28354. doi: 10.1073/pnas.2016650117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wang W., Chen J., Hu D., Pan P., Liang L., Wu W., Tang Y., Huang X.R., Yu X., Wu J., Lan H.Y. SARS-CoV-2 N protein induces acute kidney injury via smad3-dependent G1 cell cycle arrest mechanism. Adv. Sci. 2022;9 doi: 10.1002/advs.202103248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Aydemir M.N., Aydemir H.B., Korkmaz E.M., Budak M., Cekin N., Pinarbasi E. Computationally predicted SARS-COV-2 encoded microRNAs target NFKB, JAK/STAT and TGFB signaling pathways. Gene Rep. 2021;22 doi: 10.1016/j.genrep.2020.101012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wang R., Yang X., Chang M., Xue Z., Wang W., Bai L., Zhao S., Liu E. ORF3a protein of severe acute respiratory syndrome coronavirus 2 inhibits interferon-activated Janus kinase/signal transducer and activator of transcription signaling via elevating suppressor of cytokine signaling 1. Front. Microbiol. 2021;12 doi: 10.3389/fmicb.2021.752597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Chuang P.Y., He J.C. JAK/STAT signaling in renal diseases. Kidney Int. 2010;78:231–234. doi: 10.1038/ki.2010.158. [DOI] [PubMed] [Google Scholar]
- 18.Pace J., Paladugu P., Das B., He J.C., Mallipattu S.K. Targeting STAT3 signaling in kidney disease. Am. J. Physiol. Ren. Physiol. 2019;316:F1151–F1161. doi: 10.1152/ajprenal.00034.2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Jafarzadeh A., Nemati M., Jafarzadeh S. Contribution of STAT3 to the pathogenesis of COVID-19. Microb. Pathog. 2021;154 doi: 10.1016/j.micpath.2021.104836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Del Valle D.M., Kim-Schulze S., Huang H.H., Beckmann N.D., Nirenberg S., Wang B., Lavin Y., Swartz T.H., Madduri D., Stock A., et al. An inflammatory cytokine signature predicts COVID-19 severity and survival. Nat. Med. 2020;26:1636–1643. doi: 10.1038/s41591-020-1051-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Matsuyama T., Kubli S.P., Yoshinaga S.K., Pfeffer K., Mak T.W. An aberrant STAT pathway is central to COVID-19. Cell Death Differ. 2020;27:3209–3225. doi: 10.1038/s41418-020-00633-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Meliambro K., Li X., Salem F., Yi Z., Sun Z., Chan L., Chung M., Chancay J., Vy H.M.T., Nadkarni G., et al. Molecular analysis of the kidney from a patient with COVID-19-associated collapsing glomerulopathy. Kidney Med. 2021;3:653–658. doi: 10.1016/j.xkme.2021.02.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Salem F., Li X.Z., Hindi J., Casablanca N.M., Zhong F., El Jamal S.M., Haroon Al Rasheed M.R., Li L., Lee K., Chan L., He J.C. Activation of STAT3 signaling pathway in the kidney of COVID-19 patients. J. Nephrol. 2022;35:735–743. doi: 10.1007/s40620-021-01173-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zhou W., Boucher R.C., Bollig F., Englert C., Hildebrandt F. Characterization of mesonephric development and regeneration using transgenic zebrafish. Am. J. Physiol. Ren. Physiol. 2010;299:F1040–F1047. doi: 10.1152/ajprenal.00394.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wen X., Cui L., Morrisroe S., Maberry D., Jr., Emlet D., Watkins S., Hukriede N.A., Kellum J.A. A zebrafish model of infection-associated acute kidney injury. Am. J. Physiol. Ren. Physiol. 2018;315:F291–F299. doi: 10.1152/ajprenal.00328.2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.McKee R.A., Wingert R.A. Zebrafish renal pathology: emerging models of acute kidney injury. Curr. Pathobiol. Rep. 2015;3:171–181. doi: 10.1007/s40139-015-0082-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Cianciolo Cosentino C., Skrypnyk N.I., Brilli L.L., Chiba T., Novitskaya T., Woods C., West J., Korotchenko V.N., McDermott L., Day B.W., et al. Histone deacetylase inhibitor enhances recovery after AKI. J. Am. Soc. Nephrol. 2013;24:943–953. doi: 10.1681/ASN.2012111055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Yeung M.L., Teng J.L.L., Jia L., Zhang C., Huang C., Cai J.P., Zhou R., Chan K.H., Zhao H., Zhu L., et al. Soluble ACE2-mediated cell entry of SARS-CoV-2 via interaction with proteins related to the renin-angiotensin system. Cell. 2021;184:2212–2228.e12. doi: 10.1016/j.cell.2021.02.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ajay A.K., Kim T.M., Ramirez-Gonzalez V., Park P.J., Frank D.A., Vaidya V.S. A bioinformatics approach identifies signal transducer and activator of transcription-3 and checkpoint kinase 1 as upstream regulators of kidney injury molecule-1 after kidney injury. J. Am. Soc. Nephrol. 2014;25:105–118. doi: 10.1681/ASN.2013020161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Gordon D.E., Jang G.M., Bouhaddou M., Xu J., Obernier K., White K.M., O'Meara M.J., Rezelj V.V., Guo J.Z., Swaney D.L., et al. A SARS-CoV-2 protein interaction map reveals targets for drug repurposing. Nature. 2020;583:459–468. doi: 10.1038/s41586-020-2286-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sang Y., Li Y., Zhang Y., Alvarez A.A., Yu B., Zhang W., Hu B., Cheng S.Y., Feng H. CDK5-dependent phosphorylation and nuclear translocation of TRIM59 promotes macroH2A1 ubiquitination and tumorigenicity. Nat. Commun. 2019;10:4013. doi: 10.1038/s41467-019-12001-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Sang Y., Li Y., Song L., Alvarez A.A., Zhang W., Lv D., Tang J., Liu F., Chang Z., Hatakeyama S., et al. TRIM59 promotes gliomagenesis by inhibiting TC45 dephosphorylation of STAT3. Cancer Res. 2018;78:1792–1804. doi: 10.1158/0008-5472.CAN-17-2774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Braun F., Lütgehetmann M., Pfefferle S., Wong M.N., Carsten A., Lindenmeyer M.T., Nörz D., Heinrich F., Meißner K., Wichmann D., et al. SARS-CoV-2 renal tropism associates with acute kidney injury. Lancet. 2020;396:597–598. doi: 10.1016/S0140-6736(20)31759-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Puelles V.G., Lütgehetmann M., Lindenmeyer M.T., Sperhake J.P., Wong M.N., Allweiss L., Chilla S., Heinemann A., Wanner N., Liu S., et al. Multiorgan and renal tropism of SARS-CoV-2. N. Engl. J. Med. 2020;383:590–592. doi: 10.1056/NEJMc2011400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kern D.M., Sorum B., Mali S.S., Hoel C.M., Sridharan S., Remis J.P., Toso D.B., Kotecha A., Bautista D.M., Brohawn S.G. Cryo-EM structure of SARS-CoV-2 ORF3a in lipid nanodiscs. Nat. Struct. Mol. Biol. 2021;28:573–582. doi: 10.1038/s41594-021-00619-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Chen D., Zheng Q., Sun L., Ji M., Li Y., Deng H., Zhang H. ORF3a of SARS-CoV-2 promotes lysosomal exocytosis-mediated viral egress. Dev. Cell. 2021;56:3250–3263.e5. doi: 10.1016/j.devcel.2021.10.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Zhang Y., Sun H., Pei R., Mao B., Zhao Z., Li H., Lin Y., Lu K. The SARS-CoV-2 protein ORF3a inhibits fusion of autophagosomes with lysosomes. Cell Discov. 2021;7:31. doi: 10.1038/s41421-021-00268-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Qu Y., Wang X., Zhu Y., Wang W., Wang Y., Hu G., Liu C., Li J., Ren S., Xiao M.Z.X., et al. ORF3a-Mediated incomplete autophagy facilitates severe acute respiratory syndrome coronavirus-2 replication. Front. Cell Dev. Biol. 2021;9 doi: 10.3389/fcell.2021.716208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Miao G., Zhao H., Li Y., Ji M., Chen Y., Shi Y., Bi Y., Wang P., Zhang H. ORF3a of the COVID-19 virus SARS-CoV-2 blocks HOPS complex-mediated assembly of the SNARE complex required for autolysosome formation. Dev. Cell. 2021;56:427–442.e5. doi: 10.1016/j.devcel.2020.12.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ren Y., Shu T., Wu D., Mu J., Wang C., Huang M., Han Y., Zhang X.Y., Zhou W., Qiu Y., Zhou X. The ORF3a protein of SARS-CoV-2 induces apoptosis in cells. Cell. Mol. Immunol. 2020;17:881–883. doi: 10.1038/s41423-020-0485-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Yang S., Tian M., Johnson A.N. SARS-CoV-2 protein ORF3a is pathogenic in Drosophila and causes phenotypes associated with COVID-19 post-viral syndrome. bioRxiv. 2020 doi: 10.1101/2020.12.20.423533. Preprint at. [DOI] [Google Scholar]
- 42.Xu H., Chitre S.A., Akinyemi I.A., Loeb J.C., Lednicky J.A., McIntosh M.T., Bhaduri-McIntosh S. SARS-CoV-2 viroporin triggers the NLRP3 inflammatory pathway. bioRxiv. 2020 doi: 10.1101/2020.10.27.357731. Preprint at. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Sayah W., Berkane I., Guermache I., Sabri M., Lakhal F.Z., Yasmine Rahali S., Djidjeli A., Lamara Mahammed L., Merah F., Belaid B., et al. Interleukin-6, procalcitonin and neutrophil-to-lymphocyte ratio: potential immune-inflammatory parameters to identify severe and fatal forms of COVID-19. Cytokine. 2021;141 doi: 10.1016/j.cyto.2021.155428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Fujita M., Yamamoto Y., Jiang J.J., Atsumi T., Tanaka Y., Ohki T., Murao N., Funayama E., Hayashi T., Osawa M., et al. NEDD4 is involved in inflammation development during keloid formation. J. Invest. Dermatol. 2019;139:333–341. doi: 10.1016/j.jid.2018.07.044. [DOI] [PubMed] [Google Scholar]
- 45.Murakami M., Harada M., Kamimura D., Ogura H., Okuyama Y., Kumai N., Okuyama A., Singh R., Jiang J.J., Atsumi T., et al. Disease-association analysis of an inflammation-related feedback loop. Cell Rep. 2013;3:946–959. doi: 10.1016/j.celrep.2013.01.028. [DOI] [PubMed] [Google Scholar]
- 46.Morales Fénero C., Padovani B.N., do Amaral M.A., de Barros G.J.B., de Oliveira I.K.X., Hiyane M.I., Camâra N.O.S. Acute kidney injury model induced by cisplatin in adult zebrafish. JoVE. 2021;171 doi: 10.3791/61575. [DOI] [PubMed] [Google Scholar]
- 47.Wang Z., Gu Z., Hou Q., Chen W., Mu D., Zhang Y., Liu Q., Liu Z., Yang D. Zebrafish GSDMEb cleavage-gated pyroptosis drives septic acute kidney injury in vivo. J. Immunol. 2020;204:1929–1942. doi: 10.4049/jimmunol.1901456. [DOI] [PubMed] [Google Scholar]
- 48.Brilli Skvarca L., Han H.I., Espiritu E.B., Missinato M.A., Rochon E.R., McDaniels M.D., Bais A.S., Roman B.L., Waxman J.S., Watkins S.C., et al. Enhancing regeneration after acute kidney injury by promoting cellular dedifferentiation in zebrafish. Dis. Model. Mech. 2019;12:dmm037390. doi: 10.1242/dmm.037390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Outtandy P., Russell C., Kleta R., Bockenhauer D. Zebrafish as a model for kidney function and disease. Pediatr. Nephrol. 2019;34:751–762. doi: 10.1007/s00467-018-3921-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Kim D.K., Knapp J.J., Kuang D., Chawla A., Cassonnet P., Lee H., Sheykhkarimli D., Samavarchi-Tehrani P., Abdouni H., Rayhan A., et al. A comprehensive, flexible collection of SARS-CoV-2 coding regions. G3 (Bethesda) 2020;10:3399–3402. doi: 10.1534/g3.120.401554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Kwan K.M., Fujimoto E., Grabher C., Mangum B.D., Hardy M.E., Campbell D.S., Parant J.M., Yost H.J., Kanki J.P., Chien C.B. The Tol2kit: a multisite gateway-based construction kit for Tol2 transposon transgenesis constructs. Dev. Dyn. 2007;236:3088–3099. doi: 10.1002/dvdy.21343. [DOI] [PubMed] [Google Scholar]
- 52.Thisse C., Thisse B. High-resolution in situ hybridization to whole-mount zebrafish embryos. Nat. Protoc. 2008;3:59–69. doi: 10.1038/nprot.2007.514. [DOI] [PubMed] [Google Scholar]
- 53.Fu Y., Tang C., Cai J., Chen G., Zhang D., Dong Z. Rodent models of AKI-CKD transition. Am. J. Physiol. Ren. Physiol. 2018;315:F1098–F1106. doi: 10.1152/ajprenal.00199.2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Wei Q., Dong Z. Mouse model of ischemic acute kidney injury: technical notes and tricks. Am. J. Physiol. Ren. Physiol. 2012;303:F1487–F1494. doi: 10.1152/ajprenal.00352.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Nakamura A., Imaizumi A., Yanagawa Y., Kohsaka T., Johns E.J. beta(2)-Adrenoceptor activation attenuates endotoxin-induced acute renal failure. J. Am. Soc. Nephrol. 2004;15:316–325. doi: 10.1097/01.asn.0000111247.76908.59. [DOI] [PubMed] [Google Scholar]
- 56.Wang X., Liu J., Zhen J., Zhang C., Wan Q., Liu G., Wei X., Zhang Y., Wang Z., Han H., et al. Histone deacetylase 4 selectively contributes to podocyte injury in diabetic nephropathy. Kidney Int. 2014;86:712–725. doi: 10.1038/ki.2014.111. [DOI] [PubMed] [Google Scholar]
- 57.Wu J., Zheng C., Wang X., Yun S., Zhao Y., Liu L., Lu Y., Ye Y., Zhu X., Zhang C., et al. MicroRNA-30 family members regulate calcium/calcineurin signaling in podocytes. J. Clin. Invest. 2015;125:4091–4106. doi: 10.1172/JCI81061. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All the research materials and data reported in this article are available upon request to the authors.