

Abstract
Flavivirids (family Flaviviridae) are a group of positive-strand ribonucleic acid (RNA) viruses that pose serious risks to human and animal health on a global scale. Here, we use flavivirid-derived deoxyribonucleic acid (DNA) sequences, identified in animal genomes, to reconstruct the long-term evolutionary history of family Flaviviridae. We demonstrate that flavivirids are >100 million years old and show that this timing can be combined with dates inferred from co-phyletic analysis to produce a cohesive overview of their evolution, distribution, and diversity wherein the main flavivirid subgroups originate in early animals and broadly co-diverge with major animal phyla. In addition, we reveal evidence that the ‘classical flaviviruses’ of vertebrates, most of which are transmitted via blood-feeding arthropod vectors, originally evolved in haematophagous arachnids and later acquired the capacity to be transmitted by insects. Our findings imply that the biological properties of flavivirids have been acquired gradually over the course of animal evolution. Thus, broad-scale comparative analysis will likely reveal fundamental insights into their biology. We therefore published our results via an open, extensible, database (Flavivirid-GLUE), which we constructed to facilitate the wider utilisation of genomic data and evolution-related domain knowledge in flavivirid research.
Keywords: flavivirus, jingmenvirus, paleovirology, genomics, flavivirid, evolution, tamanavirus, arbovirus, vector, mosquito, tick, arthropod
Introduction
Flavivirids (family Flaviviridae) are an important group of ribonucleic acid (RNA) viruses incorporating numerous pathogens of humans and animals. Currently, four genera are recognised within the family: Pegivirus, Pestivirus, Hepacivirus, and Flavivirus (Simmonds et al. 2017). While pegiviruses are not known to be associated with disease, pestiviruses cause serious illness in domestic ungulates such as cattle and pigs (de Oliveira et al. 2020), and the Hepacivirus genus includes the blood-borne hepatitis C virus (HCV), a major cause of chronic liver disease in human populations throughout the world (Manns et al. 2017; Pierson and Diamond 2020). Moreover, the genus Flavivirus includes viruses that are transmitted between vertebrates via blood-feeding arthropod vectors (e.g. mosquitoes and ticks) and cause large-scale outbreaks resulting in millions of human infections every year—e.g. yellow fever virus (YFV), dengue viruses 1–4 (DENV 1–4), and Zika virus.
The Pegi-, Pesti-, Hepaci-, and Flavivirus genera contain viruses with monopartite genomes ∼10 kb in length and encoding one or more large polyproteins that are co- and post-translationally cleaved to generate mature virus proteins. The structural proteins of the virion—capsid (c), premembrane (prM), and envelope (E)—are encoded towards the 5ʹ end of the genome, while genes encoding non-structural (NS) proteins are located further downstream (Chambers et al. 1990). However, a diverse variety of novel ‘flavivirus-like’ viruses (flavivirids) have been identified in recent years and these viruses—most of which were identified in invertebrates and have yet to be incorporated into official taxonomy—exhibit a much greater range of variations in genome structure, with genome lengths ranging up to 20 kb (Shi et al. 2016, 2018; Parry and Asgari 2019; Porter et al. 2020; Paraskevopoulou et al. 2021). Furthermore, one novel group—‘jingmenvirus’—comprises viruses with genomes that are multipartite rather than monopartite (Qin et al. 2014). Some tick-associated jingmenviruses have been linked with disease in humans (Jia et al. 2019; Wang et al. 2019).
To prevent the spread of pathogenic viruses, it is helpful to understand their evolutionary history in as much detail as possible, because this can often provide crucial insights into virus biology and host–virus relationships (Geoghegan and Holmes 2018). Flavivirids are a taxonomically diverse group that has been extensively examined using comparative approaches, revealing uniquely clear correlations between phylogenetic relationships and ecological characteristics (Zanotto et al. 1996; Gould et al. 2003; Cook and Holmes 2006; Moureau et al. 2015). The current, rapid accumulation of genome sequence data from novel flavivirids offers unprecedented opportunities to build on these comparative studies. At present, however, there are two major obstacles to the efficient use of flavivirid genome data.
First, there is a general lack of re-use and reproducibility among comparative analysis of virus genomes, especially when deeper evolutionary relationships are being examined (Holmes and Duchêne 2019). These analyses typically entail the assembly of complex data sets composed of virus sequences, multiple sequence alignments (MSAs), and phylogenies linked to other diverse kinds of data (e.g. spatiotemporal coordinates, taxonomy, and immunity-related information). In theory, these data sets could be re-used across a wide range of analysis contents while also being collaboratively developed and refined by multiple contributors. This would likely accelerate knowledge discovery and expedite the development of expert systems utilising virus genome data. Unfortunately, however, such practices remain challenging to implement in practice, largely due to a lack of appropriate tools and data standards (Grüning et al. 2018).
Second, knowledge of the appropriate evolutionary timescale is critically lacking. So far, most studies of flavivirid evolution have proposed relatively short timelines in which individual genera and sub-groups emerge within the last 10–100 thousand years (Zanotto et al. 1996; Gould et al. 2003; Cook and Holmes 2006; Pettersson and Fiz-Palacios 2014). However, these studies were based on viruses sampled from a relatively restricted range of hosts. By contrast, recent studies utilising metagenomic techniques to sample flavivirid diversity across a broader range of animal species have prompted suggestions of a much longer timeline extending over hundreds of millions of years (Shi et al. 2018). Furthermore, for many RNA virus families, robust evidence for ancient origins has come in the form of endogenous viral elements (EVEs)—virus-derived sequences found within eukaryotic genomes (Holmes 2011). These sequences are thought to originate when infection of germline cells leads to virus-derived complementary deoxyribonucleic acid (cDNA) being incorporated into chromosomal DNA so that integrated viral genes not only are inherited as host alleles but also persist in the gene pool over many generations until they are genetically fixed (i.e. reach a frequency of 100 per cent in the species gene pool). Genome comparisons show that EVE loci are often present as orthologues in closely related host species, establishing their ancient origins (since they were incorporated into the host germline prior to species divergence). EVEs derived from flavivirids have been identified in a handful of arthropod species (Katzourakis and Gifford 2010; Lequime and Lambrechts 2017; Whitfield et al. 2017; Parry and Asgari 2019), but are relatively uncommon compared to EVEs derived from other RNA virus families (Blair, Olson, and Bonizzoni 2020). Partly reflecting this scarcity, robust calibrations of the long-term evolutionary timeline of flavivirids are lacking.
In this study, we calibrate the long-term evolutionary timeline of flavivirids, making extensive use of EVEs. In addition, we construct a cross-platform, interactive database called ‘Flavivirid-GLUE’, which we use to capture the evolution-related domain knowledge generated in our study in a way that facilitates downstream use.
Results
Creation of open resources for comparative genomic analysis of flavivirids
We previously developed a software framework called GLUE (‘Genes Linked by Underlying Evolution’) (Singer et al. 2018). Here, we used GLUE to create Flavivirid-GLUE (Gifford 2021)—a flexible, extensible, and openly accessible resource for comparative analysis of flavivirid genomes (Supplementary Fig. S1a and b). The Flavivirid-GLUE project includes the following: (i) a set of 237 reference genome sequences each representing a distinct flavivirid species and linked to isolate-associated data (Supplementary Table S1), (ii) a standardized set of 81 flavivirid genome features (Supplementary Table S2), (iii) genome annotations specifying the coordinates of genome features within selected ‘master’ reference genome sequences (Supplementary Table S3), and (iv) a set of hierarchically arranged MSAs constructed to represent distinct taxonomic ranks within the family Flaviviridae (Table 1 and Fig. 1).
Table 1.
Comprehensive mapping of flavivirid homology via hierarchically linked MSAs.
| No.a | Taxonomic scope | Name | Parentb | Childrenc | Constraining referenced | Genome coveragee | Virus count | EVE countf |
| Family | ||||||||
| 1 | Flaviviridae-like | Flaviviridae | None | 2 | YFV | ∼6% | 2 | |
| Major lineage | ||||||||
| 2 | Hepaci/Pegi-like viruses | Flaviviridae | 2 | BVDV1 | ∼60% | 2 | 1 | |
| 3 | Flavi/PL | FlaviPesti | Flaviviridae | 2 | YFV | ∼80% | 2 | 1 |
| 4 | Flavi-like | Flavi-like | FlaviPesti | 2 | YFV | ∼35% | 2 | |
| 5 | PL | PL | FlaviPesti | 3 | BVDV1 | ∼60% | 3 | |
| Minor lineage | ||||||||
| 6 | Flavi-Tamana-like | FlaviTamana | Flavi-like | 2 | YFV | ∼93% | ||
| Genus | ||||||||
| 7 | Flavivirus genus | Flavivirus | FlaviTamana | 11 | YFV | 8 | 3 | |
| 8 | Hepacivirus genus* | Hepacivirus | 0 | HCV | ∼90% | 14 | ||
| 9 | Pegivirus genus* | Pegivirus | 0 | HPgV2 | ∼60% | 9 | ||
| 10 | Insect PL * | PL2 | PL | 0 | SLV2 | ∼73% | 6 | 7 |
| 11 | SCNV + arthropod* | PL1 | PL | 0 | SCNV5 | ∼72% | 8 | |
| 12 | Pestivirus genus* | Pestivirus | PL | 0 | BVDV1 | ∼88% | 11 | |
| 13 | X2-derived EVEs | X2 | Flavi-like | 0 | NS5 | 9 | ||
| 14 | ‘Jingmenvirus’ segment 1 | Jingmen_1 | Flavi-like | 0 | JMTV Seg1 | Segment 1 | 7 | 2 |
| 14 | ‘Jingmenvirus’ segment 3 | Jingmen_3 | Flavi-like | 0 | JMTV Seg3 | Segment 3 | 4 | 1 |
| 15 | ‘Tamanavirus’ | ‘Tamanavirus’ | FlaviTamana | 0 | TABV | 100% | 6 | 5 |
| Subgenus | ||||||||
| 16 | Crustacean | Crustacean | Flavivirus | 0 | 100%Δ–genome | 2 | ||
| 17 | cISF | cISF | Flavivirus | 0 | KRV | 100% | 14 | 6 |
| 18 | NKV1 | NKV2 | Flavivirus | 0 | APOIV | 100% | 6 | |
| 19 | Tick | tick | Flavivirus | 0 | POWV | ∼91% | 15 | |
| 20 | dISF | dISF | Flavivirus | 0 | LAMV | ∼95% | 9 | |
| 21 | Mosquito-1 | Mosquito-1 | Flavivirus | 0 | DEN1 | ∼97% | 35 | |
| 22 | NKV2 | NKV2 | Flavivirus | 0 | SOKV | 100% | 3 | |
| 23 | Mosquito-2 | Mosquito-2 | Flavivirus | 0 | YFV | ∼93% | 14 | |
| Totals | 159 | 35 | ||||||
Numbers correspond to labelled nodes in Fig. 1.
The parent MSA in the hierarchy.
Children of the MSA in the hierarchy.
Reference sequence that constrains the genomic co-ordinate coordinate space in the MSA.
Percentage of the constraining reference genome spanned by the MSA. Phylogenies constructed for each of these alignment partitions are available in Flavivirid-GLUE (Gifford 2021).
Counts reflect the number of consensus EVE sequences included in the alignment—consensuses are linked to child MSAs that contain the sequences of all individual EVE loci used to create the consensus.
BVDV1=Bovine viral diarrhea virus 1; SLV2=Shuangao lacewing virus 2; SCNV=Soy bean cyst nematode virus; KRV=Kamiti River virus; APOIV=Apoi virus; POWV=Powassan virus; LAMV=Lammi virus; DEN=Dengue; SOKV=Sokoluk virus.
Figure 1.

The MSA hierarchy in Flavivirid-GLUE. The GLUE software framework defines a ‘constrained alignment tree’ data structure comprising a set of alignments that are hierarchically linked to reflect taxonomic relationships (Singer et al. 2018). To enable sequence comparisons across the entire Flaviviridae, we implemented a constrained alignment tree data structure in Flavivirid-GLUE, as shown in the cladogram. Numbers shown adjacent to nodes correspond to rows in Table 1. Jingmen, Jingmen tick virus; MBFV, mosquito-borne flavivirus.
We used hierarchically linked MSAs to enable standardised genome sequence comparisons across the entire Flaviviridae family (i.e. both within and between taxonomic ranks). For each taxonomic rank represented in the project, one reference genome was selected as the constraining ‘master’ reference that defines the genomic coordinate space. Within the MSA hierarchy, each MSA is linked to its child and/or parent MSAs via our chosen set of references. MSAs representing internal nodes (see Table 1) contain only master reference sequences but can be recursively populated with all taxa contained in child alignments via GLUE’s command layer (Singer et al. 2018). Importantly, the use of an MSA hierarchy simplifies analysis of novel taxa (since new sequences only need be aligned to the most closely related reference genome to be aligned with all other taxa included in the MSA hierarchy).
Instantiation of the Flavivirid-GLUE project (Supplementary Fig. S2) generates a relational database that contains the data items required for comparative analysis of flavivirids and represents the semantic links between them. This allows comparative analyses to be implemented in a standardised, reproducible way, wherein GLUE’s command layer is used to coordinate interactions between the Flavivirid-GLUE database and bioinformatics software tools (e.g. see Supplementary Fig. S3 and S4). Flavivirid-GLUE can be installed on all commonly used computing platforms and is fully containerised via Docker (Merkel 2014). Hosting in an openly accessible online version control system (GitHub) provides a platform for coordinating ongoing development of the resource—e.g. incorporation of additional taxa and genome annotations—following practices established in the software industry (Supplementary Fig. S1c) (Loeliger and McCullough 2012).
Mapping the distribution of flavivirid-derived DNA in animal genomes
To identify flavivirid-derived EVEs, we performed systematic in silico screening of whole genome sequence data representing 1075 animal species. This led to the identification of 374 EVE loci in 36 animal species (Table 2; Gifford 2021). We reconstructed consensus sequences representing fragments of the genomes of (presumably) extinct flavivirids, utilising EVE sequences putatively derived from a single germline incorporation event (i.e. orthologues, fragments, and duplicates) (Fig. 2 and Supplementary Fig. S5). EVE consensus sequences, MSAs, and all EVE-associated metadata were incorporated into Flavivirid-GLUE (Gifford 2021).
Table 2.
Flavivirid-derived EVEs.
| Sequence IDa | No. of Speciesb | No. of Sequencesc | lORFd | Hoste | |
| Flavivirus | |||||
| EFV-cISF.1-AedAeg* | 1 | 92 | 564 | Yellow fever mosquito | Aedes aegypti |
| EFV-cISF.2-AedAlb* | 1 | 35 | 643 | Tiger mosquito | Aedes albopictus |
| EFV-cISF.3-AnoMin* | 1 | 1 | 97 | Anopheles minimus | |
| EFV-cISF.4-AnoSin* | 1 | 1 | 138 | Anopheles sinensis | |
| EFV-cISF.5-TipOle | 1 | 4 | 226 | Marsh crane fly | Tipula oleracea |
| EFV-cISF.6-ConPat | 1 | 1 | 38 | Long-legged fly | Condylostylus patibulatus |
| EFV-Flavi.1-CraSow | 1 | 13 | 1182 | Peach blossom jellyfish | Craspedacusta sowerbyi |
| EFV-Flavi.2-DapMag* | 1 | 2 | 573 | Water flea | Daphnia magna |
| EFV-Flavi.3-LepArc* | 1 | 3 | 281 | Tadpole shrimp | Lepidurus arcticus |
| ‘Tamanavirus’ | |||||
| EFV-Tamana.1-LedTum | 1 | 1 | 55 | Meltwater stonefly | Lednia tumana |
| EFV-Tamana.2-LauKoh | 1 | 1 | 56 | Hawaiian cricket | Laupala kohalensis |
| EFV-Tamana.3-AmpSul | 1 | 2 | 44 | Spring stonefly | Amphinemura sulcicollis |
| EFV-Tamana.4-StyCho | 1 | 1 | 873 | Tube-eye | Stylephorus chordatus |
| Jingmenvirus | |||||
| EJP-Jingmen.1-Chironomus | 2 | 2 | 248 | Chironomus sp. | |
| EJP-Jingmen.2-Gerris | 1 | 1 | 45 | Water strider | Gerris buenoi |
| EJH-Jingmen.1-Gerris | 1 | 1 | 51 | ||
| X1 | |||||
| EFV-X1.1-PriCau | 1 | 2 | 347 | Penis worm | Priapulus caudatus |
| X2 | |||||
| EFV-X2.1-AusLim | 1 | 2 | 214 | Mangrove killifish | Austrofundulus limnaeus |
| EFV-X2.2-StyCho | 1 | 3 | 873 | Tube-eye | Stylephorus chordatus |
| EFV-X2.3-EurAff | 1 | 2 | 190 | Copepod | Eurytemora affinis |
| EFV-X2.4-Takifugu | 3 | 5 | 103 | Pufferfish | Takifugu |
| EFV-X2.5-Phycis | 2 | 2 | 129 | Phycid hakes | Phycis |
| EFV-X2.6-MorMor | 1 | 1 | 36 | Common mora | Mora moro |
| EFV-X2.7-BroBro | 1 | 1 | 56 | Cusk | Brosme brosme |
| EFV-X2.8-MacPee | 1 | 1 | 51 | Murray cod | Maccullochella peelii |
| EFV-X2.9-BolPec | 1 | 1 | 155 | Blue-spotted mudhopper | Boleophthalmus pectinirostris |
| PL | |||||
| EFV-PL2.1-CalCec | 1 | 5 | 78 | Red-banded hairstreak | Calycopis cecrops |
| EFV-PL2.2-EucHer | 1 | 2 | 187 | Stink bug | Eucheros histo |
| EFV-PL2.3-Apoidae | 11 | 239 | 2469 | Bees and sphecoid wasps | Superfamily Apoidea |
| EFV-PL2.4-AndCur | 2 | 6 | 961 | Cynipid gall wasp | Andricus |
| EFV-PL2.5-AnoGla | 1 | 1 | 26 | Long-horned beetle | Anoplophora glabripennis |
| EFV-PL2.6-XenBra | 1 | 16 | 2232 | Grasshopper | Xenocatantops brachycerus |
| EFV-PL2.7-OpeBru | 1 | 2 | 38 | Winter moth | Operophtera brumata |
| Pegi-Hepaci | |||||
| EFV-HepaPegi.1-ElyChl | 1 | 1 | 60 | Eastern emerald elysia | Elysia chlorotica |
Flavivirid-derived EVEs have been assigned standard IDs based on conventions established for endogenous retroviruses, wherein information about virus taxonomy and locus orthology is incorporated into the ID itself (Gifford et al. 2018). The ID comprises three elements separated by hyphens. For most EVEs characterised here, the first (i.e. leftmost) element is the classifier EFV. However, for ‘jingmenviruses’ the classifier component of the ID also specifies the gene it is derived from EJPand EJH, following conventions established for multipartite viruses and EVEs derived from mRNA sources (Kawasaki et al. 2021). The second ID element comprises two subcomponents separated by a period—the first defines the taxonomic position of the EVE in relation to established Flaviviridae taxonomy, and the second is a numeric ID that uniquely represents an EVE locus. The third ID component defines the known distribution of orthologous insertions among host species. If it is only known from a single species, a shortened version of the Latin binomial species name is used.
Number of species in which EVE locus was identified.
Number of sequences (i.e. distinct insertions) derived from this EVE that were identified via in silico screening.
Host species or species groups. *indicates EFV loci or lineages that have been reported previously.
L-ORF, longest open reading frame.
Figure 2.
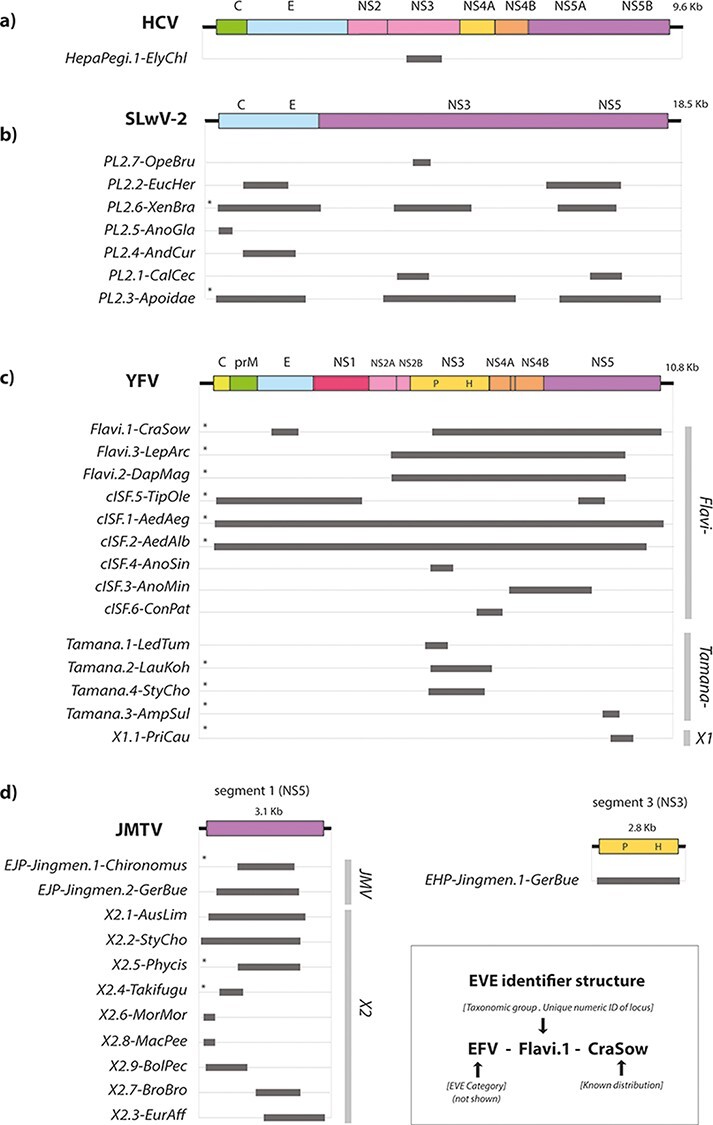
Genome structures of EFV elements. Schematic diagrams showing the genomic regions represented by flavivirid-derived EVE sequences. (a) Pegi-hepacivirus-like elements are shown relative to HCV; (b) elements derived from the ‘PL 2ʹ lineage of viruses shown relative to Shuangao lacewing virus 2; (c) elements derived from the Flavivirus genus, the ‘tamanaviruses’, and the X1 lineage shown relative to DENV type 1 (DENV-1); (d) ‘jingmenvirus’-derived EFVs shown relative to Jingmen tick virus (JMTV). Homologous regions represented by EFV sequences are shown as horizontal bars. Bars to the right show taxonomic groups. EVE IDs are shown to the left. EVE IDs were constructed as indicated in the key, following a convention established for endogenous retroviruses (Gifford et al. 2018). IDs have three components—the first is the classifier ‘EFV element’ (can be dropped when implied by context). The second comprises two subcomponents separated by a period: (i) the name of the taxonomic group of viruses from which the EFV is thought to derive and (ii) a numeric ID that uniquely identifies the integration locus. The third component describes the known taxonomic distribution of orthologous copies of the element among host species. For EVEs derived from the ‘jingmenvirus’ lineage, which contains viruses with multipartite genomes, we used classifiers that specify the gene from which the EVE is derived, in line with conventions established for EVEs derived from segmented viruses (Kawasaki et al. 2021)—endogenous ‘jingmenvirus’ helicase (EJH) and endogenous ‘jingmenvirus’ polymerase (EJP). X1, unclassified flavivirus-like lineage; X2, unclassified flavivirus-like lineage X2; P, Protease; H, Helicase. * indicates the consensus sequence.
All major flavivirid lineages are represented in the host germline
We reconstructed the evolutionary relationships between EVEs and contemporary flavivirids using maximum likelihood approaches (Fig. 3 and Supplementary Fig. S6). Bootstrapped phylogenetic trees were reconstructed from MSAs representing a range of taxonomic ranks within the Flaviviridae (Table 1), both including and excluding EVE sequences. (Gifford 2021). Consistent with official taxonomy and previously published studies (Moureau et al. 2015; Simmonds et al. 2017), phylogenetic reconstructions split flavivirids into two major lineages—‘Hepaci-Pegi’ (HP) and ‘flavi-pesti’—each of which contains several well-supported subgroups (Paraskevopoulou et al. 2021) (Fig. 3a–e and Supplementary Fig. S3). However, a divergent, flavivirid-derived EVE identified in the genome of a priapulid worm (Priapulus caudatus: Lamarck, 1816) may represent a third sub-lineage (Fig. 3b).
Figure 3.
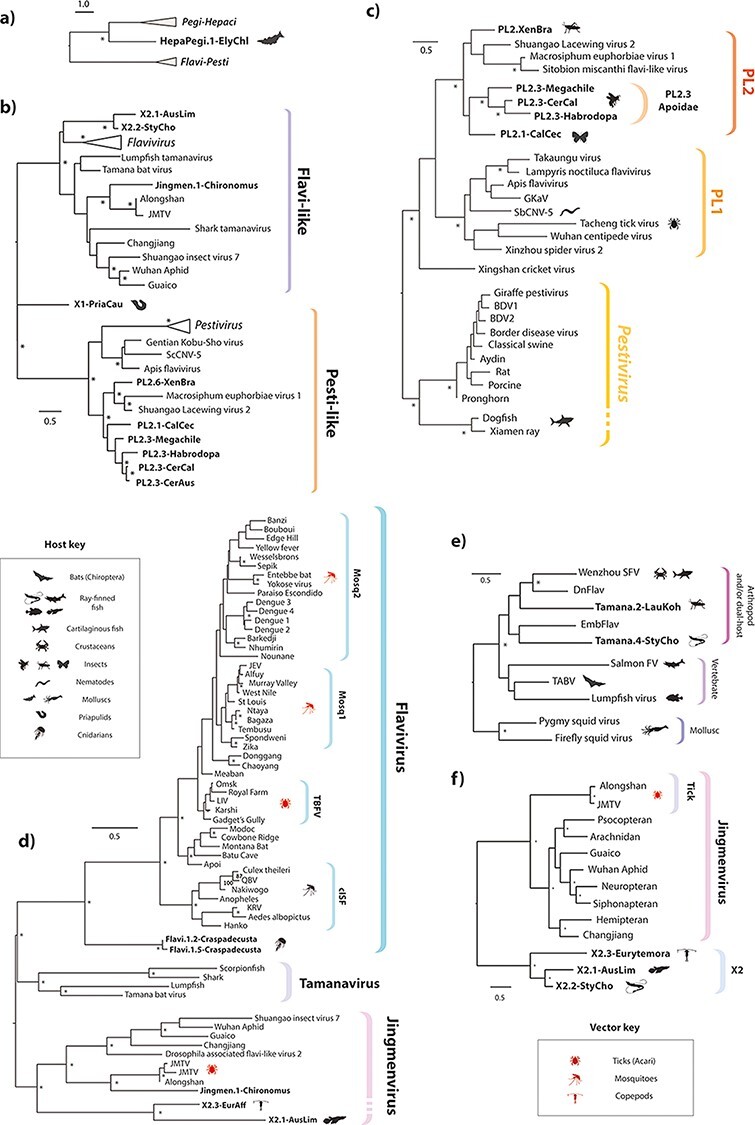
Evolutionary relationships between modern and ancient flavivirids. Bootstrapped maximum likelihood phylogenies (1000 replicates), reconstructed for viruses and EFVs across a range of taxonomic ranks, as follows: (a) Two major lineages within family Flaviviridae (‘flavi-pesti’ and ‘HP’) showing placement of Hepacivirus-derived EVE (122 aa residues in MSA spanning conserved regions in NS3, substitution model = RTREV); (b) ‘Flavi-pesti’ lineage (MSA spanning ninety-nine aa residues in NS5, substitution model = BLOSUM62); (c) ‘PL’ lineage (104 aa residues in NS5, substitution model = RTREV); (d) ‘Flavi-like’ lineage (MSA spanning 179 aa residues in NS5, substitution model = LG likelihood); (e) ‘Tamanavirus’ (MSA spanning 292 aa residues in NS5, substitution model = BLOSUM62); (f) ‘Jingmenvirus’ and related lineages (MSA spanning 727 residues in NS5, substitution model = LG likelihood). EFV names are shown in bold. Only EFVs that had sufficient coverage (>50 per cent of total MSA length) were included in the analysis. Triangular terminal branches indicate collapsed clades containing multiple taxa. Asterisks indicate bootstrap support ≥70 per cent (1000 replicates). The scale bar indicates the evolutionary distance in substitutions per site. Brackets to the right indicate genera and sub-lineages. All trees are midpoint rooted for display purposes. Host and known/suspected vector associations are indicated by animal silhouettes as shown in the key. Jingmen, Jingmen tick virus; MBFV, mosquito-borne flavivirus.
Phylogenetic reconstructions revealed that EVEs derived from a broad range of flavivirus lineages and subgroups are represented in the genomic ‘fossil record’. Only one EVE derived from the ‘HP’ lineage was identified. However, it occurs in a marine mollusc—the Eastern emerald elysia (Elysia chlorotica: Gould, 1870)—demonstrating that the host range of this major flavivirid group extends to invertebrates (Fig. 3a).
The majority of flavivirid EVEs are derived from the ‘flavi-pesti’ lineage, which is composed of robustly supported ‘flavi-like’ and ‘pesti-like’ (PL) clades. Our approach to phylogenetic reconstruction, which entails reconstructing separate phylogenies for distinct taxonomic ranks (see Table 1), supports a clean division of ‘flavi-like’ viruses into three monophyletic clades (Fig. 3d) corresponding to genus Flavivirus, the ‘jingmenviruses’, and a clade of viruses related to Tamana bat virus (TABV), which we here refer to as ‘tamanaviruses’. We identified several EVEs that grouped robustly within the diversity of contemporary ‘tamanavirus’ and ‘jingmenvirus’ isolates. We also identified EVEs derived from a more distantly related, ‘jingmenvirus-like’ lineage—here labelled X2—with no known contemporary representatives (Fig. 3f). Notably, we identified X2-derived EVEs in a Temora copepod (Eurytemora affinis: Poppe, 1880) as well as in multiple Actinopterygii fish, indicating that their host range encompasses both vertebrate and arthropod species (Fig. 3f and Table 1). The copepod element exhibited internal duplications and rearrangements typical of EVEs identified in piRNA clusters (data not shown) (Ophinni et al. 2019).
The ‘PL’ lineage contains two robustly supported clades—one composed of vertebrate viruses (including the canonical members of genus Pestivirus), while the other contains a diverse assortment of invertebrate-associated ‘large-genome flavivirids’ (Paraskevopoulou et al. 2021). The invertebrate clade contains two well-supported subclades, here labelled PL1 and PL2. The ‘PL2’ clade contains EVEs in addition to viruses (Fig. 3c).
Germline incorporation of flavivirid DNA is relatively rare
While flavivirid-derived EVEs were only identified in a small proportion of the animal species we screened, they occur at a relatively high copy number in the germline of some insect groups, including mosquitoes of genus Aedes (Meigen, 1818) as well as bees and Sphecid wasps (superfamily Apoidea: Latreille, 1802) (Table 1).
In the Aedes germline, flavivirid EVEs are closely related to contemporary flaviviruses—specifically the ‘classical insect-specific’ flaviviruses’ (cISFs). Numerous, distinct loci occur, largely representing distinct regions of the flavivirus genome (Supplementary Fig. S5). However, where they do span homologous regions of the flavivirus genome loci, are highly related (i.e. <1 per cent nucleotide sequence divergence) and are frequently arranged as tandemly repeating arrays (data not shown), suggesting recent, intra-germline amplification.
Among Apoidea EVEs, the copy number was dramatically inflated in certain species, such as the spurred ceratina (Ceratina calcarata: Robertson, 1900), which contains at least eighty-six distinct flavivirid-derived EVE sequences in its published genome sequence. However, confident estimates of EVE copy number in Apoidea species could not be obtained, due to the limitations of current genome assemblies.
Major flavivirid lineages originated in the distant evolutionary past
We used a range of approaches to calibrate the evolutionary timeline of flavirids (Table 3). Calibrations based on identification of orthologous EVEs were obtained for ‘jingmenvirus’-derived EVEs found in midges (Chironomus: Meigen, 1803), X2-derived EVEs in ray-finned fish (Class Actinopterygii: Klein, 1885), and a PL2-derived EVE in superfamily Apoidea. Orthologous, PL2-derived EVEs were identified in Apoidea species estimated to have diverged >100 million years ago (Mya) (Supplementary Fig. S7). We also derived age estimates ranging between 3 and 62 Mya for pairs of putatively duplicated EVE sequences based on the assumption a neutral molecular clock (Table 3). These calibrations, combined with the identification of flavivirid-derived EVEs in basal animal lineages such as cnidarians and priapulids, suggested that flavivirids could in fact have truly primordial origins in multicellular animals. Such an extended evolutionary timeline would be consistent with other recent data supporting the ancient origins of RNA virus families (Aiewsakun and Katzourakis 2017; Shi et al. 2018).
Table 3.
Dates and age estimates used to calibrate flavivirid evolution.
| # | Virus lineage | Host lineage(s) | MYa | Low | High |
| Codivergence A (minimum age) | |||||
| 1 | Flaviviridae | Animalia | 952 | 757 | 1147 |
| 6 | Jingmen-X2 | Insecta-Crustacea | 794 | 678 | 916 |
| 22 | Mosquito 2 subgenus | Culex-Aedes | 40 | 22 | 52 |
| 10 | Jingmenvirus | Arachnida-Insecta | 601 | 568 | 642 |
| Codivergence B (minimum age) | |||||
| 2 | Pegi-hepaci lineage | Mollusc-vertebrate | 794 | 678 | 916 |
| 3 | PL viruses | Invertebrates–vertebrates | 794 | 678 | 916 |
| 4 | Pegi-Hepaci | Chondrichthyes–Mammalia | 465 | 450 | 497 |
| 5 | Pestiviruses | Chondrichthyes–Mammalia | 465 | 450 | 497 |
| 7 | Flavivirus | Cnidaria–Arthropoda | 824 | 652 | 973 |
| 8 | Arthropod flaviviruses | Crustacea–Arachnida | 601 | 568 | 642 |
| 9 | Chelicerata flaviviruses | Pycnogonida–Arachnida | 553 | 476 | 653 |
| Orthology (minimum age) | |||||
| 13 | PL2 | Family Apoidea | 102 | 71 | 148 |
| 24 | Jingmenvirus | Genus Chironomus | n/k | n/k | n/k |
| 23 | X2 | Genus Phycis | 16.9 | 12.1 | 37.1 |
| Duplicates/molecular clock (minimum age) | |||||
| 20 | cISF | Tipula | 16 | 40 | |
| 21 | Crustacean FV | Daphnia 2 | 26 | 62 | |
| 19 | Cnidarian FV | Craspedacusta 1 | 17 | 40 | |
| 16 | X1 | Priapulus (NS5) | 12 | 29 | |
| 17 | X2 | Eurytemora (NS5) | 20 | 47 | |
| 18 | ‘Tamanavirus’ | Austrofundulus 1 (NS5) | 19 | 43 | |
| 14 | PL2 | Operophtera | 17 | 40 | |
| Origin hypothesis (maximum age) | |||||
| 11 | All vectored flaviviruses | Tick haematophagy | 300 | 90 | 400 |
| 12 | Mosquito-vectored flaviviruses | Mosquito haematophagy | 79 | 100 | |
Node numbers correspond to those shown in Fig. 5c. Haematophagy estimates obtained from Mans 2011. Culex-Aedes divergence date obtained from Sieglaff et al. 2009. All other divergence data estimates were obtained from TimeTree (Kumar et al. 2017).
Horizontal transfer of flavivirids between distantly related hosts has clearly occurred—most likely in association with parasitism (Dolja and Koonin 2018; Dheilly et al. 2022)—but when phylogenies are considered in the light of an evolutionary timescale extending back to the origin of multicellular animals (i.e. ∼500–800 Mya), a credible argument can be made for codivergence being the more common mode of evolution, at least where higher taxonomic ranks are concerned. To investigate this, we compared host and virus phylogenies. Scope for comparisons was limited due to sparse data and relatively narrow sampling across host species groups (with most flavivirids isolated from arthropods and vertebrates). We identified several host and virus clades in which the branching relationships and divergence times among animal lineages are correlated with the topology and branch lengths found in virus phylogenies (Fig. 4 and Supplementary Fig. S8). However, we were required to make strong assumptions in each case, particularly regarding the rooting of virus trees (see the legend of Fig. 4), and these deeper calibrations should therefore be taken as tentative.
Figure 4.
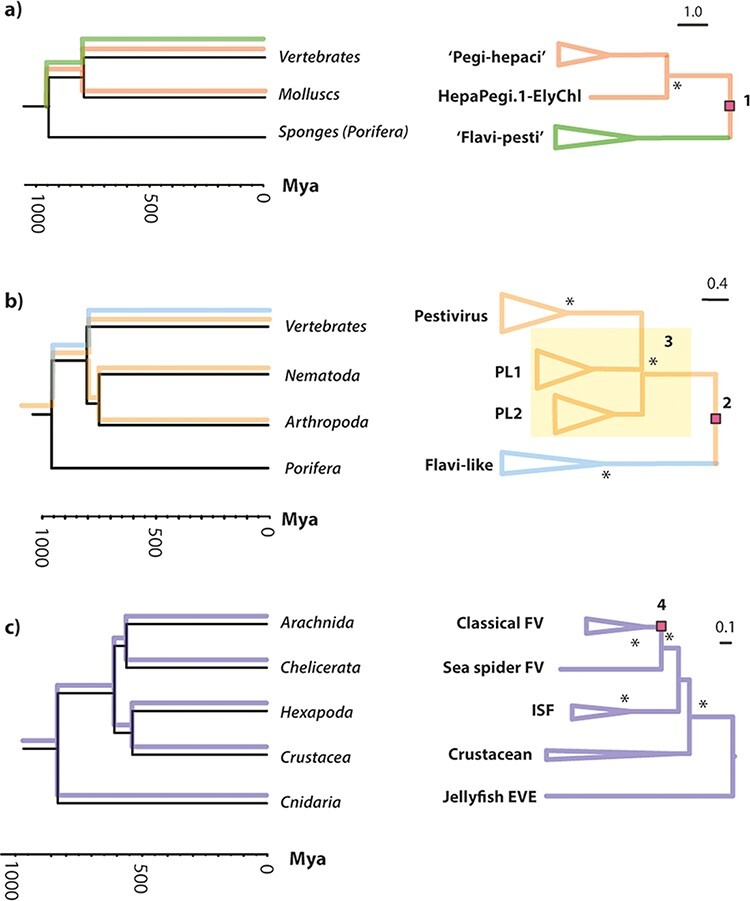
Putative codivergence of flavivirid groups and host phyla. ‘Tanglegrams’ illustrating matching topologies in animal (left) and flavivirid (right) phylogenies. Putative tracking of host lineages by viral lineages is indicated on the host phylogeny. Clades within which the branch lengths of virus phylogenies are correlated with divergence times in host animal lineages: the invertebrate and vertebrate splits in the (a) ‘HP’ lineage (R2 = 0.5); (b) ‘PL’ lineage (R2 = 0.7); (c) the cnidarian–arthropods, chelicerate–hexapoda, and crustacean–insect splits in the Flavivirus genus (R2 = 0.3). Note that, due to limited data, all comparisons rely on strong assumptions regarding virus phylogenies, indicated in the figure by numbers, as follows: (1) and (2) midpoint rooting was used, and the deepest divergence in the virus tree was approximately calibrated using the divergence date of porifera (the most basal animal lineage) in line with our hypothesis that major flavivirid lineages originated in early metazoans; (3) the splits between nematode and arthropod viruses in PL1 and PL2 clades—indicated by the yellow square—were poorly resolved in our phylogenies; (4) midpoint rooting was used, and it is assumed that the arthropod-borne classical flaviviruses originated in arachnids, as proposed in Fig. 5a.
Arthropod-vectored flaviviruses likely emerged from an arachnid source
The Flavivirus genus includes viruses that are transmitted among vertebrate hosts by arthropod vectors, as well as viruses that exclusively infect arthropods (Blitvich and Firth 2015) and others that have been identified in vertebrates but have no known arthropod vector (Blitvich and Firth 2017). The long history of association between flavivirids and their hosts implied by our investigation suggests that the largely vector-borne ‘classical flaviviruses’ (CFV) could have emerged in association with the evolution of haematophagy (blood-feeding) in arthropods. Phylogenetic reconstructions using either NS5 (Fig. 3a) or NS3 (Supplementary Fig. S9) show that flaviviruses exclusively associated with insects (Class Insecta: Linnaeus, 1758) are robustly separated from those that infect vertebrates by viruses identified in crustaceans (Subphylum Crustacea: Brünnich, 1772), sea spiders (Pycnogonida: Latreille, 1810) (Conway 2015), and arachnids (Arachnida: Lamarck, 1801). In addition, rooted phylogenies show that the most basal CFV lineages are either tick-associated (Mpulungu virus and the tick-borne CFVs) or have no known vector (NKV) (Blitvich and Firth 2017) (Fig. 3a). While much uncertainty remains, it is interesting to note that, if we parsimoniously assume that the basal lineages without known vectors are tick-borne (or were originally before subsequently losing their association with arachnids), phylogenies suggest (i) emergence of tick-borne viruses from arachnid-specific viruses followed by (ii) emergence of insect-borne viruses from tick-borne viruses (Fig. 3b). Intriguingly, we identified synapomorphic amino acid (aa) variation in the NS5 protein, wherein ancestral residues are conserved between ancestral, arachnid-associated flaviviruses but variable in the more derived, insect-vectored flaviviruses (Fig. 3a and Supplementary Fig. S10). While these patterns can of course be interpreted in alternative ways, they are consistent with positive selection accompanying the adaptation of ostensibly tick-borne viruses to newly acquired insect vectors.
Discussion
The flavivirids are a genetically and ecologically diverse group of viruses that include an unusually large number of taxonomically recognised species, many of which are associated with disease (Moureau et al. 2015). As a diverse and highly studied group, flavivirids offer unique possibilities for researchers interested in applying comparative approaches to viruses (Zanotto et al. 1996; Gould et al. 2003; Moureau et al. 2015). Species richness in this group to some extent reflects their historical importance in the development of virus research—YFV being the first human virus identified (Staples and Monath 2008)—and sampling of flavivirid diversity shows historical bias towards potential vector/reservoir species (Rosenberg et al. 2013; Gibb et al. 2022). However, with dramatic advances in DNA sequencing technology, it is now possible to investigate flavivirid distribution and diversity much more broadly, building on previous comparative investigations (Zanotto et al. 1996; Gould et al. 2003; Cook and Holmes 2006; Moureau et al. 2015).
In this report, we address two important challenges to effective use of flavivirid sequence data in comparative genomic studies. First, we implemented our analyses using a computational framework that supports re-use of underlying data sets and facilitates reproduction of comparative genomic analyses. Second, we calibrated the long-term evolutionary history of flavivirids through use of the ‘genomic fossil record’, thereby providing broad evolutionary context for interpreting their distribution and diversity.
We identify flavivirid-derived EVEs that are >100 Mya, demonstrating that the evolution of family Flaviviridae spans geological eras. Furthermore, we show that the robust calibrations obtained from EVEs can be combined with more tentative calibrations based on co-phyletic analysis to produce a cohesive overview of flavivirid evolution in which the major lineages emerged during the early evolution of multicellular animals and subsequently evolved together with major animal phyla (Fig. 5c). The timeline suggested by our analysis raises interesting questions about the ways in which animal evolution might have impacted flavivirids, since it encompasses the development of entire organ systems (e.g. the liver and vascular system) and spans the evolution of fundamental changes in animal physiology, such as the emergence of endothermy (‘warm blood’) in vertebrates. The identification of flavivirid-derived EVEs in animals lacking a circulatory system (e.g. cnidarians and priapulids) suggests that cell-to-cell transmission via exosomes—as has been reported for tick-borne flaviviruses (Zhou et al. 2018)—might represent the ancestral mode among flavivirids.
Figure 5.
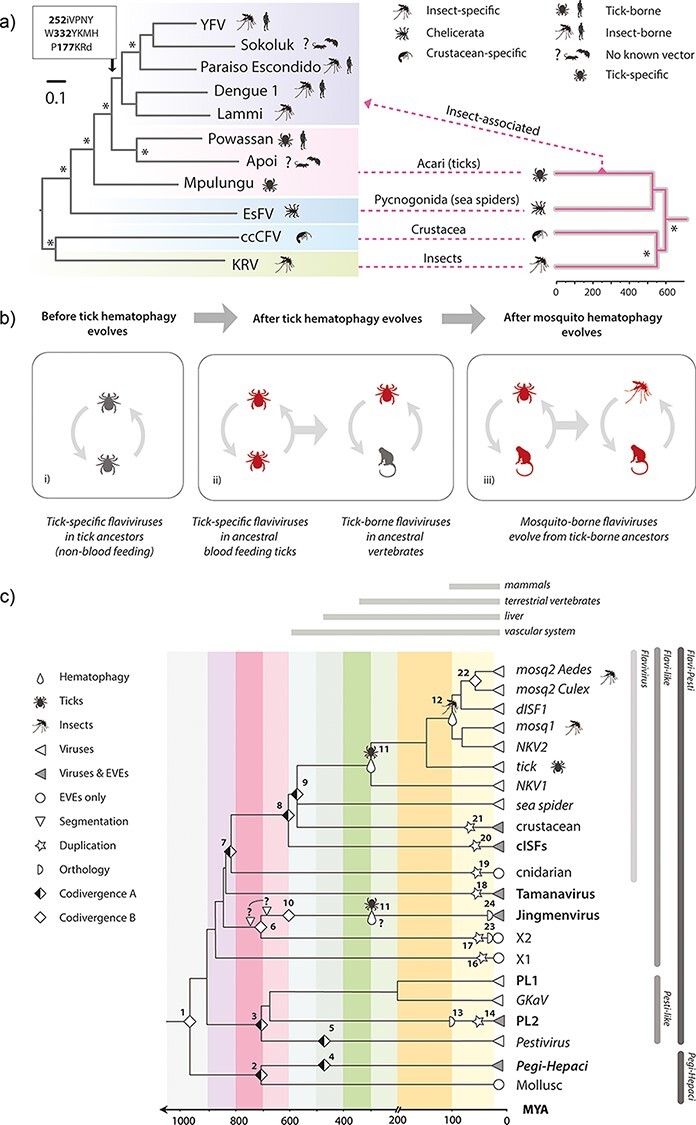
The timeline of flavivirus evolution. (a) Evolution of arthropod-borne flaviviruses. The phylogeny shown on the left was constructed from an alignment spanning 804 residues of the precursor polyprotein (substitution model = LG likelihood). The phylogeny on the right is a time-calibrated phylogeny of arthropod hosts/vectors of classical flaviviruses, obtained via TimeTree (Kumar et al. 2017). The figure shows the hypothesised relationships between the host and virus trees, with virus phylogeny broadly following host phylogeny at higher taxonomic ranks but diverging dramatically from this pattern when tick-borne viruses of vertebrates emerge. (b) Proposed model for the origin and evolution of the vector-borne flaviviruses. Three stages are shown: (i) an ancestral group of non-vectored tick viruses is present; (ii) tick haematophagy provides prolonged exposure to vertebrate blood, thereby affording tick-specific flaviviruses the opportunity, through chance events and mutation, to acquire a capacity to replicate in vertebrate cells and ultimately become tick-borne flaviviruses of vertebrates; (iii) the existence of tick-borne flaviviruses occasions viraemic vertebrate hosts, exposing mosquitoes and other haematophagous insects to flaviviruses via blood-feeding and ultimately allowing them to acquire a vector role.(c) A model for long-term evolution of flavivirids. A time-scaled tree summarising the phylogenetic relationships of flavivirids and calibrated using information obtained from analysis of EVEs, co-phyletic analysis, and the fossil record of haematophagous arthropods. Symbols on the phylogenies indicate types of calibration as shown in the key. Numbers adjacent to symbols link to rows in Table 3. ‘Codivergence type A’ = codivergence supported by co-phylogeny. Codivergence type B = potential codivergence-based calibrations without supporting evidence. Vertical lines to right of the tree show taxonomic groups.
The vertebrate circulatory system evolved >400 Mya and is thought to have been established in its basic form in endothermic vertebrates (i.e. birds and mammals) by ∼200 Mya (Monahan-Earley, Dvorak, and Aird 2013). Its role in transporting nutrients makes it a highly attractive target for parasitism, and haematophagous, arthropod parasites of vertebrates are thought to have evolved on >twenty independent occasions (Mans 2011). Whenever this occurred, it would have created new, intimate contact networks between arthropod and vertebrate species so that viruses circulating within each group could potentially encounter opportunities to expand into the other (Dolja and Koonin 2018). Interestingly, we identified closely related, X2-derived EVEs in both copepods and fish, raising the possibility that copepod parasitism has enabled X2-like arthropod viruses to expand into vertebrate hosts (Fig. 3f).
Both phylogenetic relationships within genus Flavivirus and the extended evolutionary timeline implied by our study are consistent with a parsimonious evolutionary scenario wherein the vector-borne classical flaviviruses originate in haematophagous arachnids and later acquire the capacity to be transmitted by haematophagous insects (Fig. 5a and b). This ‘ticks first’ model is quite appealing because the circumstances of tick feeding provide an obvious opportunity for tick flaviviruses capable of limited replication in vertebrate cells to emerge. Feeding entails a relatively long exposure, and ticks are thought to transmit infection to one another via host blood when multiple individuals feed in proximity (Hermance and Thangamani 2018). Once tick flaviviruses had acquired the capacity to generate sustained viremia in vertebrate hosts, opportunities for haematophagous insects to acquire vector roles would presumably arise as they become exposed to virus via blood-feeding. Interestingly, this model of flavivirus evolution is consistent with evidence that the 3ʹ untranslated region (UTR) of arthropod-borne flaviviruses—which can modulate the viral life cycle in complex and nuanced ways—evolved via duplication (Gritsun and Gould 2007; Ochsenreiter, Hofacker, and Wolfinger 2019). Duplicated UTR sequences are best preserved in tick-borne flaviviruses, consistent with an ancestral origin, while only remnants are present in the more derived, mosquito-borne flaviviruses (Gritsun et al. 2014).
Due to the overall scarcity of flavivirid-derived EVEs, our investigation provides only limited insight into the evolutionary history of certain flavivirid groups, such as the Hepaci-, Pegi-, and Pestivirus genera. However, our identification of ‘HP’-like EVEs in invertebrates, and the recent description of a pestivirus-like (PL) EVE in an insectivore (Li et al. 2022), indicates that additional, diverse flavivirid EVEs will be identified as whole genome sequencing of animal species advances, allowing for more complete perspective on the long-term evolution of flavivirids. New information obtained through broader sampling of contemporary flavivirid diversity will also allow for more rigorous testing of the evolutionary hypotheses presented here.
Phylogenetic reconstructions utilising currently available data suggest that reorganisation of the flavivirid taxonomy should be considered (Fig. 3). Currently, family Flaviviridae is placed in order Amarillovirales, class Flasuviricetes, which contains no other orders or families. Flasuviricetes could be restructured to contain groups representing the ‘HP’ and ‘flavi-pesti’ lineages and the subgroups contained therein. This would enable taxonomic classifications to reflect the deep evolutionary splits between flavivirid subgroups, such as those separating ‘jingmenviruses’, ‘tamanaviruses’, and flaviviruses (genus Flavivirus) in the ‘flavi-pesti’ lineage. The enigmatic TABV, which was isolated in 1973 from insectivorous bats and has no known arthropod vector, has puzzled flavivirologists for decades (Price 1978; Kuno et al. 1998; Blitvich and Firth 2017). It is sometimes considered a basal member of genus Flavivirus but is only distantly related to other flaviviruses and clearly distinct in signature genomic traits such as nucleotide composition (de Lamballerie et al. 2002). Notably, however, we identified an EVE in the genome of the peach blossom jellyfish (Craspedacusta sowerbii: Lankester, 1880) that is more ‘flavivirus-like’ than TABV (Fig. 3d). The identification of flavivirus-like EVEs in cnidaria (Hatschek: 1888), a basal animal lineage, combined with new information describing the extensive diversity and broad host range of TABV-related viruses (Geoghegan et al. 2018; Skoge et al. 2018; Parry and Asgari 2019; Paraskevopoulou et al. 2021) (Fig. 3e), suggests that ‘tamanaviruses’ have distinct evolutionary origins to genus Flavivirus and should be given separate taxonomic status among ‘flavi-like’ viruses. The same applies to ‘jingmenviruses’, which presumably became evolutionarily separated from other ‘flavi-like’ viruses when they evolved multipartite genomes (Qin et al. 2014).
Given the extent of uncharacterised flavivirid diversity and the accelerating rate of virus discovery, further expansion and revision of flavivirid taxonomy will no doubt be required as exploration of the virome proceeds—numerous novel and divergent flavivirids have been described since we originally submitted this report (Dong et al. 2021; Dheilly et al. 2022)—the taxonomic system proposed above would provide a relatively open structure capable of accommodating novel flavivirid diversity.
Our analysis indicates that incorporation of flavivirid-derived DNA into animal germlines is uncommon. However, in those taxa where flavivirid-derived EVEs occur, they are frequently multicopy, with some insect groups exhibiting a relatively high copy number (Table 1). Notably, these include Aedes mosquitoes in which EVEs have been shown to produce PIWI-interacting RNAs (piRNAs) that limit infection with related viruses (Suzuki et al. 2020). While recent data indicate a role for EVEs in antiviral defence (Goic et al. 2016; Whitfield et al. 2017; Ophinni et al. 2019), it remains unclear whether germline ‘capture’ of virus sequences represents a dynamic system of heritable antiviral immunity analogous to that of CRISPR-Cas. It should be noted that the distribution and diversity of cISF-derived EVEs in Aedes mosquito genomes are consistent with germline integration of genome-length cDNA, followed by intra-genomic amplification/fragmentation of the integrated sequences (e.g. mediated by transposable elements). Numerous, similar examples of amplification of virus-derived DNA within the animal germline have been described, involving a diverse range of virus families (Belshaw et al. 2005; Fischer and Suttle 2011; Inoue et al. 2018; Lytras, Arriagada, and Gifford 2021). Thus, the presence of numerous distinct, flavivirid-derived EVE loci in the Aedes germline could reflect a relatively small number of germline colonisation events, rather than a dynamic process of EVE acquisition in association with immunity.
Controlling the spread of flavivirid-associated diseases is a public health priority, and genomic data have a critical role to play in these endeavours (Pierson and Diamond 2020; Hill et al. 2021). Here, we used the GLUE software framework to capture flavivirid genome sequence data and evolution-related domain knowledge in a way that supports their future use. By following principles of ‘data-oriented programming’, wherein an explicit separation is maintained between data and the code that operates on it (Sharvit 2021), the GLUE framework can facilitate the implementation of stable data resources that make no assumptions with respect to their future usage, so they can be deployed in distinct analysis contexts (Singer et al. 2018). Besides acting as a stable repository of domain knowledge, Flavivirid-GLUE provides a broad foundation for the rapid development of tools/services (e.g. epidemiological tracking and variant analysis) focused on individual flavivirid taxa (e.g. see (Singer et al. 2020; Campbell et al. 2022)). In addition, Flavivirid-GLUE can underpin analytical procedures that require a broader taxonomic scope, such as sequence-based classification of newly identified flavivirids, or supporting empirical, laboratory-based investigations of important flavivirid traits (e.g. the capacity to replicate in both arthropod and vertebrate hosts). If—as our investigation suggests—flavivirid traits have been acquired gradually through long-term evolutionary interactions with animal hosts, comparative studies will likely yield many useful insights into their biology.
Materials and methods
Construction of sequence-based resources for comparative genomic analysis
We used the GLUE software framework (Singer et al. 2018) to create Flavivirid-GLUE (Gifford 2021), an openly accessible online resource for comparative analysis of flavivirid genomes. The advantages of GLUE include the following: (i) the organisation of virus sequence data in relation to their hypothesised evolutionary relationships (an approach that is key to the practical interpretation of sequences); (ii) integration with wider computer systems, using standard technologies such as MySQL and JSON; (iii) mechanisms to customise functionality on a usage-specific basis, for example, schema extension and scripting mechanisms; and (iv) features and functionality to streamline rapid development of bespoke analysis projects.
We extended GLUE’s core database schema to capture information specific to flavivirid reference sequences (e.g. virus isolate names, isolation species, and date and location of sampling) and EVE loci (e.g. species in which they occur, genomic coordinates within contigs/chromosomes). A library of flavivirus representative genome sequences (Supplementary Table S1) was obtained from GenBank via reference to the International Committee for Taxonomy of Viruses website (see Gifford 2021). GenBank sequence entries in XML format were imported into the Flavivirid-GLUE project using an appropriately configured version of GLUE’s GenBank importer module. We extracted isolate-specific information (e.g. date and location of isolation, isolate name, and host species) from XML files as well standard GenBank fields (e.g. submission date). Additional/missing data were loaded from tabular files using GLUE’s TextFilePopulator module. Via reference to previous studies (Moureau et al. 2015; Shi et al. 2016; Geoghegan et al. 2018; Porter et al. 2020; Paraskevopoulou et al. 2021), we assigned all flavivirus sequences included in Flavivirid-GLUE to a taxonomic group and defined a standard set of genome features for flavivirids. The coordinates of genome features (where known) within all master references were recorded within the Flavivirid-GLUE database. These reference sequences and annotations were used in combination with a codon-aware, basic local alignment search tool (BLAST)–based sequence aligner implemented in GLUE (Singer et al. 2018; Altschul et al. 1997) to generate constrained MSAs (i.e. MSAs in which the coordinate space is constrained to a selected ‘master’ reference) for each taxonomic rank within the Flaviviridae (Table 1). To address genome and gene coverage–related issues, we used GLUE to generate genome feature coverage data for member sequence. Constrained MSAs were used to infer the coordinates of genome features not explicitly defined in GenBank XML via GLUE’s ‘inherit features’ command (Singer et al. 2018).
Genome screening in silico
Systematic in silico genome screening was performed using the database-integrated genome screening (DIGS) tool (Zhu et al. 2018)—a Practical Extraction and Reporting Language (PERL)-based screening framework within which the BLAST program suite (Altschul et al. 1997) is used to perform similarity searches while the MySQL relational database management system (Community Server 8.0.26) is used to record their output. Whole genome seqeuncing (WGS) data were obtained from the National Center for Biotechnology Information genome database (Kitts et al. 2016)—we obtained all animal genomes available as of March 2020 (see (Gifford 2021)). Flavivirid reference genomes and coding feature annotations collated in Flavivirid-GLUE were used to derive polypeptide probes for tBLASTn-based screening in the DIGS framework. For virus genomes lacking detailed annotations, we created polypeptide probes based on fragments of the major polyprotein. Via screening of WGS assemblies using the DIGS tool, we generated a non-redundant database of flavivirid-derived EVE loci (Gifford 2021). We used DIGS to investigate these loci and categorise them into (i) putatively novel endogenous flaviviral (EFV) elements, (ii) orthologues of previously characterised EVEs (e.g. copies containing large indels), and (iii) non-viral sequences that cross-matched to flavivirus probes (e.g. retrotransposons). In applying identifiers (IDs) to EVE sequences (see Fig. 2 and Table 1), we conservatively assumed that the presence of multiple EVE loci generally reflects intragenomic amplification rather than multiple independent germline incorporation events.
Phylogenetic and genomic analysis
Flavivirid-GLUE was used to implement an automated process for reconstructing midpoint-rooted, bootstrapped phylogenies from MSA partitions representing each rank within the constrained MSA tree. Gene coverage data were used to condition the way in which taxa were selected into MSA partitions (Stamatakis 2014). Phylogenies were reconstructed using the maximum likelihood approach implemented in RAxML (version 8.2.12) (Stamatakis 2014). Protein substitution models were selected via the hierarchical maximum likelihood ratio test using the PROTAUTOGAMMA option in RAxML. JalView (Waterhouse et al. 2009) (version 2.11.1.4) and Se-Al (version 2.0a11) were used to inspect MSAs.
Supplementary Material
Acknowledgements
R.J.G. was funded by the Medical Research Council of the United Kingdom (MC_UU_12014/12). W.M.S. acknowledges support from the Global Virus Network Fellowship. We thank Anna Gatseva, Joseph Hughes, Alain Kohl, Spyros Lytras, Emilie Pondeville, Charles Rice, David Robertson, Greg Towers, Sam J. Wilson, and anonymous reviewers for critical reading of the manuscript.
Contributor Information
Connor G G Bamford, Wellcome-Wolfson Institute for Experimental Medicine, School of Medicine, Dentistry and Biomedical Sciences, Queen’s University Belfast, 96 Lisburn Rd, Belfast, BT9 7BL, UK.
William M de Souza, World Reference Center for Emerging Viruses and Arboviruses and Department of Microbiology and Immunology, University of Texas Medical Branch, 301 University Boulevard, Galveston, Texas, 77555, USA.
Rhys Parry, School of Chemistry and Molecular Biosciences, University of Queensland, 68 Cooper Road, St Lucia 4072, Queensland, Australia.
Robert J Gifford, MRC-University of Glasgow Centre for Virus Research, 464 Bearsden Road, Glasgow, G61 1QH, Scotland, UK.
Data availability
Data available via GitHub: https://giffordlabcvr.github.io/Flavivirus-GLUE/.
Supplementary data
Supplementary data are available at Virus Evolution online.
Conflict of interest:
None declared.
References
- Aiewsakun P., and Katzourakis A. (2017) ‘Marine Origin of Retroviruses in the Early Palaeozoic Era’, Nature Communications, 8: 13954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Altschul S. F. et al. (1997) ‘Gapped BLAST and PSI-BLAST: A New Generation of Protein Database Search Programs’, Nucleic Acids Research, 25: 3389–402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belshaw R. et al. (2005) ‘High Copy Number in Human Endogenous Retrovirus Families Is Associated with Copying Mechanisms in Addition to Reinfection’, Molecular Biology and Evolution, 22: 814–7. [DOI] [PubMed] [Google Scholar]
- Blair C. D., Olson K. E., and Bonizzoni M. (2020) ‘The Widespread Occurrence and Potential Biological Roles of Endogenous Viral Elements in Insect Genomes’, Current Issues in Molecular Biology, 34: 13–30. [DOI] [PubMed] [Google Scholar]
- Blitvich B. J., and Firth A. E. (2015) ‘Insect-specific Flaviviruses: A Systematic Review of Their Discovery, Host Range, Mode of Transmission, Superinfection Exclusion Potential and Genomic Organization’, Viruses, 7: 1927–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ——— (2017) ‘A Review of Flaviviruses that Have No Known Arthropod Vector’, Viruses, 9:154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campbell K. et al. (2022) ‘Making Genomic Surveillance Deliver: A Lineage Classification and Nomenclature System to Inform Rabies Elimination’, PLOS Pathogens, 18: e1010023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chambers T. J. et al. (1990) ‘Flavivirus Genome Organization, Expression, and Replication’, Annual Review of Microbiology, 44: 649–88. [DOI] [PubMed] [Google Scholar]
- Conway M. J. (2015) ‘Identification of a Flavivirus Sequence in a Marine Arthropod’, PLOS One, 10: e0146037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cook S., and Holmes E. C. (2006) ‘A Multigene Analysis of the Phylogenetic Relationships among the Flaviviruses (Family: Flaviviridae) and the Evolution of Vector Transmission’, Archives of Virology, 151: 309–25. [DOI] [PubMed] [Google Scholar]
- de Lamballerie X. et al. (2002) ‘Genome Sequence Analysis of Tamana Bat Virus and Its Relationship with the Genus Flavivirus’, Journal of General Virology, 83: 2443–54. [DOI] [PubMed] [Google Scholar]
- de Oliveira L. G. et al. (2020) ‘Bovine Viral Diarrhea Virus: Recent Findings about Its Occurrence in Pigs’, Viruses, 12:600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dheilly N. M. et al. (2022) ‘A World of Viruses Nested within Parasites: Unraveling Viral Diversity within Parasitic Flatworms (Platyhelminthes)’, Microbiology Spectrum, 10(3): e0013822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dolja V. V., and Koonin E. V. (2018) ‘Metagenomics Reshapes the Concepts of RNA Virus Evolution by Revealing Extensive Horizontal Virus Transfer’, Virus Research, 244: 36–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dong X. et al. (2021) ‘A Novel Virus of Flaviviridae Associated with Sexual Precocity in Macrobrachium rosenbergii’, mSystems, 6: e0000321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fischer M. G., and Suttle C. A. (2011) ‘A Virophage at the Origin of Large DNA Transposons’, Science, 332: 231–4. [DOI] [PubMed] [Google Scholar]
- Geoghegan J. L. et al. (2018) ‘Hidden Diversity and Evolution of Viruses in Market Fish’, Virus Evolution, 4: vey031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geoghegan J. L., and Holmes E. C. (2018) ‘Evolutionary Virology at 40’, Genetics, 210: 1151–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gibb R. et al. (2022) ‘Mammal Virus Diversity Estimates are Unstable Due to Accelerating Discovery Effort’, Biology Letters, 18: 20210427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gifford R. J. 2021. ‘Flavivirid-GLUE’. 10.5281/zenodo.7097745.
- ——— et al. (2018) ‘Nomenclature for Endogenous Retrovirus (ERV) Loci’, Retrovirology, 15: 59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goic B. et al. (2016) ‘Virus-derived DNA Drives Mosquito Vector Tolerance to Arboviral Infection’, Nature Communications, 7: 12410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gould E. A. et al. (2003) ‘Origins, Evolution, and Vector/host Coadaptations within the Genus Flavivirus’, Adv Virus Res, 59: 277–314. [DOI] [PubMed] [Google Scholar]
- Gritsun D. J. et al. (2014) ‘Molecular Archaeology of Flaviviridae Untranslated Regions: Duplicated RNA Structures in the Replication Enhancer of Flaviviruses and Pestiviruses Emerged via Convergent Evolution’, PLOS One, 9: e92056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gritsun T. S., and Gould E. A. (2007) ‘Direct Repeats in the Flavivirus 3’ Untranslated Region; a Strategy for Survival in the Environment?’, Virology, 358: 258–65. [DOI] [PubMed] [Google Scholar]
- Grüning B. et al. (2018) ‘Practical Computational Reproducibility in the Life Sciences’, Cell Systems, 6: 631–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hermance M. E., and Thangamani S. (2018) ‘Tick–Virus–Host Interactions at the Cutaneous Interface: The Nidus of Flavivirus Transmission’, Viruses, 10:362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hill V. et al. (2021) ‘Progress and Challenges in Virus Genomic Epidemiology’, Trends in Parasitology, 37: 1038–49. [DOI] [PubMed] [Google Scholar]
- Holmes E. C. (2011) ‘The Evolution of Endogenous Viral Elements’, Cell Host & Microbe, 10: 368–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holmes E. C., and Duchêne S. (2019) ‘Can Sequence Phylogenies Safely Infer the Origin of the Global Virome?’, mBio, 10:e00289–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inoue Y. et al. (2018) ‘Fusion of piggyBac-like Transposons and Herpesviruses Occurs Frequently in Teleosts’, Zoological Letters, 4: 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jia N. et al. (2019) ‘Emergence of Human Infection with Jingmen Tick Virus in China: A Retrospective Study’, EBioMedicine, 43: 317–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katzourakis A., and Gifford R. J. (2010) ‘Endogenous Viral Elements in Animal Genomes’, PLOS Genetics, 6: e1001191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawasaki J. et al. (2021) ‘100-My History of Bornavirus Infections Hidden in Vertebrate Genomes’, Proceedings of the National Academy of Sciences of the United States of America: 118(20):e2026235118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kitts P. A. et al. (2016) ‘Assembly: A Resource for Assembled Genomes at NCBI’, Nucleic Acids Research, 44: D73–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar S. et al. (2017) ‘TimeTree: A Resource for Timelines, Timetrees, and Divergence Times’, Molecular Biology and Evolution, 34: 1812–9. [DOI] [PubMed] [Google Scholar]
- Kuno G. et al. (1998) ‘Phylogeny of the Genus Flavivirus’, Journal of Virology, 72: 73–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lequime S., and Lambrechts L. (2017) ‘Discovery of Flavivirus-derived Endogenous Viral Elements in Anopheles Mosquito Genomes Supports the Existence of Anopheles-associated Insect-specific Flaviviruses’, Virus Evolution, 3: vew035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y. Q. et al. (2022) ‘Discovery of Flaviviridae-derived Endogenous Viral Elements in Shrew Genomes Provide Novel Insights into Pestivirus Ancient History’, bioRxiv. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loeliger J., and McCullough M. (2012) Version Control with Git: Powerful Tools and Techniques for Collaborative Software Development. Sebastopol: O’Reilly Media, Inc. [Google Scholar]
- Lytras S., Arriagada G., and Gifford R. J. (2021) ‘Ancient Evolution of Hepadnaviral Paleoviruses and Their Impact on Host Genomes’, Virus Evolution, 7: veab012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manns M. P. et al. (2017) ‘Hepatitis C Virus Infection’, Nature Reviews Disease Primers, 3: 17006. [DOI] [PubMed] [Google Scholar]
- Mans B. J. (2011) ‘Evolution of Vertebrate Hemostatic and Inflammatory Control Mechanisms in Blood-feeding Arthropods’, Journal of Innate Immunity, 3: 41–51. [DOI] [PubMed] [Google Scholar]
- Merkel D. (2014) ‘Docker: Lightweight Linux Containers for Consistent Development and Deployment’, Linux Journal, 239. [Google Scholar]
- Monahan-Earley R., Dvorak A. M., and Aird W. C. (2013) ‘Evolutionary Origins of the Blood Vascular System and Endothelium’, Journal of Thrombosis and Haemostasis, 11: 46–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moureau G. et al. (2015) ‘New Insights into Flavivirus Evolution, Taxonomy and Biogeographic History, Extended by Analysis of Canonical and Alternative Coding Sequences’, PLOS One, 10: e0117849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ochsenreiter R., Hofacker I. L., and Wolfinger M. T. (2019) ‘Functional RNA Structures in the 3’UTR of Tick-Borne, Insect-Specific and No-Known-Vector Flaviviruses’, Viruses, 11:298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ophinni Y. et al. (2019) ‘piRNA-Guided CRISPR-like Immunity in Eukaryotes’, Trends in Immunology, 40: 998–1010. [DOI] [PubMed] [Google Scholar]
- Paraskevopoulou S. et al. (2021) ‘Viromics of Extant Insect Orders Unveil the Evolution of the Flavi-like Superfamily’, Virus Evolution, 7: veab030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parry R., and Asgari S. (2019) ‘Discovery of Novel Crustacean and Cephalopod Flaviviruses: Insights into Evolution and Circulation of Flaviviruses between Marine Invertebrate and Vertebrate Hosts’, Journal of Virology, 93(14): e00432–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pettersson J. H., and Fiz-Palacios O. (2014) ‘Dating the Origin of the Genus Flavivirus in the Light of Beringian Biogeography’, Journal of General Virology, 95: 1969–82. [DOI] [PubMed] [Google Scholar]
- Pierson T. C., and Diamond M. S. (2020) ‘The Continued Threat of Emerging Flaviviruses’, Nature Microbiology, 5: 796–812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Porter A. F. et al. (2020) ‘Novel Hepaci- and Pegi-like Viruses in Native Australian Wildlife and Non-human Primates’, Virus Evolution, 6: veaa064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Price J. L. (1978) ‘Isolation of Rio Bravo and a Hitherto Undescribed Agent, Tamana Bat Virus, from Insectivorous Bats in Trinidad, with Serological Evidence of Infection in Bats and Man’, The American Journal of Tropical Medicine and Hygiene, 27: 153–61. [DOI] [PubMed] [Google Scholar]
- Qin X. C. et al. (2014) ‘A Tick-borne Segmented RNA Virus Contains Genome Segments Derived from Unsegmented Viral Ancestors’, Proceedings of the National Academy of Sciences of the United States of America, 111: 6744–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenberg R. et al. (2013) ‘Search Strategy Has Influenced the Discovery Rate of Human Viruses’, Proceedings of the National Academy of Sciences of the United States of America, 110: 13961–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharvit Y. (2021) Data-Oriented Programming - Unlearning Objects (Manning) Manning Publications, Shelter Island, New York, United States. [Google Scholar]
- Shi M. et al. (2018) ‘The Evolutionary History of Vertebrate RNA Viruses’, Nature, 556: 197–202. [DOI] [PubMed] [Google Scholar]
- ——— et al. (2016) ‘Divergent Viruses Discovered in Arthropods and Vertebrates Revise the Evolutionary History of the Flaviviridae and Related Viruses’, Journal of Virology, 90: 659–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sieglaff D. H. et al. (2009) ‘Comparative Genomics Allows the Discovery of Cis-regulatory Elements in Mosquitoes’, Proceedings of the National Academy of Sciences of the United States of America, 106: 3053–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simmonds P. et al. (2017) ‘ICTV Virus Taxonomy Profile: Flaviviridae’, Journal of General Virology, 98: 2–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singer J. et al. (2020) ‘CoV-GLUE: A Web Application for Tracking SARS-CoV-2 Genomic Variation’, Preprints, 2020060225.doi: 10.20944/preprints202006.0225.v1 [DOI] [Google Scholar]
- Singer J. B. et al. (2018) ‘GLUE: A Flexible Software System for Virus Sequence Data’, BMC Bioinformatics, 19: 532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skoge R. H. et al. (2018) ‘New Virus of the Family Flaviviridae Detected in Lumpfish (Cyclopterus lumpus)’, Archives of Virology, 163: 679–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stamatakis A. (2014) ‘RAxML Version 8: A Tool for Phylogenetic Analysis and Post-analysis of Large Phylogenies’, Bioinformatics, 30: 1312–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Staples J. E., and Monath T. P. (2008) ‘Yellow Fever: 100 Years of Discovery’, Jama, 300: 960–2. [DOI] [PubMed] [Google Scholar]
- Suzuki Y. et al. (2020) ‘Non-retroviral Endogenous Viral Element Limits Cognate Virus Replication in Aedes aegypti Ovaries’, Current Biology, 30:3495–3506.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Z. D. et al. (2019) ‘A New Segmented Virus Associated with Human Febrile Illness in China’, New England Journal of Medicine, 380: 2116–25. [DOI] [PubMed] [Google Scholar]
- Waterhouse A. M. et al. (2009) ‘Jalview Version 2—A Multiple Sequence Alignment Editor and Analysis Workbench’, Bioinformatics, 25: 1189–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whitfield Z. J. et al. (2017) ‘The Diversity, Structure, and Function of Heritable Adaptive Immunity Sequences in the Aedes aegypti Genome’, Current Biology, 27:3511–3519.e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zanotto P. M. et al. (1996) ‘Population Dynamics of Flaviviruses Revealed by Molecular Phylogenies’, Proceedings of the National Academy of Sciences of the United States of America, 93: 548–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou W. et al. (2018) ‘Exosomes Serve as Novel Modes of Tick-borne Flavivirus Transmission from Arthropod to Human Cells and Facilitates Dissemination of Viral RNA and Proteins to the Vertebrate Neuronal Cells’, PLOS Pathogens, 14: e1006764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu H. et al. (2018) ‘Database-integrated Genome Screening (DIGS): Exploring Genomes Heuristically Using Sequence Similarity Search Tools and a Relational Database’, bioRxiv. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Data available via GitHub: https://giffordlabcvr.github.io/Flavivirus-GLUE/.