

Abstract
Sirtuin1 (SIRT1) is involved in regulating substrate metabolism in the cardiovascular system. Metabolic homeostasis plays a critical role in hypertrophic heart failure. We hypothesize that cardiac SIRT1 can modulate substrate metabolism during pressure overload-induced heart failure. The inducible cardiomyocyte Sirt1 knockout (icSirt1−/−) and its wild type littermates (Sirt1f/f) C57BL/6J mice were subjected to transverse aortic constriction (TAC) surgery to induce pressure overload. The pressure overload induces upregulation of cardiac SIRT1 in Sirt1f/f but not icSirt1−/− mice. The cardiac contractile dysfunctions caused by TAC-induced pressure overload occurred in Sirt1f/f but not in icSirt1−/− mice. Intriguingly, Sirt1f/f heart showed a drastic reduction in systolic contractility and electric signals during post-TAC surgery, whereas icSirt1−/− heart demonstrated significant resistance to pathological stress by TAC-induced pressure overload as evidenced by no significant changes in systolic contractile functions and electric properties. The Seahorse data revealed that TAC-induced pressure overload causes a decrease in basal respiration and ATP generation, as well as reduced OXPHOS integrity in Sirt1f/f but not in icSirt1−/− cardiomyocytes. The targeted proteomics showed that the pressure overload triggered downregulation of the SIRT1-associated IDH2 (isocitrate dehydrogenase 2) that result in increased oxidative stress in mitochondria. Moreover, metabolic alterations were observed in Sirt1f/f but not in icSirt1−/− heart in response to TAC-induced pressure overload. Thus, SIRT1 interferes metabolic homeostasis through mitochondrial IDH2 during pressure overload. Inhibition of SIRT1 activity benefits cardiac functions under pressure overload-related pathological conditions.
Keywords: pressure overload, SIRT1, mitochondria, substrate metabolism
Graphical Abstract
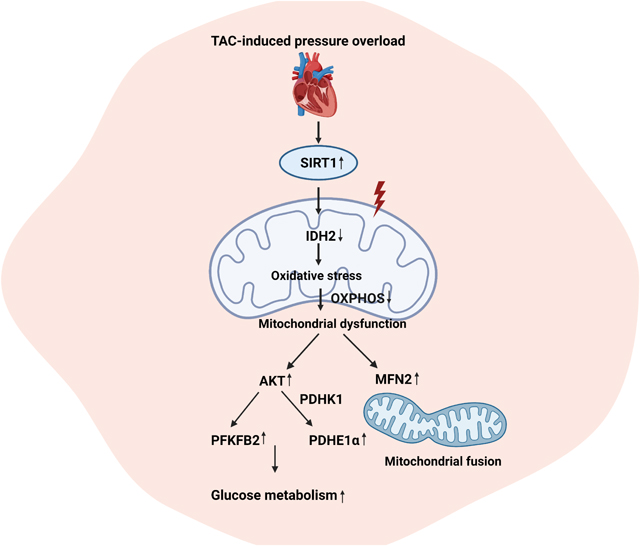
1. Introduction
The clinical hallmark of heart failure (HF) is the inability of the heart to pump adequate blood supply to the body. It is characterized by a complex phenotype that includes reduced myocardial contractility, increased apoptosis, and myocardial fibrosis. During HF, the mitochondria display morphological abnormalities and bioenergetic deficiencies, resulting in an energy-depleted state and increased oxidative stress [1]. The onset of HF is commonly preceded by hypertrophy, associated with enhanced protein synthesis and increased cardiomyocyte size. Hypertrophic growth is initially a compensatory response that serves to reduce wall stress and sustain cardiac function in the face of increased cardiac workload; however, it eventually initiates the development of HF. In this study, we utilize an experimental model, termed transverse aortic constriction (TAC), to accurately depict this progression. It has been reported that cardiac metabolism reprogramming happens early during the development of hypertrophy, with the shift from strongly relying on fatty acids (FAs) to a greater reliance on glucose for energy production. However, the increase in glucose consumption did not prevent or diminish cardiac hypertrophy apart from improving myocardial energetics [2]. It is possible that this distinct alteration in cardiac substrate metabolism could contribute to cardiac remodeling and impaired cardiac function in HF. However, the mechanism mediating pathological changes and the roles these changes play in the progression of HF is not fully elucidated.
Sirtuin1 (SIRT1) is a deacetylase located in the nucleus that could regulate transcriptional factors for cellular control [3]. Due to its critical regulatory role, SIRT1 has been implicated in cardiac hypertrophy and HF development, but the effects of SIRT1 vary depending upon the expression level. A research study showed that a low SIRT1 expression attenuated age-dependent cardiac hypertrophy, whereas a high level of SIRT1 (12.5 fold) induced it by promoting oxidative stress and cardiomyocyte growth [4]. It was also demonstrated that whole-body SIRT1-knockout mice exhibited smaller hearts compared to the wild-type littermates, indicating the resistance against the development of cardiac hypertrophy induced by hypertrophy agonists [5]. These findings are consistent with the notion that SIRT1 is needed for the induction of cardiac hypertrophy, but the specific role of SIRT1 in modulating these effects remained elusive.
Here, we utilized an inducible cardiomyocyte Sirt1 knockout (icSirt1−/−) versus its wild type littermates (Sirt1f/f) mice to investigate the role of cardiomyocyte SIRT1 in response to pressure overload-induced hypertrophic HF. The results demonstrated that the pressure overload by TAC surgery increased cardiac SIRT1 associated with alterations in mitochondrial dynamics and metabolic homeostasis. Thus, this study unveils an unrecognized role of SIRT1 in cardiac remodeling that could be a potential strategy for intervening hypertrophic HF.
2. Materials and methods
2.1. Animals
C57BL/6J wild type mice (3–5 months), SIRT1f/f mice (stock number 008041), and α-MHC-CreERTT2 (3–5 months) (stock number 005657) were supplied from Jackson Laboratory (Bar Harbor, ME). The cardiomyocyte-specific deletion of the SIRT1 gene mouse was generated by breading SIRT1f/f with transgenic mice that carried an autosomally integrated Cre gene driven by cardiac-specific alpha-myosin heavy chain promoter (α-MHC-CreERT2). The inducible cardiac-specific SIRT1 knockout (icSirt1−/−) mice were generated by Tamoxifen injection (0.08 mg/g, i.p 5 days) of α-MHC-CreERTT2- SIRT1f/f (3–5 months) mice, and SIRT1f/f mice (3–5 months) with Tamoxifen injection were used for control groups. The mouse genotype was determined using the Mouse Tail Quick Extraction kit (Bio Pioneer) to isolate genomic DNA from the tail. Both males and females were subjected to the experiments in this study. All animal protocols in this study were approved by the Institutional Animal Care and Use Committee of the University of South Florida as well as conform to the NIH Guide for the care and use of laboratory animals.
2.2. In vivo transverse aortic constriction (TAC) model
Mice were anesthetized, intubated, and ventilated as we previously described [6,7]. Mice were anesthetized with isoflurane (2%), placed on a ventilator (Harvard Rodent Ventilator, Harvard Apparatus, Holliston, MA), and put on a heating pad to maintain body temperature during surgeries. Mice were subcutaneously injected with bupivacaine (2 mg/kg) for one time before surgery. After a partial thoracotomy to the second rib was performed, the ribs were then retracted using a chest retractor. The transverse aorta was identified after separating the thymus and fat tissue from the aortic arch. Two loose knots were tied around the transverse aorta between the brachiocephalic and left common carotid artery. The loose knots were quickly tied against a 27-gauge needle, and the needle was promptly removed to yield an approximate 0.4mm aortic diameter. For the sham group, the entire procedure is identical except for the construction of the aorta. Analgesic administration was given with one single injection of buprenorphine (1 mg/kg) to relieve postoperative pain before returning to the animal facility. The whole TAC surgery procedure was performed as described [8,9].
2.3. Fatty acid/glucose oxidation analysis
The working heart system preload and afterload were set at 15 cm and 80 cm H2O, respectively [10]. The flow rate was kept at 15 mL/min. Heart function was monitored by a pressure transducer connected to the aortic outflow. [9, 10]- 3H-oleate (50 mci/L) and 14C-glucose (20 mci/L)–labeled KHB buffer was perfused into the heart via the pulmonary vein and pumped out through the aorta. Perfusate pumped out of that aorta and outflowed from the coronary artery was collected every 5 minutes to examine the radioactivity level. The fatty acid level was evaluated from 3H2O produced from [9, 10]-3H-oleate. Metabolized 3H2O was separated from [9, 10]- 3H-oleate by filtering through anion-exchange resin (BioRad, Hercules, CA). The metabolized 14CO2 was dissolved in the perfusate buffer, and the gaseous 14CO2, which was later dissolved in sodium hydroxide in the samples, were all used to measure glucose oxidation. Sulfuric acid was added to the perfusate samples to release and separate 14CO2 from 14C-glucose. 3H and 14C signals were detected to discriminate metabolic products from fatty acid and glucose, respectively.
The related common biochemical methods are listed in Supplemental Materials.
2.4. Statistics analysis
All results are reported as means ± standard error of the means (SEM). Statistical analysis was performed using the GraphPad Prism 9 software, and comparisons were performed using two-way ANOVA with Tukey’s test. A p-value of p < 0.05 was considered statistically significant.
3. Results
3.1. SIRT1 mediates cardiac dysfunctions caused by pressure-overload pathological stress
There are studies have shown that expression of SIRT1 is upregulated in dog hearts undergoing HF [4], [11]. Thus, we examined whether SIRT1 is elevated in response to the TAC-induced hypertrophy and HF model. Through immunoblotting analysis, we observed a significant increase in the level of cardiac SIRT1 in Sirt1f/f but not in icSirt1−/− mice after 6 weeks of TAC-induced pressure overload (Figure 1A). Moreover, the real-time RT-PCR data showed that pressure overload triggered Sirt1 mRNA upregulation in Sirt1f/f but not in icSirt1−/− heart (Figure 1A). It indicated that TAC-induced pressure overload stress can trigger upregulation of cardiomyocyte Sirt1 in both transcriptional and translational levels.
Figure 1. Upregulated SIRT1 exacerbated cardiac dysfunction under pressure overload-induced heart failure (HF).
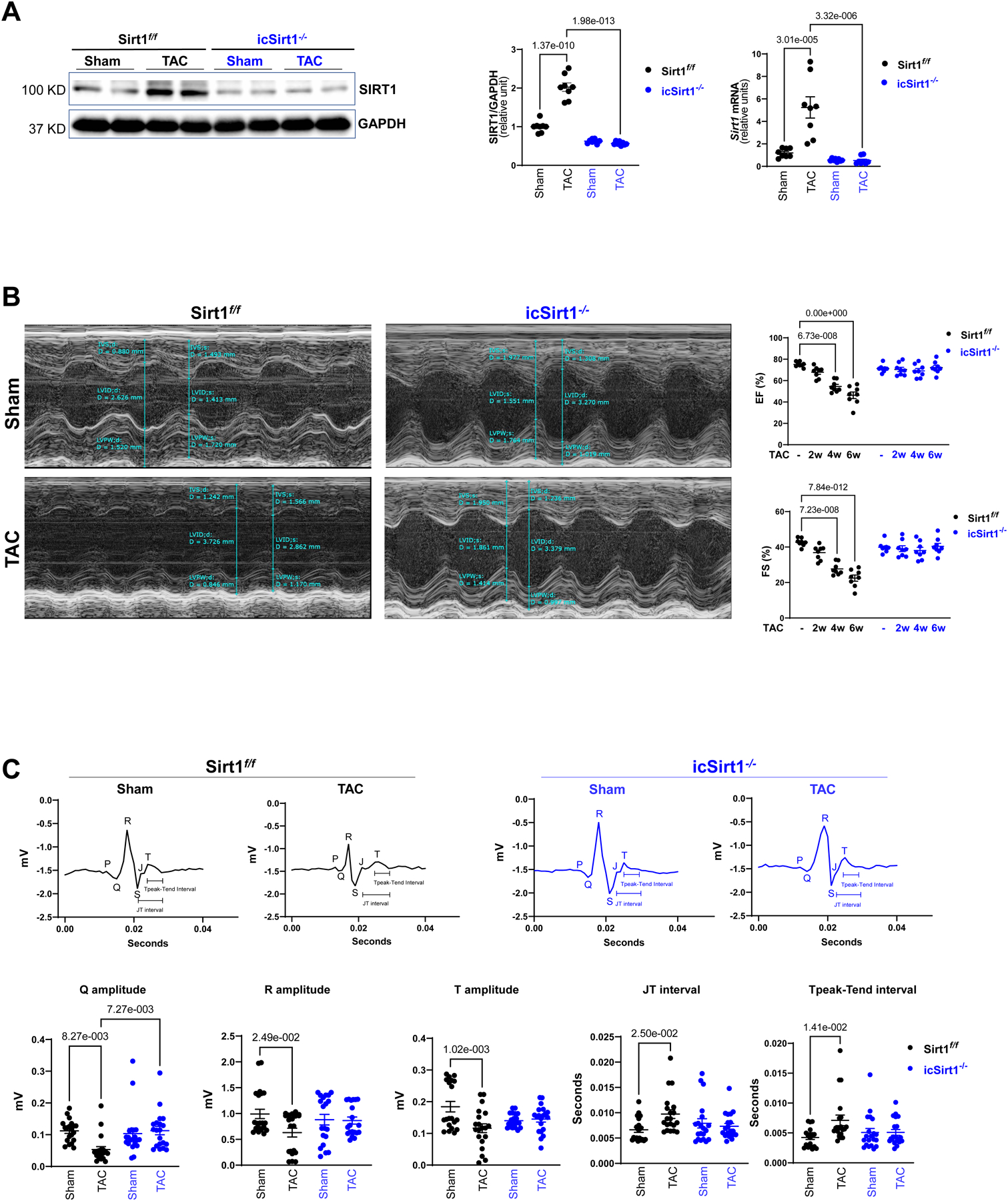
(A) The protein and mRNA expression levels of SIRT1 were upregulated in the left ventricles (LV) of Sirt1f/f heart after 6 weeks of transverse aortic constriction (TAC)-induced pressure overload. Biological replicates N=8 for each group. P value was determined by two-way ANOVA with Tukey’s post-hoc test. (B) Echocardiography showed that Sirt1f/f mice were vulnerable to TAC-induced pressure overload, as shown by decreased ejection fraction (EF) and fractional shortening (FS). Left: Representative images of M-mode echocardiography. Right: Quantification of echocardiography measurements for EF and FS. Biological replicates N=8 for each group. P value was determined by two-way ANOVA with Tukey’s post-hoc test. (C) Electrocardiography (ECG) showed that Sirt1f/f mice developed cardiac contractile dysfunction under 6 weeks of post TAC with reduced Q, R, and T amplitudes and prolonged JT and Tpeak-Tend interval. Upper: Representative images of ECG parameters. Lower: Quantification of ECG measurements. Biological replicates N=8 for each group. P value was determined by two-way ANOVA with Tukey’s post-hoc test. Biological replicates N=8 for each group. P value was determined by two-way ANOVA with Tukey’s post-hoc test.
To assess whether elevated expression of SIRT1 under TAC-induced hypertrophy and HF alters basic cardiac physiological features, we first performed echocardiography every two weeks. Two echocardiographic measurements were examined: ejection fraction (EF) and fractional shortening (FS). EF is defined by the percentage of blood exiting the heart for each contraction, and FS is referred to the reduction in length of the LV (left ventricle) towards the end of systole [12]. Both EF and FS are critical indicators to evaluate cardiac systolic function. The echocardiography measurements demonstrated that Sirt1f/f exhibited a significant reduction in EF and FS first observed at 4 weeks post-TAC and progressively decreased further at 6 weeks post-TAC compared to physiological control (Figure 1B). Of interest, icSirt1−/− 6 weeks post-TAC showed no significant changes in both EF and FS (Figure 1B). These findings suggest that the deletion of Sirt1 in cardiomyocytes had preserved cardiac function under pressure overload-induced pathological stress conditions. All other echocardiographic parameters were listed in the supplement (Table S1).
In addition to employing echocardiography to evaluate cardiac contractility, we also examined electrical signals by electrocardiography (ECG) in Sirt1f/f and icSirt1−/− mice under sham operations and TAC-surgery for 6 weeks. After 6 weeks of TAC, there were significant decreases in the amplitude of Q, R, and T waves on the ECG compared to sham operations in Sirt1f/f mice (Figure 1C). Moreover, there were increases in JT and Tpeak-Tend intervals in TAC-surgery versus sham operations in Sirt1f/f mice (Figure 1C). Intriguingly, following 6 weeks of TAC, Sirt1-deficient mice, icSirt1−/− exhibited no significant alterations in these parameters (Figure 1C). The alterations in ECG of Sirt1f/f mice in response to TAC-induced pressure overload could be attributed to the upregulation of SIRT1 in Sirt1f/f heart leading to LV enlargement, which resulted in cardiac contractility defects and abnormal electrical signals.
3.2. Cardiomyocytes Sirt1 mediates hypertrophy and fibrosis induced by pressure overload
Considering the impaired cardiac function in Sirt1f/f versus icSirt1−/− mice at 6 weeks post-TAC, we hypothesized that upregulated SIRT1 might impact the cardiac phenotype during the maladaptive response. Thus, we isolated perfused hearts from each group to conduct an initial pathological characterization. Postmortem analyses showed that Sirt1f/f but not icSirt1−/− mice at 6 weeks of post-TAC surgery showed significantly enlarged left atrium and LV (Figure 2A). Additionally, an increase in cardiomyocyte cell size has been proposed as an early hallmark that occurs before the onset of permanent myocardial fibrosis followed by cardiac dysfunction and HF [13],[14],[15]. Histological examination of LV with Masson’s trichrome revealed substantial increases in perivascular and interstitial fibrosis in Sirt1f/f 6 weeks of post-TAC surgery as compared to sham operation groups (Figure 2A). However, there were no significant differences in cardiac fibrosis of icSirt1−/− at 6 weeks of post-TAC surgery as compared to the sham groups (Figure 2A). The cardiac mRNA expression levels of collagen type 1 alpha 1 chain (Col1α1), fibronectin (Fn1), and connective tissue growth factor (Ctgf) showed a positive correlation with the observed histological analysis (Figure 2B), i.e., the mRNA levels of fibrosis-related genes (Col1α1, Fn1 and Ctgf) were elevated in Sirt1f/f but not in icSirt1−/− heart at 6 weeks of post-TAC-surgery versus sham groups (Figure 2B). Thus, these results indicated that pressure overload-induced upregulation of cardiomyocytes SIRT1 plays a role in cardiac remodeling during TAC-induced pathological conditions.
Figure 2. Deletion of cardiomyocyte SIRT1 attenuated cardiac remodeling induced by pressure overload.
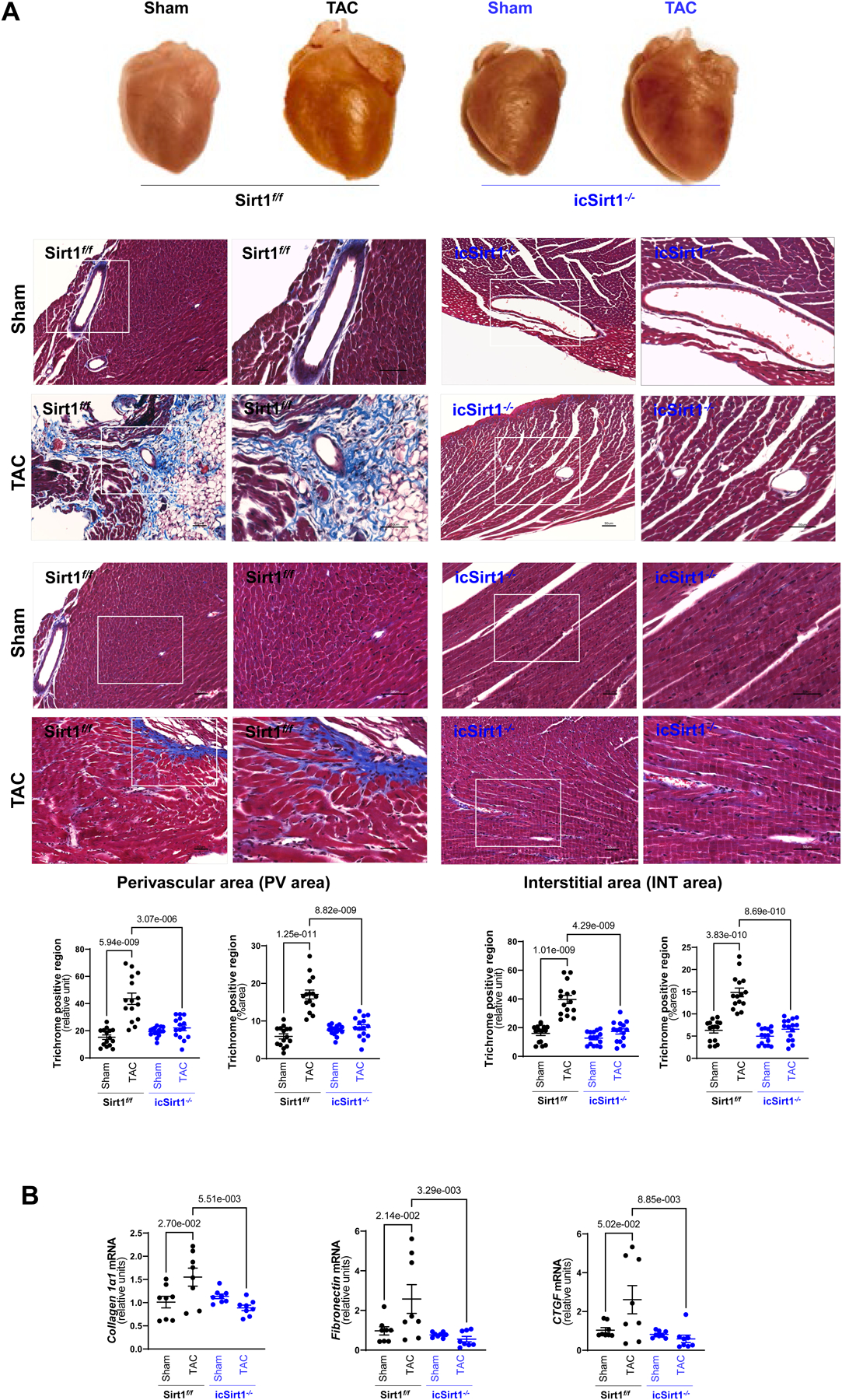
(A) Upper: Representative hearts from Sirt1f/f and icSirt1−/− under sham operations (Sham) or 6 weeks of TAC surgery-induced pressure overload (TAC). Middle: Representative images of perivascular and interstitial fibrosis measured by Masson’s trichrome staining. The scale bars are 50 μm. Lower: Quantification analysis of Masson’s trichrome staining. Biological replicates N=5 for each group. P value was determined by two-way ANOVA with Tukey’s post-hoc test. (B) Quantitation for real time RT-PCR for mRNA expression of collagen type 1 alpha 1 chain (Col1α1), fibronectin (Fn1), and connective tissue growth factor (Ctgf) in LV tissue. Biological replicates N=8 for each group. P value was determined by two-way ANOVA with Tukey’s post-hoc test.
3.3. SIRT1 associated with PPARα modulates IDH2 and mitochondrial redox homeostasis
One of the critical features of HF is the switch in substrate utilization with the decrease in fatty acid oxidation (FAO) and the enhancement in glucose oxidation metabolism [16]. The rate of FAO is partially dependent on the expression level of genes encoding for FAO enzymes, notably are the PPARs family and PPARγ co-activator 1 α (PGC-1α) [17], [18]. Additionally, studies have shown that SIRT1 could activate PGC-1α through deacetylation and interact with PPARα to regulate fatty acid (FA) metabolism [19], [20]. Thus, we performed immunoblotting and real time-PCR to detect the protein and gene expression level of those genes. The results demonstrated that there were significant upregulation of PPARα in both translational and transcriptional levels in Sirt1f/f but not in icSirt1−/− heart at 6 weeks of post-TAC surgery (Figure 3A), however there was no change in PGC-1α level across all groups (Figure S2).
Figure 3. SIRT1 plays a role in mitochondrial oxdative phosphorylation in response to TAC-induced pressure overload.
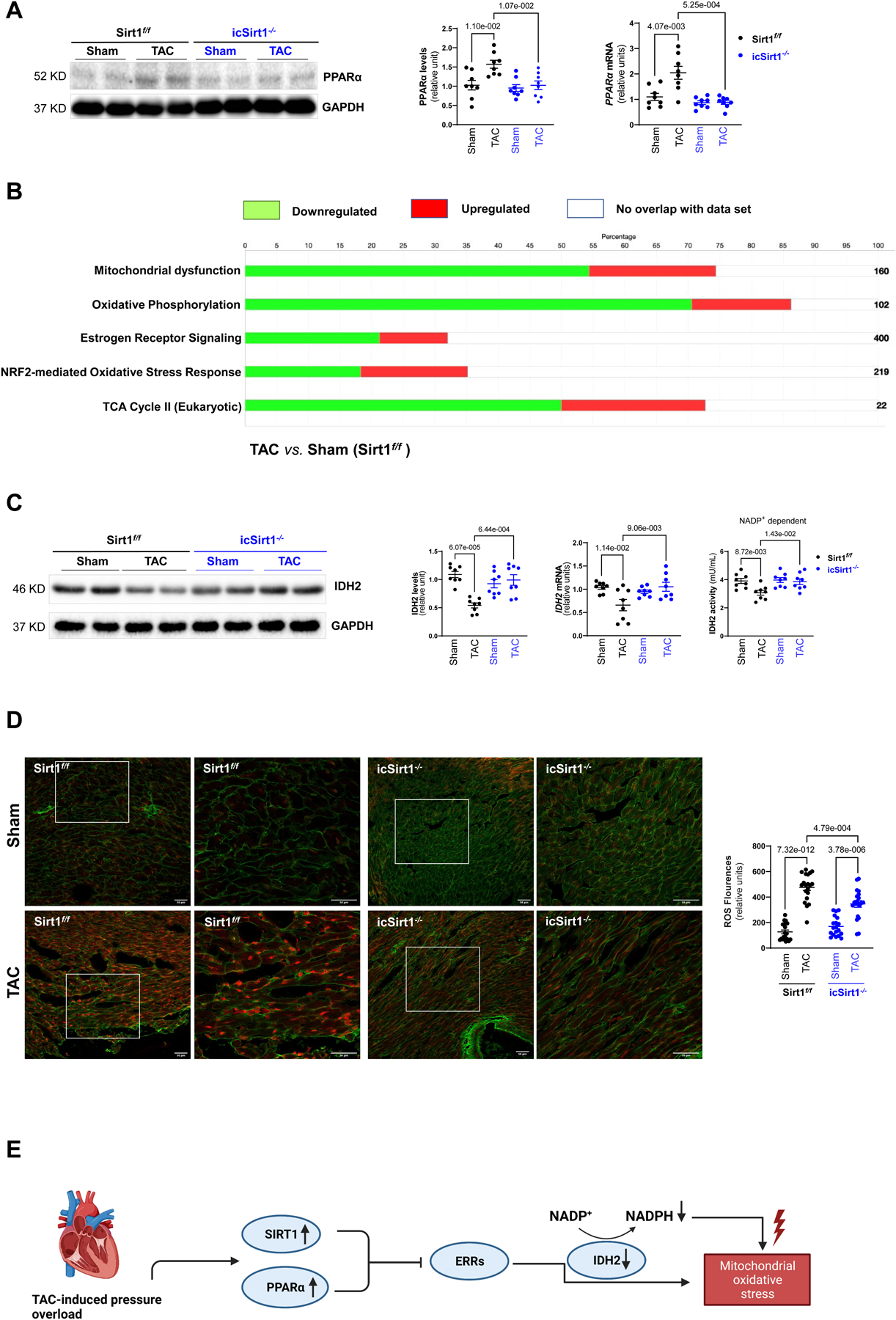
(A) TAC-induced pressure overload stress increased protein and mRNA expression levels of PPARα in Sirt1f/f mice. Biological replicates N=8 for each group. P value was determined by two-way ANOVA with Tukey’s post-hoc test. (B) Ingenuity pathway analysis (IPA) enrichment analysis of targeted proteomics of Sirt1f/f heart with 6 weeks of post TAC versus sham group. Green bars representing the percentage of genes in the pathway were downregulated in Sirt1f/f 6 weeks of post TAC versus Sham. Red bars representing the percentage of genes in the pathway were upregulated in Sirt1f/f 6 weeks of post TAC versus Sham. White bars representing the percentage of genes in the pathway were not altered in Sirt1f/f 6 weeks of post TAC versus Sham. Biological replicates N=3 for each group. (C) Reduction of the IDH2 protein, mRNA expression levels, and NADP+ dependent activity in Sirt1f/f but not in icSirt1−/− heart in response to 6 weeks of TAC-induced stress. Biological replicates N=8 for each group. P value was determined by two-way ANOVA with Tukey’s post-hoc test. (D) MitoSOX™ staining showed a substantial increase in superoxide accumulation in the heart of Sirt1f/f after 6 weeks of TAC. Left: Representative microscopy images of reactive oxygen species staining. The scale bars are 50 μm. Right: Quantification analysis of MitoSOX™ staining. Biological replicates N=5 for each group. P value was determined by two-way ANOVA with Tukey’s post-hoc test. (E) Schematic representations of the regulatory roles of SIRT1 and PPARα in mitochondrial oxidative stress during TAC-induced pathological conditions.
To characterize the role of cardiac SIRT1 and PPARα in cardiac functions during TAC-induced pressure overload pathological conditions, we performed targeted proteomics with Sirt1f/f hearts under sham operations or TAC surgery followed by Ingenuity pathway analysis (IPA). The data showed that the mitochondrial protein complex, respiratory chain, and tricarboxylic acid cycle (TCA) enzymes were downregulated at 6 weeks of TAC-surgery as compared to the sham operation groups (Figure 3B). In addition, the estrogen-related receptors (ERRs) targets, which regulate genes controlling multiple aspects of mitochondrial functions [21], were diminished during TAC-induced pressure-overload versus sham operations (Figure 3B). Indeed, we also noticed that TAC-induced pressure overload reduced both protein and mRNA expression levels of IDH2 (isocitrate dehydrogenase 2) in Sirt1f/f but not in icSirt1−/− heart (Figure 3C). Since IDH2 consumes NADP+ to convert isocitrate to α-ketoglutarate in the TCA cycle [22], we measure the NADP+ dependent IDH2 activity. The results demonstrated that there was a reduction of the IDH2 activity in Sirt1f/f but not in icSirt1−/− heart at 6 weeks of post-TAC versus sham groups; this could be due to the downregulation of IDH2 expression levels (Figure 3C). Along with the critical role in the TCA cycle, IDH2 also provides NADPH for maintaining mitochondrial redox balance [22]. As a result, the downregulation of IDH2 could disrupt mitochondrial redox homeostasis during pressure overload-induced stress conditions that can aggravate mitochondrial oxidative stress in response to TAC-induced pathological stress. We performed experiments to stain the fresh LV tissue with MitoSOX™ Red to characterize the mitochondrial redox homeostasis. The results showed a significant increase in reactive oxygen species (ROS) production in the mitochondria of Sirt1f/f and icSirt1−/− at 6 weeks of post-TAC surgery versus sham operations (Figure 3D). However, SIRT1-deficient mice icSirt1−/− versus Sirt1f/f produced significantly less ROS during TAC-induced pathological stress conditions (Figure 3D). These results suggest that co-upregulation of SIRT1 and PPARα repressed ERR target genes, particularly IDH2, resulting in mitochondrial oxidative stress (Figure 3E).
3.4. SIRT1 plays a role in the metabolic regulation in response to pressure overload
The cardiac contractility depends strongly on ATP production, provided mainly by fatty acid oxidation (FAO). As a result, defect in mitochondrial function is associated with alterations in energy metabolism [23]. Since SIRT1 hindered mitochondrial function, we further explored whether it can also modulate substrate metabolism reprogramming under pressure overload-induced pathological conditions by performing immunoprecipitation with SIRT1 specific antibodies. It was demonstrated that SIRT1 is associated with the upregulation of proteins related to glycolysis, pyruvate dehydrogenase (PDH) complex, gluconeogenesis, and aspartate biosynthesis (Figure 4A). Therefore, we performed an ex vivo working heart perfusion system to examine the oxidative consumption rate of each substrate. Glucose oxidation and FAO were analyzed by measuring [14C]-glucose incorporation into 14CO2 and 3H2O from [9, 10-3H]-oleate released in the perfusate. The results demonstrated that a shift in substrate utilization occurred in Sirt1f/f hearts from TAC-induced pressure overload group with a decrease in FAO and an increase in glucose oxidation as compared to the Sirt1f/f hearts from sham operation group (Figure 4B). On the contrary, there was no significant alterations in substrate metabolism occurred in icSirt1−/− hearts from TAC-induced pressure overload and sham operation groups (Figure 4B). Furthermore, Seahorse Mito Fuel Flex Test was utilized to characterize the preferred substrate utilization and validate the role of SIRT1 in mediating the metabolic response to TAC-induced pathological stress. The data demonstrated that the mitochondria of Sirt1f/f heart prefer to use glucose oxidation in response to TAC-induced pathological stress versus sham group, as evidenced by an increase in glucose dependency and capacity (Figure 4C). Interestingly, the mitochondria of icSirt1−/− heart showed a preserved substrate consumption with a higher dependency and capacity to utilize FAO for cardiac energetics in response to TAC-induced stress (Figure 4C). Together, the findings suggest that SIRT1 could mediate a metabolic switch with an increased reliance on glucose to disturb the adaptive metabolic response during pressure overload-induced stress conditions.
Figure 4. SIRT1-AKT signaling cascade modulates glucose metabolism in response to TAC-induced pathological stress.
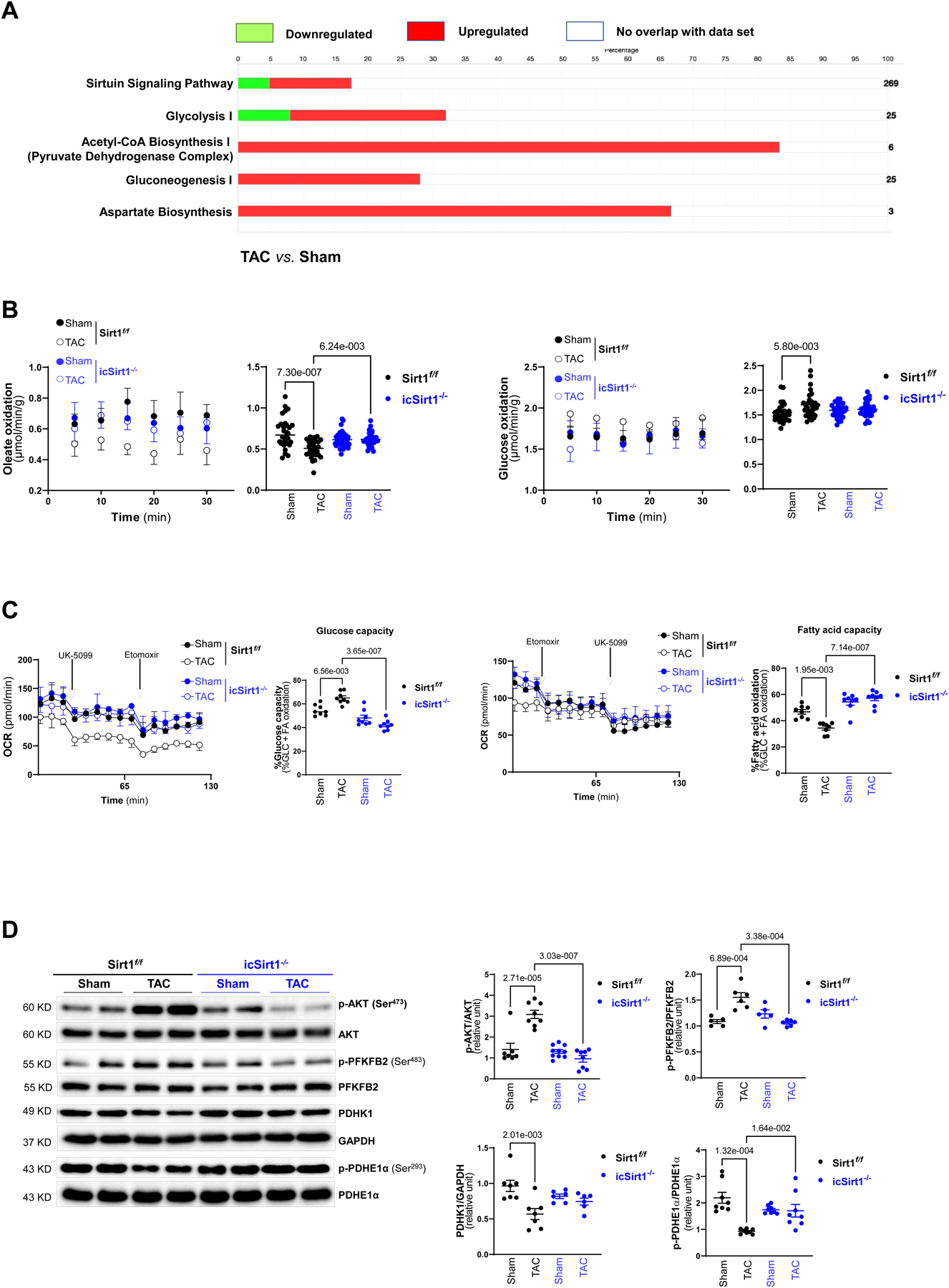
(A) Ingenuity pathway analysis (IPA) enrichment analysis of SIRT1 associated proteins in Sirt1f/f 6 weeks of post TAC versus sham. Green bars representing the percentage of genes in the pathway were downregulated in Sirt1f/f 6 weeks of post TAC versus sham. Red bars representing the percentage of genes in the pathway were upregulated in Sirt1f/f 6 weeks of post TAC versus sham. White bars representing the percentage of genes in the pathway were not altered in response to TAC or not associated with SIRT1 under physiolocal and pathological conditions. Biological replicates N=3 for each group. (B) Glucose oxidation and oleate oxidation were analyzed by measuring [14C]-glucose incorporation into 14CO2 and incorporation of [9,10-3H] oleate into 3H2O, respectively, in the ex vivo mouse working heart system for 30 minutes. Sirt1f/f 6 weeks of post TAC resulted in increased glucose utilization and decreased fatty acid oxidation. Biological replicates N=6 for each group. P value was determined by two-way ANOVA with Tukey’s post-hoc test. (C) Seahorse fuel flex assay examined substrate utilization by measuring the OCR in Sirt1f/f and icSirt1−/− hearts under sham or 6 weeks of TAC. UK5099 (8 μM) and Etomoxir (4 μM) were added to isolated cardiomyocytes of all groups. Left: Representatives of mitochondrial fuel flex assay. Right: Quantitative analysis of substrate metabolism parameters (glucose capacity and fatty acid capacity). Biological replicates N=6 for each group. P value was determined by two-way ANOVA with Tukey’s post-hoc test. (D) The immunoblotting analysis for AKT, PFKFB2, PDHK1, PDHE1α, and GAPDH signaling response of Sirt1f/f and icSirt1−/− hearts under sham or 6 weeks of TAC conditions. Biological replicates N=8 for each group. P value was determined by two-way ANOVA with Tukey’s post-hoc test.
To characterize the role of cardiac SIRT1 in glucose metabolism in response to TAC-induced pressure overload stress, we performed immunoblotting to determine the signaling pathways associated with glucose metabolic regulation under physiological or pathological conditions in Sirt1f/f and icSirt1−/− hearts. The results demonstrated that TAC-induced pressure overload triggered phosphorylation of AKT at Ser473 site, indicating enhanced AKT activation in Sirt1f/f but not in icSirt1−/− heart during TAC-induced stress as compared to sham group (Figure 4D). AKT is a master regulator of cellular metabolism, and AKT activation has been shown to be correlated with increased modulation of glucose metabolism [24], [25]. Moreover, the phosphorylation of the AKT downstream PFKFB2 (phosphofructokinase 2) associated with glycolysis was significantly increased in response to TAC-induced pressure overload stress versus sham in Sirt1f/f but not in icSirt1−/− hearts (Figure 4D). Additionally, the downregulation of PDHK1 (pyruvate dehydrogenase kinase 1) observed in Sirt1f/f under TAC-induced pathological conditions, could explain a decrease in phosphorylation of pyruvate dehydrogenase E1α (PDHE1α), reflecting a higher PDH activity in connecting glycolysis to the TCA cycle (Figure 4D). Intriguingly, there were no significant alterations occurred in PFKFB2, PDHK1, and PDHE1α in icSirt1−/− hearts during TAC-induced stress conditions (Figure 4D). Together, the results indicated that SIRT1 is essential for pressure overload-induced AKT activation, which further modulates its downstream targets associated with glucose metabolic regulations.
4. Discussion
In this study, we investigated the role of SIRT1, which was elevated in response to pressure overload-induced pathological stress. We revealed that Sirt1f/f mice at 6 weeks of post-TAC surgery showed impaired cardiac function and increased stiffness, as evidenced by cardiac examination and histological analysis. These observations were proposed to be attributed to the downregulation of IDH2, which increased mitochondrial oxidative stress, impaired mitochondrial function, and induced metabolism alterations. Furthermore, our data shed new light on the contribution of the SIRT1-AKT signaling cascade to enhanced glucose metabolism.
SIRT1 has been found to be involved in many physiological processes that regulate cellular metabolic status, stress resistance, and apoptosis [26], [27]. Previous research has shown that the SIRT1 level was upregulated in failing hearts as a compensatory mechanism [11], however, a high level of SIRT1 is not very beneficial for the hearts. Overexpression of SIRT1 makes the heart more prone to hypertrophy and HF with an increase in apoptosis, fibrosis, and oxidative stress, as well as a decrease in mitochondrial biogenesis [4]. The observations from our study also supported that elevated SIRT1 expression under pressure overload-induced stress leads to impaired cardiac function through lowering cardiac output, diminishing electrical signals, and alternating cardiac morphology with an increase in cardiomyocyte size and fibrosis. Furthermore, co-upregulation of SIRT1 and PPARα suppressed ERR signaling pathway, which is implicated in the TCA cycle, OXPHOS, and mitochondrial function. Through controlling a subset of ERR transcriptional pathway, SIRT1/PPARα complex plays an essential role in fasting response as it restricts nutritional consumption to increment endurance and survival.[28] Indeed, our findings demonstrated that Sirt1f/f heart under TAC-induced stress had a lower OCR and lower ATP production of mitochondria.. This fasting response is deemed maladaptive during the pressure overload stress with high energy demand since the energy production could not keep up with the need. Thus, Sirt1f/f under pressure overload-induced stress mediated mitochondrial proliferation and displayed elongated mitochondria by triggering the expression of MFN2, which may compensate for the diminished energy generation. We also observed a high MFN2 level in icSirt1−/− versus Sirt1f/f but with normal mitochondrial morphology. This could be due to a higher rate of the counterbalance in mitochondrial dynamics between mitochondrial fusion and fission in icSirt1−/− versus Sirt1f/f hearts.
The low expression levels and activity of IDH2 were identified in TAC-induced Sirt1f/f hearts. IDH2 is a major producer of mitochondrial NADPH, which is required for glutathione-associated mitochondrial antioxidants.[29] Therefore, IDH2 is an essential enzyme in regulating redox homeostasis and protecting cells from oxidative stress [30]. As expected, Sirt1f/f heart in response to TAC-induced stress had a drastic increase in ROS generation and accumulation, whereas icSirt1−/− heart under TAC-induced stress showed a significantly lower level of ROS. The increased mitochondrial ROS could be an early event that initiates the mitochondrial structural dysfunctions. Overall, it appeared that SIRT1 cooperated with PPARα to repress ERR target genes, particularly IDH2, which results in altered mitochondrial morphology and increased mitochondrial oxidative stress. Moreover, this study advances the understanding of SIRT1’s roles in metabolic alterations by unveiling a novel SIRT1-AKT signaling pathway in response to pressure overload stress. In light of AKT activation in Sirt1f/f hearts at 6 weeks of post-TAC, we observed increased phosphorylation of PFKFB2 (Ser483), which indicated the promotion of glycolysis processes [31]. Additionally, we detected the protein level of PDHK1 and phosphorylated PDHE1α Ser293. PDHK1 regulates the conversion of pyruvate to acetyl-coenzyme A through phosphorylation of the PDH enzyme complex. The decrease in PDHK1 level in TAC-induced Sirt1f/f hearts was consistent with the reduced phosphorylation of PDHE1α, indicating the elevated PDH complex activity. PDHE1α is important in linking glycolysis and the TCA cycle, which implies the increased glucose-derived substrate into the TCA cycle. The experimental data from Sirt1f/f versus icSirt1−/− provided clear evidence supporting the role of SIRT1 in modulating glucose metabolism via AKT activation during pressure overload stress conditions.
5. Conclusions
An upregulation of cardiac SIRT1 in response to pressure overload pathological stress exacerbates cardiac dysfunction. The association of SIRT1 with PPARα suppressed the proteins involved in the TCA cycle and mitochondrial function, particularly through downregulating IDH2. SIRT1-AKT signaling cascade plays a role in glucose metabolic regulation in response to pressure overload-induced pathological conditions. The alterations in metabolic homeostasis caused by SIRT1 mediated pressure overload stress can result in defects in mitochondrial function with a higher level of oxidative stress in mitochondria. Thus, upregulation of SIRT1 by pressure overload stress could render the heart more susceptible to hypertrophic stress by promoting mitochondrial dysfunction and metabolic remodelings.
Supplementary Material
Highlights.
Deletion of SIRT1 in cardiomyocytes attenuates adverse cardiac remodeling through preserving mitochondrial metabolic homeostasis.
Inhibiting SIRT1 activity can be a potential treatment for hypertrophic heart failure.
Controlling metabolic homeostasis under pressure overload-related pathological stress conditions is a potential intervention for hypertrophic heart failure patients.
Funding Information
This work was supported by NIH R01HL158515 and R01GM124108, Department of Veterans Affairs Merit Award I01BX005625, and the James ans Esther King Biomedical Research Program of The Florida Department of Health 22K08.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declaration of competing interest
The authors declare no conflict of interest.
Data Availability Statement
The datasets generated and/or analyzed during the current study are available from the corresponding author on reasonable request.
REFERENCES
- [1].Russell LK, Finck BN, Kelly DP, Mouse models of mitochondrial dysfunction and heart failure, J Mol Cell Cardiol 38 (2005) 81–91. 10.1016/j.yjmcc.2004.10.010. [DOI] [PubMed] [Google Scholar]
- [2].Ritterhoff J, Young S, Villet O, Shao D, Neto FC, Bettcher LF, Hsu YA, Kolwicz SC Jr., Raftery D, Tian R, Metabolic Remodeling Promotes Cardiac Hypertrophy by Directing Glucose to Aspartate Biosynthesis, Circ Res 126 (2020) 182–196. 10.1161/CIRCRESAHA.119.315483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Gu C, Xing Y, Jiang L, Chen M, Xu M, Yin Y, Li C, Yang Z, Yu L, Ma H, Impaired cardiac SIRT1 activity by carbonyl stress contributes to aging-related ischemic intolerance, PLoS One 8 (2013) e74050. 10.1371/journal.pone.0074050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Alcendor RR, Gao S, Zhai P, Zablocki D, Holle E, Yu X, Tian B, Wagner T, Vatner SF, Sadoshima J, Sirt1 regulates aging and resistance to oxidative stress in the heart, Circ Res 100 (2007) 1512–1521. 01.RES.0000267723.65696.4a [pii] 10.1161/01.RES.0000267723.65696.4a. [DOI] [PubMed] [Google Scholar]
- [5].Sundaresan NR, Pillai VB, Gupta MP, Emerging roles of SIRT1 deacetylase in regulating cardiomyocyte survival and hypertrophy, J Mol Cell Cardiol 51 (2011) 614–618. 10.1016/j.yjmcc.2011.01.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Quan N, Sun W, Wang L, Chen X, Bogan JS, Zhou X, Cates C, Liu Q, Zheng Y, Li J, Sestrin2 prevents age-related intolerance to ischemia and reperfusion injury by modulating substrate metabolism, FASEB J 31 (2017) 4153–4167. 10.1096/fj.201700063R. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Wang J, Yang L, Rezaie AR, Li J, Activated protein C protects against myocardial ischemic/reperfusion injury through AMP-activated protein kinase signaling, J Thromb Haemost 9 (2011) 1308–1317. 10.1111/j.1538-7836.2011.04331.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Richards DA, Aronovitz MJ, Calamaras TD, Tam K, Martin GL, Liu P, Bowditch HK, Zhang P, Huggins GS, Blanton RM, Distinct Phenotypes Induced by Three Degrees of Transverse Aortic Constriction in Mice, Sci Rep 9 (2019) 5844. 10.1038/s41598-019-42209-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].deAlmeida AC, van Oort RJ, Wehrens XH, Transverse aortic constriction in mice, J Vis Exp (2010). 10.3791/1729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Ren D, Fedorova J, Davitt K, Van Le TN, Griffin JH, Liaw PC, Esmon CT, Rezaie AR, Li J, Activated Protein C Strengthens Cardiac Tolerance to Ischemic Insults in Aging, Circ Res 130 (2022) 252–272. 10.1161/CIRCRESAHA.121.319044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Alcendor RR, Kirshenbaum LA, Imai S, Vatner SF, Sadoshima J, Silent information regulator 2alpha, a longevity factor and class III histone deacetylase, is an essential endogenous apoptosis inhibitor in cardiac myocytes, Circ Res 95 (2004) 971–980. 10.1161/01.RES.0000147557.75257.ff. [DOI] [PubMed] [Google Scholar]
- [12].Mattiazzi AR, Cingolani HE, Montenegro H, Shortening fraction: its dependence on the Starling mechanism, Cardiovasc Res 15 (1981) 475–482. 10.1093/cvr/15.8.475. [DOI] [PubMed] [Google Scholar]
- [13].Boluyt MO, O’Neill L, Meredith AL, Bing OH, Brooks WW, Conrad CH, Crow MT, Lakatta EG, Alterations in cardiac gene expression during the transition from stable hypertrophy to heart failure. Marked upregulation of genes encoding extracellular matrix components, Circ Res 75 (1994) 23–32. 10.1161/01.res.75.1.23. [DOI] [PubMed] [Google Scholar]
- [14].Lorell BH, Carabello BA, Left ventricular hypertrophy: pathogenesis, detection, and prognosis, Circulation 102 (2000) 470–479. 10.1161/01.cir.102.4.470. [DOI] [PubMed] [Google Scholar]
- [15].Scimia MC, Hurtado C, Ray S, Metzler S, Wei K, Wang J, Woods CE, Purcell NH, Catalucci D, Akasaka T, Bueno OF, Vlasuk GP, Kaliman P, Bodmer R, Smith LH, Ashley E, Mercola M, Brown JH, Ruiz-Lozano P, APJ acts as a dual receptor in cardiac hypertrophy, Nature 488 (2012) 394–398. 10.1038/nature11263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Lopaschuk GD, Ussher JR, Folmes CD, Jaswal JS, Stanley WC, Myocardial fatty acid metabolism in health and disease, Physiol Rev 90 (2010) 207–258. 10.1152/physrev.00015.2009. [DOI] [PubMed] [Google Scholar]
- [17].Finck BN, Kelly DP, Peroxisome proliferator-activated receptor gamma coactivator-1 (PGC-1) regulatory cascade in cardiac physiology and disease, Circulation 115 (2007) 2540–2548. 10.1161/CIRCULATIONAHA.107.670588. [DOI] [PubMed] [Google Scholar]
- [18].Spiegelman BM, PPAR-gamma: adipogenic regulator and thiazolidinedione receptor, Diabetes 47 (1998) 507–514. 10.2337/diabetes.47.4.507. [DOI] [PubMed] [Google Scholar]
- [19].Feige JN, Lagouge M, Canto C, Strehle A, Houten SM, Milne JC, Lambert PD, Mataki C, Elliott PJ, Auwerx J, Specific SIRT1 activation mimics low energy levels and protects against diet-induced metabolic disorders by enhancing fat oxidation, Cell Metab 8 (2008) 347–358. 10.1016/j.cmet.2008.08.017. [DOI] [PubMed] [Google Scholar]
- [20].Purushotham A, Schug TT, Xu Q, Surapureddi S, Guo X, Li X, Hepatocyte-specific deletion of SIRT1 alters fatty acid metabolism and results in hepatic steatosis and inflammation, Cell Metab 9 (2009) 327–338. 10.1016/j.cmet.2009.02.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Eichner LJ, Giguere V, Estrogen related receptors (ERRs): a new dawn in transcriptional control of mitochondrial gene networks, Mitochondrion 11 (2011) 544–552. 10.1016/j.mito.2011.03.121. [DOI] [PubMed] [Google Scholar]
- [22].Smolkova K, Jezek P, The Role of Mitochondrial NADPH-Dependent Isocitrate Dehydrogenase in Cancer Cells, Int J Cell Biol 2012 (2012) 273947. 10.1155/2012/273947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Lightowlers RN, Taylor RW, Turnbull DM, Mutations causing mitochondrial disease: What is new and what challenges remain?, Science 349 (2015) 1494–1499. 10.1126/science.aac7516. [DOI] [PubMed] [Google Scholar]
- [24].Robey RB, Hay N, Is Akt the “Warburg kinase”?-Akt-energy metabolism interactions and oncogenesis, Semin Cancer Biol 19 (2009) 25–31. 10.1016/j.semcancer.2008.11.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Kohn AD, Summers SA, Birnbaum MJ, Roth RA, Expression of a constitutively active Akt Ser/Thr kinase in 3T3-L1 adipocytes stimulates glucose uptake and glucose transporter 4 translocation, J Biol Chem 271 (1996) 31372–31378. 10.1074/jbc.271.49.31372. [DOI] [PubMed] [Google Scholar]
- [26].Davis PA, Pagnin E, Dal Maso L, Caielli P, Maiolino G, Fusaro M, Paolo Rossi G, Calo LA, SIRT1, heme oxygenase-1 and NO-mediated vasodilation in a human model of endogenous angiotensin II type 1 receptor antagonism: implications for hypertension, Hypertens Res 36 (2013) 873–878. 10.1038/hr.2013.48. [DOI] [PubMed] [Google Scholar]
- [27].Zhang T, Kraus WL, SIRT1-dependent regulation of chromatin and transcription: linking NAD(+) metabolism and signaling to the control of cellular functions, Biochim Biophys Acta 1804 (2010) 1666–1675. 10.1016/j.bbapap.2009.10.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Oka S, Alcendor R, Zhai P, Park JY, Shao D, Cho J, Yamamoto T, Tian B, Sadoshima J, PPARalpha-Sirt1 complex mediates cardiac hypertrophy and failure through suppression of the ERR transcriptional pathway, Cell Metab 14 (2011) 598–611. 10.1016/j.cmet.2011.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Han SJ, Choi HS, Kim JI, Park JW, Park KM, IDH2 deficiency increases the liver susceptibility to ischemia-reperfusion injury via increased mitochondrial oxidative injury, Redox Biol 14 (2018) 142–153. 10.1016/j.redox.2017.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Park JB, Nagar H, Choi S, Jung SB, Kim HW, Kang SK, Lee JW, Lee JH, Park JW, Irani K, Jeon BH, Song HJ, Kim CS, IDH2 deficiency impairs mitochondrial function in endothelial cells and endothelium-dependent vasomotor function, Free Radic Biol Med 94 (2016) 36–46. 10.1016/j.freeradbiomed.2016.02.017. [DOI] [PubMed] [Google Scholar]
- [31].Ros S, Schulze A, Balancing glycolytic flux: the role of 6-phosphofructo-2-kinase/fructose 2,6-bisphosphatases in cancer metabolism, Cancer Metab 1 (2013) 8. 10.1186/2049-3002-1-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The datasets generated and/or analyzed during the current study are available from the corresponding author on reasonable request.