

Abstract
A 58-year-old man presented with painful edema of the extremities, and a diagnosis of eosinophilic fasciitis (EF) was confirmed. He also met the criteria for hypereosinophilic syndrome (HES), but there were no findings suggestive of malignancies or hematologic neoplasms despite a close examination. He was started on steroid therapy but subsequently developed severe liver dysfunction, hemophagocytic lymphohistiocytosis, hepatosplenomegaly, and renal involvement. The diagnosis of peripheral T-cell lymphoma, not otherwise specified was finally established by a bone marrow reexamination and liver biopsy. In cases of eosinophilia, EF, and/or HES, it is important to suspect an intrinsic abnormality, including potential T-cell lymphoma.
Keywords: eosinophilic fasciitis, hypereosinophilic syndrome, eosinophilia, hematologic neoplasms, T-cell lymphoma
Introduction
Eosinophilia may be caused by a variety of factors, including allergies, infections, autoimmune disorders, and malignancies. Most of them are reactive changes to the underlying disease, and eosinophils themselves do not often cause organ damage. However, eosinophilia or eosinophilic disorders can sometimes present as an isolated sign of a serious acute illness and require a difficult differential diagnosis.
We herein report a rare but instructive case of eosinophilic fasciitis (EF) with hypereosinophilia as the initial clinical manifestation of occult T-cell lymphoma that was difficult to identify.
Case Report
A 58-year-old Japanese man complained of painful edema of his extremities that had appeared one month prior and was referred to our department for marked eosinophilia. He had no pre-existing medical conditions, including asthma or other allergic diseases, no abnormalities at his last annual checkup, and no family history of note. He was not taking any medications and had had no episodes of suspected exposure to drugs or allergens. He had a fever, and a physical examination revealed marked angioedema of the extremities with 10% weight gain, but findings were otherwise unremarkable.
His laboratory findings were as follows: white blood cells 32.8×109/L (76% eosinophils, and no pathological cells); hemoglobin 12.9 g/dL; platelets 475×109/L; lactate dehydrogenase (LDH) 580 IU/L (normal, <222); alanine aminotransferase (ALT) 15 IU/L (normal, <42); aspartate aminotransferase (AST) 19 IU/L (normal, <30); C-reactive protein 0.76 mg/dL; iron 17 μg/dL (normal, 40-188); ferritin 219 ng/mL (normal, 50-200); immunoglobulin (Ig) G 1,222 mg/dL; IgA 131 mg/dL; IgM 38 mg/dL; total IgE 13.9 IU/mL (normal, <173); C3 126 mg/dL (normal, 73-138); C4 28 mg/dL (normal, 11-31); and total functional hemolytic complement (CH50) 59.8 U/mL (normal, 30-50). Anti-nuclear and anti-neutrophil cytoplasmic antibodies were not detected. His thyroid function was normal, and no parasite eggs were detected in his feces.
Computed tomography findings were unremarkable. A bone marrow specimen showed a marked increase in eosinophils, but no proliferation of blastoid cells, and no abnormal cell populations were identified by immunohistochemistry or flow cytometric analyses. Myeloid/lymphoid neoplasms with eosinophilia were suspected, but a chromosomal analysis indicated a normal karyotype, with no 4q12 deletion, PDGFRB or FGFR1 rearrangement, or BCR/ABL1 fusion genes detected by fluorescent in situ hybridization, and JAK2 gene mutations were not found. A skin biopsy showed eosinophil infiltrates into the deep dermis and subcutaneous tissue with edema but no inflammatory cell infiltrates into vessel walls. A subsequent fascial biopsy revealed inflammatory infiltrates of lymphocytes, eosinophils, and histiocytes into the fascia (Fig. 1A). Magnetic resonance imaging (MRI) showed an increase in the T2 signal intensity within the fascia, accompanied by thickening and enhancement of the fascia (Fig. 1B). Based on these findings, a diagnosis of EF was made.
Figure 1.
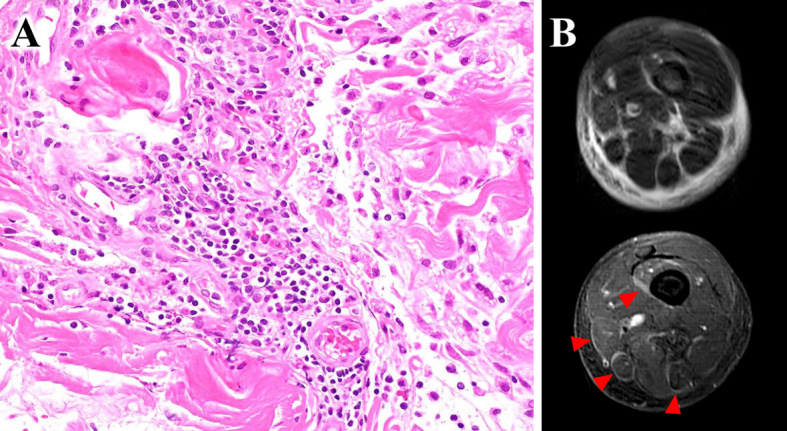
(A) Diffuse inflammatory infiltrates in the fascia, composed mainly of lymphocytes with some eosinophils and histiocytes (Hematoxylin and Eosin staining, ×400 magnification). (B) Thigh muscle magnetic resonance imaging (MRI): Axial fat-suppressed, T2-weighted MRI showing an increased signal intensity within the superficial and deep fascia and enhanced T1-weighted MRI showing fascial enhancement corresponding to locations of T2 signal abnormalities.
A daily dose of 60 mg of prednisone improved his fever and edema, and the eosinophils in his peripheral blood disappeared. However, when the prednisone was reduced to 50 mg on the 8th day, the fever re-emerged, followed by severe thrombocytopenia and hepatic dysfunction (ALT 186 IU/L, AST 229 IU/L) with markedly elevated LDH at 1,424 IU/L, ferritin at 20,192 ng/mL, and soluble interleukin-2 receptor at 8,512 U/mL (normal, <474), suggesting hemophagocytic lymphohistiocytosis associated with lymphoid malignancy. Computed tomography performed again revealed hepatosplenomegaly, bilateral renal enlargement, and multiple round low-density lesions in the liver and kidneys, which had not been observed in the previous scan (Fig. 2). A second bone marrow examination revealed platelet hemophagocytosis and clusters of large abnormal lymphocytes with the following immunophenotypes: CD3+, CD5+, CD7+, CD8+, CD4-, CD30-, CD56-, T-cell receptor (TCR)-alpha/beta+, TCR-gamma/delta-, granzyme B+, T-cell intracellular antigen-1-, and perforin- (Fig. 3). These abnormal cells were partially positive for BCL6, but negative for CD10, PD1, and CXCL13. No Epstein-Barr virus-encoded small RNA (EBER)-positive cells were detected by in situ hybridization. A chromosomal analysis indicated the following aberrations: 46,XY,der(11)t(6;11)(?;q24)[10]/46,XY[10]. A Southern blot analysis revealed rearrangements of TCR-β chain genes. Furthermore, a transjugular liver biopsy revealed infiltrates of atypical lymphocytes with an identical phenotype, along the Glisson sheath and within the sinusoids. Based on these findings, a final diagnosis of Ann Arbor stage IV peripheral T-cell lymphoma, not otherwise specified (PTCL-NOS) was established.
Figure 2.

(A, B) The first computed tomography scan with no obvious abnormality. (C, D) The second computed tomography scan, in which hepatosplenomegaly, splenic infarction, renal enlargement, and multiple round low-density lesions in the liver and kidneys (arrowheads) were newly observed.
Figure 3.

(A, B) Large abnormal lymphocytes with a basophilic cytoplasm and azurophilic granules as well as hemophagocytosis in the patient’s bone marrow (May-Giemsa stain, ×1,000 magnification). (C) Bone marrow infiltrates of neoplastic cells with pleomorphic nuclei and prominent nucleoli (Hematoxylin and Eosin staining, ×400 magnification). (D) Neoplastic cells were positive for CD3, (E) CD8, and (F) granzyme B (G) but negative for CD4 (×400 magnification).
Following the treatment with CHOEP (cyclophosphamide, doxorubicin, vincristine, etoposide, and prednisone), his fever resolved, and the laboratory findings temporarily improved, but he subsequently had an early relapse. His disease was refractory to multidrug chemotherapy, and he passed away two months after the diagnosis.
Discussion
Eosinophilia in peripheral T-cell lymphoma is one of the more commonly observed clinical manifestations, occurring in up to 30% of patients (1). Eosinophilopoietic cytokines, including interleukin-3, granulocyte-macrophage colony-stimulating factor, and/or interleukin-5, are reportedly produced by neoplastic T-cells in these patients (2). Although eosinophilia is common in angioimmunoblastic T-cell lymphoma, the tumor cells in this patient did not express the T-follicular helper cell phenotype consistent with this category but rather the CD8 positive cytotoxic T-cell phenotype, which led to the diagnosis of PTCL-NOS according to the revised 2017 WHO classification (3). However, severe hypereosinophilia and EF are uncommon initial presentations of peripheral T-cell lymphoma. Furthermore, in this case, the fact that the tumor cells were initially cryptic and undetectable and that the patient met the diagnostic criteria for hypereosinophilic syndrome (HES) made a correct understanding and diagnosis difficult.
EF is a rare connective tissue disease characterized by symmetrical, painful edematous swelling of the extremities, skin induration, and collagenous thickening of the fascia. There are no standardized diagnostic criteria for EF, and its diagnosis is often based on a full skin-to-muscle skin biopsy and MRI findings (4,5). Pathologically, inflammatory infiltrates of lymphocytes, eosinophils, histiocytes, and plasma cells into the deep fascia and subcutaneous tissues, as well as fascial thickening, are evident. MRI findings show abnormal T2 signal hyperintensity within the fascia and fascial enhancement with gadolinium. The nature and pathogenesis of EF remain poorly understood. While autoimmune disorders and certain hematological neoplasms, including myelomonocytic leukemia, chronic lymphocytic leukemia, and malignant lymphoma, are known to trigger the onset of EF (4), most cases are idiopathic with no clear association. Indeed, there are few reports of T-cell lymphoma preceded or accompanied by EF (6-9). According to what reports do exist, EF can occur in association with T-cell lymphoma, at either the same time as the lymphoma or months or more in advance. Due to the scarcity of these cases, the exact causal relationships and secondary mechanisms of T-cell lymphoma and EF remain unclear. In one report, elevated transforming growth factor-β1 derived from activated T-cells and connective tissue growth factor production in fibroblasts were postulated to be involved in the pathogenesis of EF (9). Tumor-derived abnormal cytokines, mainly interleukin-5, and the secondarily increased eosinophils may also have been responsible for the EF in our case.
EF can appear as a complication of HES, which may have been the case in our patient. HES, a highly heterogeneous disorder, is currently defined by the presence of hypereosinophilia (>1.5 eosinophils ×109/L in the peripheral blood on 2 examinations and/or tissue hypereosinophilia), eosinophil-mediated organ damage and/or dysfunction, and the exclusion of other potential causes for organ damage (10). Two main mechanisms of dysregulated overproduction of eosinophils are known in the pathogenesis of HES: clonal eosinophilic proliferation and overexpression of eosinophilopoietic cytokines (11). Clinically, HES can be classified into several variants, including myeloproliferative variants, where the eosinophils are part of the neoplastic clone; lymphocytic variants (L-HES), where eosinophils proliferate polyclonally secondary to cytokines overproduced from aberrant T-cell subsets; idiopathic HES; and familial HES (11). L-HES, a secondary/reactive eosinophilia, is characterized by abnormal type 2 helper T-cells in the peripheral blood or bone marrow that induce excess production of certain cytokines, including interleukin-5 (12,13). This T-cell population commonly has aberrant phenotypes of CD3-, CD4+, and TCR-alpha/beta+, and often lacks some T-cell markers such as CD7. The TCR-gene rearrangement suggests that these T-cells are a monoclonal population, but this population is often inconspicuous, accounting for less than 2% of total lymphocytes in nearly half of L-HES cases (14). The majority of L-HES cases are benign, respond to steroid therapy, and have an indolent clinical course. In previous cohort reviews, a small number - approximately 10% of those diagnosed with L-HES - progressed to overt T-cell lymphoma within several years (14-16). Of note, there is a small subset of patients who are refractory to steroid therapy, and they seem to be more likely to progress to T-cell lymphoma than others.
The present patient had common cutaneous lesions in L-HES (13,14), such as angioedema with weight gain and fasciitis at the first onset, but the aberrant T-cell population was not evident, and the initial bone marrow examination failed to identify the neoplastic cells by either cytogenetic or immunohistochemical approaches, which may have delayed his precise diagnosis. Based on the diagnosis of EF, he was started on 60 mg of prednisone. His edema and eosinophilia improved quickly with no further exacerbations thereafter, but he then developed severe hemophagocytic lymphohistiocytosis, suggesting the presence of latent lymphoma. Although the results of the second bone marrow chromosomal analysis and Southern blotting indicated the presence of a clonal abnormal cell population, it is unclear how this chromosomal anomaly contributes to tumor development, growth, or eosinophilia. Given that we were unable to detect the aberrant T-cells pathognomonic for L-HES, and the clinical course was not consistent with the previously reported slow course of L-HES overt T-cell lymphoma, we believe that he most likely developed de novo PTCL-NOS and presented with paraneoplastic EF caused by certain cytokines produced by lymphoma cells. The discrepancy between the surface markers of his lymphoma cells and those of the aberrant T-cells commonly seen in L-HES also supports this assumption. However, we were unable to completely rule out the possibility that the patient was in a chronic phase of L-HES before the first presentation and the eosinophilia was caused by an undetectable, aberrant T-cell clone.
In conclusion, this is a unique case of PTCL-NOS that was difficult to diagnose due to the rare initial presentation of EF and hypereosinophilia with no identifiable abnormal cells. This case highlights the issue that in T-cell lymphoma, which can be protean, an overlap of eosinophilic disorders may exist, or essential abnormalities may be obscured by other manifestations. Eosinophilia and HES raise broad differential diagnoses, but in practice, it is sometimes difficult to identify the underlying cause. Clinicians should be reminded that EF or eosinophilia may be the only diagnostic clue for T-cell lymphoma, and careful attention should be paid to organ damage, with repeated histologic examinations potentially helping to confirm the diagnosis.
The authors state that they have no Conflict of Interest (COI).
Acknowledgements
The authors would like to thank the pathologist Ichiro Ito for his helpful advice on the pathological diagnosis.
References
- 1. Greer JP, York JC, Cousar JB, et al. Peripheral T-cell lymphoma: a clinicopathologic study of 42 cases. J Clin Oncol 2: 788-798, 1984. [DOI] [PubMed] [Google Scholar]
- 2. Kitano K, Ichikawa N, Shimodaira S, Ito T, Ishida F, Kiyosawa K. Eosinophilia associated with clonal T-cell proliferation. Leuk Lymphoma 27: 335-342, 1997. [DOI] [PubMed] [Google Scholar]
- 3. Swerdlow SH, Campo E, Pileri SA, et al. The 2016 revision of the World Health Organization classification of lymphoid neoplasms. Blood 127: 2375-2390, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Lebeaux D, Sène D. Eosinophilic fasciitis (Shulman disease). Best Pract Res Clin Rheumatol 26: 449-458, 2012. [DOI] [PubMed] [Google Scholar]
- 5. Moulton SJ, Kransdorf MJ, Ginsburg WW, Abril A, Persellin S. Eosinophilic fasciitis: spectrum of MRI findings. AJR Am J Roentgenol 184: 975-978, 2005. [DOI] [PubMed] [Google Scholar]
- 6. Castellanos-González M, Velasco Rodriguez D, Blanco Echevarría A, et al. Eosinophilic fasciitis as a manifestation of a cutaneous T-cell lymphoma not otherwise specified. Am J Dermatopathol 35: 666-670, 2013. [DOI] [PubMed] [Google Scholar]
- 7. Eklund KK, Anttila P, Leirisalo-Repo M. Eosinophilic fasciitis, myositis and arthritis as early manifestations of peripheral T-cell lymphoma. Scand J Rheumatol 32: 376-377, 2003. [DOI] [PubMed] [Google Scholar]
- 8. Kim H, Kim MO, Ahn MJ, et al. Eosinophilic fasciitis preceding relapse of peripheral T-cell lymphoma. J Korean Med Sci 15: 346-350, 2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Masuoka H, Kikuchi K, Takahashi S, Kakinuma T, Hayashi N, Furue M. Eosinophilic fasciitis associated with low-grade T-cell lymphoma. Br J Dermatol 139: 928-930, 1998. [DOI] [PubMed] [Google Scholar]
- 10. Valent P, Klion AD, Horny HP, et al. Contemporary consensus proposal on criteria and classification of eosinophilic disorders and related syndromes. J Allergy Clin Immunol 130: 607-612.e609, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Simon HU, Rothenberg ME, Bochner BS, et al. Refining the definition of hypereosinophilic syndrome. J Allergy Clin Immunol 126: 45-49, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Cogan E, Schandené L, Crusiaux A, Cochaux P, Velu T, Goldman M. Brief report: clonal proliferation of type 2 helper T cells in a man with the hypereosinophilic syndrome. N Engl J Med 330: 535-538, 1994. [DOI] [PubMed] [Google Scholar]
- 13. Roufosse F, Cogan E, Goldman M. Lymphocytic variant hypereosinophilic syndromes. Immunol Allergy Clin North Am 27: 389-413, 2007. [DOI] [PubMed] [Google Scholar]
- 14. Carpentier C, Verbanck S, Schandené L, et al. Eosinophilia associated with CD3-CD4+ T cells: characterization and outcome of a single-center cohort of 26 patients. Front Immunol 11: 1765, 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Lefèvre G, Copin MC, Staumont-Sallé D, et al. The lymphoid variant of hypereosinophilic syndrome: study of 21 patients with CD3-CD4+ aberrant T-cell phenotype. Medicine (Baltimore) 93: 255-266, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Lefèvre G, Copin MC, Roumier C, et al. CD3-CD4+ lymphoid variant of hypereosinophilic syndrome: nodal and extranodal histopathological and immunophenotypic features of a peripheral indolent clonal T-cell lymphoproliferative disorder. Haematologica 100: 1086-1095, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]