

Abstract
Despite a rapid expansion in the number of documented viruses following the advent of metagenomic sequencing, the identification and annotation of highly divergent RNA viruses remain challenging, particularly from poorly characterized hosts and environmental samples. Protein structures are more conserved than primary sequence data, such that structure-based comparisons provide an opportunity to reveal the viral ‘dusk matter’: viral sequences with low, but detectable, levels of sequence identity to known viruses with available protein structures. Here, we present a new open computational resource—RdRp-scan—that contains a standardized bioinformatic toolkit to identify and annotate divergent RNA viruses in metagenomic sequence data based on the detection of RNA-dependent RNA polymerase (RdRp) sequences. By combining RdRp-specific hidden Markov models (HMMs) and structural comparisons, we show that RdRp-scan can efficiently detect RdRp sequences with identity levels as low as 10 per cent to those from known viruses and not identifiable using standard sequence-to-sequence comparisons. In addition, to facilitate the annotation and placement of newly detected and divergent virus-like sequences into the diversity of RNA viruses, RdRp-scan provides new custom and curated databases of viral RdRp sequences and core motifs, as well as pre-built RdRp multiple sequence alignments. In parallel, our analysis of the sequence diversity detected by the RdRp-scan revealed that while most of the taxonomically unassigned RdRps fell into pre-established clusters, some fell into potentially new orders of RNA viruses related to the Wolframvirales and Tolivirales. Finally, a survey of the conserved A, B, and C RdRp motifs within the RdRp-scan sequence database revealed additional variations of both sequence and position that might provide new insights into the structure, function, and evolution of viral polymerases.
Keywords: viral dusk matter, metagenomics, evolution, phylogeny, RNA-dependent RNA polymerase, HMM-based homology detection
1. Introduction
The explosion of viral metagenomic projects and associated high-throughput sequencing data over the last decade have paved the way for a reappraisal of the diversity and evolution of RNA viruses (Shi et al. 2016; Krishnamurthy and Wang 2017; Wolf et al. 2020). The expansion of the RNA virus discovery into overlooked habitats, environments, and organisms has led to the recognition that RNA viruses are ubiquitous and likely infect all types of cellular organisms (Culley, Lang, and Suttle 2006; Bolduc et al. 2012; Shi et al. 2016; Charon et al. 2019; Sutela et al. 2020; Wolf et al. 2020; Charon, Murray, and Holmes 2021). Although there is an understandable focus on the potential health impact of RNA viruses, their importance goes far beyond their role as pathogenic agents in humans, domestic animals, and livestock. For example, investigating RNA virus diversity in marine habitats has revealed their importance in fundamental ecological and biogeochemical processes (Suttle 2005, 2007). Attempts to illuminate the RNA virus world are also expected to provide fundamental insights into the origins of RNA viruses and the long-standing evolutionary processes that have shaped their diversity.
RNA viruses exhibit evolutionary rates ∼104 to 106 higher than those of cellular organisms (Duffy, Shackelton, and Holmes 2008; Sanjuán et al. 2010) and do not possess universally conserved and easily interpretable gene sequences equivalent to the 16S and 18S ribosomal RNAs (rRNAs) used to classify other microorganisms The identification of RNA viruses from metagenomic data sets largely relies on sequence similarity-based comparisons. While such studies have enriched the RNA virus phylogeny, they are only able to detect sequences with relatively high levels of sequence similarity to existing sequences, with detection usually limited to proteins sharing at least 30 per cent sequence identity (Rost 1999). As a consequence, it is highly likely that a vast world of undiscovered RNA virus sequences—the so-called ‘viral dark matter’ (Youle, Haynes, and Rohwer 2012; Krishnamurthy and Wang 2017)—is present within the sequence data generated to date but which are too divergent to identify with currently available bioinformatic tools.
Protein structures are up to ten times more conserved than nucleotide sequences (Illergård, Ardell, and Elofsson 2009). Integrating protein structural comparison steps into metagenomic pipelines is therefore expected to greatly extend our limit of detection (Charon et al. 2020; Cobbin et al. 2021), potentially enabling the identification of sequences in the range of 10–30 per cent identity (the protein ‘twilight zone’; Rost 1999) and for which corresponding viruses might be referred to as the ‘viral dusk matter’. The viral RNA-dependent RNA polymerase (RdRp or replicase) is the most conserved protein in RNA viruses (Venkataraman, Prasad, and Selvarajan 2018; Ferrero, Falqui, and Verdaguer 2021; Mönttinen, Ravantti, and Poranen 2021). As a consequence, the RdRp is widely used as a reference gene in the estimation of RNA virus phylogenies, which in turn forms the basis of the newly established classification scheme for RNA viruses (Koonin et al. 2020). Because of its central role in genome replication and transcription, the RdRp is essential from the earliest stages of the virus infection cycle, and RdRps are relatively well characterized at both the functional and structural levels (Venkataraman, Prasad, and Selvarajan 2018; Ferrero, Falqui, and Verdaguer 2021; Mönttinen, Ravantti, and Poranen 2021).
Viral RdRp structures can be envisaged as comprising a ‘right hand’ shape with thumb, palm, and finger subdomains (Hansen, Long, and Schultz 1997; Bruenn 2003; Te Velthuis 2014; Ferrero, Falqui, and Verdaguer 2021). The thumb region helps in complex interactions and stabilization between the RNA template and free nucleotides, while the finger subdomain is involved in recognition and binding to nucleic acids and positioning the template for polymerization catalysis. The palm domain is the most conserved and forms the catalytic core of RdRp. Crucial residues and secondary structures are involved in this catalytic function, and three main amino acid motifs—denoted A, B, and C—have been identified and consistently conserved at both the primary and secondary structure levels. The catalytic A motif is formed by an invariant aspartate as well as a second (Dx2-4D) in most RdRps reported to date (Venkataraman, Prasad, and Selvarajan 2018). The second aspartate can be replaced by a lysine residue in single-strand negative-sense (ss−) RNA viruses (Venkataraman, Prasad, and Selvarajan 2018). The B motif is involved in template binding and displays a conserved glycine to potentially allow the conformational changes required to accommodate template and substrate interaction, and its sequence varies among the major groups of RNA viruses. The C motif is the most conserved. It comprises a loop that most commonly contains a triplet GDD (Gly-Asp-Asp) motif flanked by two beta strands. Along with the aspartate of the A motif, it forms the catalytic triad essential for metal ion binding and coordinating the elongation reaction of the newly synthesized strand. Despite its strong conservation, sequence variation has been reported in some viral families with, for example, a GDD or ADN (Ala-Asp-Asn) C motif preceding the A motif in the Permutotetraviridae and Birnaviridae, respectively (Gorbalenya et al. 2002; Pan, Vakharia, and Tao 2007; Ferrero et al. 2015; Ferrero, Falqui, and Verdaguer 2021). Similarly, in some other single-strand positive-sense (ss+) and ss(−)RNA viruses, the glycine in the GDD motif has been replaced by a serine (S) (Poch et al. 1990; Stevaert and Naesens 2016), while the second aspartate has been replaced by an asparagine (N) in some ss(−)RNA viruses (Poch et al. 1989, 1990).
Recent studies have emphasized the conserved nature of RdRp protein structures and associated mechanisms of catalysis among RNA viruses as a whole, reinforcing the idea that they share an ancient common ancestry despite their very low levels of amino acid sequence similarity (Venkataraman, Prasad, and Selvarajan 2018; Peersen 2019; Ferrero, Falqui, and Verdaguer 2021; Mönttinen, Ravantti, and Poranen 2021). The structural conservation of the RNA virus RdRp and the resulting ‘evolutionary fingerprints’ at the sequence level therefore provide a valuable tool to infer remote protein homologies and relatedness, such that the RdRp appears to be the candidate of choice for the structure-based exploration of the viral dusk matter.
The use of hidden Markov models (HMMs), rather than primary sequences, also provides a powerful way to identify highly divergent viral sequences (Skewes-Cox et al. 2014). By converting protein multiple sequence alignments (MSAs) into statistical models with position-specific scores, HMMs integrate evolutionary information and have been shown to be particularly powerful in the detection of remote protein homologies (Chen et al. 2018). Importantly, HMMs tend to outperform classical sequence-based profiles or sequence alignment approaches, providing a more sensitive performance at minimum computational cost (Eddy 1998; Chen et al. 2018). As such, they constitute a promising approach to improve the detection of divergent RNA viruses (Cobbin et al. 2021).
Herein, we outline a new set of computational methodologies and resources to identify and annotate remote viral RdRp sequences from metagenomic data sets. To do so, we first inferred the existing diversity of viral RdRp contained in the non-redundant (nr) database from the National Centre for Biotechnology Information (NCBI), constituting the largest protein sequence database currently available (NCBI; https://www.ncbi.nlm.nih.gov/) as well as in some of the most recent RNA virus metagenomic surveys. By conducting filtering and curation steps, we developed a global and non-redundant RdRp core database that was then used for the description of the RdRp diversity and functional annotation among the already established clades of RNA viruses. We then evaluated the performance of some current tools to identify remote RdRp signals from primary sequence data and created a new custom RdRp profile database to improve profile-based homology detection. Finally, an overall profile and structural-based workflow was proposed.
2. Methods
2.1. RdRp sequence database
2.1.2. Retrieval of viral RdRps using keywords
Annotated RdRps from the Riboviria (i.e. RNA viruses) contained in the NCBI non-redundant protein (nr) database (https://www.ncbi.nlm.nih.gov/protein) were searched using the Entrez Programming Utilities from the NCBI (https://www.ncbi.nlm.nih.gov/books/NBK25501/) and retained based on a 300 amino acid length cut-off and using a list of RdRp-related keywords available at https://github.com/JustineCharon/RdRp-scan/rdrp_keyword.list. Proteins containing no-RdRp description were filtered out based on keywords (‘polymerase cofactor’; ‘glycoprotein’; ‘capsid protein’; ‘subunit’; ‘nucleocapsid’; ‘NS5a’; ‘matrix’; ‘gglutinin’; ‘coat’; ‘reverse’). A 90 per cent sequence identity clustering was performed using CD-HIT (CD-HIT v4.6.1) (Fu et al. 2012). Manual curation was conducted based on the presence of the three canonical A, B, and C motifs identified using the Geneious software (v11.1.4) (Kearse et al. (2012).
2.1.3. Addition of RdRp sequences from metagenomic studies
Additional metagenomic-based RdRp sequences were retrieved from recent studies (Wolf et al. 2020; Chen et al. 2021) and added to the nr RdRp using CD-hit 90 per cent (v4.6.1) (Fu et al. 2012).
2.1.4. Blast analysis of the nr database
A comparison against the nr NCBI database was performed using Diamond BLASTp (v2.0.9) with an e-value cut-off of 1e−05 and employing the ‘very-sensitive’ option (Buchfink, Xie, and Huson 2015).
2.1.5. Taxonomic assignment of RdRps
NCBI taxonomy assignments were retrieved using the option lineage from the taxonkit tool available at https://github.com/shenwei356/taxonkit (Shen and Ren 2021). All NCBI RdRp sequences were grouped according to their placement within the realm Riboviria and kingdom Orthornavirae (which includes all the RdRp-encoding RNA viruses), using both order and phylum ranks according to the current International Committee on Taxonomy of Viruses (ICTV) virus classification (https://talk.ictvonline.org/taxonomy/). The clustering and automatic assignment was performed using cd-hit-2d with 60, 50, and 40 per cent successive clustering (v4.6.1) (Fu et al. 2012). The 30 per cent clustering of unclassified sequences was performed using hierarchical clustering, with the h3-cd-hit option of CD-HIT using the WebMGA server available at http://weizhong-lab.ucsd.edu/webMGA/server/psi-cd-hit-protein/. Briefly, h3-cd-hit performs three iterative runs of CD-HIT clustering at 90, 60, and 30 per cent sequence identity using a neighbour-joining method.
2.1.6. Host assignment
Whenever available, the putative host information of each RdRp-corresponding virus was retrieved using taxonkit name2taxid, available at https://github.com/shenwei356/taxonkit (Shen and Ren 2021).
2.2. RdRp profile database
2.2.1. PALMdb RdRp core sequences
PALMdb RdRp sequences (uniques.fa—version 02/03/2021) and those generated by the Serratus project update (serratus.fa—version 14/03/2021) were both retrieved from the PALMdb github repository (https://github.com/rcedgar/palmdb).
2.2.2. RdRp MSA and profile construction
For each viral order and phylum as well as unassigned clusters, RdRp sequences were enriched with the Serratus/PALMdb RdRp sequences sharing more than 40 per cent identity using CD-HIT-2D (v4.6.1) (Fu et al. 2012). Redundancy was removed at the 40 per cent identity level using CD-HIT, and MSAs of various sizes were obtained using Clustal Omega (—auto option) (v1.2.4) (Sievers et al. 2014; Figure S2). The resulting sequence alignments were manually curated to remove partial RdRp core sequences using Geneious software (v11.1.4) (Kearse et al. 2012). HMM profiles were built from each MSA using HMMer3 using standard parameters (v3.3) (Eddy and Pearson 2011). The resulting HMM profiles were combined and converted into one final HMM profile database used in the subsequent profile analysis with the HMMer3 hmmpress option.
2.2.3. Profile construction of the unassigned viral RdRp
Clusters of ‘unassigned’ sequences (i.e. sharing <30 per cent sequence identity with RdRp members of pre-established viral groups) containing more than ten sequences were enriched with the Serratus/Palmdb RdRp sequences sharing more than 40 per cent sequence identity using CD-HIT-2D (v4.6.1) (Fu et al. 2012).
2.3. Constructing an RdRp A, B, and C motif database and phylogenetic analysis
RdRp alignments were built for each virus phylum using Clustal Omega (—auto option) (v1.2.4) (Sievers et al. 2014) and the 40 per cent sequence identity sequence files identified previously, but depleted of the Serratus and PALMdb sequences. RdRp A, B, and C motif sequences were extracted from RdRp alignments, with the corresponding logos being obtained using WebLogo (v3.7.8) (Crooks et al. 2004). For phylogenetic analysis, iterative alignments of the RdRp were processed using the -p1 option, employing a previous structural alignment (Mönttinen, Ravantti, and Poranen 2021) as a backbone. Unclassified sequences were then aligned to the intermediary alignment, resulting in a final alignment of ∼3,300 sequences (Fig. S2). Intermediary and final alignments were manually inspected using Geneious software to check for the presence of aligned motif blocks. Finally, phylogenetic trees were inferred from this alignment using FastTree2 (v2.1.9) (Price, Dehal, and Arkin 2010), an approximate maximum likelihood-based method, applying the default options. The resulting phylogenies were mid-point rooted and represented using FigTree software.
As the RNA virus RdRp is distantly related to the reverse transcriptase (RT) protein that is present in other viruses—Kingdom Pararnavirae—we also retrieved the A, B, and C motifs from all the Pararnavirae RT amino acid sequences available in the RefSeq database (version of August 2022). Accordingly, the RT domains from 166 amino acid sequences were validated and extracted using InterProScan (v5.52–86.0) (PROSITE RT_POL; PFAM RVT_1) and aligned using Clustal Omega (—auto option) (v1.2.4) (Sievers et al. 2014). The RT motifs logo was then determined using WebLogo (v3.7.8) (Crooks et al. 2004).
2.4. InterProScan analysis
InterProScan annotations were performed by comparing RdRp-like sequences to the European Bioinformatics Institute (EBI) InterPro-embedded PANTHER, Pfam, PIRSF, PRINTS, PROSITEPATTERNS, PROSITEPROFILES, SUPERFAMILY, and TIGRFAM protein profile databases using InterProScan (v5.52–86.0) (Jones et al. 2014). RdRp-like profile entries contained in the EBI databases are available at https://github.com/JustineCharon/RdRp-scan /RdRp_InterPro_keyword.list.
2.5. Phyre2 analysis
Analyses of structural homology were conducted using the batch mode of the Phyre2 server (Kelley et al. 2015), available at http://www.sbg.bio.ic.ac.uk/phyre2/html/page.cgi?id=index. A pre-clustering of RdRp sequences at 30 per cent identity was performed using the h3-cd-hit option of the WebMGA server, available at http://weizhong-lab.ucsd.edu/webMGA/server/psi-cd-hit-protein/.
2.6. RdRp-scan workflow
Open reading frames of orphan contigs were obtained using the GetORF tool from the EMBOSS package (v6.6.0) (Rice, Longden, and Bleasby 2000) using the -find 0 option (defining an Open reading frame (ORF) as a sequence between two STOP codons). The list of genetic codes used by viruses was retrieved from the NCBI taxonomy resource (https://www.ncbi.nlm.nih.gov/Taxonomy/CommonTree/wwwcmt.cgi) and corresponding translation tables 1, 3, 4, 5, 6, 11, and 16 were used to translate potential viral ORFs.
2.7. Evaluation of RdRp-scan
Microbial eukaryote transcriptome data sets used to evaluate RdRp-scan sensitivity and specificity were retrieved from Keeling et al. (2014). ROC curves were built by calculating both the true-positive and false-positive rates for various e-value ranges based on the following formulas: true-positive rates = TP/(TP + FN); false-positive rates = FP/(FP + TN). A recently derived data set of 44,779 divergent RdRp sequences (Zayed et al. 2022) was used to assess the performance of RdRp-scan prediction and determine false-negative rates.
3. Results and discussion
3.1. A new curated viral RdRp database
Revealing the diversity of RNA viruses in metagenomic data sets relies on our ability to compare sequenced and assembled contigs to pre-existing viral nucleotide and protein databases. However, current general protein databases are either too large (NCBI nr) or not comprehensive (NCBI RefSeq), while viral-specific ones such as the Reference Viral Database (Goodacre et al. 2018; Regnault et al. 2021) also contain DNA viruses and endogenous virus sequences. In addition, many RdRp-like sequences obtained from viral metagenomic studies are mis-annotated or not assigned to any function and/or viral clades. Such factors can compromise the detection of more divergent viral sequences and slow analyses by requiring extensive computational resources or providing high numbers of false-positive results (e.g. non-RNA virus hits). To facilitate and speed the specific detection of RNA virus signals, we provide a comprehensive non-redundant and manually curated viral RdRp protein sequence database. Specifically, we compiled, at a single location, all the RdRp and RdRp-like sequences contained in the nr NCBI protein database, as well as from two major viral metatranscriptomic studies that identified thousands of new RNA viruses (Wolf et al. 2020; Chen et al. 2021).
3.1.1. A new viral RdRp database
To build our viral RdRp-specific database, we first manually retrieved all the RNA virus-encoded RdRps from the nr NCBI database (https://www.ncbi.nlm.nih.gov/protein) based on taxonomy (Realm: Riboviria—Kingdom: Orthornavirae—excluding Pararnavirae retro-viruses), keywords, and a length cut-off of 300 amino acids (Fig. 1). The RdRp sequences from recent metagenomic studies (Wolf et al. 2020; Chen et al. 2021) were added, and a filtering and manual curation step was employed to remove all non-RdRp sequences. Briefly, the ‘core’ region of the RdRp—containing the three motifs A, B, and C—was located and extracted. RdRps without identifiable C motifs or displaying non-RdRp domains were excluded. Sequences in which the A and B motifs could not be directly identified were functionally annotated using the EBI InterProScan package (Jones et al. 2014) to check for the presence and position of an RdRp-like sequence. To retrieve additional unannotated RdRp and facilitate further alignment and annotation steps, only the RdRp core region and partial N-terminal and C-terminal flanking regions were retained, using an arbitrary length cut-off of 600 amino acids. Finally, sequences with >90 per cent full-length sequence identity to each other or known sequences were excluded to facilitate manual curation (Fig. 1).
Figure 1.
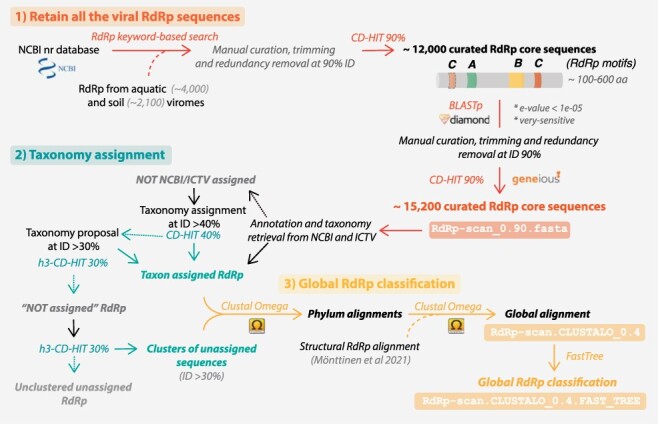
RdRp-scan sequence database workflow. Description of the procedure used to build the RdRp-scan RdRp sequence database and associated annotations. The file names are indicated in boxes and available at https://github.com/JustineCharon/RdRp-scan.
A BLAST search against the nr database was then conducted using the curated RdRp core sequences to ensure database completeness. All RdRp-like homologs were retrieved, manually checked, and trimmed, and their redundancy was removed as described previously. The NCBI information (taxonomy and host when available) of every nr-based sequence was retrieved and used to classify sequences based on the corresponding RNA virus phyla or orders (Fig. 1). An attempt was made to automatically assign viral RdRp sequences to pre-established virus orders based on an arbitrary 40 per cent full-length sequence identity cut-off, widely used in protein sequence analysis (Jumper et al. 2021). A second clustering at 30 per cent full-length sequence identity was also used to suggest a taxonomic assignment at the order rank level for viruses that contain more divergent RdRps. Whenever possible, the final non-assigned RdRp core sequences (i.e. sharing <30 per cent identity with any of the assigned RdRp) were clustered into ‘unassigned clusters’ using a 30 per cent sequence identity cut-off (Fig. 1). The resulting sequence database of RdRp clustered at 90 per cent full-length sequence identity is available at https://github.com/JustineCharon/RdRp-scan/.
To describe newly identified RdRps and to potentially suggest a taxonomic placement, intermediary alignments of the Riboviria were built at both phylum and order levels. Considering the very high level of sequence divergence between the phyla of RNA viruses, we used a previous structural alignment (Mönttinen, Ravantti, and Poranen 2021) as a backbone, and pre-aligned RdRps were iteratively added to build the final alignment (Fig. 1). Finally, unassigned sequences were added to the master RdRp alignments. Importantly, at this level of sequence divergence, the quality of the resulting alignments cannot be used to accurately infer RNA virus phylogenies. Rather, such phylogenies should be regarded as broad indicators of how the unassigned sequences, and ultimately any newly identified sequences, might fit into global virus diversity.
3.1.2. Overall RNA virus diversity
The RdRp database provided in this study was also designed to infer the diversity of RdRp sequences covered by the current nr database. The distribution of all the curated RdRp sequences obtained at the 90 per cent sequence identity level reveals that our global knowledge of RdRp diversity is heterogeneous, partial, and subject to profound sampling biases. This is particularly obvious at the level of virus hosts, with mammals and land plant-infecting virus RdRps accounting for almost 80 per cent of all the host-assigned viruses (Fig. 2). The very limited proportion of RdRp from RNA viruses infecting microbial eukaryotes and Archaea (in which bona fide RNA viruses have yet to be identified) highlights the clear need to investigate viral diversity in these hosts (Fig. 2). The bias in the distribution of RdRp diversity is also apparent in phylogenetic analyses in which those viral clades associated with mammals (e.g. Picornavirales) and land plants (e.g. Tolivirales) are over-represented (Fig. 3).
Figure 2.
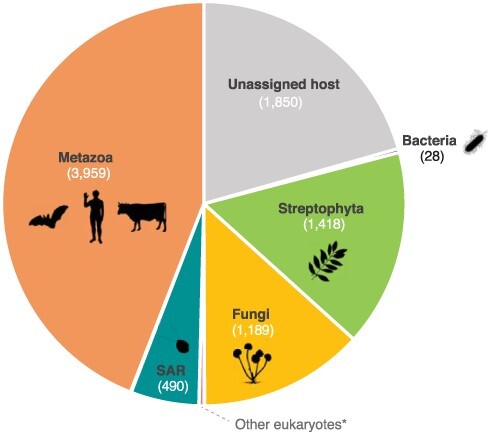
Host assignment of RdRp corresponding viruses. Host information was retrieved for each RdRp viral entry at the NCBI present in the 90 per cent redundancy RdRp-scan database, using either the VirusHostdb (Mihara et al. 2016) or NCBI resources (https://www.ncbi.nlm.nih.gov/). SAR: Stramenopile–Alveolate–Rhizaria supergroup. Archaea were not represented as no archaea-infecting RNA viruses have been formally reported. *Other eukaryotic clades: Picozoa, Chlorophyta, Rhodophyta, Metamonada, Kinetoplastea, Amoebozoa, and Haptophyta. Icons were retrieved from Phylopic (http://phylopic.org/). Credits: Matt Crook (Bacteria), Sergio A. Muñoz-Gómez (SAR), Ville Koistinen, and T. Michael Keesey (Streptophyta), under the Creative Commons Attribution-ShareAlike 3.0 Unported license (https://creativecommons.org/licenses/by-sa/3.0/).
Figure 3.
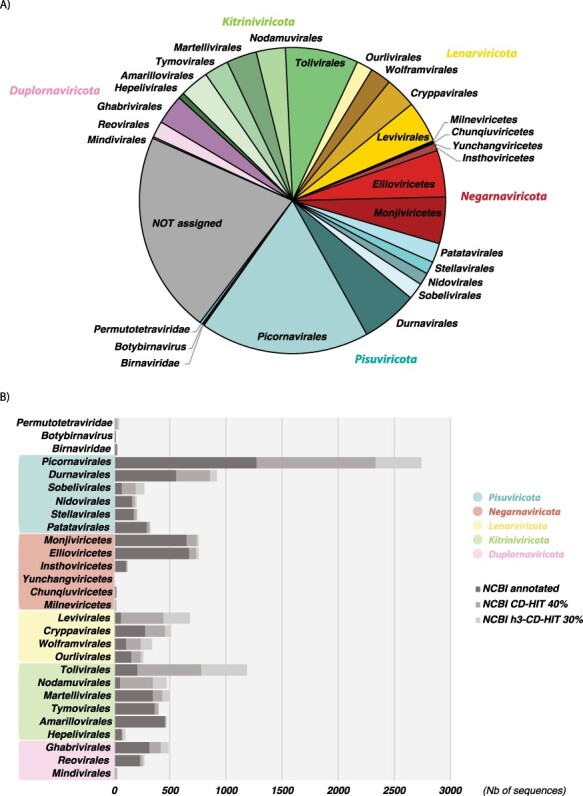
Taxonomic distribution of RdRps among the Riboviria phyla and clades. (A) Distribution of taxonomically assigned RdRp sequences (at 90 per cent redundancy) among the taxonomy of the Riboviria versus those RdRps that are currently unassigned. (B) Distribution of NCBI-annotated RdRp (dark grey) and automatically assigned RdRps at the 40 and 30 per cent sequence identity levels (grey and light grey, respectively) among the current ICTV taxonomy of the Riboviria.
Interestingly, the phylum Lenarviricota encompassing viruses identified as infecting bacteria, fungi, and unicellular eukaryotes (‘protists’) from families, such as the Leviviridae, Mitoviridae, and Narnaviridae, contains a high number of sequences, most of which are automatically assigned based on 30–40 per cent sequence identity. Indeed, our automatic classification of previously unassigned sequences (i.e. showing >30 per cent identity with assigned taxa) shows that phyla like the Lenarviricota are particularly enriched with such ‘assignable’ sequences and that their real diversity/importance may have been substantially underestimated.
We next attempted to integrate the unassigned sequences (i.e. those with <30 per cent sequence identity with assigned RdRps) into the global RdRp phylogenies. This revealed that a large majority of the unassigned RdRps fell into pre-established families or clades with a reasonable degree of certainty (Fig. 4). Moreover, the sequences that remain unassigned were distributed throughout global RdRp diversity, and their assignment may considerably enrich the diversity inside each RNA virus phylum. Only twenty-four RdRp were positioned outside of the currently established Riboviria phyla and without close assigned relatives. Such a small number is presumably a consequence of the biased sampling of viruses and the use of sequence-to-sequence tools in the metagenomic studies (Fig. 4). Nevertheless, some major unassigned clusters were largely obtained from metagenomic studies conducted on poorly studied environments (marine and soils) or hosts (unicellular eukaryotes and fungi), can be identified as distant relatives to the orders Wolframvirales (clusters 724, 295, 561, 197, 1,028, 297, and 298) and Tolivirales (clusters 1279, 652, and more distantly clusters 1,260, 731, 1,282, 955, 589, 1,135, 441, 373, 504, and 1,039), and might constitute new orders within the phyla Lenarviricota and Kitrinoviricota, respectively (Fig. 4). This strongly supports the notion that most of the RNA virus diversity remains uncharacterized, particularly at levels <30 per cent sequence identity.
Figure 4.
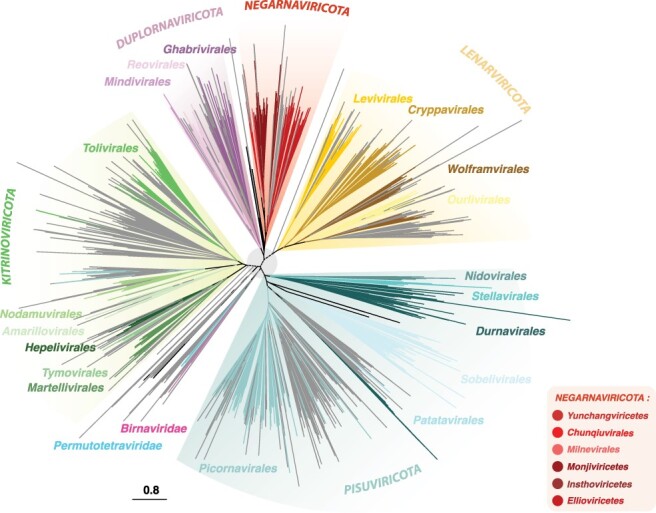
Phylogenetic tree from the RdRp-scan protein sequence database. Given the high level of sequence divergence, the tree is unrooted and a light grey circle (centre node) is presented to highlight the uncertainty of the phylogeny at the inter-phylum level. Riboviria phyla Pisuviricota, Kitrinoviricota, Duplornaviricota, Negarnaviricota, and Lenarnaviricota are indicated in capital letters.
3.2. Identifying divergent viruses using profiles and structure-based homology
3.2.1. Ability of InterProScan and Phyre2 to detect divergent RdRps using our database
Two complementary approaches were used to evaluate the capacity to detect RdRps using the currently available profile and structural databases. First, InterProScan was run on our new custom RdRp database, and the total proportion and the number of detected RdRps were reported for each viral order (Fig. 5). This revealed that InterProScan and integrated profile databases are readily able to detect RdRp sequences, with 97 per cent of total RdRp identified regardless of viral taxonomy. Nevertheless, some viral orders were not well covered, with 18–85 per cent of undetected RdRps in the Wolframvirales, Ourlivirales, Reovirales, and Chunqiuvirales—or even completely missed in the case of the Yunchangviricetes.
Figure 5.
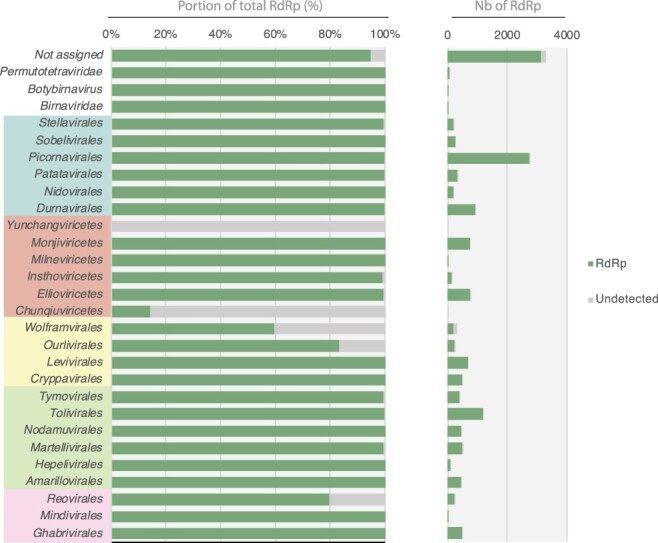
Detection of viral RdRp homologies using InterProScan. The proportion (left) and total numbers (right) of RdRp detected/undetected by InterProScan are represented in green and grey, respectively. Viral orders are grouped according to their corresponding phyla (from top to bottom, Pisuviricota, Negarnaviricota, Lenarviricota, Kitrinoviricota, Duplornaviricota).
Second, to estimate the power of currently available tools to detect structural homology, we performed a comparison against the Protein Data Bank (PDB) (https://www.rcsb.org/) (Burley et al. 2021) using the Phyre2 server (Kelley et al. 2015). Given the computational constraints inherent in Phyre2, a preliminary clustering of the RdRp at 30 per cent identity was performed, and only one representative sequence per cluster was submitted to Phyre2, corresponding to approximately 1,500 sequences in total. We assume that protein sequences sharing more than 30 per cent identity would have the same result at the level of protein structure comparison and that such clustering step does not affect the final results.
Remarkably, >96 per cent of the RdRps submitted to Phyre2 were detected as homologous to the RdRp deposited in the PDB, regardless of viral phyla (Fig. 6A). A limited number of RdRps could not be detected in few viral orders (Wolframvirales, Ourlivirales, Durnavirales, Tolivirales, and Reovirales) or among the unassigned sequences (Fig. 6A). Importantly, therefore, this analysis shows that Phyre2 can confidently detect homology at very low levels of sequence identity, with some of the PDB RdRp hits sharing as little as 10 per cent identity with the RdRp sequences (Fig. 6B). Despite the very limited representation of viral RdRps in the current PDB database (Cobbin et al. 2021), Phyre2 is still capable of detecting RdRps by confidently identifying remote homologies between proteins that share very low levels of sequence identity, with most <20 per cent. As such, it constitutes a useful tool for the analysis of viral dusk matter, although computational constraints make it difficult to use for most metagenomic data. In contrast, InterProScan processes thousands of RdRp sequences but fails to detect RdRp signals in some viral lineages. In addition, as it is not specifically designed for RNA viruses, the retrieval of all the different viral RdRp-specific signals needs to be performed manually using different keywords covering all the diverse viral RdRp-like profile names. Nevertheless, the use of protein HMMs is expected to help reveal deep evolutionary fingerprints shared between distant RdRp based on amino acid sequences.
Figure 6.
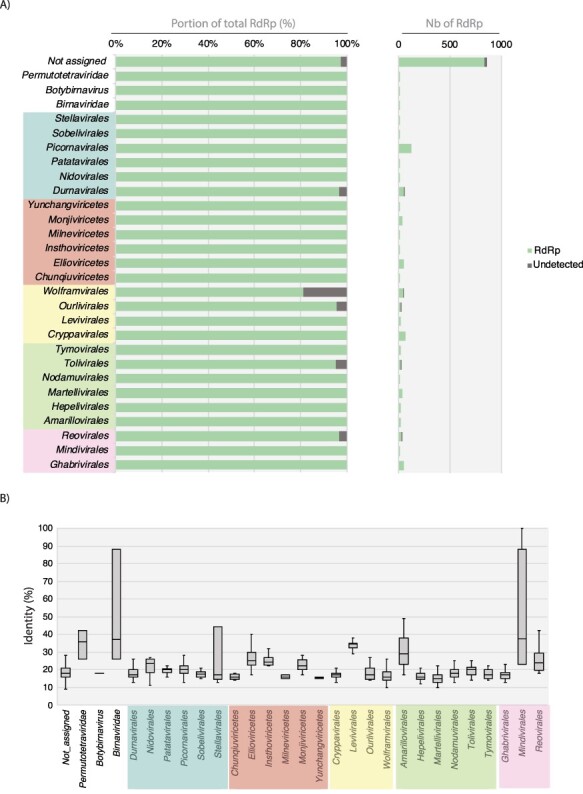
Detection of viral RdRp homologies using Phyre2. (A) Proportion (left) and total numbers (right) of RdRps with PDB RdRp homologs detected at >80 per cent of confidence levels for each order of the Riboviria. (B) Levels of sequence identity between Phyre2-detected homologs. The proportion (left) and total numbers (right) of RdRp detected/undetected by Phyre2 are represented in green and dark grey, respectively. Viral orders are grouped according to their corresponding phyla (from top to bottom and left to right, Pisuviricota, Negarnaviricota, Lenarviricota, Kitrinoviricota, Duplornaviricota).
3.2.2. Profile construction from the new RdRp database
To increase the diversity and specificity covered by RdRp profile analyses and facilitate the detection of divergent viral RdRp homology signals, we built new RdRp profiles specific to each order or phylum of Riboviria-based RdRp diversity obtained from both our RdRp database and those retrieved from unassigned RNA virus RdRp sequences. To maximize the sequence diversity covered by those new RdRp HMM profiles, RdRp sequences recently obtained from the ultra-wide Serratus SRA mining project and members of the PalmDB resource (Babaian and Edgar 2021; Edgar et al. 2022) were also integrated into pre-existing taxa using a 30 per cent ID cut-off (Fig. 7).
Figure 7.
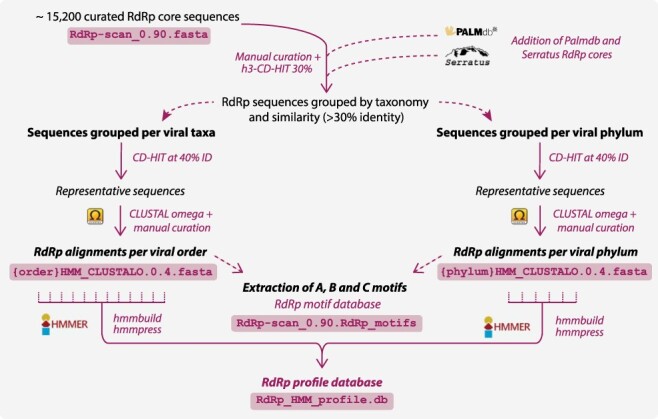
RdRp-scan profile database workflow. The procedure used to obtain the HMM-based RdRp profiles. Corresponding file names are indicated in boxes and available at https://github.com/JustineCharon/RdRp-scan.
Each RdRp from our database, as well as those from the Serratus SRA mining, was grouped into viral orders or phyla according to their NCBI taxonomy assignment or their level of similarity with taxonomically assigned RdRps, as described previously. Sequence alignments were produced for each group as well as for unassigned sequences after removing the redundancy at 40 per cent of sequence identity. Each alignment was manually checked for the presence of A, B, and C motifs, and whenever identified, sequences containing permuted C motifs in N-terminal position were extracted and re-aligned separately. In total, sixty-eight HMM profiles were obtained (eight and twenty-nine from established phyla and orders, respectively, and thirty-one from unassigned clusters) and then pooled into one single HMM profile database to use with the HMMer3 package available at https://github.com/JustineCharon/RdRp-scan (Fig. 7).
3.2.3. Catalytic motif diversity from the RdRp profiles
Covering the full diversity of RdRp motifs is crucial to validate newly identified divergent viral candidates based on structure and profile-based homology. The A, B, and C motifs were, therefore, manually identified from alignments used to build HMM profiles (Fig. 7) and the corresponding motif sequence conservations were visualized using logo representations (Fig. 8, Figure S2). To prevent mis-annotation, motif characterization and RdRp validation were conducted exclusively based on those reported and/or validated from previous studies. Therefore, the existence of additional A, B, and C motifs in viral RdRps not present in our database cannot be excluded.
Figure 8.
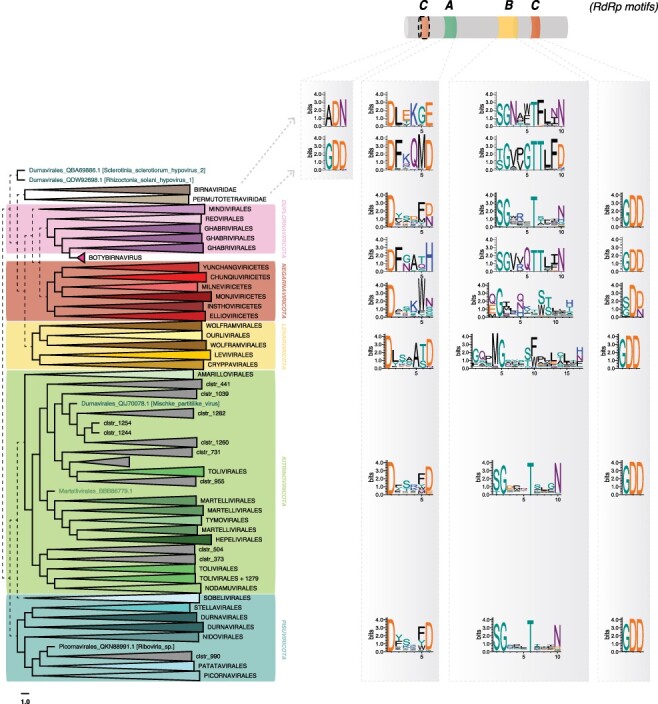
A, B, and C RdRp motif diversity within the Riboviria (Orthornavirae) phyla. Motifs are represented as logos obtained from the alignment of RdRp within each phylum, using WebLogo (Crooks et al. 2004). Dashed lines represent the uncertain placement/phylogenies due to the high level of sequence divergence.
This analysis provided a number of notable observations. The A motifs show slight variation to the commonly described DxxxxD motif at both the phylum and order levels (Figs. 8 and S2). While most of the Pisuviricota and Kitrinoviricota display a double aspartate residue, the second aspartate is replaced by E or H in the Birnaviridae and in the genus Botybirnavirus. Members of the Lenarviricota commonly display an additional residue between the two aspartates (DxxxxxD), while the Negarnaviricota have a conserved tryptophan downstream of the critical aspartate residue (DxxxW) (Figs. 8 and S2). The B motifs could be easily identified, and B motif sequences were retrieved from the Birnaviridae, Permutotetraviridae, Botybirnavirus, Duplornaviricota, Kitrinoviricota, and Pisuviricota based on the canonical SGxxxTxxxN with only a few variations at the level of virus order (Fig. S2). Conversely, the Lenarvicota display a very different and low-conserved motif at the primary sequence level, with a GQxMGxxxF/WxxL/ExxxF/H/N consensus. Similarly, the Negarnaviricota also displays alternative B motif sequences, with a high diversity of residues arranged around the conserved glycine, crucial for the RdRp structure and function (Venkataraman, Prasad, and Selvarajan 2018). The organization and structural relevance of such alternative B motifs requires additional investigation but may provide insights into the structural and evolution of viral RdRps.
Of most note, we observed variations in the C motif even though this is considered to be the most conserved RdRp motif. The GDD motif contains two of the three aspartate residues needed for RdRp catalytic functions (Ferrero et al. 2015). As confirmed here, the C motif is most commonly located downstream of the B motif, although exceptions have been reported with Birnavirus and the permutotetraviruses that display a C–A–B motif order (Gorbalenya et al. 2002; Shwed et al. 2002). Moreover, the Birnavirus C motif comprises the ADN motif instead of the widely conserved GDD, replacing the third aspartate of the catalytic triad, which decreases the replicase activity (Pan, Vakharia, and Tao 2007). Similar permuted C–A–B motifs are newly reported here in the Tolivirales, Martellivirales (Kitrinoviricota), and Patatavirales, suggesting a wider use of this uncommon C motif organization. While the GDN and SDD alternative sequences were already reported for the C motif of the ss(−)RNA virus RdRps (Poch et al. 1989, 1990), those from the Chunqiuviricetes show a conserved IDD (Ile-Asp-Asp). Importantly, however, these new variant consensus sequences or motif arrangements need to be experimentally validated, and their role in RdRp structure and function was investigated more in detail. Despite this, their identification will assist in the manual annotation of divergent viral RdRps and discriminate exogenous viruses from endogenous viral elements (EVEs) or other non-RdRp false-positive signals. Accordingly, a database of all the motifs identified in the RdRP db is provided at https://github.com/JustineCharon/RdRp-scan to help with such identification and the annotation of new viral candidates (Fig. 7).
Viral RTs share structural properties with RdRps and also display A, B, and C motifs in their core sequence (Te Velthuis 2014). This leads to a potential risk of mis-assignment at very low levels of sequence similarity. To assess this possibility, we examined the diversity of 163 viral RT proteins, representative of the major groups within the Kingdom Pararnavirae (the Caulimoviridae, Hepadnaviridae, Metaviridae, and Retroviridae) (Fig. S3). The viral RT motifs detected were substantially different from those observed in the RdRps (Figs. 8 and S2). Hence, a manual curation step to remove sequences containing any RT-like motifs should be sufficient to prevent incorporation of RT-like sequences into RdRp db and the subsequent mis-identification of RdRp sequences from metagenomic data.
3.2.4. Proof-of-concept of RdRp detection using microbial eukaryote RNAseq data
To evaluate the performance of the new RdRp-scan HMMs in the detection of remote RdRp signals in metatranscriptomic data, we utilized unicellular eukaryotic transcriptomes from the Marine Microbial Eukaryote Transcriptome Sequencing Project (MMETSP) (Keeling et al. 2014). The MMETSP-based orphan ORFs (i.e. without detectable match in nr protein database using BLAST), combined with thirty divergent RNA viruses obtained previously (Charon, Murray, and Holmes 2021) but not included in the current RdRp-scan database, representing 3,291,862 contigs in total, were submitted in parallel to HMMer3 using RdRp-scan and InterProScan HMMs. The resulting hits—702 and 93 for RdRp-scan and InterProScan, respectively—were then submitted to Phyre2, and a further validation using motif detection was performed. To limit the number of sequences to be submitted to Phyre2, RdRp-scan hits with high values (>1e−05) and not containing any of the potential C motifs (GDD/SDD/IDD/GDN) were discarded. To evaluate the sensitivity versus specificity of the RdRp-scan compared to InterProScan, an ROC curve was obtained by calculating the true-positive versus false-positive rates (Fig. S4). This confirmed that an e-value value cut-off between 1e−05 and 1e−06 was appropriate for RdRp-scan HMM analysis. Sequences with e-values > 1e−05 were, therefore, discarded from the analysis. Sequences without motif similarities to known RdRps and/or detected as non-RdRp in Phyre2 with a confidence score higher than 90 per cent were considered as false positives. Previously identified divergent viruses that could not be detected using InterProScan or RdRp-scan profiles were similarly reported as false negatives. Previously identified viruses were detected as true hits in this study, and true-like RdRp hits originally discovered using profiles were counted as true positives. Finally, the false-positive hits detected using one method but not detected using the other method were reported as ‘true negatives’.
Overall, HMM profile-based detection using our newly built RdRp-scan profile exhibited a greater sensitivity with a significantly lower number of false positives than InterProScan (Fig. 9). Importantly, InterProScan was unable to detect five of the thirty previously reported RdRp sequences (Charon, Murray, and Holmes 2021). Conversely, by using HMMer3 combined to our new HMM RdRp profile database, we were able to identify all the divergent viruses as well as new RdRp-like sequences. This constitutes a proof-of-concept of the relevance of using a specific RdRp profile to detect remote viral signals. Among the ten RdRp-like hits identified only using the HMM profile database, five were similar to Wolframvirales, three were similar to Ghabrivirales, one matched with Permutotetraviridae, and one was similar to Chunqiuviricetes. This is in accordance with the previously assessed InterProScan coverage of the RdRp diversity (Fig. 5).
Figure 9.
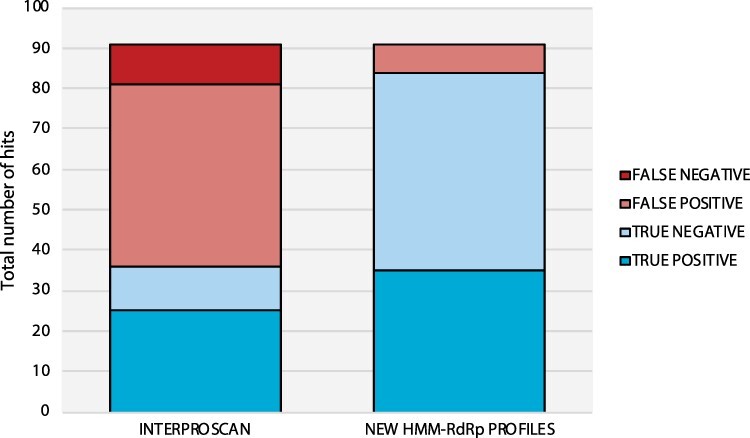
Comparison of RdRp detection from unicellular eukaryote transcriptomes using RdRp-scan HMM profiles versus InterProScan. Data sets obtained from MMETSP transcriptomes (Charon, Murray, and Holmes 2021) were submitted to HMMer3 using either RdRp-scan or InterProScan profiles. False positives are indicated in light red and refer to sequences without RdRp A, B, and C motifs detected and/or detected as non-RdRp in Phyre2 with a confidence score higher than 90 per cent. False negatives are indicated in dark red and refer to previously detected viruses (Charon, Murray, and Holmes 2021) detected using InterProScan or RdRp-scan profiles. True positives are indicated in dark blue and refer to both previously detected viruses (Charon, Murray, and Holmes 2021), and true RdRp-like hits originally discovered using RdRp-scan and InterProScan profiles. True negatives are indicated in light blue and refer to false-positive hits detected using one method but not detected using the other method.
3.2.5. Evaluation of the ability of RdRp-scan to detect new divergent viral RdRps
A second evaluation of RdRp-scan capacity to detect divergent RdRp was conducted on a new RdRp-data set recently obtained from an expansive metagenomic study of marine viruses comprising 44,779 RdRp-like sequences (Zayed et al. 2022). All sequences were submitted to both InterProScan and RdRp-scan using an e-value cut-off of 1e−06. Strikingly, the number of sequences detected was highly dependent on the length of the RdRp sequence submitted, with a drastic reduction in the level of detection for amino acid sequence queries shorter than 100–200 amino acids (Fig. S5). Assuming that RdRps shorter than 200 amino acids are inviable for phylogenetic and/or annotation studies and hence are not considered in the RdRp-scan workflow, only input sequences longer than 200 amino acids were used in the RdRp-scan and InterProScan comparison. Of the 17,211 RdRps submitted, 94 per cent could be detected with an RdRp-scan with only 6 per cent false negatives, against 75 per cent detected and 25 per cent false negatives with InterProScan (Figure S5B). This illustrates the improvement resulting from the use of a specific viral RdRp HMM database in the detection of viral RdRps. Despite this, the fact that a proportion of RdRps were not detected highlights the need for the RdRp-scan database to be regularly updated.
3.3. Analysis of new divergent RdRps using RdRp-scan
Together with the identification of divergent RdRp-like signals from metagenomic data, our study assists in the validation and characterization of newly identified RdRp-like sequences. Sequence comparisons with pre-existing RdRp can be challenging at such very low levels of similarity. Particular care should be exercised when submitting new sequences, especially concerning the annotation of amino sequences and the taxonomic placement of the corresponding viruses within documented viral diversity. While sequence annotation and taxonomic placement is relatively straightforward for well-established families, it becomes a major challenge for new divergent sequences identified either from overlooked hosts or likely belonging to new and/or undescribed viral taxa that are prone to mis-classification.
Motif annotation using the motif database extracted from our custom database
To help in the annotation and detection of RdRp putative functional motifs from those divergent sequences, we have made available the RdRp motif data set extracted from the whole RdRp database, which can be mapped directly onto candidate sequences using classical sequence visualization software (such as CLC Bio, Geneious, and DNASTAR). The three RdRp motifs A, B, and C have strong conservation and consistency among current RdRp diversity at both phylum and order levels (Figs. 8 and S2). It is of obvious importance to verify the presence of the three motifs to prevent the mis-annotation of host genes as RdRps. Such RdRp-like signals identified in hosts always miss at least one of the three motifs, and this can be used to identify such false positives. The profile alignments used to build the HMM RdRp alignment are available at https://github.com/JustineCharon/RdRp-scan and can be used to align the candidate sequence to help identify conserved regions and motifs.
Taxonomic placement of new viral candidates
RdRp amino acid sequences are commonly used to infer RNA virus phylogenies. When attempting to taxonomically assign newly discovered sequences, it is therefore tempting to put new virus candidates into the global diversity of RdRps. Importantly, however, such large-scale RdRp phylogenies are not based on robust alignments; pairwise genetic distances between divergent sequences will be large underestimates because of large-scale multiple substitution at single sites, and hence, deeper topological arrangements may be inaccurate. Such phylogenies should therefore be treated with great caution. Hence, we utilized the meta-RdRp alignment at https://github.com/JustineCharon/RdRp-scan only as an intermediary step to provisionally place candidate sequence(s) within global RdRp diversity and identify their closest related sequences (Fig. 10). This should not be considered an accurate phylogeny in itself. Following this initial phylogeny, the scale of analysis can be narrowed, and the candidate sequences compared to their closest homologs in a more refined manner, utilizing more robust alignments and phylogenetic trees (Fig. 10).
Figure 10.
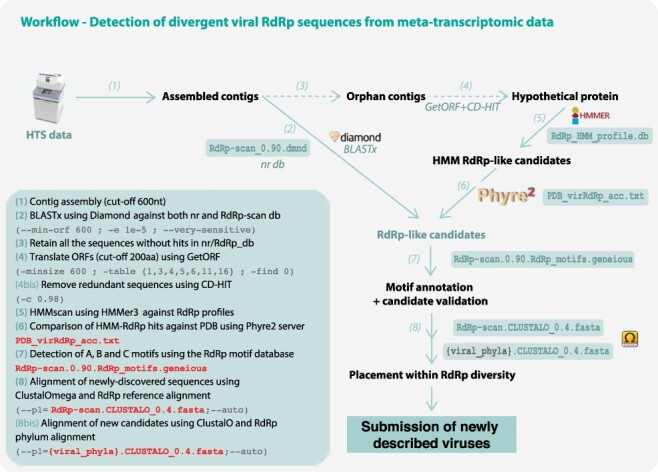
RdRp-scan workflow. (1) Contigs are assembled from trimmed read files using assembler software using a 600-nt cut-off. (2) Assembled contigs are compared in parallel to nr and our new RdRp-scan database using Diamond BLASTx (Buchfink, Xie, and Huson 2015), applying an e-value cut-off of 1e−05. All the nr matches of the sequences identified as RdRp-like are checked to remove false-positive sequences based on the e-value and BLAST scores. (3) All ‘orphan’ contigs—i.e. those without any match in nr or RdRp-scan database—are retrieved. (4) The open reading frames or orphan contigs are then translated using getORF (Rice, Longden, and Bleasby 2000) by applying a 200 amino acid length cut-off and according to all the genetic codes described in viruses (i.e. translation tables 1, 3, 4, 5, 6, 11, and 16). Redundant sequences are then removed using CD-HIT (Fu et al. 2012) at 90 per cent sequence identity. (5) Non-redundant candidate protein sequences are then compared to the RdRp HMM profile database using HMMer3 (Eddy and Pearson 2011), with an e-value cut-off of 1e−06. (6) Retained hits are then submitted to the Phyre2 server (Kelley et al. 2015) using the batch option, and results are parsed manually. To assist with this manual identification of RdRp-like hits, a list of PDB-RdRp-like structures is made available. Best hits matching with PDB non-viral entries with a confidence score >90 per cent are considered as false positives and discarded. Hits matching with RdRp PDB entries or without any confident match with PDB (at 90 per cent confidence cut-off) are retained for further validation and characterization steps. (7) A, B, and C motifs are then screened in the corresponding candidates using the RdRp motif database. The motif sequence and position in the sequence are manually inspected and candidate validated as true putative viral RdRp based on the presence of the three RdRp-like motifs. (8) Confirmed candidates are then aligned with the meta-RdRp alignment using ClustalOmega, (Sievers et al. 2014) and their position in the global RdRp diversity is assessed using FastTREE (Price, Dehal, and Arkin 2010). According to their phylogenetic position, new RdRp-like sequences are then aligned with the closest viral phyla or order, and the corresponding alignment is checked and used for a more accurate phylogenetic analysis. Steps prior to the contig assembly are not described.
Workflow to identify, annotate, and assign new viral sequences.
Finally, we propose a workflow that integrates the newly proposed resources as well as pre-existing open-access tools to detect new and divergent RNA virus sequences from metagenomic data (Fig. 10). Briefly, the workflow consists of identifying highly divergent RdRps by combining HMMs and structural homology detection using the newly built RdRp-scan RdRp and Phyre2 server, respectively. Candidates can thus be confirmed and annotated using the alignments and motif databases made available in the RdRp-scan package. Importantly, the whole analysis—from the assembled contigs to the new virus RdRp annotation—can handle millions of contig sequences simultaneously with relatively limited computational resources.
The viral sequences identified using RdRp-scan can be used as queries for a second round of BLASTp/HMM profile searches that in turn may illuminate new parts of the RNA virus phylogeny. The use of RdRp-scan in combination with the complementary and recently developed PalmScan approach (Babaian and Edgar 2021) could also help validate and annotate the RdRp sequences identified using RdRp-scan. Finally, the addition of de novo prediction-based structures, such as those made using AlphaFold2 (Jumper et al. 2021) and potentially available in the AlphaFold protein structure database (https://alphafold.ebi.ac.uk/), could also be integrated into the structural-based comparison steps and is expected to enlarge the scale of structural comparison across the RNA virus phylogeny (Cobbin et al. 2021). Building a custom PDB database, restricted to viral RdRp structures, could also accelerate Phyre2 analyses and accommodate larger numbers of sequences.
Conclusion and perspectives
The detection of divergent RNA viruses relies heavily on the de novo discovery, annotation, and validation of newly described functional features such as new functional motifs and domains. By combining sequence, profile, and structural-based analyses into a single workflow, our study shows that it is possible to detect RdRp sequences sharing as little as 10 per cent sequence identity with known RdRps and that this can be realistically conducted at a metagenomic scale. This work provides resources to ease the challenging steps that lie beyond the detection of new divergent viral sequences, particularly the identification and annotation of functional RdRp motifs and taxonomy placement within the diversity of Riboviria. The resources and workflow generated here will, therefore, facilitate the detection of divergent RNA virus sequences and expand our current knowledge of RNA virus diversity. With thousands of new viral RNA species regularly described, RdRp-scan is intended to be regularly updated to integrate the most up-to-date picture of RNA virus diversity. Indeed, an updated version is currently being developed to accommodate recent large-scale descriptions of RNA viruses in nature (Neri et al. 2022; Zayed et al. 2022). Ongoing progress in both the accessibility and accuracy of de novo structural predictors (Baek et al. 2021; Jumper et al. 2021) is also expected to provide a new perspective on the discovery of highly divergent RNAs and will be integrated into such workflows.
Supplementary Material
Acknowledgements
We thank Ayda Susana Ortiz-Baez, Callum Le Lay, and Dr Robert Edgar for helpful comments and Mathilde Chagneaud for her help in designing the RdRp-scan logo.
Contributor Information
Justine Charon, Sydney Institute for Infectious Diseases, School of Life and Environmental Sciences and Sydney Medical School, The University of Sydney, Camperdown, NSW 2006, Australia.
Jan P Buchmann, Institute for Biological Data Science, Heinrich-Heine-University, Universitätsstrasse 1, Düsseldorf D-40225, Germany.
Sabrina Sadiq, Sydney Institute for Infectious Diseases, School of Life and Environmental Sciences and Sydney Medical School, The University of Sydney, Camperdown, NSW 2006, Australia.
Edward C Holmes, Sydney Institute for Infectious Diseases, School of Life and Environmental Sciences and Sydney Medical School, The University of Sydney, Camperdown, NSW 2006, Australia.
Data availability
All the data produced in this study (alignment, database, and phylogenetic tree files) are available at https://github.com/JustineCharon/RdRp-scan.
Supplementary data
Supplementary data are available at Virus Evolution online.
Funding
E.C.H. is funded by an Australian Research Council Australian Laureate Fellowship (FL170100022) and by AIR@InnoHK administered by the Innovation and Technology Commission, Hong Kong Special Administrative Region, China.
Conflict of interest:
None declared.
References
- Babaian A., and Edgar R. C. (2021) ‘Ribovirus Classification by a Polymerase Barcode Sequence’, bioRxiv.doi: 10.1101/2021.03.02.433648 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baek M. et al. (2021) ‘Accurate Prediction of Protein Structures and Interactions Using a Three-Track Neural Network’, Science, 373: 871–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bolduc B. et al. (2012) ‘Identification of Novel Positive-Strand RNA Viruses by Metagenomic Analysis of Archaea-Dominated Yellowstone Hot Springs’, Journal of Virology, 86: 5562–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bruenn J. A. (2003) ‘A Structural and Primary Sequence Comparison of the Viral RNA-Dependent RNA Polymerases’, Nucleic Acids Research, 31: 1821–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buchfink B., Xie C., and Huson D. H. (2015) ‘Fast and Sensitive Protein Alignment Using DIAMOND’, Nature Methods, 12: 59–60. [DOI] [PubMed] [Google Scholar]
- Burley S. K. et al. (2021) ‘RCSB Protein Data Bank: Powerful New Tools for Exploring 3D Structures of Biological Macromolecules for Basic and Applied Research and Education in Fundamental Biology, Biomedicine, Biotechnology, Bioengineering and Energy Sciences’, Nucleic Acids Research, 49: D437–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Charon J. et al. (2019) ‘Novel RNA Viruses Associated with Plasmodium vivax in Human Malaria and Leucocytozoon Parasites in Avian Disease’, PLoS Pathogens, 15: e1008216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ——— et al. (2020) ‘Metatranscriptomic Identification of Diverse and Divergent RNA Viruses in Green and Chlorarachniophyte Algae Cultures’, Viruses, 12: 1180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Charon J., Murray S., and Holmes E. C. (2021) ‘Revealing RNA Virus Diversity and Evolution in Unicellular Algae Transcriptomes’, Virus Evolution, 7: veab070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen J. et al. (2018) ‘A Comprehensive Review and Comparison of Different Computational Methods for Protein Remote Homology Detection’, Briefings in Bioinformatics, 19: 231–44. [DOI] [PubMed] [Google Scholar]
- Chen Y. M., Sadiq S., Tian J. H., et al. (2022), ‘RNA viromes from terrestrial sites across China expand environmental viral diversity’, Nat Microbiol, 7: 1312–23. [DOI] [PubMed] [Google Scholar]
- Cobbin J. C. et al. (2021) ‘Current Challenges to Virus Discovery by Meta-transcriptomics’, Current Opinion in Virology, 51: 48–55. [DOI] [PubMed] [Google Scholar]
- Crooks G. E. et al. (2004) ‘WebLogo: A Sequence Logo Generator’, Genome Research, 14: 1188–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Culley A. I., Lang A. S., and Suttle C. A. (2006) ‘Metagenomic Analysis of Coastal RNA Virus Communities’, Science, 312: 1795–8. [DOI] [PubMed] [Google Scholar]
- Duffy S., Shackelton L. A., and Holmes E. C. (2008) ‘Rates of Evolutionary Change in Viruses: Patterns and Determinants’, Nature Reviews. Genetics, 9: 267–76. [DOI] [PubMed] [Google Scholar]
- Eddy S. R. (1998) ‘Profile Hidden Markov Models’, Bioinformatics, 14: 755–63. [DOI] [PubMed] [Google Scholar]
- Eddy S. R., and Pearson W. R. (2011) ‘Accelerated Profile HMM Searches’, PLoS Computational Biology, 7: e1002195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edgar R. C. et al. (2022) ‘Petabase-Scale Sequence Alignment Catalyses Viral Discovery’, Nature, 602: 142–7. [DOI] [PubMed] [Google Scholar]
- Ferrero D. S. et al. (2015) ‘The Structure of the RNA-Dependent RNA Polymerase of a Permutotetravirus Suggests a Link between Primer-Dependent and Primer-Independent Polymerases’, PLoS Pathogens, 11: e1005265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferrero D. S., Falqui M., and Verdaguer N. (2021) ‘Snapshots of a Non-Canonical RdRp in Action’, Viruses, 13: 1260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu L. et al. (2012) ‘CD-HIT: Accelerated for Clustering the Next-Generation Sequencing Data’, Bioinformatics, 28: 3150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goodacre N. et al. (2018) ‘A Reference Viral Database (RVDB) to Enhance Bioinformatics Analysis of High-Throughput Sequencing for Novel Virus Detection’, mSphere, 3: e00069–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gorbalenya A. E. et al. (2002) ‘The Palm Subdomain-Based Active Site Is Internally Permuted in Viral RNA-Dependent RNA Polymerases of an Ancient Lineage’, Journal of Molecular Biology, 324: 47–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hansen J. L., Long A. M., and Schultz S. C. (1997) ‘Structure of the RNA-Dependent RNA Polymerase of Poliovirus’, Structure, 5: 1109–22. [DOI] [PubMed] [Google Scholar]
- Illergård K., Ardell D. H., and Elofsson A. (2009) ‘Structure Is Three to Ten Times More Conserved than Sequence—A Study of Structural Response in Protein Cores’, Proteins: Structure, Function, and Bioinformatics, 77: 499–508. [DOI] [PubMed] [Google Scholar]
- Jones P. et al. (2014) ‘InterProScan 5: Genome-Scale Protein Function Classification’, Bioinformatics, 30: 1236–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jumper J. et al. (2021) ‘Highly Accurate Protein Structure Prediction with AlphaFold’, Nature, 596: 583–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kearse M. et al. (2012) ‘Geneious Basic: An Integrated and Extendable Desktop Software Platform for the Organization and Analysis of Sequence Data’, Bioinformatics, 28: 1647–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keeling P. J. et al. (2014) ‘The Marine Microbial Eukaryote Transcriptome Sequencing Project (MMETSP): Illuminating the Functional Diversity of Eukaryotic Life in the Oceans through Transcriptome Sequencing’, PLoS Biology, 12: e1001889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelley L. A. et al. (2015) ‘The Phyre2 Web Portal for Protein Modeling, Prediction and Analysis’, Nature Protocols, 10: 845–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koonin E. V. et al. (2020) ‘Global Organization and Proposed Megataxonomy of the Virus World’, Microbiology and Molecular Biology Reviews, 84: e00061–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krishnamurthy S. R., and Wang D. (2017) ‘Origins and Challenges of Viral Dark Matter’, Virus Research, 239: 136–42. [DOI] [PubMed] [Google Scholar]
- Mihara T. et al. (2016) ‘Linking Virus Genomes with Host Taxonomy’, Viruses, 8: 66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mönttinen H. A. M., Ravantti J. J., and Poranen M. M. (2021) ‘Structure Unveils Relationships between RNA Virus Polymerases’, Viruses, 13: 313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neri U. et al. (2022) A Five-Fold Expansion of the Global RNA Virome Reveals Multiple New Clades of RNA Bacteriophages. bioRxiv.doi: 10.1101/2022.02.15.480533 [DOI] [PubMed] [Google Scholar]
- Pan J., Vakharia V. N., and Tao Y. J. (2007) ‘The Structure of a Birnavirus Polymerase Reveals a Distinct Active Site Topology’, Proceedings of the National Academy of Sciences of the United States of America, 104: 7385–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peersen O. B. A. (2019) ‘A Comprehensive Superposition of Viral Polymerase Structures’, Viruses, 11: 745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poch O. et al. (1990) ‘Sequence Comparison of Five Polymerases (L Proteins) of Unsegmented Negative-Strand RNA Viruses: Theoretical Assignment of Functional Domains’, Journal of General Virology, 71: 1153–62. [DOI] [PubMed] [Google Scholar]
- ——— et al. (1989) ‘Identification of Four Conserved Motifs among the RNA-Dependent Polymerase Encoding Elements’, The EMBO Journal, 8: 3867–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Price M. N., Dehal P. S., and Arkin A. P. (2010) ‘FastTree 2 – Approximately Maximum-Likelihood Trees for Large Alignments’, PLoS One, 5: e9490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Regnault B. et al. (2021) ‘Deep Impact of Random Amplification and Library Construction Methods on Viral Metagenomics Results’, Viruses, 13: 253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rice P., Longden L., and Bleasby A. (2000) ‘EMBOSS: The European Molecular Biology Open Software Suite’, Trends in Genetics, 16: 276–7. [DOI] [PubMed] [Google Scholar]
- Rost B. (1999) ‘Twilight Zone of Protein Sequence Alignments’, Protein Engineering, Design and Selection, 12: 85–94. [DOI] [PubMed] [Google Scholar]
- Sanjuán R. et al. (2010) ‘Viral Mutation Rates’, Journal of Virology, 84: 9733–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen W., and Ren H. (2021) ‘TaxonKit: A Practical and Efficient NCBI Taxonomy Toolkit’, Journal of Genetics and Genomics, 48: 844–50. [DOI] [PubMed] [Google Scholar]
- Shi M. et al. (2016) ‘Redefining the Invertebrate RNA Virosphere’, Nature, 540: 539–43. [DOI] [PubMed] [Google Scholar]
- Shwed P. S. et al. (2002) ‘Birnavirus VP1 Proteins Form a Distinct Subgroup of RNA-Dependent RNA Polymerases Lacking a GDD Motif’, Virology, 296: 241–50. [DOI] [PubMed] [Google Scholar]
- Sievers F. et al. (2014) ‘Fast, Scalable Generation of High-Quality Protein Multiple Sequence Alignments using Clustal Omega’, Molecular Systems Biology, 7: 539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skewes-Cox P. et al. (2014) ‘Profile Hidden Markov Models for the Detection of Viruses within Metagenomic Sequence Data’, PLoS One, 9: e105067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stevaert A., and Naesens L. (2016) ‘The Influenza Virus Polymerase Complex: An Update on Its Structure, Functions, and Significance for Antiviral Drug Design’, Medicinal Research Reviews, 36: 1127–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sutela S. et al. (2020) ‘The Virome from a Collection of Endomycorrhizal Fungi Reveals New Viral Taxa with Unprecedented Genome Organization’, Virus Evolution, 6: veaa076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suttle C. A. (2005) ‘Viruses in the Sea’, Nature, 437: 356–61. [DOI] [PubMed] [Google Scholar]
- ——— (2007) ‘Marine Viruses - Major Players in the Global Ecosystem’, Nature Reviews. Microbiology, 5: 801–12. [DOI] [PubMed] [Google Scholar]
- Te Velthuis A. J. W. (2014) ‘Common and Unique Features of Viral RNA-Dependent Polymerases’, Cellular and Molecular Life Sciences, 71: 4403–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Venkataraman S., Prasad B. V. L. S., and Selvarajan R. (2018) ‘RNA Dependent RNA Polymerases: Insights from Structure, Function and Evolution’, Viruses, 10: 76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolf Y. I. et al. (2020) ‘‘Doubling of the Known Set of RNA Viruses by Metagenomic Analysis of an Aquatic Virome’’, Nature Microbiology, 5: 1262–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Youle M., Haynes M., and Rohwer F. (2012) ‘Scratching the Surface of Biology’s Dark Matter’. In: Viruses: Essential Agents of Life, pp. 61–81. Springer: Dordrecht. [Google Scholar]
- Zayed A. A. et al. (2022) ‘Cryptic and Abundant Marine Viruses at the Evolutionary Origins of Earth’s RNA Virome’, Science, 376: 156–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All the data produced in this study (alignment, database, and phylogenetic tree files) are available at https://github.com/JustineCharon/RdRp-scan.