

Abstract
Purpose:
The low mutational load of some cancers is considered one reason for the difficulty to develop effective tumor vaccines. To overcome this problem, we developed a strategy to design neopeptides through single amino acid mutations to enhance their immunogenicity.
Experimental Design:
Exome and RNA sequencing as well as in silico HLA-binding predictions to autologous HLA molecules were used to identify candidate neopeptides. Subsequently, in silico HLA-anchor placements were used to deduce putative T-cell receptor (TCR) contacts of peptides. Single amino acids of TCR contacting residues were then mutated by amino acid replacements. Overall, 175 peptides were synthesized and sets of 25 each containing both peptides designed to bind to HLA class I and II molecules applied in the vaccination. Upon development of a tumor recurrence, the tumor-infiltrating lymphocytes (TIL) were characterized in detail both at the bulk and clonal level.
Results:
The immune response of peripheral blood T cells to vaccine peptides, including natural peptides and designed neopeptides, gradually increased with repetitive vaccination, but remained low. In contrast, at the time of tumor recurrence, CD8+ TILs and CD4+ TILs responded to 45% and 100%, respectively, of the vaccine peptides. Furthermore, TIL-derived CD4+ T-cell clones showed strong responses and tumor cell lysis not only against the designed neopeptide but also against the unmutated natural peptides of the tumor.
Conclusions:
Turning tumor self-peptides into foreign antigens by introduction of designed mutations is a promising strategy to induce strong intratumoral CD4+ T-cell responses in a cold tumor like glioblastoma.
Translational Relevance.
Low mutational load is considered the main reason for the difficulty to develop effective tumor vaccines. To overcome this problem in a tumor with low mutational load, we performed a highly personalized vaccination with neopeptides containing artificially designed single amino acid substitutions, that is, designed neopeptides. The immune response of peripheral blood T cells to vaccine peptides gradually increased with repetitive vaccination, and tumor-infiltrating lymphocytes (TIL) responded strongly to all vaccine peptides designed for HLA class II molecules and 45% designed for HLA class I molecules. Furthermore, TIL-derived CD4+ T-cell clones showed that designed neopeptides elicited strong CD4+ T-cell responses against the unmutated natural peptides and tumor cell lysis. Thus, turning tumor self-peptides into foreign antigens is a promising strategy to induce intratumoral CD4+ T-cell responses in a cold tumor.
Introduction
Glioblastoma (GBM) is the most frequent malignant primary brain tumor in adults. Standard-of-care treatment for newly diagnosed GBM includes tumor resection, radiation, and chemotherapy with temozolomide (1, 2). Despite intensive efforts, the median survival of patients with GBM is still in the range of only 16–20 months in clinical trial populations (1). Because of the still poor prognosis, numerous studies are testing approaches that aim at unspecific or antigen-specific activation of immune responses by immune checkpoint inhibitors (3, 4), different vaccination strategies including peptide-pulsed or tumor lysate–pulsed dendritic cells (5–7), mRNAs encoding tumor antigens (8), and peptides alone or in combination with adjuvants (9–11). While successes toward increased progression-free survival and quality of life have been seen in some patients, no immunotherapeutic approach prolonged the survival of patients with GBM in a randomized trial. The disappointing outcome of most trials indicates that several key aspects are so far poorly understood and need to be improved.
Because of intratumoral heterogeneity with differences in mutations, expression profiles, and cellular characteristics of GBM among patients as well as variation in the individual HLA background, it is desirable to develop truly personalized vaccination approaches. These should be optimized with respect to the tumor characteristics of each patient, including mutations, gene and protein expression profiles, and HLA types. A key aspect for inducing efficient immune responses is to identify putative tumor antigens that can be employed in the respective vaccination approach (12). Mutations and proteins that are highly expressed in the tumor are the most interesting targets. CD8+ T cells are best known for their cytotoxic function, which is so far considered critical for the tumor immune response. However, it is becoming increasingly recognized that CD4+ T-cell responses are also indispensable for tumor control or eradication (13–16). On the basis of data from animal models as well as human tumors, a personalized vaccination approach could benefit from inducing both tumor-specific CD4+ and CD8+ T cells (17–19). Recent promising data with a mutation-specific peptide vaccine for IDH1-mutant glioma underscore the importance of tumor antigen-specific CD4+ T cells (11, 20).
Because peptides can relatively easily be synthesized in large quantities at GMP grade and have low risks for antigen-induced anaphylaxis, they are particularly suited to develop personalized tumor vaccines compared with whole proteins and tumor lysates (21). Therefore, we reasoned that a personalized vaccine should contain peptides designed to stimulate both CD4+ and CD8+ T-cell responses against multiple tumor targets. Toward this aim, we first identified tumor-specific antigens (TSA) and tumor-associated antigens (TAA) based on somatic mutations and overexpressed/highly expressed genes of the autologous tumor respectively from a patient with GBM and some previously reported GBM antigens (7, 22, 23). Peptides from TSAs and TAAs were then chosen based on predicted binding to the HLA class I and II molecules of the patient. Most importantly, considering the low mutational load of GBM and the low immunogenicity of self-proteins, we designed neopeptides by targeted mutation of putative T-cell receptor (TCR) contact amino acids (AA) in the selected peptides. With this, we followed two aims: (i) to increase the number of possible target antigens, and (ii), to increase immunogenicity rather than relying on the often poorly immunogenic and/or poorly HLA-binding peptides derived from somatic mutations of the tumor. This concept was based on our prior findings with an altered peptide ligand (APL) of myelin basic protein (MBP), which was designed to block disease activity by partial agonist or antagonist activity in patients with multiple sclerosis (MS; ref. 24). However, we observed a strong increase of disease activity and inflammation in the brain in 3 of 8 patients receiving the highest dose of the APL peptide. Mechanistic studies showed that APL immunization activated CD4+ T cells that cross-recognized both the APL, the equivalent of a neopeptide, but also the natural unmutated MBP peptide. Interestingly, the responses of some T-cell clones (TCC) against the natural peptide (N-peptides) were significantly higher than against the “foreign” APL peptide. On the basis of these findings, we adapted this concept as an attractive approach toward personalized tumor vaccination in the current study.
Materials and Methods
Ethical considerations
The current study was performed with the goal to increase the possibility for improving the clinical outcome. A new treatment method was used because the patient was aware of the poor prognosis of standard therapy and wanted to try the vaccination. By definition, this is an individual medical treatment (“compassionate use”), which is not subject to special regulation for medical research according to the Therapeutic Products Act (TPA) or Swiss Federal Human Research Act (HRA). Consequently, individual medical treatments are not approved by the Ethics Committee or Swiss Agency for Therapeutic Product (Swissmedic), as they do not constitute clinical trials for the purpose of systematically gaining knowledge. Such an activity falls within the scope of the physician's professional freedom and requires comprehensive and documented patient information. The patient was fully informed about and consented to the steps of the personalized vaccination throughout the treatment period.
Patient information
A 55-year-old man reported headache and memory impairment. A cerebral mass was diagnosed by MRI, and the diagnosis of an isocitrate dehydrogenase wild-type GBM was made following surgical resection (first surgical resection). The patient received standard of care with temozolomide-based radiochemotherapy and maintenance with temozolomide. Comprehensive genomic profiling (CGP; F1CDx assay) detected KRAS amplification (G12D), ERRFI1 loss, CDKN2A/B loss, LZTR1 loss, and TERT promoter methylation (−124C>T). Approved therapies for tumors with KRAS amplification are cobimetinib or trametinib, but these therapies are not approved for GBM. Because of further tumor progression, treatment with pembrolizumab, bevacizumab, and an experimental, personalized peptide cocktail vaccination was initiated. Because of further tumor recurrence, the patient underwent a second surgical resection 14 months after diagnosis. Unfortunately, the patient died 4 months later.
CGP
Tumor DNA was extracted from formalin-fixed paraffin-embedded (FFPE) tissue of the primary tumor and analyzed by FoundationOneCDx (F1CDx) assay. The F1CDx assay detects genomic alterations in a panel of 324 genes (all exons). For analysis, the Illumina HiSeq 2500 platform is used; hybrid capture-selected libraries were sequenced (targeting >500× median coverage with >99% of exons). Sequence data were analyzed by a customized pipeline to detect all classes of genomic alterations, including base substitutions, insertions/deletions (indel), genomic rearrangements (e.g., gene fusions), and copy-number alterations (i.e., gene amplifications and homozygous gene deletions).
GBM tissue, leukapheresis, and peripheral blood mononuclear cell samples
Part of the primary GBM tissue from the first surgical resection was immediately frozen at −80°C, which was used for RNA sequencing (RNA-seq) and whole-exome sequencing (WES). The recurrent GBM tissue from the second surgical resection was partly used for cryopreservation, which was subjected to imaging mass cytometry (IMC), and partly for TIL isolation. Leukapheresis was collected prior to treatment to isolate sufficient antigen-presenting cells (APC). PBMCs were collected before and after peptide vaccination, which were used for WES, HLA genotyping, or vaccine peptide testing.
DNA extraction, HLA genotyping, and WES
DNA was extracted from PBMCs and primary GBM tissue using DNeasy Blood & Tissue Kits (Qiagen). The genotypes of HLA class I (A and B) and HLA class II (DRB1, DRB3, DRB4, DRB5, DPA1, DPB1, DQA1, and DQB1) alleles were determined by high-resolution HLA sequence-based typing (Histogenetics). The WES of PBMCs and the primary tumor was performed at Genomics Facility Basel [Illumina HiSeq 2500, 2×101 bp, Capture kit: Agilent SureSelect XT v6 (Virginia Tech Biocomplexity Institute Genomics Sequencing Center Core Facility, RRID:SCR_017958) +COSMIC]. WES reads for tumor and PBMCs were aligned to the human reference genome (hg19) using bwa (arXiv:1303.3997). Secondary alignments as well as duplicate reads were removed using samtools (25) and Picard, RRID:SCR_006525 (http://broadinstitute.github.io/picard/), respectively. Subsequently, we performed Base Quality Score Recalibration and Indel realignment using GATK (26). The average coverage for the primary tumor analysis was 255x and 85x for tumor and normal, respectively. On the resulting WES alignments, Strelka (27), Mutect (28), and Varscan 2 (29) were used to call somatic variants. Only variants called by at least two of the callers were considered. The WES raw data have been deposited to the European Nucleotide Archive (ENA) with the accession codes PRJEB56517.
RNA extraction and RNA-seq
RNA from primary GBM (RNeasy Mini Kit, Qiagen) were used for RNA-seq using HiSeq 4000 System (Illumina), 2×150 bp, at the Functional Genomics Center Zurich and yielded 154,153,378 read pairs. STAR, RRID:SCR_004463 (version 2.4.2) was used to map (2-pass) the RNA-seq reads against the human reference genome (hg19). On the basis of TCGA-RNAseqv2 pipeline (https://webshare.bioinf.unc.edu/public/mRNAseq_TCGA/UNC_mRNAseq_summary.pdf) and the STAR alignment, quantile normalized gene counts were determined. These quantile normalized gene counts were used to compare the gene expression in the primary tumor with The Cancer Genome Atlas (TCGA) GBM cohort. Gene expression data for TCGA GBM cohort were downloaded from the Broad GDAC Firehose [Broad Institute TCGA Genome Data Analysis Center (2016): Analysis Overview for GBM (Primary solid tumor cohort) - 28 January 2016. Broad Institute of MIT and Harvard. doi:10.7908/C16T0M0N]. Gene expression up to 1.5 times the interquartile range greater than the 75th percentile in the cohort gene expression distribution is considered high expression. Gene expression greater than that is considered overexpression. The RNA-seq raw data have been deposited to the ENA with the accession code PRJEB56507.
Identification of potential TSAs and TAAs
To determine TSAs containing somatic mutations, genomic DNA was extracted from the primary GBM and PBMCs and analyzed using WES. A total of 40 somatic mutants were found and used as TSAs for vaccine peptide prediction (Supplementary Table S1). To identify TAAs with high transcription levels in the tumor, RNA was extracted from the primary GBM tissue and analyzed using RNA-seq. Overexpressed/highly expressed genes in the primary GBM, when comparing with GBM cohort in TCGA Database, were selected and used as TAAs for vaccine peptide prediction (Supplementary Tables S2 and S5). In addition, a total of 12 GBM-associated TAAs (7, 22, 23) that had been reported previously by immunopeptidome or sequencing analyses were also used for vaccine peptide prediction (Supplementary Table S3). Manual curation of the TSA/TAA based on transcription and prior knowledge was performed to exclude peptides with putative high risk of causing damage to vital organs, although we are aware of the limitations of such estimates.
Prediction of TSA/TAA-derived N-peptides
Given the HLA genotype of the patient, TSA/TAA-derived peptides were predicted using NetMHCpan 4.0 (30) and the MHC-I and MHC-II binding prediction tools (IEDB_recommended mode) of the IEDB Analysis Resource (31–33). According to the binding motif and predicted binding affinity of the peptides for HLA class I and II molecules, 78 N-peptides from TSAs or TAAs were selected, which included peptides with a length of 9 AAs for HLA class I molecules (N-class I-peptides, I_MUT/RNA/GBM_X_0) and peptides with a length of 11–13 AAs for HLA class II molecules (N-class II-peptides, II_MUT/RNA/GBM_X_0) as shown in Supplementary Table S4.
Artificially designed mutation of predicted TSA/TAA-derived N-peptides
To potentially strengthen the stimulatory effect, single AA substitutions were introduced at putative peptide-TCR contact positions of the N-class I/II-peptides, which were deduced on the basis of SYFPEITHI database (34), as shown in Fig. 1. These peptides are referred to as designed neopeptides (D-neopeptides) as shown in Supplementary Table S4.
Figure 1.

Strategy for predicting TSA/TAA-derived peptides and for designing D-neopeptides by mutations of peptide-to-TCR contact positions. A, Primary GBM tissue and PBMCs were collected for WES to identify somatic mutants in the tumor, which were considered as TSAs. Primary GBM tissue was also used for RNA-seq to identify overexpressed/highly expressed genes in the tumor, which were considered as TAAs. PBMCs were used for DNA sequencing to identify the HLA genotype of the patient. HLA binding was predicted for TSA/TAA-associated peptides from somatic mutants, overexpressed/highly expressed molecules, and reported GBM antigens using NetMHCpan 4.0 Server and IEDB Analysis Resource and for the patient's HLA class I and II molecules. A total of 78 N-peptides, including N-class I-peptides and N-peptides with 11–13 AAs for HLA class II molecules (N-class II-peptides), were selected on the basis of predicted HLA binding affinities. Designed mutations with the intent to increase the stimulatory strength of N-peptides by single AA substitutions of putative peptide-to-TCR contact positions for TCR were introduced. These are referred to as D-neopeptides. A total of 175 peptides, including N-peptides (n = 78) and D-neopeptides (n = 97), were selected as the peptide library for preparing vaccine peptide cocktails. B, Mutation strategy for designing D-neopeptides for HLA class I molecules (D-class I-neopeptide). Using HLA-A*26:01 as an example, putative peptide-anchor positions 2, 6, and 9 as well as preferred AAs for HLA-A*26:01 are reported in the database of SYFPEITHI, which means that other positions, in particular positions 3, 4, and 5, are potential peptide-to-TCR contact positions. Conservative or nonconservative AA substitutions were implemented in one of these positions for each N-class I-peptides. C, Mutation strategy for designing D-neopeptides for HLA class II molecules (D-class II-neopeptide). The strategy is similar to designing D-class I-neopeptides; however, before analyzing the peptide-anchor/contact positions for HLA/TCR, the 9 AA binding motifs of N-class II-peptides were analyzed using NetMHCII 2.3 Server and/or SYFPHEITI.
Design of peptide cocktails for vaccination
Before the first vaccination, the 78 N-peptides and their corresponding D-neopeptides were tested with untouched PBMCs to examine their stimulatory effects to T cells. PBMCs of the patient responded low or had no response to all peptides, so we chose approximately 25 peptides from somatic mutants and reported GBM antigens for the first and second vaccination (Supplementary Table S6). After the second vaccination, the PBMCs of the patient were collected and tested with the vaccine peptides (v-peptides) to examine the effect of the peptide immunotherapy. Peptides that did not elicit a T-cell response after several rounds of vaccination were taken out of the peptide cocktail and replaced by others from the overall set of peptides (Supplementary Table S4) in the subsequent vaccination.
Histologic analyses
Primary and recurrent GBMs were FFPE at the Institute of Pathology and Molecular Pathology, University Hospital Zurich. Paraffin-embedded tissue sections were used for hematoxylin and eosin (H&E) staining, IHC staining for CD3 (Clone: LN10; Dilution: 1:500; Leica Biosystems), and IMC. For IMC, an antibody panel (Supplementary Table S7) was designed and used to target markers specific for GBM, immune cell types/subsets, epithelial cells, activation, and proliferation. The preparation and staining of paraffin-embedded tissue sections, acquirement of images, and image data processing were performed as described before (35).
Isolation and expansion of TILs
Resected recurrent GBM tissue was washed [Iscove's modified Dulbecco's medium (IMDM, GE Healthcare)] to remove blood clots and collagen in a 100 mm Petri dish (Sigma-Aldrich) and then mechanically minced in 5 mL digestion solution: IMDM with 1 mg/mL collagenase A (Roche) and 50 U DNase (Roche). Small tissue pieces were digested at 37°C for 1 hour with pipetting up and down every 10 minutes. The digested product was washed twice with IMDM and passed through a 70 μm Cell Strainer (Corning) to obtain a single-cell suspension, which was centrifuged at 1,200 rpm for 10 minutes at 20°C, the supernatant removed, and cells resuspended with 6 mL 30% Percoll solution (GE Healthcare). TILs were isolated using 30%/78% Percoll gradients. For expansion, TILs were seeded at 1.5 × 103 cells/well in 96-well U-bottom plates (Greiner Bio-One) together with 1.5 × 105 irradiated autologous PBMCs (45 Gy), 1 μg/mL PHA (REMEL, Thermo Fisher Scientific), and 20 U/mL human IL2 (hIL2 containing supernatant produced by T6 cell line kindly provided by F. Sallusto, IRB, Bellinzona, Switzerland) in RPMI1640 medium (Sigma-Aldrich) containing 5% human serum (Blood Bank Basel, Switzerland), 2 mmol/L glutamine (Thermo Fisher Scientific), 1% (vol/vol) nonessential AAs, 1% (vol/vol) sodium pyruvate, 5 μmol/L β-Mercaptoethanol (all Gibco), 100 U/mL penicillin, and 100 μg/mL streptomycin (Corning). Medium was changed by aspirating half of the medium and adding the same volume of fresh medium containing hIL2 every 3–4 days for 14 days until cells were fully rested.
Proliferation assay
All peptides used in this study for stimulation purposes were synthesized with C-terminal amide (Peptides & Elephants). For peptide stimulation assay of PBMCs, untouched or CD45RA− PBMCs were tested in the indicated conditions. CD45RA− PBMCs were isolated by negative selection using CD45RA microbeads, human (Miltenyi Biotec). In the tests, PBMCs were seeded at 2 × 105 cells/well in 200 μL X-VIVO 15 medium (Lonza) in 96-well U-bottom plates (Greiner Bio-One), and peptides were then added at a final concentration of 5 μmol/L. Proliferations were measured at day 7 by 3H-thymidine (Hartmann Analytic) incorporation assay. Proliferation strength is displayed as stimulation index (SI). SI indicates the ratio of counts per minute (cpm) in the presence of the peptide versus cpm of the no peptide control.
For peptide stimulation, TILs were seeded at 5 × 104 cells/well with 2 × 105 irradiated autologous PBMCs (45 Gy) as APCs in 200 μL X-VIVO 15 medium (Lonza) in 96-well U-bottom plates (Greiner Bio-One) and stimulated with peptides at 5 or 10 μmol/L. The anti-human HLA-DR/DP/DQ antibody (TÜ39, kindly provided by Stefan Stevanovic) was added at a final concentration of 10 μg/mL to block the function of HLA class II molecules. Proliferation was measured at day 5 by 3H-thymidine (Hartmann Analytic) incorporation assay.
For peptide stimulation assay of TCCs, TCCs were seeded at 2 × 104 cells/well with 1 × 105 irradiated autologous PBMCs (45 Gy) in 200 μL X-VIVO 15 medium (Lonza) in 96-well U-bottom plates (Greiner Bio-One) with peptides at 5 μmol/L.
Cytotoxicity assay
Effector TILs or TCCs were stimulated with pooled peptides for 5 days or 4 days, respectively, using irradiated autologous PBMCs as APCs. TILs and TCCs were then harvested, washed, and resuspended with X-VIVO 15 medium (Lonza). Tumor target cells were detached with accutase (Thermo Fisher Scientific), washed, and labeled with a PKH26 Red Fluorescent Cell Linker Kit (Sigma-Aldrich) for 3 minutes. After labeling, cells were washed three times with serum-containing medium, resuspended with X-VIVO 15 medium (Lonza), adjusted to 2 × 105 cells/mL, and added in 100 μL/well in 96-well U-bottom plates (Greiner Bio-One). Effector TILs or TCCs were added to respective wells containing PKH26-labeled target tumor cells at 1:1, 2:1, 5:1, and 10:1 effector-to-target ratios. Plates were centrifuged at 80 × g for 1 minute to improve effector-target cell contact and incubated for 24 hours. After incubation, plates were centrifuged at 250 × g for 5 minutes, supernatants removed, and cells detached with accutase (Thermo Fisher Scientific). Cells were harvested, stained with a LIVE/DEAD Fixable Near-IR Dead Cell Stain Kit (Thermo Fisher Scientific), and analyzed using a BD LSR Fortessa to analyze PKH26+Near-IR+ dead target cells.
Flow cytometric analysis
For surface marker staining, cells were incubated with human IgG (Sigma-Aldrich) and Live/Dead Aqua (Invitrogen) at 4°C for 30 minutes and then stained using fluorochrome-conjugated antibodies, including FITC anti-human CD69 (FN50), PerCP/Cyanine5.5 anti-human CD3 (HIT3a), PerCP/Cyanine5.5 anti-human CD19 (HIB19), APC anti-human CD25 (BC96), APC/Cyanine7 anti-human CD4 (A161A1), Pacific Blue anti-human CD8 (SK1), Alexa Fluor 700 anti-human CD3 (HIT3a), Brilliant Violet 510 anti-human CD8 (SK1), APC anti-human CD194 (CCR4) (L291H4), Brilliant Violet 785 anti-human CD196 (CCR6) (G034E3), Brilliant Violet 711 anti-human CD45RA (HI100), PE anti-human CD294 (CRTH2) (BM16), Brilliant Violet 421 anti-human CD197 (CCR7) (G043H7) antibodies (BioLegend), and CD4 mAb (S3.5), PE-Texas Red (Thermo Fisher Scientific), at 4°C for 30 minutes. For intracellular staining, cells were treated with fixation/permeabilization kits (eBioscience) and then stained after surface marker staining for intracellular markers using fluorochrome-conjugated antibodies (BioLegend), including FITC anti-human/mouse granzyme B Antibody (GB11), at 4°C overnight. Cells were then washed twice and resuspended with 1× PBS containing 2 mmol/L ethylenediamine tetraacetic acid (AppliChem) and 2% FCS (Eurobio Scientific). Analyses were performed with LSR Fortessa Flow Cytometer (BD Biosciences) and FlowJo software, RRID:SCR_008520 (Tree Star).
T-cell cloning, specificity, and cross-reactivity
To generate CD4+ TCCs that recognize vaccine peptides, TILs were stained with carboxyfluorescein succinimidyl ester (CFSE, Sigma-Aldrich) and stimulated with pooled peptides, including II_GBM_14_1, II_GBM_12_1, and II_GBM_24_1. Proliferating (CFSElow) CD4+ T cells were sorted at day 9 as single cells into 96-well U-bottom plates using a Sorter (SH800S, Sony) and expanded using irradiated allogeneic PBMCs (45 Gy), 1 μg/mL PHA (Remel), and hIL2 in RPMI1640 medium containing 5% human serum (Blood Bank Basel, Switzerland), 2 mmol/L l-glutamine (Thermo Fisher Scientific), 100 U/mL penicillin (Corning), 100 μg/mL streptomycin (Corning), and 50 μg/mL gentamicin (Sigma-Aldrich). Medium was changed by aspirating half of the old medium and adding the same amount of fresh medium containing hIL2 every 3–4 days for 14 days. The TCR β-chain phenotype of the CD4+ TCCs was analyzed using fluorochrome-conjugated antibodies (TCR Vβ1, 2, 3, 5.1, 5.2, 5.3, 6.7, 7, 8, 9, 11, 12, 13.1, 13.6, 17, 18, 20, 21.3, 22, 23, Beckman Coulter) by FACS.
To identify the target peptides of CD4+ TCCs, TCCs were seeded at 2 × 104 cells/well with 1 × 105 irradiated autologous PBMCs (45 Gy) in 200 μL X-VIVO 15 medium (Lonza) in 96-well U-bottom plates (Greiner Bio-One) and stimulated with peptides II_GBM_14_0, II_GBM_14_1, II_GBM_12_0, II_GBM_12_1, II_GBM_24_0, or II_GBM_24_1 at 5 μmol/L. Proliferation was measured at day 3 by 3H-thymidine (Hartmann Analytic) incorporation and supernatants collected for testing IFNγ using ELISA.
TCRVβ sequencing
To analyze the TCRVβ repertoire, DNA samples were extracted from the primary and recurrent tumors. The TCRVβ sequencing was conducted using the immunoSEQ Platform (Adaptive Biotechnologies). The sequencing data were analyzed using immunoSEQ analyzer 3.0 of Adaptive Biotechnologies based on CDR3, V-chain, and J-chain sequences. The frequencies of Vβ-Jβ pairing in the primary and recurrent tumors were visualized using Circos (36).
Cytokine measurement
Supernatants were harvested from TILs at day 5 after stimulation with anti-CD2/CD3/CD28 antibody-loaded MACSibead particles (Miltenyi Biotec) or peptides. Cytokines in the supernatants were measured using LEGENDplex Multi-analyte Flow Assay kit (#740722, BioLegend). Supernatants were harvested from TCCs at day 3 after stimulation with peptides. Cytokines in the supernatants were measured using ELISA MAX Standard Set Human IFNγ (BioLegend).
Statistical analysis
Statistical analyses were performed with GraphPad Prism 8.0, RRID:SCR_002798. Unpaired t tests were used for statistical analyses. Data are expressed as mean or mean ± SEM, and the data were considered statistically significant when differences achieved values of P < 0.05.
Data availability statement
The data generated in this study are not publicly available due to information that could compromise patient consent but are available upon reasonable request from the corresponding author.
Results
Prediction strategy and designed mutations of TSA/TAA-derived peptides
We performed a personalized vaccination (37) in a 55-year-old patient with GBM (Supplementary Fig. S1A). We first identified TSAs including somatic mutants and TAAs that were overexpressed/highly expressed in the primary tumor. We found 40 somatic mutants (Supplementary Table S1). Furthermore, overexpressed/highly expressed genes were identified by comparison with GBMs in TCGA database (Supplementary Table S2). In addition, 12 TAAs that had been reported in the context of GBM (7, 22, 23, 38) were also included for peptide prediction (Supplementary Table S3).
Next, we identified TSA/TAA-derived N-peptides with predicted high binding affinity and HLA motifs for the autologous HLA class I and II molecules (Fig. 1A; Supplementary Fig. S1B). A total of 78 TSA/TAA-derived N-peptides, including N-class I-peptides and peptides with 11–13 AAs for HLA class II molecules (N-class II-peptides; Supplementary Table S4), were selected from 10 somatic mutants (Supplementary Table S1), 22 overexpressed/highly expressed genes (Supplementary Table S5), and 12 reported GBM antigens (Supplementary Table S3). N-class I-peptides included: (i) four neopeptides that contain naturally mutated AA from TSAs (I_MUT_X_0′), (ii) one unmutated peptide from a TSA (I_MUT_X_0), (iii) 15 unmutated peptides from overexpressed/highly expressed TAAs (I_RNA_X_0), and (iv) 11 unmutated peptides from reported GBM antigens (I_GBM_X_0). N-class II-peptides included: (i) five neopeptides that contain naturally mutated AA from TSAs (II_MUT_X_0′), (ii) three unmutated peptides from TSAs (II_MUT_X_0), (iii) 13 unmutated peptides from overexpressed/highly expressed TAAs (II_RNA_X_0), and (iv) 26 unmutated peptides from reported GBM antigens (II_GBM_X_0; Supplementary Table S4). The peptide nomenclature is depicted in Supplementary Fig. S2A.
As key step of this study, we designed mutations in critical AA positions of N-peptides to increase the number of neopeptides available for vaccination and to enhance immunogenicity. Designed mutations were introduced by first positioning peptides in the peptide-binding grooves of HLA class I and II molecules using known binding motifs in the database of SYFPEITHI and then introducing single conservative, neutral, or nonconservative AA exchanges in putative TCR contact positions (Fig. 1B and C). These artificially mutated peptides are referred to as D-neopeptides. The nomenclature of the D-neopeptides is as follows: I/II_MUT/RNA/GBM_X_1/2 (Supplementary Fig. S2A; Supplementary Table S4). Suffixes 1 or 2 are used to distinguish between the up to two D-neopeptides of the respective N-peptide. Overall, 175 peptides from the above sources were synthesized (Supplementary Table S4).
Vaccination strategy
The vaccination schedule after surgical resection and radiochemotherapy and parallel treatment with pembrolizumab and bevacizumab is schematically depicted (Supplementary Fig. S1A). PBMCs collected before vaccination were tested with all N-peptides (n = 78) and D-neopeptides (n = 97). PBMCs did not or only poorly respond to N-peptides and D-neopeptides (Supplementary Fig. S2B–S2D). Therefore, we only chose 25 peptides from somatic mutants or reported GBM antigens for the first and second vaccinations, and the ratio of N-peptide and D-neopeptide is approximately 1:2 (Supplementary Table S6). Peptide cocktails were subcutaneously injected for vaccination, and the adjuvants were used to augment peptide-specific immune responses.
Peripheral blood T-cell responses to vaccine peptides increase with repetitive vaccination
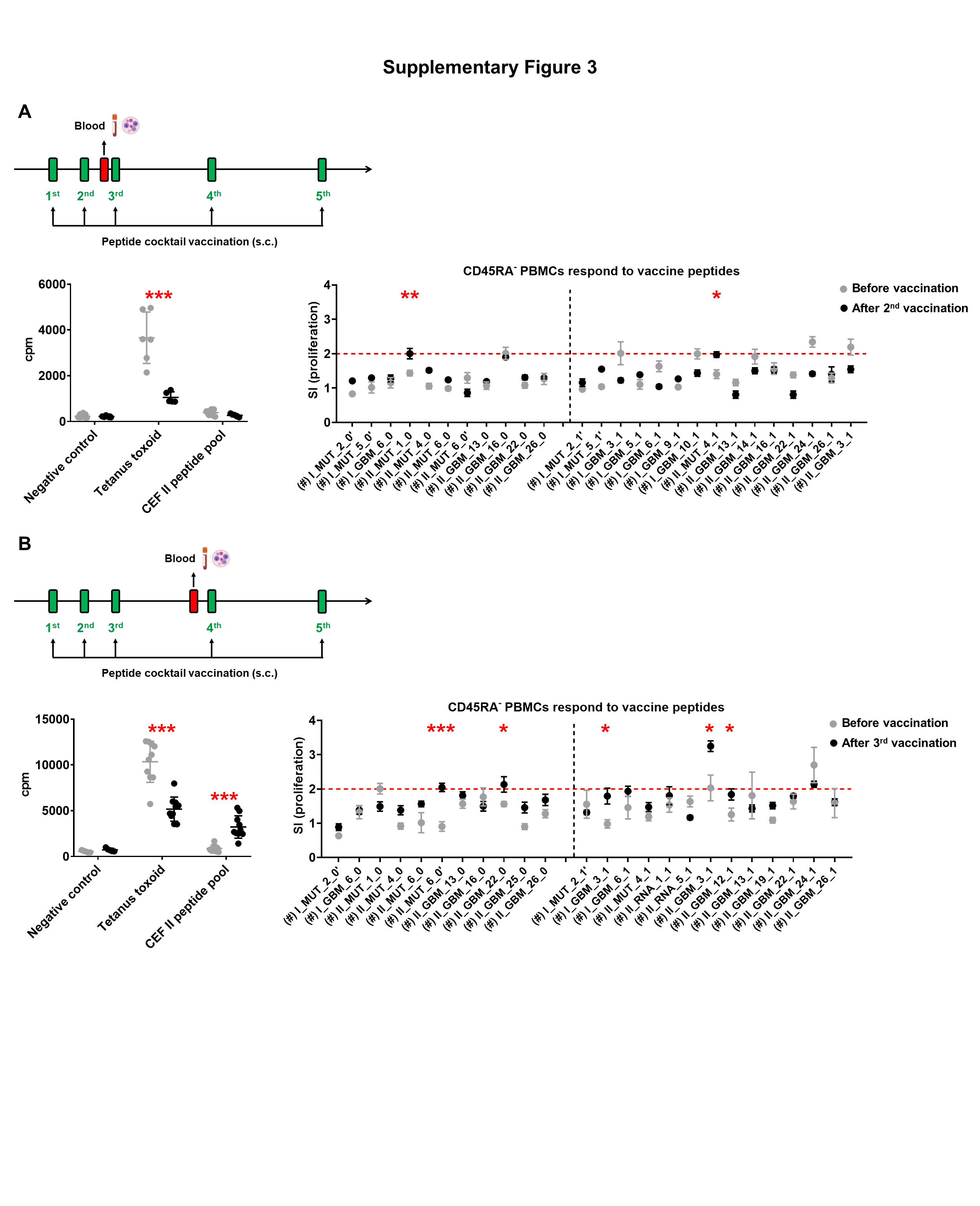
To optimize the vaccine cocktail to induce strong immune responses, we assessed the response of PBMCs to v-peptides after vaccination. We used CD45RA-depleted (CD45RA−) PBMCs as a source of memory T cells. Cells collected 20 days after the second vaccination did not show stronger responses to v-peptides than CD45RA− PBMCs collected before vaccination. Some responses were even lower (Supplementary Fig. S3A). Therefore, we removed the latter peptides, because they might have been inhibitory, replaced them with others from the overall set of peptides, and finally used 24 peptides for the third vaccination (Supplementary Table S6). CD45RA− PBMCs collected 40 days after the third vaccination showed increased responses to some v-peptides compared with CD45RA− PBMCs collected before vaccination or after the second vaccination (Supplementary Fig. S3B). However, responses to v-peptides remained overall modest (Supplementary Fig. S3), which might be due to the low precursor frequency of v-peptide–specific T cells in blood, migration of vaccine-activated T cells into the tumor, or technical aspects, that is, low sensitivity of the 3H-thymidine proliferation assay. Similarly, some peptides were replaced or added, and 27 peptides were used for the fourth vaccination (Supplementary Table S6).
Increased T-cell infiltration in perivascular areas of the recurrent tumor after peptide vaccination
One month after the fourth vaccination, a recurrent tumor was resected, which allowed us to examine the potential effects of peptide vaccination on TILs. TILs were studied in the primary and recurrent tumor by H&E staining (Fig. 2A and B), CD3 IHC staining (Fig. 2A and B; Supplementary Fig. S4A), and IMC for multimarker analysis (Fig. 2C and D; Supplementary Fig. S5; Supplementary Table S7). Few CD3+ TILs were found in the parenchymal areas of both primary and recurrent tumor (Fig. 2A_i and B_i). However, compared with the primary tumor (Fig. 2A_ii and C_top; Supplementary Fig. S4B_top), the recurrent tumor showed increased CD3+ TILs in the perivascular areas (Fig. 2B_ii and C_bottom; Supplementary Fig. S4B_middle) and peritumoral tissue (Fig. 2B_iii and D; Supplementary Fig. S4B_bottom). Furthermore, few CD8+ T cells were seen in the perivascular areas of the primary tumor (Fig. 2C_top), while the number of infiltrating CD8+ T cells increased in the recurrent tumor (Fig. 2C_bottom and D). In particular, CD4+ T cells (CD3+CD8− T cells) were relatively abundant and infiltrated both perivascular and peritumoral areas of the recurrent tumor but not the primary tumor (Fig. 2C and D), and the tumor-infiltrating CD4+ T cells displayed an activated (Supplementary Fig. S5A) and proliferative phenotype (Fig. 2C and D). Natural killer cells were also increased in the recurrent tumor (Supplementary Fig. S5B), and infiltrating CD68+ macrophages that displayed a proinflammatory phenotype (Supplementary Fig. S5C) might function as APCs to activate and/or retain the TILs in the recurrent tumor (Supplementary Fig. S5D). These results indicate that v-peptides might have stimulated and expanded TILs after vaccination.
Figure 2.

Increased T lymphocyte infiltration in the recurrent compared with the primary tumor. H&E and CD3 IHC staining in the primary (A) and recurrent tumor (B). The regions that are selected to show the CD3 IHC staining are indicated in the H&E-stained tissue sections. Expressions of GFAP (GBM cells), CD31 (vascular epithelial cells), CD3 (T lymphocytes), CD8 (CD8+ T cells), and Ki67 (proliferating marker) in the perivascular areas of primary and recurrent tumor (C) and peritumoral areas of the recurrent tumor (D) were detected by IMC. Scale bars are indicated in the figures.
Composition, phenotype, and response to vaccine peptides of TILs
Next, we isolated TILs from the recurrent tumor and characterized them by immunophenotyping. Two-thirds of infiltrated T cells were CD4+, one-third CD8+ T cells, and all CD45RA− memory T cells (Fig. 3A), and there were very few CD19+ B cells (Fig. 3B). CD4+ T cells mainly showed a Th1 (CRTh2−CCR6−CCR4−)/Th2 (CRTh2− CCR6−CCR4+)-multifunctional phenotype, and CD8+ T cells were mainly Tc1 (CRTh2−CCR6−CCR4−) cells (Fig. 3C). Accordingly, TILs released IFNγ, IL5, and IL13 after stimulation (Fig. 3D). We then tested TILs against all N-peptides. TILs did not respond to N-class I-peptides in proliferation assays (Supplementary Fig. S6) but reacted strongly and in a dose-dependent manner to most N-class II-peptides, including both v-peptides (labeled with #) and non-vaccine (nv)-peptides (Fig. 3E). However, compared with nv-peptides, TIL responses to v-peptides were much stronger (Fig. 3F). These results indicate that repetitive vaccination with a complex peptide cocktail containing D-neopeptides likely led to infiltration of proinflammatory CD4+ and CD8+ T cells in the tumor.
Figure 3.

TILs display a Th1/Th2-mixed phenotype and respond strongly to N-class II-peptides. A, Phenotypes and frequencies of naïve and memory CD4+ and CD8+ T-cell subsets in PBMCs and TILs, which were collected after the fourth peptide cocktail vaccination, were detected by FACS. B, Frequencies of CD19+ B cells in PBMCs and TILs were detected by FACS. C, Functional phenotypes of naïve and memory CD4+ and CD8+ T cells in PBMCs and TILs were detected by FACS. D, TILs were stimulated with anti-CD2/CD3/CD28 beads for 5 days, and cytokines in the supernatants measured with a bead-based immunoassay (LEGENDplex multianalyte flow assay kit). E, TILs were stimulated with 5 or 10
 N-class II-peptides for 5 days, and proliferation was measured by 3H-thymidine incorporation assay. Five replicate wells were tested for proliferation, and responses were depicted as SI. The red dotted line indicates a stimulatory response of SI = 2, and values above the red dotted line were considered positive. F, Comparison of the proliferation levels of TILs after stimulation with positive nv- and v-N-class II-peptides at the concentrations of 5 or 10 μmol/L. Five replicate wells were tested for proliferation. (#) indicates the peptide that was used in the first to fourth vaccinations. Data are expressed as mean ± SEM, and P values were determined using an unpaired t test.
N-class II-peptides for 5 days, and proliferation was measured by 3H-thymidine incorporation assay. Five replicate wells were tested for proliferation, and responses were depicted as SI. The red dotted line indicates a stimulatory response of SI = 2, and values above the red dotted line were considered positive. F, Comparison of the proliferation levels of TILs after stimulation with positive nv- and v-N-class II-peptides at the concentrations of 5 or 10 μmol/L. Five replicate wells were tested for proliferation. (#) indicates the peptide that was used in the first to fourth vaccinations. Data are expressed as mean ± SEM, and P values were determined using an unpaired t test.
Peptide vaccination induces robust intratumoral CD4+ T-cell responses
To address the above point in more detail, we compared the response of the TILs with v-peptides used in the first to fourth vaccination rounds (Supplementary Table S6), including both N-peptides and D-neopeptides, with nv-peptides for HLA class I and II molecules (Supplementary Fig. S7). For class I-peptides, the response rate to nv-peptides was low (5.36%), and an almost nine times higher rate was observed to v-peptides (45.46%; Fig. 4A), although the overall response was still low (Fig. 4B). For class II-peptides, TILs responded to 43.53% of the nv-peptides, and strongly to 100% of the v-peptides (Fig. 4C and D). Importantly, v-D-class II-neopeptides stimulated much higher TIL responses compared with nv-D-class II-neopeptides (Fig. 4E), which also held for v-N-class II-peptides (Fig. 3F). Hence, TILs preferentially respond to v-peptides and more strongly to class II-peptides.
Figure 4.

TILs preferentially respond to v-peptides. A, Percentages of peptides among all predicted class I-peptides, nv-class I-peptides, and v-class I-peptides that stimulated the TILs. B, Proliferations of TILs after stimulation with 5
 v-class I-peptides used in the first to fourth vaccinations for 5 days. Five replicate wells were tested for proliferation. C, Percentages of peptides among all predicted class II-peptides, nv-class II-peptides, and v-class II-peptides that stimulated the TILs. D, Proliferations of TILs were stimulated with 5
v-class I-peptides used in the first to fourth vaccinations for 5 days. Five replicate wells were tested for proliferation. C, Percentages of peptides among all predicted class II-peptides, nv-class II-peptides, and v-class II-peptides that stimulated the TILs. D, Proliferations of TILs were stimulated with 5
 v-class II-peptides used in the first to fourth vaccinations for 5 days. Five replicate wells were tested for proliferation. E, Comparison of the proliferation levels of TILs after stimulation with positive nv- and v-D-class I/II-neopeptides. TILs were stimulated with v-class II-peptides in the presence of a blocking anti-HLA-DR/DP/DQ (TÜ39) antibody or isotype control. Proliferations of TILs (F) and concentrations of IFNγ in the supernatant (G) were detected 5 days after stimulation. Five replicate wells were set up for each condition, and cells were collected and pooled from replicate wells for proliferation test. H, Expression levels of activation markers CD69 and CD25 as well as cytotoxicity marker granzyme B on/in CD4+ TILs after stimulation with v-class II-peptides for 5 days. Five replicate wells were set up for each condition, and cells were collected and pooled from replicate wells for testing. (#) indicates the peptide that was used in the first to fourth vaccinations. Data are expressed as mean ± SEM, and P values were determined using an unpaired t test.
v-class II-peptides used in the first to fourth vaccinations for 5 days. Five replicate wells were tested for proliferation. E, Comparison of the proliferation levels of TILs after stimulation with positive nv- and v-D-class I/II-neopeptides. TILs were stimulated with v-class II-peptides in the presence of a blocking anti-HLA-DR/DP/DQ (TÜ39) antibody or isotype control. Proliferations of TILs (F) and concentrations of IFNγ in the supernatant (G) were detected 5 days after stimulation. Five replicate wells were set up for each condition, and cells were collected and pooled from replicate wells for proliferation test. H, Expression levels of activation markers CD69 and CD25 as well as cytotoxicity marker granzyme B on/in CD4+ TILs after stimulation with v-class II-peptides for 5 days. Five replicate wells were set up for each condition, and cells were collected and pooled from replicate wells for testing. (#) indicates the peptide that was used in the first to fourth vaccinations. Data are expressed as mean ± SEM, and P values were determined using an unpaired t test.
To confirm that peptides stimulated the TILs in an HLA-dependent manner, we performed blocking studies with the anti-HLA-DR/DP/DQ antibody (TÜ39), which inhibited both proliferation and IFNγ secretion of TILs after stimulation with v-class II-peptides (Fig. 4F and G) and confirmed HLA class II restriction of the TIL responses. Furthermore, activation with v-class II-peptides not only led to upregulation of activation markers, but also significantly increased the fraction of cytotoxic granzyme B+ CD4+ T cells (Fig. 4H).
D-neopeptides stimulate stronger TIL responses than N-peptides
The purpose of artificially designing D-neopeptides was to induce strong or even more robust T-cell responses against N-peptides with the hope to foster T-cell infiltration and lysis of tumor cells. Comparing the stimulatory strength of N-peptides with the corresponding D-neopeptides for TILs disclosed that the latter elicited often stronger but sometimes also weaker responses (Supplementary Fig. S7). For class I-peptides, some v-D-neopeptides, such as I_GBM_6_1 (Fig. 5A), I_MUT_1_1, I_GBM_3_1, and I_GBM_10_1 (Fig. 5B), induced positive TIL responses, but TILs did not respond to any of the corresponding N-peptides, indicating that vaccination with D-class I-neopeptides did not activate CD8+ T cells sufficiently against N-class I-peptides, and/or the precursor frequencies were too low for detection in the assays that we applied. This was different for TIL responses to N-class II-peptides and D-class II-neopeptides (Fig. 5C and D). D-class II-neopeptides elicited robust TIL responses, which applied to both peptides with conservative (Fig. 5C), that is, AA exchanges with similar physicochemical properties, including charge, hydrophobicity, and size, but also neutral or nonconservative substitutions (Fig. 5D). CD4+ TILs responded better to D-class II-neopeptides compared with N-class II-peptides based on upregulation of activation markers (Fig. 5E) and cytotoxicity against autologous tumor cells (Fig. 5F). The overall stronger TIL responses to D-neopeptides lends support to the hypothesis that mutations in critical AA positions are likely to stimulate T cells that have escaped negative selection and are recognized with higher avidity than natural self-peptides.
Figure 5.

Single AA substitutions increase the stimulatory effect of D-neopeptides. Comparison of the stimulatory strengths of N-class I-peptides (A and B) or N-class II-peptides (C and D) and their corresponding D-neopeptides by conservative (A, C) or nonconservative (B, D) AA substitution to TILs. Five replicate wells were tested for proliferation, and responses were depicted as SI. E, Comparison of the expression levels of activation markers CD69 and CD25 as well as cytotoxicity marker granzyme B on/in CD4+ TILs after stimulation with N-class II-peptides and their corresponding D-neopeptides. Five replicate wells were set up for each condition, and cells were collected and pooled from replicate wells for testing. F, Comparison of the cytotoxicity of TILs to tumor cells after stimulation with N-class II-peptide and D-class II-neopeptide pools for 5 days. Three replicate wells were set up for each condition. (#) indicates the peptide that was used in the first to fourth vaccinations. Data are expressed as mean ± SEM, and P values were determined using an unpaired t test.
To elicit cross-reactivity of T cells against both D-neopeptides and N-peptides is a key aim of our approach. If T cells responding to D-neopeptides did not cross-react with the N-peptides that the tumor expresses, they would likely not be useful for the tumor defense. On the other hand, if D-neopeptides induced cross-reactive T-cell responses against the N-peptides, especially against the unmutated N-peptides, it would open the possibility to target many more antigens in the tumor than through naturally occurring mutations. Following this idea, we focused our analyses on the peptide groups in which D-neopeptides were used for vaccination but the corresponding N-peptides were not, such as II_GBM_24_0 and II_GBM_24_1 (Fig. 5C), II_GBM_12_0 and II_GBM_12_1, II_GBM_14_0 and II_GBM_14_1, and II_GBM_3_0 and II_GBM_3_1 (Fig. 5D). In these peptide groups, TIL responses to both N-peptides and D-neopeptides were positive. We further noticed that for some peptide pairs, including II_RNA_8_0 and II_RNA_8_1, II_GBM_15_0 and II_GBM_15_1, II_GBM_23_0 and II_GBM_23_1 (Fig. 5C), and II_GBM_17_0 and II_GBM_17_1 (Fig. 5D), when the D-neopeptide stimulated a positive TIL response but was not used for vaccination, TILs had no or a low response to the N-peptides. These data indicate, similar to the abovementioned experience in the MS trial (24), that D-class II-neopeptide vaccination activates CD4+ T cells, which are cross-reactive against their unmutated counterpart.
Vaccination with D-neopeptides induces cross-reactive T-cell responses against N-peptides
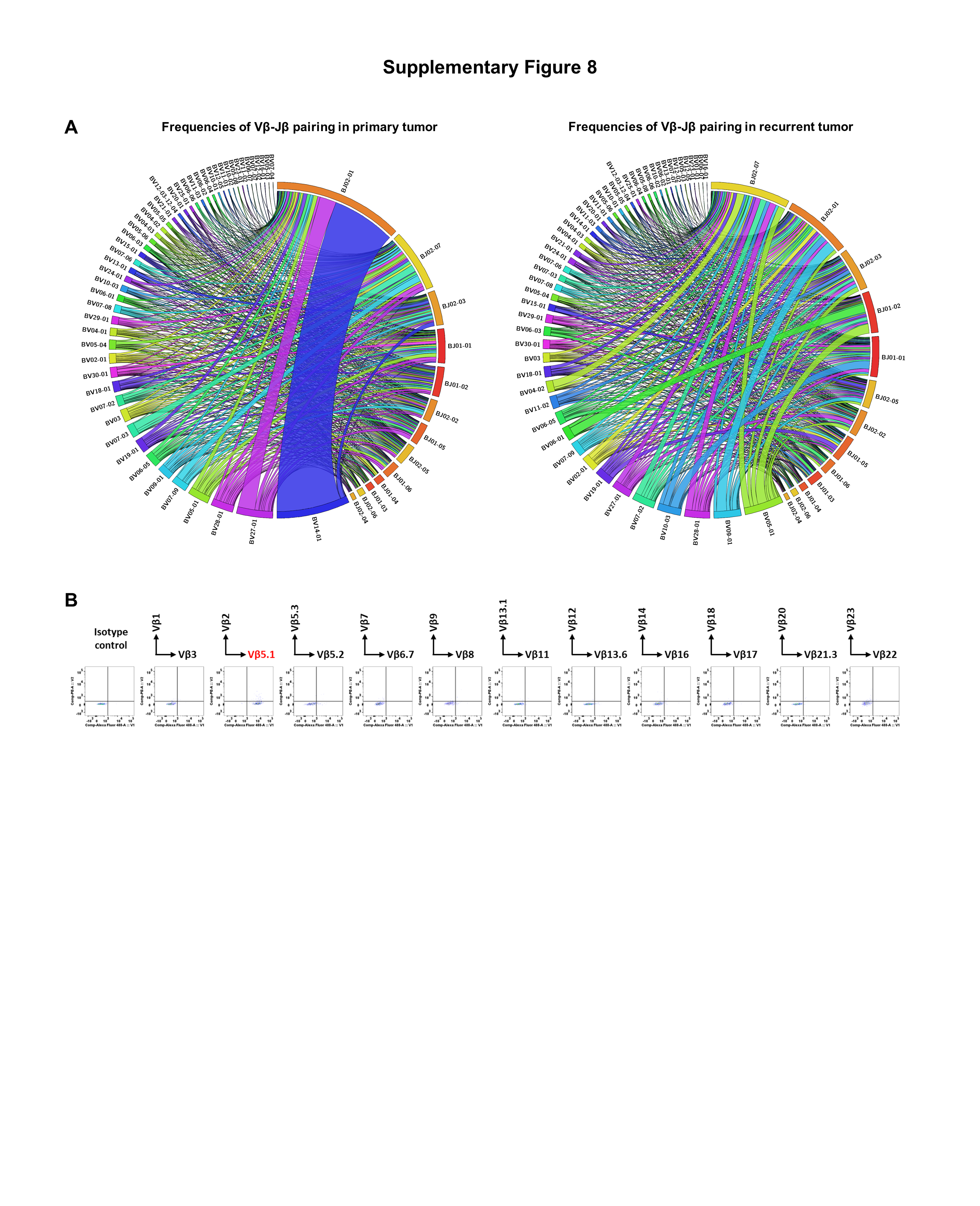
Cross-reactivity against D-neopeptides and N-peptides cannot be addressed adequately at the level of bulk TILs. We therefore generated TCCs using a pool of three exemplary D-class II-neopeptides, including II_GBM_12_1 from the dopachrome tautomerase (DCT), II_GBM_14_1 from the receptor tyrosine-protein kinase erbB-2 (ERBB2), and II_GBM_24_1 from the receptor-type tyrosine-protein phosphatase zeta 1 (PTPRZ1). CD4+ TILs responded strongly to the peptide pool and proliferated, while the proliferation of CD8+ TILs was substantially lower. Proliferating (CFSElow) CD4+ T cells were isolated by single cell sorting and expanded to generate CD4+ TCCs (Fig. 6A). Specificity testing of the TCCs showed that 61 CD4+ TCCs recognized II_GBM_12_1, 48 CD4+ TCCs recognized II_GBM_14_1, and 31 CD4+ TCCs recognized II_GBM_24_1 (Fig. 6B). When testing cross-reactivity against D-neopeptides and their corresponding N-peptides, we observed that most of the D-neopeptide–specific CD4+ TCCs cross-reacted with the N-peptides. This cross-reactivity was seen with all three D-neopeptides (Fig. 6C). Although most D-neopeptide–specific CD4+ TCCs responded slightly less against the N-peptides (lower avidity groups), some CD4+ TCCs showed similar (similar avidity groups) or even higher (higher avidity groups) responses to the N-peptides based on proliferation (Fig. 6C), IFNγ secretion (Fig. 6D), and dose titration of the peptides (Fig. 6E). Importantly, after stimulation with N-peptides, CD4+ TCCs showed cytotoxic activity against the autologous tumor cells comparable with those after stimulation with D-neopeptides (Fig. 6F). Furthermore, we compared the TCR repertoire in the primary and recurrent tumors and found that the dominant TCRs significantly changed in the recurrent tumor after vaccine treatment (Supplementary Fig. S8A). The Vβ-Jβ pairing of BV05-01/BJ01-02 was dominant in the recurrent tumor, and a TCRVβ5.1+CD4+ TCC (Supplementary Fig. S8B), which responded to the v-D-neopeptide II_GBM (DCT)_12_1 (Fig. 6A and C) and corresponding N-peptides (Fig. 6C), was isolated from the TILs of the recurrent tumor. Thus, vaccination with D-neopeptides can lead to strongly cross-reactive, intratumoral CD4+ T-cell responses that are directed against the N-peptides. Furthermore, these cross-reactive responses show the desired proinflammatory phenotype and induction of tumor cell lysis (Fig. 6G).
Figure 6.

D-neopeptide–specific CD4+ T cells cross-react with the corresponding N-peptides. A, Procedure of generating D-class II-neopeptide–specific CD4+ TCCs. B, CD4+ TCCs were stimulated with 5
 of the indicated individual peptides for 3 days, and then proliferation was measured by 3H-thymidine incorporation assay. Number of CD4+ TCCs responding to II_GBM_14_1, II_GBM_12_1, or II_GBM_24_1. C–E, CD4+ TCCs were stimulated with 5
of the indicated individual peptides for 3 days, and then proliferation was measured by 3H-thymidine incorporation assay. Number of CD4+ TCCs responding to II_GBM_14_1, II_GBM_12_1, or II_GBM_24_1. C–E, CD4+ TCCs were stimulated with 5
 of the indicated individual peptides for 3 days. Three replicate wells were tested for proliferation, and responses were depicted as SI. C, Three different patterns of cross-reactivity between N-peptides and D-neopeptides for CD4+ TCCs: CD4+ TCCs with lower avidity for N-peptides as compared with D-neopeptides (lower avidity); CD4+ TCCs with similar avidity for N-peptides and D-neopeptides (similar avidity); and CD4+ TCCs with higher avidity for N-peptides as compared with D-neopeptides (higher avidity). D, Concentrations of IFNγ in the supernatant were detected by ELISA. E, Dose–response curves of CD4+ TCCs to N-peptides and D-neopeptides. F, Comparison of the cytotoxicity of CD4+ TCCs to tumor cells after stimulation with N-peptides or D-neopeptides for 4 days. Three replicate wells were set up for each condition. G, Working model of how designed D-neopeptides elicit cross-reactive CD4+ T cells that also respond to the natural neopeptides and unmutated N-peptides and mediate tumor cell lysis. (#) indicates the peptide that was used in the first to fourth vaccinations.
of the indicated individual peptides for 3 days. Three replicate wells were tested for proliferation, and responses were depicted as SI. C, Three different patterns of cross-reactivity between N-peptides and D-neopeptides for CD4+ TCCs: CD4+ TCCs with lower avidity for N-peptides as compared with D-neopeptides (lower avidity); CD4+ TCCs with similar avidity for N-peptides and D-neopeptides (similar avidity); and CD4+ TCCs with higher avidity for N-peptides as compared with D-neopeptides (higher avidity). D, Concentrations of IFNγ in the supernatant were detected by ELISA. E, Dose–response curves of CD4+ TCCs to N-peptides and D-neopeptides. F, Comparison of the cytotoxicity of CD4+ TCCs to tumor cells after stimulation with N-peptides or D-neopeptides for 4 days. Three replicate wells were set up for each condition. G, Working model of how designed D-neopeptides elicit cross-reactive CD4+ T cells that also respond to the natural neopeptides and unmutated N-peptides and mediate tumor cell lysis. (#) indicates the peptide that was used in the first to fourth vaccinations.
Discussion
Recent peptide vaccination approaches to treat GBM or other gliomas have shown promising results with respect to inducing intratumoral T-cell responses (9–11), which suggest that individualized vaccination approaches can increase antiglioma immune responses, and, although preliminary, may also improve clinical outcomes. On this basis, we hypothesized that the introduction of designed AA modifications into unmutated TAAs may increase immunogenicity against tumor self-peptides, particularly in a tumor with low mutational load, and improve the effect of treatment. This study confirms that vaccination with designed neopeptides could be an interesting strategy to increase the number of tumor targets in cold tumors and that it elicits robust intratumoral CD4+ T-cell responses not only against the designed neopeptides, but also the unmutated targets expressed by the tumor.
Although we observed a gradual increase of memory T-cell responses with successive vaccination rounds in the peripheral blood, T-cell responses remained modest, and therefore were only useful to a limited extent with respect to assessing which peptides are optimal for the first and successive rounds of vaccination. This may again in part be due to the readout, that is, proliferation, which is less sensitive regarding detection of low-frequency autoreactive T cells than for example ELISPOT testing. Furthermore, memory T cells with specificity for v-N-peptides and v-D-neopeptides may have homed into the tumor, which is supported by the IMC data and the very strong responses of TILs.
Furthermore, while it is not clear why the induction of CD8+ T-cell proliferative responses was weaker than those of CD4+ T cells, several factors may have contributed. CD8+ T cells proliferate less vigorously than CD4+ T cells, and antigen-specific proliferation as a readout to test responses is likely not optimal. While the novel, water-soluble adjuvant XS15 had previously been shown to induce robust CD4+ and CD8+ T-cell responses (39), in our study, the effects on CD4+ T cells were stronger than on CD8+ T cells. Route of administration, peptide doses, and the intervals of administration are other important factors. Despite these limitations, our concept of introducing designed AA mutations into tumor-associated peptides as outlined above was remarkably effective with respect to inducing strong intratumoral CD4+ T-cell responses to 100% of the v-class II-peptides.
At the level of bulk TILs, activation with D-neopeptides led to efficient lysis of autologous tumor cells. The examination of bulk TILs, however, did not permit to assess whether the responses to both N-peptides and D-neopeptides were due to different T cells in the bulk populations responding to one or both peptides or due to cross-reactivity at the level of single TCCs. The latter case was our objective based on prior experience with the APL in the MS trial (24). To address this aspect, we established over 100 CD4+ TCCs from bulk TILs with three exemplary D-neopeptides that had been included in the vaccination and assessed their cross-reactivity against both D-neopeptides and corresponding N-peptides. These experiments led to several interesting observations. In most cases, TCCs recognized both the N-peptides and D-neopeptides albeit with different strengths fulfilling the most important requirement. Specifically, the goal was to design a mutated or foreign peptide, which presumably should be more immunogenic than the unmutated self-peptide and activate an immune response that is also directed against the unmutated antigen of the tumor. We encountered three different reactivity patterns: lower, equal or even better activation by the N-peptides versus the D-neopeptides. These reactivity patterns are therefore almost identical to what we found previously with the APL peptide that had been used in the MS trial to induce downmodulation of disease activity (24). Furthermore, it is promising that the functional responses included not only activation of proliferation, but also the release of proinflammatory cytokines and cytolysis of autologous tumor cells upon activation with both D-neopeptide and N-peptides.
We replaced putative TCR contact positions by AA with either conservative (similar), neutral, or nonconservative (different in charge, size, and side chains) physicochemical characteristics. Of note, some D-neopeptides stimulated bulk TILs efficiently and much better than the N-peptides. However, the number of D-neopeptides and the different types of mutations were too small to systematically compare the efficiency of conservative versus nonconservative AA replacements. In both cases, we observed low/moderate or strong stimulatory capacity.
An important question that remains is how to apply this complicated and multipronged approach for personalized tumor vaccination to more patients and a broader range of cancers. Several prerequisites would need to be met. The different sequencing steps, HLA genotyping, target predictions, and choices of peptides including the design of the neopeptides would need to be performed rapidly in patients with GBM, pancreatic cancer, or other tumors with rapid progression. Next, GMP peptides would have to be used and need to be synthesized fast, at large numbers and at an acceptable cost to accommodate clinical trials with larger patient numbers. Furthermore, it would be important to optimize peptide doses, vaccination intervals, and adjuvants, for example, the inclusion of GM-CSF in addition to currently available adjuvants. We anticipate that most of these steps can be overcome either already now or in the near future.
Supplementary Material
Supplementary Table 1. Somatic mutations of the patient's glioblastoma; Supplementary Table 2. See Excel File; Supplementary Table 3. Literature-derived, glioblastoma-associated TAAs; Supplementary Table 4. See Excel File; Supplementary Table 5. Over-/highly expressed genes that were chosen for the design of vaccine peptides; Supplementary Table 6. Peptide cocktails for vaccination; Supplementary Table 7. Antibodies for imaging mass cytometry.
Over-/highly expressed genes in the primary glioblastoma when compared with glioblastoma cohort in TCGA Database.
Predicted TSA/TAA-derived N-peptides and D-neopeptides
Schematic outline of the personalized peptide vaccination. (A) Timeline of clinical events for a patient with glioblastoma in whom we applied a highly personalized peptide vaccination. I. Vaccine peptide design. Surgically resected primary glioblastoma and PBMCs were collected. Somatic mutants and over-/highly expressed genes in glioblastoma were identified as TSAs and TAAs by WES and RNA-seq, and the HLA genotype were identified with PBMCs by DNA sequencing. Potential vaccine peptides were predicted and selected from TSAs and TAAs based on HLA binding prediction. Peptide cocktails containing ~25 peptides for HLA class I or II molecules were prepared for vaccination. II. Peptide cocktail vaccination and testing. The patient was s.c. vaccinated with the peptide cocktails, and Imiquimod or XS15 + MontanideTM were used as the adjuvant. Meanwhile, the patient received treatment with pembrolizumab and bevacizumab. No dexamethasone was given throughout the periods of vaccination. PBMCs were collected after vaccination at the indicated time points and tested with vaccine peptides to examine the effect of immunotherapy. Surgically resected recurrent glioblastoma was collected after four vaccinations, and tumor-infiltrating lymphocytes (TILs) were isolated and tested with vaccine peptides. (B) HLA genotypes of the patient with glioblastoma, including HLA-A, HLA-B, HLA-DR, HLA-DP, and HLA-DQ alleles. n.a. indicates not available.
{kind=link}
PBMCs collected before vaccination show low/no response to candidate vaccine peptides. (A) The nomenclature of the N-peptides and D-neopeptides. I or II indicate if the peptides were chosen/designed for HLA class I or -II binding; MUT/RNA/GBM indicates a naturally mutated peptide (MUT), derivation from the over-/highly expressed genes (RNA) or from known glioblastoma (GBM) targets; X indicates the number of the peptide; 0 or 1 indicate if the peptides had been artificially mutated (1) or not (0); (’) indicates that peptides contained the naturally mutated amino acid. (B) PBMCs collected before vaccination were stimulated with 4 μg/mL Tetanus toxoid or 2 μM CEF II peptide pool for 7 days, and proliferation was measured by ³H-thymidine incorporation assay. The proliferation strength is depicted as counts per minute (cpm). In each group, the N-peptides and their corresponding D-neopeptides are included, and the designed mutated positions are highlighted in the red box. The blue dotted line indicates the mean value of the no peptide control. The red dotted line indicates the mean value plus three standard deviations of the no peptide control. Values above the red dotted line were considered positive. (C) PBMCs collected before vaccination were stimulated with 5 μM predicted class I-peptides, including N-peptides and D-neopeptides, for 7 days, and proliferation was measured by ³H-thymidine incorporation assay. 10-15 replicate wells were tested for proliferation, and responses were depicted as cpm. (D) PBMCs collected before vaccination were stimulated with 5 μM predicted class II-peptides, including N-peptides and D-neopeptides, for 7 days, and proliferation was measured by ³H-thymidine incorporation assay. 10-15 replicate wells were tested for proliferation, and responses were depicted as cpm.
{kind=link}
Peripheral blood T cell responses to v-peptides increase with repetitive vaccination. (A) PBMCs were collected 3 weeks after the 2nd peptide cocktail vaccination, and CD45RA-negative PBMCs were stimulated with 4 μg/mL Tetanus toxoid, 2 μM CEF II peptide pool or 5 μM v-peptides for 7 days. 5-10 replicate wells were tested for proliferation, and proliferation measured by 3H-thymidine incorporation assay. CD45RA-negative PBMCs collected before vaccination were used as reference. The proliferation strength is depicted as stimulation index (SI). The red dotted line indicates a stimulatory response of SI = 2, and values above the red dotted line were considered positive. (#) indicates the peptide that was used in the vaccinations. (B) PBMCs were collected 5 weeks after the 3rd peptide cocktail vaccination, and CD45RA-negative PBMCs were stimulated with 4 μg/mL Tetanus toxoid, 2 μM CEF II peptide pool or 5 μM v-peptides for 7 days. 5-10 replicate wells were tested for proliferation, and proliferation was measured by 3H-thymidine incorporation assay. CD45RA-negative PBMCs collected before vaccination were used as the control. Data are expressed as mean ± SEM, and p values were determined using an unpaired t-test. * p < 0.05, ** p < 0.01, *** p < 0.001.
{kind=link}
Increased T lymphocyte infiltration in the recurrent compared to the primary tumor. (A) CD3 immunohistochemical staining in the primary and recurrent tumor. (B) Three fields of view were selected and counted CD3+ T cells in both perivascular and peritumoral areas of the primary and recurrent tumors using ImageJ. Scale bars are indicated in the figures.
{kind=link}
Activated T lymphocytes in recurrent tumor interact with macrophages. (A) Expression of T cell activating markers HLA-DR and Tim-3 in the perivascular areas and peritumoral areas of the primary and recurrent tumor was detected by IMC. (B) CD3-CD7+ NK cells infiltrated in the perivascular areas and peritumoral areas of the primary and recurrent tumor was detected by IMC. (C) Pro-inflammatory CD11c+ CD68+ macrophages, which had infiltrated the perivascular areas and peritumoral areas of the primary and recurrent tumor, were detected by IMC. (D) Colocalization analysis of CD68+ macrophages and CD3+ T cells in the perivascular areas and peritumoral areas of the primary and recurrent tumor were detected by IMC. Scale bars are indicated in the figures.
{kind=link}
TILs do not respond to N-class I-peptides. TILs were stimulated with 5- or 10 μM N-class I-peptides for 5 days. 5 replicate wells were tested for proliferation, and proliferation was measured by 3H-thymidine incorporation assay. The proliferation strength is depicted as SI. (#) indicates the peptide that was used in the 1st to 4th vaccinations. Data are expressed as mean ± SEM.
{kind=link}
Designed mutations increase the stimulatory strength of D-neopeptides above N-peptides. (A) TILs were stimulated with 5 μM class I-peptides, including N-peptides and D-neopeptides, for 5 days. 5 replicate wells were tested for proliferation, and proliferation was measured by 3H-thymidine incorporation assay. In each group, the N-peptides and their corresponding D-neopeptides are included, and the designed mutated positions are highlighted in the red box. The blue dotted line indicates the mean value of the no peptide control. The red dotted line indicates the mean value plus three standard deviations of the no peptide control. Values above the red dotted line were considered positive. (#) indicates the peptide that was used in the 1st to 4th vaccinations. (B) TILs were stimulated with 5 μM class II-peptides, including N-peptides and D-neopeptides, for 5 days. 5 replicate wells were tested for proliferation, and proliferation was measured by 3H-thymidine incorporation assay.
{kind=link}
Vaccinated D-neopeptides activate tumor-infiltrating antitumor T cells. (A) Circos plots provide an overview of the frequencies of Vβ-Jβ pairing in the primary and recurrent tumors. (B) Surface TCR β chain expression of the D-neopeptide-specific CD4+ TCCs was analyzed using FACs.
{kind=link}
Acknowledgments
We thank the patient for his interest to participate in this one-patient trial, Magdalena Foege (Neuroimmunology and MS Research, Neurology Clinic, University Hospital Zurich) for assisting with preparation of the regulatory documentation, Karl-Heinz Wiesmüller for preparing and providing the adjuvant XS15, and Hans-Georg Rammensee for discussing the vaccination strategy and optimal use of adjuvants. The project was supported by a Swiss National Science Foundation grant (SNF 320030_153213, to R. Martin and M. Weller), by a European Research Council Advanced grant (ERC-2013- ADG 340733, to R. Martin), and by the Clinical Research Priority Projects (CRPP) of the University of Zurich: the project of MS heterogeneity- and Precision-MS to R. Martin and the project of ImmunoCure to P. Roth and M. Weller.
The publication costs of this article were defrayed in part by the payment of publication fees. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Footnotes
Note: Supplementary data for this article are available at Clinical Cancer Research Online (http://clincancerres.aacrjournals.org/).
Authors' Disclosures
J. Wang reports a patent for EP22185038.1 pending. T. Weiss reports personal fees from Philogen and non-financial support from Cellis outside the submitted work. M.C. Neidert reports grants from Novocure outside the submitted work. N.C. Toussaint reports other support from University Hospital Zurich during the conduct of the study. B. Schrörs reports personal fees from BioNTech SE during the conduct of the study, as well as personal fees from TRON gGmbH and BioNTech SE outside the submitted work; in addition, B. Schrörs has a patent for WO2017194170A1;WO2017194610A1 licensed, a patent for WO2016128060A1;WO2016128376A1 licensed, and a patent for WO2018224162A1;WO2018224406A1 licensed. C.J. Wu reports other support from BioNTech and Pharmacyclics outside the submitted work. L. Regli reports personal fees from BBraun, Germany outside the submitted work. M. Weller reports grants from Quercis and Versameb, as well as personal fees from Novocure, Bayer, Merck (EMD), Novartis, Orbus, and Philogen outside the submitted work. P. Roth reports grants from University of Zurich, CRPP ImmunoCure during the conduct of the study; P. Roth also reports personal fees from BMS, Boehringer Ingelheim, Debiopharm, Medac, Novartis, QED, Roche, and Midatech Pharma, as well as grants and personal fees from MSD and Novocure outside the submitted work. R. Martin reports grants from Biogen, Novartis, Roche, and Third Rock outside the submitted work; a patent for EP22185038.1 pending; advisory roles and lectures for Roche, Novartis, Biogen, Genzyme, Neuway, CellProtect, and Third Rock; co-founder of Abata; and co-founder and co-owner of Cellerys. Finally, R. Martin is co-holder on patents of: daclizumab in MS, JCV VP1 for vaccination against PML; JCV-specific neutralizing antibodies to treat PML; antigen-specific tolerization with peptide-coupled cells and novel autoantigens in MS. No disclosures were reported by the other authors.
Authors' Contributions
J. Wang: Investigation, methodology, writing–original draft, project administration. T. Weiss: Data curation, formal analysis. M.C. Neidert: Resources. N.C. Toussaint: Formal analysis. R. Naghavian: Investigation. C. Sellés Moreno: Formal analysis, investigation. M. Foege: Data curation. P. Tomas Ojer: Investigation. G. Medici: Resources. I. Jelcic: Data curation. D. Schulz: Investigation, visualization. E. Rushing: Investigation, visualization. S. Dettwiler: Investigation, visualization. B. Schrörs: Resources. J.H. Shin: Resources. R. McKay: Resources. C.J. Wu: Supervision. A. Lutterotti: Supervision. M. Sospedra: Supervision, investigation. H. Moch: Resources, investigation. E.F. Greiner: Resources, data curation. B. Bodenmiller: Validation, investigation. L. Regli: Formal analysis. M. Weller: Resources. P. Roth: Resources, data curation, formal analysis. R. Martin: Conceptualization, supervision, funding acquisition, writing–original draft, project administration.
References
- 1. Weller M, van den Bent M, Preusser M, Le Rhun E, Tonn JC, Minniti G, et al. EANO guidelines on the diagnosis and treatment of diffuse gliomas of adulthood. Nat Rev Clin Oncol 2021;18:170–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Wen PY, Weller M, Lee EQ, Alexander BM, Barnholtz-Sloan JS, Barthel FP, et al. Glioblastoma in adults: a Society for Neuro-Oncology (SNO) and European Society of Neuro-Oncology (EANO) consensus review on current management and future directions. Neuro Oncol 2020;22:1073–113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Preusser M, Lim M, Hafler DA, Reardon DA, Sampson JH. Prospects of immune checkpoint modulators in the treatment of glioblastoma. Nat Rev Neurol 2015;11:504–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Reardon DA, Brandes AA, Omuro A, Mulholland P, Lim M, Wick A, et al. Effect of nivolumab vs bevacizumab in patients with recurrent glioblastoma: the CheckMate 143 phase 3 randomized clinical trial. JAMA Oncol 2020;6:1003–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Fadul CE, Fisher JL, Hampton TH, Lallana EC, Li Z, Gui J, et al. Immune response in patients with newly diagnosed glioblastoma multiforme treated with intranodal autologous tumor lysate-dendritic cell vaccination after radiation chemotherapy. J Immunother 2011;34:382–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Inoges S, Tejada S, de Cerio AL, Gallego Perez-Larraya J, Espinos J, Idoate MA, et al. A phase II trial of autologous dendritic cell vaccination and radiochemotherapy following fluorescence-guided surgery in newly diagnosed glioblastoma patients. J Transl Med 2017;15:104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Phuphanich S, Wheeler CJ, Rudnick JD, Mazer M, Wang H, Nuno MA, et al. Phase I trial of a multi-epitope-pulsed dendritic cell vaccine for patients with newly diagnosed glioblastoma. Cancer Immunol Immunother 2013;62:125–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Vik-Mo EO, Nyakas M, Mikkelsen BV, Moe MC, Due-Tonnesen P, Suso EM, et al. Therapeutic vaccination against autologous cancer stem cells with mRNA-transfected dendritic cells in patients with glioblastoma. Cancer Immunol Immunother 2013;62:1499–509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Hilf N, Kuttruff-Coqui S, Frenzel K, Bukur V, Stevanovic S, Gouttefangeas C, et al. Actively personalized vaccination trial for newly diagnosed glioblastoma. Nature 2019;565:240–5. [DOI] [PubMed] [Google Scholar]
- 10. Keskin DB, Anandappa AJ, Sun J, Tirosh I, Mathewson ND, Li S, et al. Neoantigen vaccine generates intratumoral T cell responses in phase Ib glioblastoma trial. Nature 2019;565:234–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Platten M, Bunse L, Wick A, Bunse T, Le Cornet L, Harting I, et al. A vaccine targeting mutant IDH1 in newly diagnosed glioma. Nature 2021;592:463–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Romero P, Banchereau J, Bhardwaj N, Cockett M, Disis ML, Dranoff G, et al. The human vaccines project: a roadmap for cancer vaccine development. Sci Transl Med 2016;8:334ps9. [DOI] [PubMed] [Google Scholar]
- 13. Hunder NN, Wallen H, Cao J, Hendricks DW, Reilly JZ, Rodmyre R, et al. Treatment of metastatic melanoma with autologous CD4+ T cells against NY-ESO-1. N Engl J Med 2008;358:2698–703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Quezada SA, Simpson TR, Peggs KS, Merghoub T, Vider J, Fan X, et al. Tumor-reactive CD4(+) T cells develop cytotoxic activity and eradicate large established melanoma after transfer into lymphopenic hosts. J Exp Med 2010;207:637–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Kreiter S, Vormehr M, van de Roemer N, Diken M, Lower M, Diekmann J, et al. Mutant MHC class II epitopes drive therapeutic immune responses to cancer. Nature 2015;520:692–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Costa-Nunes C, Cachot A, Bobisse S, Arnaud M, Genolet R, Baumgaertner P, et al. High-throughput screening of human tumor antigen-specific CD4 T cells, including neoantigen-reactive T cells. Clin Cancer Res 2019;25:4320–31. [DOI] [PubMed] [Google Scholar]
- 17. Dobrzanski MJ. Expanding roles for CD4 T cells and their subpopulations in tumor immunity and therapy. Front Oncol 2013;3:63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Melief CJ, van der Burg SH. Immunotherapy of established (pre)malignant disease by synthetic long peptide vaccines. Nat Rev Cancer 2008;8:351–60. [DOI] [PubMed] [Google Scholar]
- 19. Antonarelli G, Corti C, Tarantino P, Ascione L, Cortes J, Romero P, et al. Therapeutic cancer vaccines revamping: technology advancements and pitfalls. Ann Oncol 2021;32:1537–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Schumacher T, Bunse L, Pusch S, Sahm F, Wiestler B, Quandt J, et al. A vaccine targeting mutant IDH1 induces antitumour immunity. Nature 2014;512:324–7. [DOI] [PubMed] [Google Scholar]
- 21. Kumai T, Kobayashi H, Harabuchi Y, Celis E. Peptide vaccines in cancer-old concept revisited. Curr Opin Immunol 2017;45:1–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Dutoit V, Migliorini D, Ranzanici G, Marinari E, Widmer V, Lobrinus JA, et al. Antigenic expression and spontaneous immune responses support the use of a selected peptide set from the IMA950 glioblastoma vaccine for immunotherapy of grade II and III glioma. Oncoimmunology 2018;7:e1391972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Dutoit V, Herold-Mende C, Hilf N, Schoor O, Beckhove P, Bucher J, et al. Exploiting the glioblastoma peptidome to discover novel tumour-associated antigens for immunotherapy. Brain 2012;135:1042–54. [DOI] [PubMed] [Google Scholar]
- 24. Bielekova B, Goodwin B, Richert N, Cortese I, Kondo T, Afshar G, et al. Encephalitogenic potential of the myelin basic protein peptide (amino acids 83–99) in multiple sclerosis: results of a phase II clinical trial with an altered peptide ligand. Nat Med 2000;6:1167–75. [DOI] [PubMed] [Google Scholar]
- 25. Li H. A statistical framework for SNP calling, mutation discovery, association mapping and population genetical parameter estimation from sequencing data. Bioinformatics 2011;27:2987–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. McKenna A, Hanna M, Banks E, Sivachenko A, Cibulskis K, Kernytsky A, et al. The genome analysis toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res 2010;20:1297–303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Saunders CT, Wong WS, Swamy S, Becq J, Murray LJ, Cheetham RK. Strelka: accurate somatic small-variant calling from sequenced tumor-normal sample pairs. Bioinformatics 2012;28:1811–7. [DOI] [PubMed] [Google Scholar]
- 28. Cibulskis K, Lawrence MS, Carter SL, Sivachenko A, Jaffe D, Sougnez C, et al. Sensitive detection of somatic point mutations in impure and heterogeneous cancer samples. Nat Biotechnol 2013;31:213–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Koboldt DC, Zhang Q, Larson DE, Shen D, McLellan MD, Lin L, et al. VarScan 2: somatic mutation and copy number alteration discovery in cancer by exome sequencing. Genome Res 2012;22:568–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Jurtz V, Paul S, Andreatta M, Marcatili P, Peters B, Nielsen M. NetMHCpan-4.0: improved peptide-MHC class I interaction predictions integrating eluted ligand and peptide binding affinity data. J Immunol 2017;199:3360–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Wang P, Sidney J, Kim Y, Sette A, Lund O, Nielsen M, et al. Peptide binding predictions for HLA DR, DP and DQ molecules. BMC Bioinformatics 2010;11:568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Sturniolo T, Bono E, Ding J, Raddrizzani L, Tuereci O, Sahin U, et al. Generation of tissue-specific and promiscuous HLA ligand databases using DNA microarrays and virtual HLA class II matrices. Nat Biotechnol 1999;17:555–61. [DOI] [PubMed] [Google Scholar]
- 33. Reynisson B, Alvarez B, Paul S, Peters B, Nielsen M. NetMHCpan-4.1 and NetMHCIIpan-4.0: improved predictions of MHC antigen presentation by concurrent motif deconvolution and integration of MS MHC eluted ligand data. Nucleic Acids Res 2020;48:W449–W54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Rammensee H, Bachmann J, Emmerich NP, Bachor OA, Stevanovic S. SYFPEITHI: database for MHC ligands and peptide motifs. Immunogenetics 1999;50:213–9. [DOI] [PubMed] [Google Scholar]
- 35. Jackson HW, Fischer JR, Zanotelli VRT, Ali HR, Mechera R, Soysal SD, et al. The single-cell pathology landscape of breast cancer. Nature 2020;578:615–20. [DOI] [PubMed] [Google Scholar]
- 36. Krzywinski M, Schein J, Birol I, Connors J, Gascoyne R, Horsman D, et al. Circos: an information aesthetic for comparative genomics. Genome Res 2009;19:1639–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Schork NJ. Personalized medicine: time for one-person trials. Nature 2015;520:609–11. [DOI] [PubMed] [Google Scholar]
- 38. Nair SK, De Leon G, Boczkowski D, Schmittling R, Xie W, Staats J, et al. Recognition and killing of autologous, primary glioblastoma tumor cells by human cytomegalovirus pp65-specific cytotoxic T cells. Clin Cancer Res 2014;20:2684–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Rammensee HG, Wiesmuller KH, Chandran PA, Zelba H, Rusch E, Gouttefangeas C, et al. A new synthetic toll-like receptor 1/2 ligand is an efficient adjuvant for peptide vaccination in a human volunteer. J Immunother Cancer 2019;7:307. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Table 1. Somatic mutations of the patient's glioblastoma; Supplementary Table 2. See Excel File; Supplementary Table 3. Literature-derived, glioblastoma-associated TAAs; Supplementary Table 4. See Excel File; Supplementary Table 5. Over-/highly expressed genes that were chosen for the design of vaccine peptides; Supplementary Table 6. Peptide cocktails for vaccination; Supplementary Table 7. Antibodies for imaging mass cytometry.
Over-/highly expressed genes in the primary glioblastoma when compared with glioblastoma cohort in TCGA Database.
Predicted TSA/TAA-derived N-peptides and D-neopeptides
Schematic outline of the personalized peptide vaccination. (A) Timeline of clinical events for a patient with glioblastoma in whom we applied a highly personalized peptide vaccination. I. Vaccine peptide design. Surgically resected primary glioblastoma and PBMCs were collected. Somatic mutants and over-/highly expressed genes in glioblastoma were identified as TSAs and TAAs by WES and RNA-seq, and the HLA genotype were identified with PBMCs by DNA sequencing. Potential vaccine peptides were predicted and selected from TSAs and TAAs based on HLA binding prediction. Peptide cocktails containing ~25 peptides for HLA class I or II molecules were prepared for vaccination. II. Peptide cocktail vaccination and testing. The patient was s.c. vaccinated with the peptide cocktails, and Imiquimod or XS15 + MontanideTM were used as the adjuvant. Meanwhile, the patient received treatment with pembrolizumab and bevacizumab. No dexamethasone was given throughout the periods of vaccination. PBMCs were collected after vaccination at the indicated time points and tested with vaccine peptides to examine the effect of immunotherapy. Surgically resected recurrent glioblastoma was collected after four vaccinations, and tumor-infiltrating lymphocytes (TILs) were isolated and tested with vaccine peptides. (B) HLA genotypes of the patient with glioblastoma, including HLA-A, HLA-B, HLA-DR, HLA-DP, and HLA-DQ alleles. n.a. indicates not available.
PBMCs collected before vaccination show low/no response to candidate vaccine peptides. (A) The nomenclature of the N-peptides and D-neopeptides. I or II indicate if the peptides were chosen/designed for HLA class I or -II binding; MUT/RNA/GBM indicates a naturally mutated peptide (MUT), derivation from the over-/highly expressed genes (RNA) or from known glioblastoma (GBM) targets; X indicates the number of the peptide; 0 or 1 indicate if the peptides had been artificially mutated (1) or not (0); (’) indicates that peptides contained the naturally mutated amino acid. (B) PBMCs collected before vaccination were stimulated with 4 μg/mL Tetanus toxoid or 2 μM CEF II peptide pool for 7 days, and proliferation was measured by ³H-thymidine incorporation assay. The proliferation strength is depicted as counts per minute (cpm). In each group, the N-peptides and their corresponding D-neopeptides are included, and the designed mutated positions are highlighted in the red box. The blue dotted line indicates the mean value of the no peptide control. The red dotted line indicates the mean value plus three standard deviations of the no peptide control. Values above the red dotted line were considered positive. (C) PBMCs collected before vaccination were stimulated with 5 μM predicted class I-peptides, including N-peptides and D-neopeptides, for 7 days, and proliferation was measured by ³H-thymidine incorporation assay. 10-15 replicate wells were tested for proliferation, and responses were depicted as cpm. (D) PBMCs collected before vaccination were stimulated with 5 μM predicted class II-peptides, including N-peptides and D-neopeptides, for 7 days, and proliferation was measured by ³H-thymidine incorporation assay. 10-15 replicate wells were tested for proliferation, and responses were depicted as cpm.
Peripheral blood T cell responses to v-peptides increase with repetitive vaccination. (A) PBMCs were collected 3 weeks after the 2nd peptide cocktail vaccination, and CD45RA-negative PBMCs were stimulated with 4 μg/mL Tetanus toxoid, 2 μM CEF II peptide pool or 5 μM v-peptides for 7 days. 5-10 replicate wells were tested for proliferation, and proliferation measured by 3H-thymidine incorporation assay. CD45RA-negative PBMCs collected before vaccination were used as reference. The proliferation strength is depicted as stimulation index (SI). The red dotted line indicates a stimulatory response of SI = 2, and values above the red dotted line were considered positive. (#) indicates the peptide that was used in the vaccinations. (B) PBMCs were collected 5 weeks after the 3rd peptide cocktail vaccination, and CD45RA-negative PBMCs were stimulated with 4 μg/mL Tetanus toxoid, 2 μM CEF II peptide pool or 5 μM v-peptides for 7 days. 5-10 replicate wells were tested for proliferation, and proliferation was measured by 3H-thymidine incorporation assay. CD45RA-negative PBMCs collected before vaccination were used as the control. Data are expressed as mean ± SEM, and p values were determined using an unpaired t-test. * p < 0.05, ** p < 0.01, *** p < 0.001.
Increased T lymphocyte infiltration in the recurrent compared to the primary tumor. (A) CD3 immunohistochemical staining in the primary and recurrent tumor. (B) Three fields of view were selected and counted CD3+ T cells in both perivascular and peritumoral areas of the primary and recurrent tumors using ImageJ. Scale bars are indicated in the figures.
Activated T lymphocytes in recurrent tumor interact with macrophages. (A) Expression of T cell activating markers HLA-DR and Tim-3 in the perivascular areas and peritumoral areas of the primary and recurrent tumor was detected by IMC. (B) CD3-CD7+ NK cells infiltrated in the perivascular areas and peritumoral areas of the primary and recurrent tumor was detected by IMC. (C) Pro-inflammatory CD11c+ CD68+ macrophages, which had infiltrated the perivascular areas and peritumoral areas of the primary and recurrent tumor, were detected by IMC. (D) Colocalization analysis of CD68+ macrophages and CD3+ T cells in the perivascular areas and peritumoral areas of the primary and recurrent tumor were detected by IMC. Scale bars are indicated in the figures.
TILs do not respond to N-class I-peptides. TILs were stimulated with 5- or 10 μM N-class I-peptides for 5 days. 5 replicate wells were tested for proliferation, and proliferation was measured by 3H-thymidine incorporation assay. The proliferation strength is depicted as SI. (#) indicates the peptide that was used in the 1st to 4th vaccinations. Data are expressed as mean ± SEM.
Designed mutations increase the stimulatory strength of D-neopeptides above N-peptides. (A) TILs were stimulated with 5 μM class I-peptides, including N-peptides and D-neopeptides, for 5 days. 5 replicate wells were tested for proliferation, and proliferation was measured by 3H-thymidine incorporation assay. In each group, the N-peptides and their corresponding D-neopeptides are included, and the designed mutated positions are highlighted in the red box. The blue dotted line indicates the mean value of the no peptide control. The red dotted line indicates the mean value plus three standard deviations of the no peptide control. Values above the red dotted line were considered positive. (#) indicates the peptide that was used in the 1st to 4th vaccinations. (B) TILs were stimulated with 5 μM class II-peptides, including N-peptides and D-neopeptides, for 5 days. 5 replicate wells were tested for proliferation, and proliferation was measured by 3H-thymidine incorporation assay.
Vaccinated D-neopeptides activate tumor-infiltrating antitumor T cells. (A) Circos plots provide an overview of the frequencies of Vβ-Jβ pairing in the primary and recurrent tumors. (B) Surface TCR β chain expression of the D-neopeptide-specific CD4+ TCCs was analyzed using FACs.
Data Availability Statement
The data generated in this study are not publicly available due to information that could compromise patient consent but are available upon reasonable request from the corresponding author.