

Abstract
The first SARS-CoV-2 variant of concern (VOC) to be designated was lineage B.1.1.7, later labelled by the World Health Organization as Alpha. Originating in early autumn but discovered in December 2020, it spread rapidly and caused large waves of infections worldwide. The Alpha variant is notable for being defined by a long ancestral phylogenetic branch with an increased evolutionary rate, along which only two sequences have been sampled. Alpha genomes comprise a well-supported monophyletic clade within which the evolutionary rate is typical of SARS-CoV-2. The Alpha epidemic continued to grow despite the continued restrictions on social mixing across the UK and the imposition of new restrictions, in particular, the English national lockdown in November 2020. While these interventions succeeded in reducing the absolute number of cases, the impact of these non-pharmaceutical interventions was predominantly to drive the decline of the SARS-CoV-2 lineages that preceded Alpha. We investigate the only two sampled sequences that fall on the branch ancestral to Alpha. We find that one is likely to be a true intermediate sequence, providing information about the order of mutational events that led to Alpha. We explore alternate hypotheses that can explain how Alpha acquired a large number of mutations yet remained largely unobserved in a region of high genomic surveillance: an under-sampled geographical location, a non-human animal population, or a chronically infected individual. We conclude that the latter provides the best explanation of the observed behaviour and dynamics of the variant, although the individual need not be immunocompromised, as persistently infected immunocompetent hosts also display a higher within-host rate of evolution. Finally, we compare the ancestral branches and mutation profiles of other VOCs and find that Delta appears to be an outlier both in terms of the genomic locations of its defining mutations and a lack of the rapid evolutionary rate on its ancestral branch. As new variants, such as Omicron, continue to evolve (potentially through similar mechanisms), it remains important to investigate the origins of other variants to identify ways to potentially disrupt their evolution and emergence.
Keywords: Virus evolution, SARS-COV-2, Within-host evolution, variants of concern
Introduction
In early December 2020, one of the four UK public health agencies (Public Health England (PHE) now known as UK Health Security Agency (UKHSA)) began tracking and investigating a rapid increase in COVID-19 incidence in South East England, centred on Kent and East London. The number of new cases had grown more rapidly than expected over the previous 4 weeks, despite an elevated level of non-pharmaceutical interventions (NPIs) in the region, and increased incidence had begun to be observed in other locations in the UK, indicating further spread (Public Health England 2020). A corresponding genomic cluster was detected separately within the COVID-19 Genomics UK (COG-UK) Consortium (COVID-19 Genomics UK (COG-UK) 2020) severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) genomic surveillance data set, and the genome sequences carried a substantially larger than usual number of genetic changes (Rambaut et al. 2020a). At a routine PHE meeting on the 8th December 2020, the link between the genomic cluster and the Kent epidemiological situation was made, and investigations were initiated rapidly to characterise the mutations and estimate the growth rate of the cluster. Evidence accumulated that this cluster was growing rapidly and had expanded throughout November, during a national lockdown in England. The cluster was designated B.1.1.7 under the Pango lineage naming system (Rambaut et al. 2020b) and was later labelled as variant of concern (VOC) Alpha under the World Health Organisation (WHO) variant nomenclature (Konings et al. 2021).
Since its discovery, substantial analytical effort has been put into teasing apart the contributions of human behavioural factors and true virological effects on the rapid growth of the lineage (Kraemer et al. 2021). It is now clear that Alpha was associated with a higher transmission rate than the background D614G lineages that dominated in the UK at the time (Davies et al. 2021a; Leung et al. 2021; Volz et al. 2021), as well as a higher case fatality rate (Davies et al. 2021b).
Alpha contained fourteen lineage-specific amino acid replacements and three deletions compared to its contemporaneous lineages (Rambaut et al. 2020a), which at the time of its emergence was unprecedented in the global SARS-CoV-2 virus genomic data set (Supplementary Table S1). This mutational constellation included several mutations that have arisen independently in other VOCs. For example, N501Y in the Spike protein is also found in Beta (B.1.351), Gamma (P.1), and Omicron (B.1.1.529 descendants) and is a key contact residue in the receptor-binding domain; experimental data has determined that it increases binding affinity to human and murine angiotensisn converting enzyme 2 (ACE2) (Starr et al. 2020; Tian et al. 2021) and it has been associated with increased infectivity and virulence in a mouse model (Hongjing et al. 2020). N501Y alone has also been associated with higher infectivity and transmissibility of SARS-CoV-2 (Liu et al. 2021). There are also two deletions of interest in Alpha’s Spike gene: six base pairs at position 21765 (amino acid Positions 69–70) and three base pairs at Position 21991 (amino acid Position 144). Both have previously arisen in chronically infected individuals (Avanzato et al. 2020; Choi et al. 2020; Kemp et al. 2021; McCarthy et al. 2021). The former was also associated with a rapid outbreak of mink in Denmark (Oude Mennink et al. 2021) and has been shown in vitro to increase infectivity (Meng et al. 2021). The latter has been shown to prevent monoclonal antibody and, to a lesser extent, convalescent antisera binding (Andreano et al. 2020; Collier et al. 2021), as well as exhibiting decreased neutralisation efficiency (Weigang et al. 2021). Furthermore, Alpha contains a nine base pair deletion in Non-structural protein 6 (NSP6), also found in the VOCs Beta, Gamma, and Omicron, which is on the outside of the autophagy vesicle, theoretically limiting autophagosome expansion (Benvenuto et al. 2020). There is also a mutation in the accessory protein Open reading frame 8 (ORF8), which truncates the protein from 121 to only 27 amino acids in length, likely resulting in the loss of function and allowing further downstream mutations to accrue. However, these mutations and deletions have arisen multiple times during the pandemic and are not always associated with rapid growth or VOCs. This suggests that there are epistatic effects between many of the mutations present in Alpha that together lead to its increased fitness, as well as some hitchhiking mutations that are selectively neutral.
While the constellation of mutations observed in Alpha appears to have arisen in one evolutionary leap, two sequences have been identified in the COG-UK genomic surveillance data set that contain some, but not all, of the Alpha-defining mutations, and hence they may represent intermediate steps in the evolution of the Alpha lineage. These sequences could provide clues to the evolutionary processes underlying the evolution and emergence of VOCs and information on the timing of mutational events.
The existence of only two potential intermediate samples also requires some explanation. Due to the high level of SARS-CoV-2 genomic surveillance in the UK, it is unlikely that Alpha would transmit and evolve in a conventional manner (i.e. transmitting between individuals in the general UK population) without numerous intermediate genomes being sampled. Instead, the lineage may have evolved in an unsampled population before being detected in the general population in the UK, with the potential intermediates indicating early introductions from this population to the general population. We propose three possible alternatives for the nature of this unsampled population: first, Alpha may have evolved in a conventional manner in a location with little or no virus genomic surveillance before being introduced into the UK in Kent; second, it may have evolved in a non-human animal population before a zoonotic event reintroduced it into the human population in the UK; or finally, it may have evolved in a single or small number of chronically infected individuals, who were not sampled, before a single transmission event into the general population.
In late 2020 and early 2021, other VOCs began to be detected and appeared to have arisen through evolutionary jumps similar to that first characterised for Alpha: Beta (B.1.351) discovered in South Africa (Tegally et al. 2021a), Gamma (P.1) discovered in Japan and Brazil (Faria et al. 2021; Fujino et al. 2021), and Delta (B.1.617.2) discovered in India (Vaidyanathan 2021). The sudden appearance, in late 2021, of the VOC Omicron, designated as a descendant of B.1.1.529, has renewed interest in the processes underlying the emergence of variants exhibiting major leaps in evolution: Omicron is defined by forty-five non-synonymous mutations and exhibits increased transmissibility, increased ability to bind to ACE2 compared to Delta, and marked changes in its antigenic profile, enabling antibody to escape from much of the pre-existing population immunity (World Health Organisation 2021; Meng et al. 2022; Viana et al. 2022).
In this study, we use Bayesian phylogenetic analysis to explore the rate and nature of evolutionary processes on the phylogenetic branch ancestral to the B.1.1.7 lineage, as well as examining the two sequences that appear to be evolutionary intermediates. We then conduct coalescent and birth–death analyses to explore any differences in growth rates between the Alpha and background lineages. Finally, in order to identify any common patterns among VOCs, we perform similar evolutionary rate analyses on all VOCs and Variants of interest (VOIs) and compare their mutation profiles.
Results
Characterising the ancestral branch of B.1.1.7
While the first sequence of B.1.1.7 was sampled on the 20th September 2020 (GISAID Accession ID: EPI_ISL_601443), the lineage diverged from other concurrently circulating background lineages in the UK in early March 2020 (time of most recent common ancestor (TMRCA): 5th March, 25th January, 3rd May 2020, 95 % highest posterior density (HPD): 25th January 2020 to 3rd May 2020). However, it appears that sustained human-to-human transmission of Alpha in the UK began later in the year, with the TMRCA of the Alpha clade estimated at 28 August 2020 (95 % HPD: 15th August 2020 to 9th September 2020, Fig. 1A).
Figure 1.
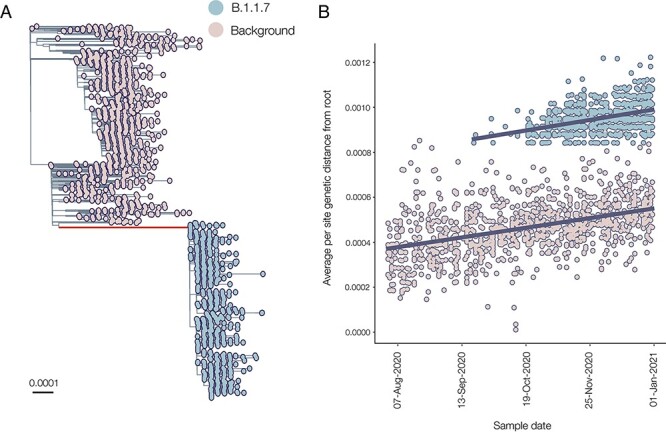
(A) Maximum likelihood phylogeny showing the well-supported monophyletic clade that constitutes B.1.1.7. The ancestral branch with the higher rate of evolution is highlighted in red, and branch lengths represent substitutions/sites. (B) Regression of root-to-tip genetic distances against sampling dates, for sequences belonging to lineage B.1.1.7 (blue) and those in its immediate out-group in the global phylogenetic tree (pink). The regression lines are fitted to the two data sets independently. The regression gradient is an estimate of the rate of sequence evolution. These rates are 4.6 × 10−4 and 4.3 × 10−4 nucleotide changes/site/year for the B.1.1.7 and out-group data sets, respectively.
The ancestral branch leading to the B.1.1.7 lineage is exceptionally long, both in terms of time (mean = 175 days, 95 % HPD: 104–213 days) and genetic changes: there are twenty-three nucleotide changes, with the majority being amino acid-altering (fourteen non-synonymous mutations and three deletions). We found that the evolutionary rate of the ancestral branch was an average of 2.77 times higher than the background rate (95 % HPD: 1.58 to 4.95). There is, however, little evidence for an increased rate of evolution within the B.1.1.7 clade itself: a regression of root-to-tip of genetic distances against genome sampling date (Fig. 1B) shows that the rates within the B.1.1.7 clade are very similar to those of the background lineage (4.6 × 10−4 and 4.3 × 10−4 nucleotide changes/site/year, respectively).
Two sequences lie along the branch leading to the B.1.1.7 clade, and both contain some, but not all, of the Alpha-defining mutations (not shown in Fig. 1A). The earlier of the two (COG-UK identifier: CAMC-946506, GISAID ID: EPI_ISL_556680) was sampled on 15th July 2020, and the more recent genome (MILK-B154B6, GISAID ID: EPI_ISL_2735517) was sampled on 23rd October 2020. If these two sequences are truly intermediate—i.e. they represent midpoints in the accrual of the twenty-three lineage-defining mutations for Alpha—then they may provide insight into the order of mutational accumulation during VOC evolution.
The more recent sequence, MILK-B154B6, is ambiguous at two Alpha-defining sites (see Supplementary Table S2), including at Position 501 in Spike, with 75 % of reads encoding N (asparagine, found in the background lineages) and 25 % Y (tyrosine, found in Alpha). These ambiguous sites imply either a coinfection of two different virus populations or laboratory contamination. If it was a coinfection, the sample could have been an individual who was infected by an early Alpha sequence and by a background lineage (possible in late October 2020 in the South East of England, as both lineages were circulating there at the time). MILK-B154B6 contains the synonymous mutation C5986T, which is also found in 971 of the 976 early Alpha sequences, but none of the 1,100 background sequences used in this study. It also carries two more mutations (C15279T and C913T) that are, respectively, found in 974 and 970 (out of 976) sequences in the Alpha data set, but only once in the background data set. As this sequence contains mutations that are shared by most Alpha sequences but not frequently found in earlier clades, it suggests that its intermediate status is due to a coinfection of an Alpha sequence (which contains these mutations) and a background clade that was co-circulating, and so the consensus sequence contains only some of the Alpha-defining mutations or cross-contamination in a laboratory handling samples from both the Alpha and background lineages. MILK-B154B6 can therefore not be definitively considered to be an evolutionary intermediate.
The older sequence, CAMC-946506, contains no ambiguous sites and carries four of the Alpha-defining mutations: N501Y in Spike, the nine base pair deletion in NSP6, as well as R52I and Q27 to stop codon mutation (Q27*) in ORF8. In the UK, prior to 1st September 2020, thirty-seven sequences were sampled with Q27*, five with R52I (all in July and August, one of which also had Q27*), and none with the NSP6 deletion or N501Y (n = 34,291). This makes it unlikely either that a virus containing all of these mutations existed in early 2020, prior to when Alpha diverged from the background lineages, or that this sequence has convergently acquired these mutations and has been placed erroneously in the tree. Instead, the evidence suggests that CAMC-94506 could be a true intermediate sequence resulting from a short transmission chain originating from the chronic population (Fig. 2A, Scenario 1); however, it must be noted that it may also simply contain mutations shared by the common ancestor of the hypothesised cryptic population and CAMC-94506 (Fig. 2A, Scenario 2).
Figure 2.
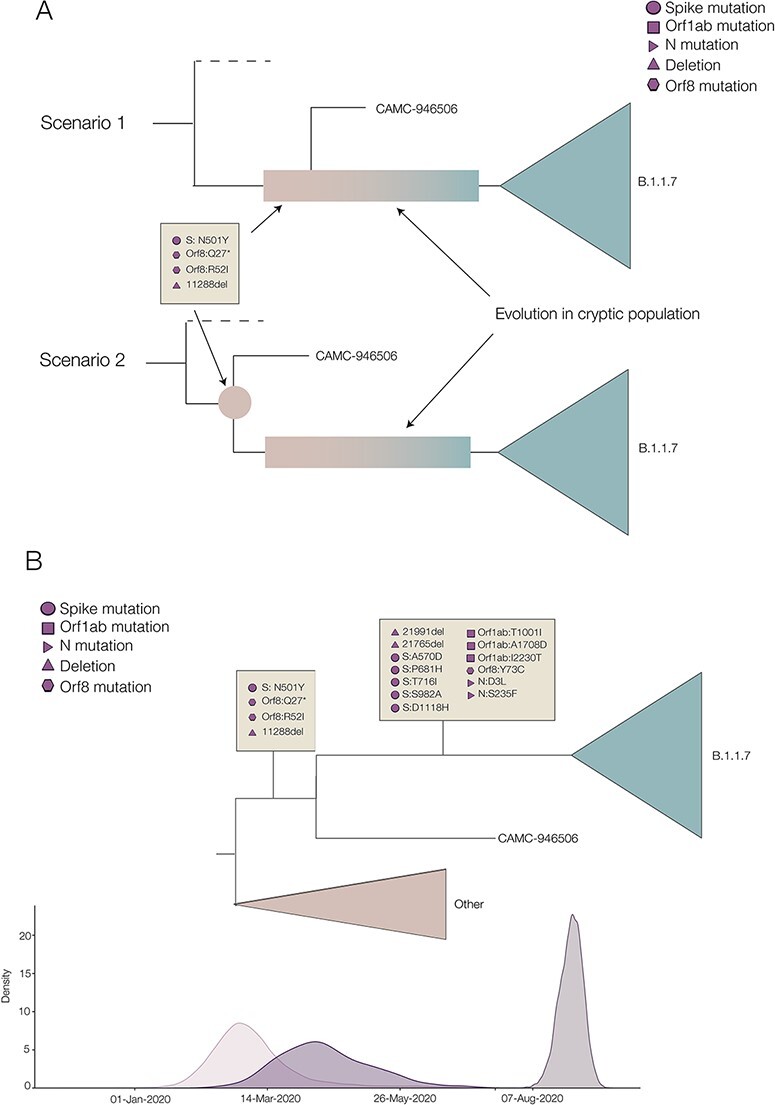
(A) Two different scenarios of how the shared mutations between CAMC-946506 and the B.1.1.7 clade could have arisen. Scenario 1 shows CAMC-946506 as resulting from a transmission chain spilling over from an isolated cryptic population, such as a chronically infected individual, and the mutations arising early in the infection. Scenario 2 shows the mutations as being shared by the common ancestor of CAMC-946506 and a cryptic population. (B) Schematic of the time tree showing possible timings for B.1.1.7 lineage-defining mutations. Densities of the most recent common ancestors for, respectively, the background lineages and all B.1.1.7, the intermediate sequence and B.1.1.7, and all B.1.1.7 are shown along the bottom.
This intermediate sequence provides evidence that the Spike mutation N501Y, the two ORF8 mutations Q27* and R52I, and the nine base pair deletion in NSP6 all evolved early in the evolutionary history of Alpha (see Fig. 2B), between 5th March 2020 (95 % HPD: 25th January 2020 to 3rd May 2020) and 15th April 2020 (95 % HPD: 28th February 2020 to 6th June 2020), i.e. between the TMRCA of B.1.1.7 and all background sequences, and the TMRCA of CAMC-934506 and B.1.1.7.
Early growth rate of B.1.1.7 in the UK and interaction with the November lockdown in England
Using a non-parametric coalescent model, we found that the growth of B.1.1.7 in England in the second half of 2020 was rapid compared to the background lineages present at the time (Fig. 3A). At the start of 2021, B.1.1.7 continued to grow, while other lineages began to decrease. These trends are broadly similar when comparing B.1.1.7 to just the B.1.177 lineage (Supplementary Fig. S1B), which spread rapidly across the UK and became the dominant lineage over the summer of 2020 (Hodcroft et al. 2021).
Figure 3.
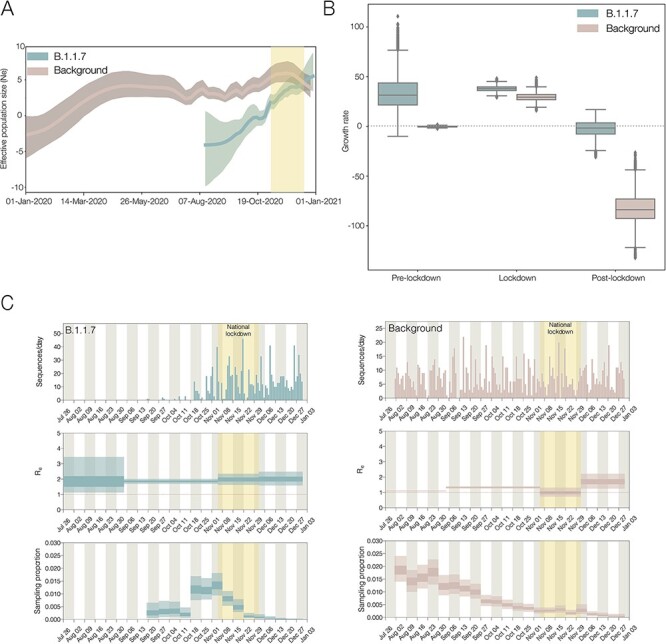
(A) Effective population sizes for the background lineages (pink) and B.1.1.7 (blue), generated from independent BEAST analyses. About 95 % of HPDs are shown as shaded areas. (B) Growth rate estimates with fixed transition times at pre-lockdown, lockdown, and post-lockdown, split by the background lineage and B.1.1.7. (C) Independent birth–death skyline analyses showing the number of sequences per day, the effective reproduction number (Re), and sampling proportion (which is allowed to vary on a weekly basis) for B.1.1.7 and the background. About 95 % of HPDs are shown as light-shaded areas and 50 % HPDs as dark-shaded areas. The English national lockdown in November is highlighted in all plots.
To further investigate the growth of B.1.1.7, we tested the difference between three standard population growth models: logistic, exponential, and epoch-based. For the last model (in which different epochs are permitted to have different growth rates), we estimated growth rates in three periods: pre-lockdown (1st September 2020 to 4th November 2020), during lockdown (5th November 2020 to 4th December 2020), and post-lockdown (5th December 2020 to 31st December 2020). Using a marginal likelihood estimation (MLE) approach, we found that the three-epoch model provided the best fit to the genomic data (Supplementary Table S3). For the second time period, B.1.1.7 has a positive growth rate, and the post-lockdown period estimation includes zero, whereas the background lineages have a very strong negative growth rate in the most recent time period (Fig. 3B). This suggests that while the national lockdown in the England in November significantly reduced the growth rates of both lineages, it was not sufficiently strict to push the growth rate of B.1.1.7 below zero. This reduced, but non-negative, growth rate for B.1.1.7 during the November lockdown has also been shown on a spatial level in Kraemer et al. (2021).
In order to explore this further, we also estimated the effective reproduction number (Re), using a birth–death approach, which allowed the sampling proportion to vary to account for changes in genomic surveillance intensity across time (Fig. 3C). This showed that while both the background lineages and B.1.1.7 had an Re value above 1 (i.e. the epidemic was growing) in September and October, the English national lockdown in November was sufficient to push the Re of the background lineages to around 1 (i.e. the epidemic was stable), and once they were released in December, the Re rebounded slightly. However, the Re of B.1.1.7 remained above 1, matching epidemiological information that showed growth of S-gene target failure (SGTF) positive cases despite the November lockdown (Kraemer et al. 2021; Volz et al. 2021).
Other VOCs
Under current WHO designations, there are four VOCs other than Alpha: Beta, discovered in South Africa at the end of 2020; Gamma, discovered in Brazil at the start of 2021; Delta, discovered in India at the start of 2021; and Omicron, discovered in South Africa and Botswana at the end of 2021. There are also two VOIs: Lambda, discovered in Peru in mid-2021, and Mu, discovered in Colombia at the start of 2021. Each of these variants has had differing impacts across different regions, but the current Omicron wave has displaced almost all other lineages (outbreak.info).
Similar to B.1.1.7, Omicron has a long ancestral branch (Fig. 4A), and the root-to-tip plot shows that Omicron sequences are distinct from the background diversity (Fig. 4B). However, as Omicron did not evolve out of the dominant circulating variant (i.e. Delta, B.1.617.2, and its descendants), it is more difficult to identify the clear pattern that can be observed in B.1.1.7, which evolved out of the dominant lineage at the time (i.e. B.1.1). Furthermore, Omicron contains five distinct sibling clades, BA.1, BA.2, BA.3, BA.4, and BA.5, which may represent multiple independent introductions into the general population. There is also some evidence of recombination involving BA.1, BA.2, and BA.3, but the evidence is unclear (Viana et al. 2022). The circumstances under which Omicron arose are clearly more complex than those that led to the evolution of Alpha. This could indicate a chronically infected individual or individuals with more contact with the general population (Maponga et al. 2022), or perhaps a non-human animal population. In addition, the Gamma variant has a long ancestral branch (Faria et al. 2021; Fujino et al. 2021) and evidence of an increased evolutionary rate (Supplementary Fig. S3; Gräf et al. 2021). The Beta variant shows some evidence of being distinct from the background lineages, although this is more inconclusive. The Beta and Gamma variants may therefore have a similar process of emergence involving a potential chronic infection. Finally, there is no clear increase in evolutionary rate on the ancestral branches leading to Delta, Lambda, or Mu (Supplementary Fig. S3), suggesting that these variants may have arisen under more traditional evolutionary processes involving intense between-host transmission.
Figure 4.
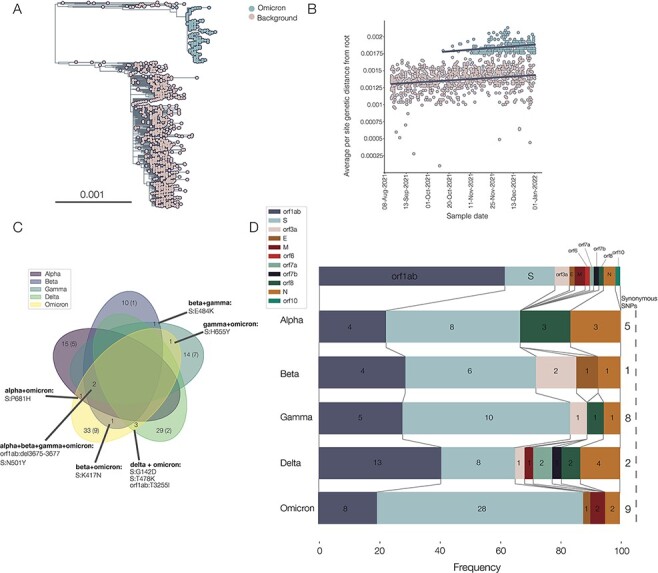
(A) Phylogeny showing Omicron in blue and background sequences in pink. The large background group is the Delta variant, the dominant variant globally in the second half of 2021. (B) Separate regressions of distance from the root against sample time for background sequences and Omicron sequences. Note that the parallel lines indicate similar rates of evolution within each clade (5.03 × 10–4 and 3.88 × 10–4 for the Omicron and background lineages, respectively). (C) Venn diagram showing numbers of mutations shared between different VOCs, with synonymous mutations in brackets. Zeroes, denoting no shared mutations acquired on the ancestral branch, are omitted. (D) Frequency of non-synonymous mutations acquired on the ancestral branch in different parts of the genome between VOCs. A schematic of the genome is shown along the top, numbers on each slice represent the absolute numbers of non-synonymous mutations or deletions in that gene, and numbers of synonymous mutations are shown along the right-hand side.
Looking at mutations or patterns shared by VOCs could provide evidence of common emergence routes or evolutionary pressures. We therefore collated mutations for each VOC (Alpha, Beta, Gamma, Delta, and Omicron) compared to the background that they emerged from (see Methods) to identify any similarities. In general, there were very few shared mutations, and in particular, none shared by all variants. No synonymous mutations were shared between any variants (Fig. 4C).
The most commonly shared mutations were N501Y in Spike and the nine base pair deletion in NSP6, which were both found in Alpha, Beta, Gamma, and Omicron (Fig. 4C): all four of these VOCs show evidence of an increased evolutionary rate prior to their emergence. Notably, the intermediate genome for B.1.1.7 (CAMC-94506) also exhibits both N501Y and the NSP6 deletion. N501Y has been monitored throughout the pandemic due to its ability to increase binding to the ACE2 of human and murine cells. However, it appears that by itself, it is not necessarily enough to create a VOC, as there was a cluster in Wales in late 2020 defined by N501Y, but without the NSP6 deletion, which was rapidly outcompeted by Alpha (Supplementary Fig. S2B).
There is only one shared mutation that was acquired during the evolution of Alpha and Omicron that is not shared by Beta, Gamma, and Delta: P681H in the furin cleavage site of the Spike protein (Fig. 4C), which enhances Spike cleavage (Peacock et al. 2022). It must also be noted that Delta carries P681R but shares no other mutations compared to the background lineages with Alpha and Omicron. Furthermore, sub-lineages of the Gamma variant also contain P681H (P.1.6 and P.1.7) and P681R (P.1.8; Naveca et al. 2022). The polybasic furin cleavage site is found in other coronaviruses, although not in any other sarbecovirus, and it is required for SARS-CoV-2 virus entry into human lung cells (Hoffmann, Kleine-Weber, and Pöhlmann 2020). Mutations in this gene region may be an adaptation to the human host, providing evidence for evolution in a human cryptic population. Of note, Peacock et al. (2021) found that mutants with deletions in the furin cleavage site were rare; we speculate that this could be part of the fitness valley that Alpha (and other variants) had to cross, as the furin cleavage site appears to be relatively conserved and so possibly many mutations are deleterious. It is also worth noting that the 69–70 deletion in the Spike protein, while not a defining mutation of all sub-lineages of Omicron, is present in BA.1, BA.4, and BA.5, all of which have caused significant waves of infection across the world, but absent from BA.2, which was also an important source of infection.
Finally, Beta and Omicron are the variants with the most evidence for immune evasion (Cele et al. 2021; Hu et al. 2022), and both variants share the common mutation K417N in the Spike protein (a similar mutation, K417T, is found in Gamma, Fig. 4C). K417N has been found to confer reduced susceptibility to neutralisation by specific monoclonal antibody therapies (Starr et al. 2021). This mutation also arose in AY.1, the so-called ‘Delta plus’ variant descended from B.1.617.2 (Kannan et al. 2021), but this variant did not appear to acquire any noticeable advantage compared to the background Delta wave (outbreak.info).
In terms of the frequency of regions of mutations (Fig. 4D), all bar Delta have the highest frequency of non-synonymous mutations in the Spike protein, and Delta has the highest frequency in orf1ab (∼38 % of its mutations). Omicron has the highest frequency of Spike mutations (56 %), Gamma has the highest frequency of synonymous mutations (28 %), and Alpha and Beta have the highest frequency of deletions (∼13 %). Overall, there does not appear to be a discernible pattern in the types or locations of deletions.
Discussion
B.1.1.7/Alpha was first sampled in Kent, in South East England on 20 September 2020, and spread quickly across the rest of the UK (Kraemer et al. 2021; Volz et al. 2021). It was able to grow rapidly in the context of the NPIs applied in November 2020, which did not include school closures and were not as strict in restricting mixing, although these measures were sufficient to reduce the background lineage growth rate significantly. While these trends have been investigated previously (Kraemer et al. 2021; Volz et al. 2021), we have here replicated them using only phylodynamic techniques and a small but representative genomic data set. This confirmation is useful for future studies that may wish to investigate VOCs in areas with less genomic sequencing or for tracking those without clear genetic markers (e.g. SGTF drop-out).
Any proposed origin of B.1.1.7 must explain three observations: first, the long branch leading to the B.1.1.7 clade with at most one intermediate sequence, despite high genomic surveillance; second, an increased evolutionary rate along this branch; and third, a single geographical and evolutionary origin of B.1.1.7 (Kraemer et al. 2021).
In a country with an extensive virus genomic surveillance programme like the UK, which includes random and relatively dense sampling (an average of 7.9 % of weekly reported cases in Kent and Medway between 24th April 2020 and 19th September 2020 was sequenced), it is unlikely that a precursor lineage was circulating in Kent over the summer of 2020 and was not detected. It is also worth noting that B.1.1.7 was captured by this surveillance programme within approximately 3 weeks of its origin—the MRCA of the clade is 28th August 2020 (see Results), and the first sample was taken on 20th September.
One possible explanation for the lack of detection of the precursor lineage to B.1.1.7 in the UK surveillance data is simply that it was not in the UK prior to its expansion in South East England, but in a region of the world with little or no genomic surveillance. However, this hypothesis requires that the lineage was introduced twice in the UK (first for the intermediate sequence and second for the B.1.1.7 lineage) without being exported and establishing transmission anywhere else. Genomic surveillance has since been scaled up in many regions, and the fact that no descendants of such a cryptic source population have been sampled to date indicates either that this population went extinct (unlikely given the fitness advantages conferred by the lineage-defining mutations) or that no such population existed. Furthermore, transmission between humans, even if rapid, would not explain the higher rate of evolution observed along the branch. These arguments would also apply to a population in the UK, which is disproportionately under-sampled, for example vulnerable communities, such as individuals experiencing homelessness, who are unlikely or unable to seek healthcare.
An alternative explanation is a zoonotic event, as SARS-CoV-2 has been shown to spread in non-human animals, for example in mink (Oreshkova et al. 2020; Oude Mennink et al. 2021), white-tailed deer (Chandler et al. 2021), and Syrian hamsters (Yen et al. 2022). In this hypothesis, there would have been a reverse zoonosis from humans, an increased rate of molecular evolution among animals, perhaps due to natural selection for the new host species, followed by at most two zoonotic events in the course of several months (the intermediate and the final clade). For the former, in an animal population that had sufficient contact with humans for a reverse zoonotic event and then two later zoonoses, it is unlikely that there would be only two spillovers in five months: in mink farms in the Netherlands, it was estimated that there were forty-three spillovers between April and November 2020 (Lu et al. 2021); in a pet shop in Hong Kong, there were at least two spillovers in the space of a few weeks (Yen et al. 2022). Furthermore, transmission between animals has not been observed to lead to a higher rate of evolution: even in a large outbreak among densely farmed mink, the rate of evolution was estimated to be similar to what is expected between humans, at approximately 7.9 × 10−4 nucleotide changes/site/year (Lu et al. 2021). Furthermore, it is unlikely that a variant so effective at spreading in the human population would have evolved in a non-human population; the mutations required to be successful in a human population may well be different due to differences in cell receptors, as well as behaviour. Common mutations appear in animal populations, such as N501T and Y453F in mink across different continents (Eckstrand et al. 2021; Lu et al. 2021), but are rarely found in human infections, and not in any of the VOCs. While it would be possible for a two-step evolutionary process to have occurred, whereby there is first some adaptation in an animal population, allowing the crossing of a fitness valley, followed by spillover and human adaptation through conventional transmission, it would once again be difficult to explain the intermediate sample we observe through under such a scenario.
We propose that the most likely explanation for the emergence of B.1.1.7 is that an individual was chronically infected with SARS-CoV-2 over the course of months, providing an evolutionary environment conducive to the virus making adaptive jumps. The evolutionary environment within a single host is different from that at the between-host scale, with a large effective population size and the opportunity for recombination (Jackson et al. 2021). This large effective population size can be established and maintained partially due to the different compartments that a respiratory virus can establish infection in, for example, the upper and lower respiratory tract, as well as deeper into the lung (e.g. Lakdawala et al. 2015; Richard et al. 2020). Conversely, the effective population size at the between-host level is small due to tight bottlenecks occurring in transmission (Lythgoe et al. 2021). Furthermore, although a persistent infection will provide the time and selective environment for a period of adaptive evolution, the exact cause of the persistence may affect the traits of the virus that are selected.
There are now a number of studies on chronically or persistently infected individuals, which report longitudinal sampling of the viruses present (Avanzato et al. 2020; Choi et al. 2020; Voloch et al. 2020; Clark et al. 2021; Karim et al. 2021; Kemp et al. 2021; Ramírez et al. 2021; Stanevich et al. 2021; Weigang et al. 2021; Williamson et al. 2021). Across these studies, there was an average of 4.0 (95 % confidence interval: 0.63–9.76) evolutionary events (i.e. gaining or losing a mutation compared to the individual’s first sample) per week (Supplementary Table S4), compared to the rate of approximately 0.5 mutations per week expected during between-host transmission. Furthermore, in these studies, deletions (notably, the 69/70 deletion found in Alpha and the BA.1 sub-lineage of Omicron) have been found to both increase and decrease in frequency during the period of infection (Kemp et al. 2021; Stanevich et al. 2021). As it is unlikely that a deletion could be reverted, this is evidence of multiple coexisting viral populations (Lythgoe et al. 2021).
Much of the discussion of variant emergence from within-host evolution has focussed on the idea of individuals with compromised immune systems, either pathologically (e.g. a lymphoma) or medically (e.g. post-transplant suppression or chemotherapy) induced (Karim et al. 2021; Maponga et al. 2022). Persistent infections have also been recorded in apparently immunocompetent individuals (Voloch et al. 2020; Ramírez et al. 2021), although the average length of these infections is shorter than in immunocompromised cases—from the aforementioned studies, an average of 32 and 174 days, respectively (Supplementary Fig. S2B). However, in one case, an immunocompetent individual was infected for approximately 64 days (Voloch et al. 2020). Furthermore, there may be an observation bias towards data from immunocompromised hosts, and long-term infections in immunocompetent hosts are less likely to be recorded.
The degree of immunocompromisation is variable and will depend on whether antibody or cellular immunity (or both) is affected by the condition of the individual. Grenfell et al. (2004) used a simple population genetic model to posit that the rate of viral adaptation is a non-linear function of immune (selection) pressure, because of the opposing effects of raised immune pressure on virus population size and average selection coefficients. Hence, the highest viral adaptation rate is predicted to arise from intermediate immune pressures. Although there is currently no evidence of a difference in the numbers of evolutionary events between immunocompromised and immunocompetent hosts (Supplementary Fig. S2A), we propose that this may be because these cases lie on either side of the peak of viral adaptation rate in the Grenfell et al. model. Thus, within-host virus evolution in both healthy and immunocompromised hosts could lead to an increased evolutionary rate as observed in this study, but we have not yet observed an individual at the part of the immune spectrum which lies at the Grenfell et al. peak of evolutionary rate, leading to the explosive adaptation seen in Alpha and Omicron. It must be noted however that the measures of evolutionary rates from this literature search are crude and have a small sample size and therefore should only be taken as preliminary. Further research into immunocompetent chronically infected individuals would elucidate this issue and provide evidence to support the evolutionary theory.
The absence of more than one sampled transmission chain arising from intermediate combinations of the Alpha mutations, even on the background of mild NPIs in the summer of 2020 in the UK, as well as this constellation not evolving elsewhere across the phylogeny, suggests that the fitness peak is difficult to reach, despite its large selective advantage compared to the background lineages. This implies a complex fitness landscape, with a large fitness valley prior to the peak of between-host fitness that the Alpha variant has reached.
The different selective pressures inside a host could enable the crossing of such a valley due to a relaxation of selection for transmission in favour of selection for factors such as evading neutralising antibodies, as found in longitudinal samples by Weigang et al. (2021), or focussing on cell entry. The intermediate sequence is likely part of a transmission chain that was ultimately very short, either due to stochastic extinction or because the virus was still not particularly well adapted to transmitting between hosts at this time point. It must be noted that while the intermediate sequence contains apparently crucial mutations such as N501Y, it is missing P681H (Peacock et al. 2022), potentially providing some explanation as to why it may not have had a sufficient transmission advantage over the background lineages at this time point.
However, Alpha and other VOCs have clearly become well adapted to spreading between hosts as well as within a host. While a transmission advantage would not be explicitly selected for within hosts, traits that are useful within a host could also lead to better among-host transmission. For example, increased ACE2 binding, which increases the efficiency of cell entry (Ozono et al. 2021), would allow a virion to enter and use a host cell faster than its competitor within a host, leading to faster growth, and would also make it easier for a virus population to establish an infection in a new host. The evasion of a host immune response is another clear advantage within a host, allowing fewer virions to be destroyed by the immune system, and on a population level, especially in the immune context of widespread previous infection and vaccination. Finally, and non-exclusively, an efficient and host-adapted infection could lead to a large amount of viral shedding, increasing transmissibility.
In comparing all of the VOCs, the Delta variant appears to be an outlier. Alpha, Gamma, and Omicron share the most evidence for a faster rate of evolution along their ancestral branch and Beta remains somewhat inconclusive, but with some evidence of the lineage having distinctly more root-to-tip divergence than expected. However, the VOIs and Delta show no evidence of a faster rate of evolution along their ancestral branches. Furthermore, Delta carries mutations distributed more evenly along its genome than the other VOCs and, consequently, has relatively fewer Spike mutations, and it does not contain N501Y or the deletion in NSP6. We therefore hypothesise that Delta may have followed an alternative route of emergence, perhaps simply intense between-host transmission in an under-sampled location. Given that the estimate of the TMRCA of Delta is several months prior to the first sample (McCrone et al. 2021), it is plausible that the lineage-defining mutations could have been acquired sequentially, prior to a larger explosion of cases once the full constellation of mutations in Delta was present.
The common patterns found in the emergence of Alpha, Beta, Gamma, and Omicron provide some evidence that Beta, Gamma, and Omicron may also have arisen through chronically infected individuals. It is notable that Southern Africa, where Beta and Omicron were first sampled, has a high prevalence of people living with HIV-1. An individual with a highly viraemic HIV infection would provide another avenue for a large viral population to be maintained in an individual over a long period of time and, therefore, new between-host fitness peaks to be explored. Indeed, an individual with an uncontrolled HIV infection accumulated more than twenty mutations in the course of nine months (Maponga et al. 2022); in a different HIV-positive individual, a beta-like virus evolved during a 6-month persistent infection (Cele et al. 2022). In both cases, the HIV infection was controlled and the SARS-CoV-2 cleared through antiretroviral treatment. However, in other circumstances, the progression of an HIV infection could allow the partially controlled SARS-CoV-2 infection to proliferate significantly, allowing for shedding and, therefore, transmission. Accessible antiretroviral therapy is, therefore, a key element in mitigating the risk of further SARS-CoV-2 variant emergence in countries with significant numbers of people living with HIV, as called for by Maponga et al. (2022). More generally, equitable and universal access to SARS-CoV-2 vaccination and antiviral drugs will be a critical strategy, given the apparent diversity of circumstances in which VOCs have emerged thus far. Extensive evolution during long-term infection has also been observed for Omicron: a long-term infection of the BA.1 sub-lineage of Omicron has led to the accumulation of eight non-synonymous mutations in 12 weeks, and it has been transmitted at least five times (Gonzalez-Reiche et al. 2022).
Chronic COVID-19 cases are relatively rare, but as another wave of transmission sweeps across the world, there will be many more as has been seen recently with the Omicron variant (Viana et al. 2022). If all persistent infections present a risk of a new, highly transmissible, or immune-evasive variant, then simply shielding the vulnerable and selective vaccination will not be sufficient to prevent the emergence of another wave of morbidity and mortality. Without urgent and truly widespread vaccination efforts and dispersal of antiviral medication, we expect to see the delayed impacts of uncontrolled transmission resulting from vaccine and antiviral inequity in the future.
Methods
Genomic data set
The COG-UK alignment and metadata of all SARS-CoV-2 genomes from 21th April 2021 was restricted to between 1th August 2020 and 31 December 2020 and surveillance (i.e. pillar 2) sequences from the England. This data set was then subsampled in a time-homogenous way to generate approximately 1,000 sequences per data set, which comprised 50 per week for non-B.1.1.7 sequences and 100 per week for B.1.1.7 sequences. The B.1.1.7 data set was checked for molecular clock outliers (sequences that have disproportionately too much or too little root-to-tip divergence for its sampling time (Hill and Baele 2019)) using TempEST, and one sequence was identified and removed (England/CAMC-BBDA45/2020). The resulting data set contained 1,100 background sequences and 976 B.1.1.7 sequences.
For mutation scanning, the sequences were aligned to the reference sequence Wuhan-Hu-1 using minimap2 (Li 2018) and gofasta (https://github.com/cov-ert/gofasta). Variants were determined using type_variants.py (https://github.com/cov-ert/type_variants).
Evolutionary rate calculation
First, a maximum likelihood tree was generated using IQTree v2.1.2 (Minh et al. 2020) and an Hasegawa-Kishino-Yano (HKY) substitution model (Hasegawa, Kishino, and Yano 1985). This was used to generate the plots showing a linear regression of root-to-tip genetic distance against sampling date and provide estimates of the rate of evolution in background sequences and within the B.1.1.7 clade (slope of the linear regression).
To estimate the rate of evolution in the branch leading up to B.1.1.7, a local clock model in BEAST v1.10.4 (Suchard et al. 2018) was used. Briefly, a strict clock model was applied to the B.1.1.7 lineage, the background sequences, and the branch leading up to B.1.1.7 separately so that their clock rates could be estimated independently, each with a Gamma prior. Preliminary analyses suggested that the within-B.1.1.7 evolutionary rate was similar to the background rate, and so the same clock rate model was placed on both the background and within-B.1.1.7 clade. Nested taxon sets containing one plausible intermediate sequence (CAMC-946506) were used to estimate the dates of the most recent common ancestor of B.1.1.7 and the intermediate sequence. A non-parametric coalescent Skygrid model (Gill et al. 2012) with sixty-four change points spanning 15 months was placed on the background sequences, including the intermediate sequence, and an exponential growth coalescent model was placed on B.1.1.7. For both sequence alignments, an HKY substitution model was used. All of these parameters were jointly estimated in a single analysis. Two replicate chains with 100 million states were run to check for convergence, and following assessment via Tracer (Rambaut et al. 2018), 45 million states from each were removed as burn-in.
Growth rate calculations
To describe general patterns in the growth of B.1.1.7 compared to the background rate, as well as B.1.177 (N = 1,069) and a Welsh cluster containing N501Y (N = 478), we ran a series of non-parametric Skygrid analyses (Gill et al. 2012). These were run independently in BEAST, each for two chains of 100 million states. For B.1.1.7, B.1.177, and the Welsh cluster, seventy-seven change points were used, spaced equidistantly between the most recent tip and 0.75 of a year before it (approximately 9 months). For the background data set, sixty-four change points were used with 1.25 of a year as the cut-off. All analyses assumed a strict molecular clock model and the HKY substitution model (Hasegawa, Kishino, and Yano 1985).
To compare parametric growth models, a logistic, exponential, and three-epoch growth model were run only on the B.1.1.7 data set. The three-epoch model used fixed transition times between epochs and estimated different exponential growth rates within each epoch. Transition times were fixed at the start and end of the lockdown in England (5 November 2020 and 2 December 2020), and an earlier transition time was also placed on 1 September 2020 (the TMRCA of B.1.1.7) at the start of the study period to focus on the growth after B.1.1.7 began to diversify. Each of the growth models used an HKY substitution model and a strict clock model. An MLE analysis, a commonly used form of Bayesian model selection integrated into the BEAST software package, was used to distinguish between these three models (Suchard, Weiss, and Sinsheimer 2001).
To directly compare growth rates between the B.1.1.7 and background lineage data sets, we used the three-epoch model to simultaneously estimate growth rates for both lineages (N = 2,076). For all the preceding analyses, transition times were fixed as described earlier, and two chains were run independently for 100 million states. Chains were combined after removing 10 % of states as burn-in, and convergence was assessed using Tracer (Rambaut et al. 2018).
To estimate differences in the effective reproduction number (Re) between the B.1.1.7 and background lineages, a Bayesian birth–death skyline (Stadler et al. 2013) model was run independently on the B.1.1.7 and background data sets. An HKY substitution model was used along with a strict clock model, and a Gamma prior with α = 0.001 and β = 1,000 was placed on the clock rate. A lognormal prior with a mean of 0.8 and a standard deviation of 0.5 was placed on Re and a Beta prior with α = 2 and β = 1,000 on the sampling proportion. Re was parameterised into four epochs with transition times fixed at the start and end of the lockdown in England (5 November 2020 and 2 December 2020), and an earlier transition time placed on 4 September 2020. The sampling proportion was fixed to 0 before the first week containing a sample and then estimated for each week thereafter, resulting in sixteen epochs for B.1.1.7 (from 19 September 2020) and twenty-three for the background data set (from 1 August 2020). The becoming-uninfectious rate was assumed to be constant and fixed at 36.5, which is equivalent to a mean period of 10 days from infection to loss of infectiousness (through recovery, isolation, or death). Analyses were started from initial trees estimated in IQTree v2.1.2 (Minh et al. 2020) and scaled to calendar time using TreeTime (Sagulenko, Puller, and Neher 2018). For both data sets, four chains of 100 million iterations were run independently, sampling states and trees every 10,000 iterations. Convergence was assessed using the R-package coda (Plummer et al. 2006), and 10 % of states were removed to account for burn-in.
Sequencing proportion
For calculating the percentage of cases that were sequenced over the summer of 2020, we took case data from the UK government dashboard (https://coronavirus.data.gov.uk/). Cases and sequences with sampling dates between 24 April 2020 and 19 September 2020 (the first day of the week of the most common recent ancestors of the stem of B.1.1.7 and the clade of B.1.1.7, respectively) were aggregated by week. Only cases and sequences with the locations of ‘Kent’ or ‘Medway’, a county surrounded by Kent, were included.
Rates of evolution in chronically infected individuals
For each of the eight studies identified containing longitudinal samples of chronically infected individuals, we counted the number of mutations present per individual in the paper. A mutation to the derived state and back to the reference allele each counted as a separate evolutionary event, relative to the first individual sample available, which was within the first week of infection start for all papers other than Karim et al.(Day 12) and Avanzato et al.(Day 49). For papers (e.g. Kemp et al.) where proportions of variants were given, a 25 % cut-off was used for the presence/absence of a mutation. Ambiguous or missing data were treated as the reference allele.
The rate of mutation events was calculated by dividing the number of events by the number of days the individual was followed up, divided by 7 to change the denominator to weeks. Note that for Karim et al., only non-synonymous substitutions were provided in the paper, meaning that the estimate of 2.04 events per week is an underestimate.
The start of infection was taken to be the start of symptoms or the date of the first positive PCR test, depending on what was available for each study. The end of infection was the date of the final negative PCR test in the study (Avanzato et al. 2020; Voloch et al. 2020; Karim et al. 2021; Stanevich et al. 2021; Weigang et al. 2021; Williamson et al. 2021), death (Choi et al. 2020), or when the individual was lost to follow up (Ramírez et al. 2021).
Other variant analyses
For performing the linear regression of root-to-tip genetic distance against sampling date to provide estimates of the rate of evolution in background sequences and within each VOC lineage, background data sets were obtained from the master COG-UK alignment. For Delta, Lambda and Mu, we took sequences sampled globally between 1st January 2021 and the 1st June 2021 that were not any of the analysed variants, and downsampled them to fifty per week where possible. For Gamma and Beta, the same background data set as Alpha was used (see above). Then, sequences from the correct time period for each variant were taken (the same as the background data set, other than Delta, which was 1 March 2021 to 1 June 2021 due to data quality issues, see below), and all were downsampled to fifty sequences per week, where possible. Finally, all sequences were run through metadata and sequence quality control. The final data set sizes were 1,145 for the 2021 background set, 703 for Beta, 95 for Gamma, 678 for Delta, 708 for Lambda, and 412 for Mu.
For Omicron, B.1 sequences were sampled between 1 September 2021 and 1 January 2022, and all Omicron sequences until the same cut-off were taken. These were then also downsampled to fifty per week, resulting in 900 background sequences and 390 Omicron sequences. We excluded any Omicron sequences with lineage-defining single nucleotide polymorphisms that were either missing entirely (i.e. an N), the same as the reference sequence (e.g. E for E484K), or had any of the Delta-defining mutations that are also not Omicron-defining. Finally, two molecular clock outliers in the background data set were also excluded. The final data set comprised 898 background and 328 Omicron sequences.
To undertake the mutation analysis for each variant, the first ten sequences for each variant other than Delta were taken from the oldest reported sample in the original paper or report describing the variant. These were from 20 September 2020 for Alpha (Rambaut et al. 2020a), 15 October 2020 for Beta (Tegally et al. 2021b), 4 December 2020 for Gamma (Faria et al. 2021; Naveca et al. 2021), and 15 November 2021 for Omicron (Viana et al. 2022). For Delta, due to an unclear starting point and large amounts of missing data in the sequences, all Delta sequences in March 2021 (following the estimate of the start of Delta expansion; McCrone et al. 2021) were extracted from the COG-UK alignment and run through Scorpio (https://github.com/cov-lineages/scorpio), filtering to allow no reference alleles (from Wuhan-Hu-1) and a maximum of two missing alleles in lineage-defining positions. This resulted in ninety-eight sequences, ten of which were from India, which were taken as the representative group. For all variants, mutations that were common to all representative ten sequences were taken and supplemented with any lineage-defining mutations that were missing (due to a small amount of missing data), based again on the original defining publication for each variant.
These were then compared to a representative background set. For each variant, this entailed up to ten sequences from the month of the first reported sample of the parent lineage: B.1.1 for Alpha and Omicron, B.1 for Beta and Omicron, and B.1.1.28 for Gamma.
Supplementary Material
Acknowledgements
J.T.M., R.C., and A.R. acknowledge support from the Wellcome Trust (Collaborator Award 206298/Z/17/Z—ARTIC network). O.G.P. and L.D.P. acknowledge support from the Oxford Martin School.
Contributor Information
Verity Hill, Ashworth Laboratories, Institute of Evolutionary Biology, University of Edinburgh, Edinburgh EH9 3FL, UK; Department of Epidemiology of Microbial Diseases, Yale School of Public Health, New Haven, CT, USA.
Louis Du Plessis, Department of Biology, University of Oxford, 11a Mansfield Rd, Oxford OX1 3SZ, UK; Department of Biosystems Science and Engineering, ETH Zürich, Zürich, Switzerland.
Thomas P Peacock, Department of Infectious Disease, Imperial College London, London W2 1PG, UK.
Dinesh Aggarwal, Wellcome Sanger Institute, Wellcome Genome Campus, Hinxton CB10 1RQ, UK; UK Health Security Agency, 61 Colindale Avenue, London NW9 5EQ, UK; Department of Medicine, University of Cambridge, Cambridge, UK; Cambridge University Hospital NHS Foundation Trust, Cambridge, UK.
Rachel Colquhoun, Ashworth Laboratories, Institute of Evolutionary Biology, University of Edinburgh, Edinburgh EH9 3FL, UK.
Alesandro M Carabelli, Department of Medicine, University of Cambridge, Cambridge, UK.
Nicholas Ellaby, UK Health Security Agency, 61 Colindale Avenue, London NW9 5EQ, UK.
Eileen Gallagher, UK Health Security Agency, 61 Colindale Avenue, London NW9 5EQ, UK.
Natalie Groves, UK Health Security Agency, 61 Colindale Avenue, London NW9 5EQ, UK.
Ben Jackson, Ashworth Laboratories, Institute of Evolutionary Biology, University of Edinburgh, Edinburgh EH9 3FL, UK.
J T McCrone, Ashworth Laboratories, Institute of Evolutionary Biology, University of Edinburgh, Edinburgh EH9 3FL, UK.
Áine O’Toole, Ashworth Laboratories, Institute of Evolutionary Biology, University of Edinburgh, Edinburgh EH9 3FL, UK.
Anna Price, School of Biosciences, The Sir Martin Evans Building, Cardiff University, Cardiff CF10 AX, UK.
Theo Sanderson, Wellcome Sanger Institute, Wellcome Genome Campus, Hinxton CB10 1RQ, UK; The Francis Crick Institute, 1 Midland Rd, London NW1 1AT, UK.
Emily Scher, Ashworth Laboratories, Institute of Evolutionary Biology, University of Edinburgh, Edinburgh EH9 3FL, UK.
Joel Southgate, School of Biosciences, The Sir Martin Evans Building, Cardiff University, Cardiff CF10 AX, UK.
Erik Volz, MRC Unit for Global Infectious Disease Analysis, School of Public Health, Imperial College London, London, UK.
Wendy S Barclay, Department of Infectious Disease, Imperial College London, London W2 1PG, UK.
Jeffrey C Barrett, Wellcome Sanger Institute, Wellcome Genome Campus, Hinxton CB10 1RQ, UK.
Meera Chand, UK Health Security Agency, 61 Colindale Avenue, London NW9 5EQ, UK; Guy’s and St Thomas’ Hospital NHS Trust, St Thomas’ Hospital, Westminster Bridge Rd, London SE1 7EH, UK.
Thomas Connor, School of Biosciences, The Sir Martin Evans Building, Cardiff University, Cardiff CF10 AX, UK; Pathogen Genomics Unit, Public Health Wales NHS Trust, Cardiff CF14 4XW, UK.
Ian Goodfellow, Department of Pathology, University of Cambridge, Cambridge CB2 1QP, UK.
Ravindra K Gupta, Department of Medicine, University of Cambridge, Cambridge, UK; Africa Health Research Institute, Durban, South Africa.
Ewan M Harrison, Wellcome Sanger Institute, Wellcome Genome Campus, Hinxton CB10 1RQ, UK; Department of Medicine, University of Cambridge, Cambridge, UK; Department of Public Health and Primary Care, University of Cambridge, Cambridge, UK.
Nicholas Loman, Institute of Microbiology and Infection, University of Birmingham, Birmingham B15 2TT, UK.
Richard Myers, UK Health Security Agency, 61 Colindale Avenue, London NW9 5EQ, UK.
David L Robertson, MRC-University of Glasgow Centre for Virus Research, 464 Bearsden Road, Glasgow G61 1QH, Scotland, UK.
Oliver G Pybus, Department of Biology, University of Oxford, 11a Mansfield Rd, Oxford OX1 3SZ, UK; Department of Pathobiology and Population Science, The Royal Veterinary College, London, UK.
Andrew Rambaut, Ashworth Laboratories, Institute of Evolutionary Biology, University of Edinburgh, Edinburgh EH9 3FL, UK.
Data availability
UK genome sequences used were generated by the COG-UK (https://www.cogconsortium.uk/). Non-UK data were from GISAID (gisaid.org), and an acknowledgement table for the sequences used can be found in the Supplementary material. Short-read data for the two putative intermediate sequences can be found on the NCBI Short Read Archive using accession numbers ERX4738666 and ERX4562166 for MILK-B154B6 and CAMC-946506, respectively. XML for evolutionary and growth rate analyses (with alignments removed for data protection reasons) can be found in the Supplementary material.
Supplementary data
Supplementary data are available at Virus Evolution online.
Funding
COG-UK is supported by funding from the Medical Research Council (MRC) part of UK Research & Innovation, the National Institute of Health Research (grant code: MC_PC_19027), and Genome Research Limited, operating as the Wellcome Sanger Institute. V.H. was supported by the Biotechnology and Biological Sciences Research Council (grant number: BB/M010996/1). T.P.P. and W.S.B. are supported by the G2P-UK National Virology Consortium funded by the MRC (grant number: MR/W005611/1). A.R. is also supported by the European Research Council (grant agreement number: 725422—ReservoirDOCs) and the Bill & Melinda Gates Foundation (OPP1175094—HIV-PANGEA II). A.O.T. is supported by the Wellcome Trust Hosts, Pathogens & Global Health Programme (grant number: grant.203783/Z16/Z), and Fast Grants (award number: 2236). D.A. is a Wellcome Clinical PhD Fellow and gratefully supported by the Wellcome Trust (grant number: 222903/Z/21/Z). I.G. is a Wellcome Senior Fellow and is supported by the Wellcome Trust (grant number: 207498/Z/17/Z). E.V. is supported by the Wellcome Trust (Grant number: 220885/Z/20/Z).
Conflict of interest
None declared.
References
- Andreano E. et al. (2020) ‘SARS-CoV-2 Escape in Vitro from a Highly Neutralizing COVID-19 Convalescent Plasma’, bioRxiv.doi: 10.1101/2020.12.28.424451 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Avanzato V. A. et al. (2020) ‘Case Study: Prolonged Infectious SARS-CoV-2 Shedding from an Asymptomatic Immunocompromised Individual with Cancer’, Cell, 183: 1901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benvenuto D. et al. (2020) ‘Evolutionary Analysis of SARS-CoV-2: How Mutation of Non-structural Protein 6 (NSP6) Could Affect Viral Autophagy’, The Journal of Infection, 81: e24–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cele S. et al. (2021) ‘Escape of SARS-CoV-2 501Y.V2 from Neutralization by Convalescent Plasma’, Nature, 593: 7857.doi: 10.1038/s41586-021-03471-w [DOI] [PMC free article] [PubMed] [Google Scholar]
- ——— et al. (2022) ‘SARS-CoV-2 Prolonged Infection during Advanced HIV Disease Evolves Extensive Immune Escape’, Cell Host & Microbe, 30: 154–62.e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chandler J. C. et al. (2021) ‘SARS-CoV-2 Exposure in Wild White-Tailed Deer (Odocoileus virginianus)’, Proceedings of the National Academy of Sciences of the United States of America, 118: 47.doi: 10.1073/pnas.2114828118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi B. et al. (2020) ‘Persistence and Evolution of SARS-CoV-2 in an Immunocompromised Host’, The New England Journal of Medicine, 383: 2291–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clark S. A. et al. (2021) ‘SARS-CoV-2 Evolution in an Immunocompromised Host Reveals Shared Neutralization Escape Mechanisms’, Cell, 184: 2605–17.e18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collier D. A. et al. (2021) ‘Sensitivity of SARS-CoV-2 B.1.1.7 to mRNA Vaccine-Elicited Antibodies’, Nature, 593: 136–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- COVID-19 Genomics UK (COG-UK) . (2020) ‘An Integrated National Scale SARS-CoV-2 Genomic Surveillance Network’, The Lancet Microbe, 1: e99–100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davies N. G. et al. (2021a) ‘Estimated Transmissibility and Impact of SARS-CoV-2 Lineage B.1.1.7 in England’, Science, 372: 6538.doi: 10.1126/science.abg3055 [DOI] [PMC free article] [PubMed] [Google Scholar]
- ——— et al. (2021b) ‘Increased Mortality in Community-Tested Cases of SARS-CoV-2 Lineage B.1.1.7’, Nature, 593: 270–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eckstrand C. D. et al. (2021) ‘An Outbreak of SARS-CoV-2 with High Mortality in Mink (Neovison Vison) on Multiple Utah Farms’, PLoS Pathogens, 17: e1009952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faria N. R. et al. (2021) ‘Genomics and Epidemiology of the P.1 SARS-CoV-2 Lineage in Manaus, Brazil’, Science, 372: 815–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujino T. et al. (2021) ‘Novel SARS-CoV-2 Variant in Travelers from Brazil to Japan’, Emerging Infectious Diseases, 27: 4.doi: 10.3201/eid2704.210138 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gill M. S. et al. (2012) ‘Improving Bayesian Population Dynamics Inference: A Coalescent-Based Model for Multiple Loci’, Molecular Biology and Evolution, 30: 713–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonzalez-Reiche A. S. et al. (2022) ‘Intrahost Evolution and Forward Transmission of a Novel SARS-CoV-2 Omicron BA.1 Subvariant’, medRxiv.doi: 10.1101/2022.05.25.22275533 [DOI] [Google Scholar]
- Gräf T. et al. (2021) ‘Identification of a Novel SARS-CoV-2 P.1 Sub-lineage in Brazil Provides New Insights about the Mechanisms of Emergence of Variants of Concern’, Virus Evolution, 7: veab091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grenfell B. T. et al. (2004) ‘Unifying the Epidemiological and Evolutionary Dynamics of Pathogens’, Science, 303: 327–32. [DOI] [PubMed] [Google Scholar]
- Hasegawa M., Kishino H., and Yano T.-A. (1985) ‘Dating of the Human-Ape Splitting by a Molecular Clock of Mitochondrial DNA’, Journal of Molecular Evolution, 22: 160–74. [DOI] [PubMed] [Google Scholar]
- Hill V., and Baele G. (2019) ‘Bayesian Estimation of past Population Dynamics in BEAST 1.10 Using the Skygrid Coalescent Model’, Molecular Biology and Evolution, 36: 2620–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hodcroft E. B. et al. (2021) ‘Spread of a SARS-CoV-2 Variant Through Europe in the Summer of 2020’, Nature, 595: 707–12. [DOI] [PubMed] [Google Scholar]
- Hoffmann M., Kleine-Weber H., and Pöhlmann S. (2020) ‘A Multibasic Cleavage Site in the Spike Protein of SARS-CoV-2 Is Essential for Infection of Human Lung Cells’, Molecular Cell, 78: 779–84.e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hongjing G. et al. (2020) ‘Adaptation of SARS-CoV-2 in BALB/c Mice for Testing Vaccine Efficacy’, Science, 369: 1603–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu J. et al. (2022) ‘Increased Immune Escape of the New SARS-CoV-2 Variant of Concern Omicron’, Cellular & Molecular Immunology, 19: 293–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jackson B. et al. (2021) ‘Generation and Transmission of Interlineage Recombinants in the SARS-CoV-2 Pandemic’, Cell.doi: 10.1016/j.cell.2021.08.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kannan S. R. et al. (2021) ‘Evolutionary Analysis of the Delta and Delta Plus Variants of the SARS-CoV-2 Viruses’, Journal of Autoimmunity, 124.doi: 10.1016/j.jaut.2021.102715 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karim F. et al. (2021) ‘Persistent SARS-CoV-2 Infection and Intra-Host Evolution in Association with Advanced HIV Infection’, medRxiv. [Google Scholar]
- Kemp S. A. et al. (2021) ‘SARS-CoV-2 Evolution during Treatment of Chronic Infection’, Nature, 592: 277–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Konings F. et al. (2021) ‘SARS-CoV-2 Variants of Interest and Concern Naming Scheme Conducive for Global Discourse’, Nature Microbiology, 6: 821–3. [DOI] [PubMed] [Google Scholar]
- Kraemer M. U. G. et al. (2021) ‘Spatiotemporal Invasion Dynamics of SARS-CoV-2 Lineage B.1.1.7 Emergence’, Science, 373: 889–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lakdawala S. S. et al. (2015) ‘The Soft Palate Is an Important Site of Adaptation for Transmissible Influenza Viruses’, Nature, 526: 122–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leung K. et al. (2021) ‘Early Transmissibility Assessment of the N501Y Mutant Strains of SARS-CoV-2 in the United Kingdom, October to November 2020’, Eurosurveillance, 26: 2002106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H. (2018) ‘Minimap2: Pairwise Alignment for Nucleotide Sequences’, Bioinformatics, 34: 3094–100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y. et al. (2021) ‘The N501Y Spike Substitution Enhances SARS-CoV-2 Infection and Transmission’, Nature, 602: 294–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu L. et al. (2021) ‘Adaptation, Spread and Transmission of SARS-CoV-2 in Farmed Minks and Associated Humans in the Netherlands’, Nature Communications, 12: 1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lythgoe K. A. et al. (2021) ‘SARS-CoV-2 Within-Host Diversity and Transmission’, Science, 372: 6539.doi: 10.1126/science.abg0821 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maponga T. G. et al. (2022) ‘Persistent SARS-CoV-2 Infection with Accumulation of Mutations in a Patient with Poorly Controlled HIV Infection’, Clinical Infectious Diseases.doi: 10.2139/ssrn.4014499 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCarthy K. R. et al. (2021) ‘Recurrent Deletions in the SARS-CoV-2 Spike Glycoprotein Drive Antibody Escape’, Science, 371: 1139–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCrone J. T. et al. (2021) ‘Context-Specific Emergence and Growth of the SARS-CoV-2 Delta Variant’, medRxiv. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meng B. et al. (2022) ‘Altered TMPRSS2 Usage by SARS-CoV-2 Omicron Impacts Tropism and Fusogenicity’, Nature, 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ——— et al. (2021) ‘Recurrent Emergence of SARS-CoV-2 Spike Deletion H69/V70 and Its Role in the Alpha Variant B.1.1.7’, Cell Reports, 35: 109292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minh B. Q. et al. (2020) ‘IQ-TREE 2: New Models and Efficient Methods for Phylogenetic Inference in the Genomic Era’, Molecular Biology and Evolution, 37: 1530–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oude Mennink O. et al. (2021) ‘Transmission of SARS-CoV-2 on Mink Farms between Humans and Mink and Back to Humans’, Science, 371: 172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naveca F. G. et al. (2022) ‘Spread of Gamma (P.1) Sub-lineages Carrying Spike Mutations Close to the Furin Cleavage Site and Deletions in the N-Terminal Domain Drives Ongoing Transmission of SARS-CoV-2 in Amazonas, Brazil’, Microbiology Spectrum, 10: e0236621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ——— et al. (2021) ‘COVID-19 in Amazonas, Brazil, Was Driven by the Persistence of Endemic Lineages and P.1 Emergence’, Nature Medicine, 27: 1230–8. [DOI] [PubMed] [Google Scholar]
- Oreshkova N. et al. (2020) ‘SARS-CoV-2 Infection in Farmed Minks, the Netherlands, April and May 2020’, Eurosurveillance, 25.doi: 10.2807/1560-7917.ES.2020.25.23.2001005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ozono S. et al. (2021) ‘SARS-CoV-2 D614G Spike Mutation Increases Entry Efficiency with Enhanced ACE2-Binding Affinity’, Nature Communications, 12.doi: 10.1038/s41467-021-21118-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peacock T. P. et al. (2022) ‘The SARS-CoV-2 Variant, Omicron, Shows Rapid Replication in Human Primary Nasal Epithelial Cultures and Efficiently Uses the Endosomal Route of Entry’, bioRxiv.doi: 10.1101/2021.12.31.474653 [DOI] [Google Scholar]
- ——— et al. (2021) ‘The Furin Cleavage Site in the SARS-CoV-2 Spike Protein Is Required for Transmission in Ferrets’, Nature Microbiology, 6: 899–909. [DOI] [PubMed] [Google Scholar]
- Plummer M. et al. (2006) ‘CODA: Convergence Diagnosis and Output Analysis for MCMC’, R News, 6: 7–11. [Google Scholar]
- Public Health England . 2020. “Public Health England Investigation of Novel SARS-COV-2 Variant 202012/01: Technical Briefing 1.”
- Rambaut A. et al. (2018) ‘Posterior Summarization in Bayesian Phylogenetics Using Tracer 1.7’, Systematic Biology, 67: 5.doi: 10.1093/sysbio/syy032 [DOI] [PMC free article] [PubMed] [Google Scholar]
- ——— et al. (2020a) ‘A Dynamic Nomenclature Proposal for SARS-CoV-2 Lineages to Assist Genomic Epidemiology’, Nature Microbiology, 5: 1403–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ——— et al. (2020b) Preliminary Genomic Characterisation of an Emergent SARS-CoV-2 Lineage in the UK Defined by a Novel Set of Spike Mutations. <https://virological.org/t/preliminary-genomic-characterisation-of-an-emergent-sars-cov-2-lineage-in-the-uk-defined-by-a-novel-set-of-spike-mutations/563> accessed 21 Sep 2021.
- Ramírez J. D. et al. (2021) ‘Phylogenomic Evidence of Reinfection and Persistence of SARS-CoV-2: First Report from Colombia’, Vaccines, 9: 282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richard M. et al. (2020) ‘Influenza A Viruses Are Transmitted via the Air from the Nasal Respiratory Epithelium of Ferrets’, Nature Communications, 11: 1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sagulenko P., Puller V., and Neher R. A. (2018) ‘TreeTime: Maximum-Likelihood Phylodynamic Analysis’, Virus Evolution, 4: 1.doi: 10.1093/ve/vex042 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stadler T. et al. (2013) ‘Birth–Death Skyline Plot Reveals Temporal Changes of Epidemic Spread in HIV and Hepatitis C Virus (HCV)’, Proceedings of the National Academy of Sciences of the United States of America, 110: 228–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stanevich O. et al. (2021) ‘SARS-CoV-2 Escape from Cytotoxic T Cells during Long-Term COVID-19’, Research Square.doi: 10.21203/rs.3.rs-750741/v1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Starr T. N. et al. (2021) ‘Complete Map of SARS-CoV-2 RBD Mutations That Escape the Monoclonal Antibody LY-CoV555 and Its Cocktail with LY-CoV016’, Cell Reports Medicine, 2: 4.doi: 10.1016/j.xcrm.2021.100255 [DOI] [PMC free article] [PubMed] [Google Scholar]
- ——— et al. (2020) ‘Deep Mutational Scanning of SARS-CoV-2 Receptor Binding Domain Reveals Constraints on Folding and ACE2 Binding’, Cell, 182: 1295–1310.e20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suchard M. A. et al. (2018) ‘Bayesian Phylogenetic and Phylodynamic Data Integration Using BEAST 1.10’, Virus Evolution, 4: 1.doi: 10.1093/ve/vey016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suchard M. A., Weiss R. E., and Sinsheimer J. S. (2001) ‘Bayesian Selection of Continuous-Time Markov Chain Evolutionary Models’, Molecular Biology and Evolution, 18: 1001–13. [DOI] [PubMed] [Google Scholar]
- Tegally H. et al. (2021a) ‘Detection of a SARS-CoV-2 Variant of Concern in South Africa’, Nature, 592: 438–43. [DOI] [PubMed] [Google Scholar]
- ——— et al. (2021b) ‘Detection of a SARS-CoV-2 Variant of Concern in South Africa’, Nature, 592: 438–43. [DOI] [PubMed] [Google Scholar]
- Tian F. et al. (2021) ‘N501Y Mutation of Spike Protein in SARS-CoV-2 Strengthens Its Binding to Receptor ACE2’, eLife, 10.doi: 10.7554/eLife.69091 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vaidyanathan G. (2021) ‘Coronavirus Variants are Spreading in India - What Scientists Know So Far’, Nature, 593: 7859.doi: 10.1038/d41586-021-01274-7 [DOI] [PubMed] [Google Scholar]
- Viana R. et al. (2022) ‘Rapid Epidemic Expansion of the SARS-CoV-2 Omicron Variant in Southern Africa’, Nature.doi: 10.1038/d41586-021-03832-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Voloch C. M. et al. (2020) ‘Intra-Host Evolution during SARS-CoV-2 Persistent Infection’, medRxiv. [Google Scholar]
- Volz E. et al. (2021) ‘Assessing Transmissibility of SARS-CoV-2 Lineage B.1.1.7 in England’, Nature, 593: 266–9. [DOI] [PubMed] [Google Scholar]
- Weigang S. et al. (2021) ‘Within-Host Evolution of SARS-CoV-2 in an Immunosuppressed COVID-19 Patient as a Source of Immune Escape Variants’, Nature Communications, 12: 1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williamson M. K. et al. (2021) ‘Chronic SARS-CoV-2 Infection and Viral Evolution in a Hypogammaglobulinaemic Individual’, medRxiv. [Google Scholar]
- World Health Organization . (2021) Classification of Omicron (B.1.1.529): SARS-CoV-2 Variant of Concern. <https://www.who.int/news/item/26-11-2021-classification-of-omicron-(b.1.1.529)-sars-cov-2-variant-of-concern> accessed 30 Nov 2021.
- Yen H.-L. et al. HKU-SPH study team. (2022) ‘Transmission of SARS-CoV-2 delta variant (AY.127) from pet hamsters to humans, leading to onward human-to-human transmission: a case study’, Lancet, 399: 1070–1078. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
UK genome sequences used were generated by the COG-UK (https://www.cogconsortium.uk/). Non-UK data were from GISAID (gisaid.org), and an acknowledgement table for the sequences used can be found in the Supplementary material. Short-read data for the two putative intermediate sequences can be found on the NCBI Short Read Archive using accession numbers ERX4738666 and ERX4562166 for MILK-B154B6 and CAMC-946506, respectively. XML for evolutionary and growth rate analyses (with alignments removed for data protection reasons) can be found in the Supplementary material.