

Abstract
The Microviridae family represents one of the major clades of single-stranded DNA (ssDNA) phages. Their cultivated members are lytic and infect Proteobacteria, Bacteroidetes, and Chlamydiae. Prophages have been predicted in the genomes from Bacteroidales, Hyphomicrobiales, and Enterobacteriaceae and cluster within the ‘Alpavirinae’, ‘Amoyvirinae’, and Gokushovirinae. We have isolated ‘Ascunsovirus oldenburgi’ ICBM5, a novel phage distantly related to known Microviridae. It infects Sulfitobacter dubius SH24-1b and uses both a lytic and a carrier-state life strategy. Using ICBM5 proteins as a query, we uncovered in publicly available resources sixty-five new Microviridae prophages and episomes in bacterial genomes and retrieved forty-seven environmental viral genomes (EVGs) from various viromes. Genome clustering based on protein content and phylogenetic analysis showed that ICBM5, together with Rhizobium phages, new prophages, episomes, and EVGs cluster within two new phylogenetic clades, here tentatively assigned the rank of subfamily and named ‘Tainavirinae’ and ‘Occultatumvirinae’. They both infect Rhodobacterales. Occultatumviruses also infect Hyphomicrobiales, including nitrogen-fixing endosymbionts from cosmopolitan legumes. A biogeographical assessment showed that tainaviruses and occultatumviruses are spread worldwide, in terrestrial and marine environments. The new phage isolated here sheds light onto new and diverse branches of the Microviridae tree, suggesting that much of the ssDNA phage diversity remains in the dark.
Keywords: ssDNA phages, Microviridae, Tainavirinae, Occultatumvirinae, Ascunsovirus oldenburgi ICBM5, prophages, phage carrier-state
Introduction
Viruses infecting bacteria and archaea are highly abundant in the environment. For example, in the oceans, viruses outnumber their hosts by an order of magnitude (Bergh et al. 1989; Wommack and Colwell 2000). Through microbial cell lysis, modification of the host metabolism, and horizontal gene transfer, viruses emerge as major drivers of marine biogeochemical cycles (Suttle 2005; Roux et al. 2016; Touchon, Bernheim, and Rocha 2016).
Single-stranded DNA (ssDNA) phages have been discovered quite early in the history of virology (Sertic and Boulgakov 1935; Loeb 1960) and have been extensively used as model systems and molecular biology tools (Sanger, Nicklen, and Coulson 1977; reviewed in (Székely and Breitbart 2016)). Nevertheless, most of the phages known to date, either cultivated or environmental, have double-stranded DNA (dsDNA) genomes.
A recent overhaul of the viral taxonomy (Koonin et al. 2020) has placed most ssDNA viruses, including filamentous and icosahedral phages, in the realm of Monodnaviria. But not all ssDNA phages are classified within the Monodnaviria. The Obscuriviridae family, with representatives infecting marine Cellulophaga species, is as yet unassigned to any kingdom (Bartlau et al. 2021). Furthermore, the Cellulophaga phage phi48:2 (Holmfeldt et al. 2013) has a protein content completely different from any of the above-mentioned phages and remains so far unclassified (Bartlau et al. 2021). Within Monodnaviria, filamentous phages are part of the Loebvirae kingdom. They have long, filamentous capsids, a circular ssDNA genome of 4.5–15 kb, and a chronic life cycle (Rakonjac, Bennet, and Spagnuolo 2011; Hay and Lithgow 2019). Inovirus-like prophages have been predicted bioinformatically in a broad range of bacterial and archaeal phyla (Roux et al. 2019).
Microviridae is the other phage family in Monodnaviria, and it was recently placed in the Sangervirae kingdom. This family is very common and diverse. Its members are found in most ecosystems and are known to infect very different bacterial hosts. Its phages have small icosahedral capsids (∼30 nm in diameter) and a circular ssDNA of 4.4–6.3 kb, and all cultivated representatives are strictly lytic (Doore and Fane 2016). Viruses in the Microviridae family are further classified according to their genome composition and capsid structure into two subfamilies currently recognized by the International Committee on Taxonomy of Viruses (ICTV), the Bullavirinae and the Gokushovirinae. The Bullavirinae (former Microvirinae) infect enterobacteria. Gokushoviruses infect obligate parasitic bacteria, such as Spiroplasma, Chlamydia, and Bdellovibrio (Chipman et al. 1998; Brentlinger et al. 2002; Everson et al. 2002). A distinguishing feature of the gokushoviruses is the mushroom-like protrusion within their major capsid protein (MCP) (Chipman et al. 1998).
Due to their small and circular genomes, hundreds of Microviridae-complete genomes were assembled in metagenomic studies from diverse environments: in marine habitats (Angly et al. 2006; Tucker et al. 2011; Labonté and Suttle 2013; Székely and Breitbart 2016), freshwater habitats (López-Bueno et al. 2009; Roux et al. 2012), human gut or feces (Roux et al. 2012), stromatolites (Desnues et al. 2008), dragonflies (Rosario et al. 2012), and sewage and sediments (Hopkins et al. 2014; Quaiser et al. 2015). These environmental microviruses greatly improve our understanding of the diversity of this phage family. First, the well-studied PhiX-like phages are rare in nature, and only a few genomes were detected to form a group, named pequeñoviruses, related to the Bullavirinae (Bryson et al. 2015; Doore and Fane 2016). Second, about half of the new genomes were affiliated to the other known subfamily, the Gokushovirinae. Finally, potential new subfamilies were also identified in several studies, for example, ‘Alpavirinae’, ‘Pichovirinae’, ‘Aravirinae’ and ‘Stokavirinae’ (Krupovic and Forterre 2011; Roux et al. 2012; Quaiser et al. 2015).
Recently cultivated ssDNA phages, infecting the marine bacteria Citromicrobium bathyomarinum RCC1878, a Sphingomonadaceae, and Ruegeria pomeroyi DSS-3, a Rhodobacteraceae, reveal further diversity of the Microviridae (Zheng et al. 2018; Zhan and Chen 2019a). The Citromicrobium phage was suggested to belong to a new subfamily in the Microviridae—the ‘Amoyvirinae’ (Zheng et al. 2018), whereas the two ssDNA Ruegeria phages vB_RpoMi-Mini and vB_RpoMi-V15 are considered as unclassified Microviridae (Zhan and Chen 2019a).
For a long time, Microviridae were believed to be strictly lytic and incapable of lysogeny (Fane et al. 2006), until prophages were predicted bioinformatically in the genomes of Bacteroidetes (Krupovic and Forterre 2011). Further studies predicted Microviridae-like prophages in other Bacteroidetes (Roux et al. 2012; Holmfeldt et al. 2013; Quaiser et al. 2015), in a Caenibius tardaugens strain, an alphaproteobacterium (Zheng et al. 2018), and in Enterobacteriaceae (Kirchberger and Ochman 2020). Even more, the ability of a gokushovirus prophage to form viable virus particles was recently demonstrated by leveraging molecular cloning techniques (Kirchberger and Ochman 2020). Lacking integrases, these phages integrate into the host genome using its chromosome dimer resolution system (Krupovic and Forterre 2011; Kirchberger and Ochman 2020).
In our laboratory, we have established a large collection of marine phage isolates from the North Sea, infecting environmentally relevant heterotrophic bacteria belonging to the Roseobacter group. Through this work, we have screened for the presence of ssDNA phages, based on the hypothesis that the use of new hosts for phage isolation could reveal new Microviridae diversity. One of our phage isolates infecting Sulfitobacter dubius SH24-1b and named ICBM5 was a lytic, icosahedral ssDNA phage, distantly related to known Microviridae. Further, we wanted to know how does ICBM5 and its relatives compare with other Microviridae in terms of lifestyle, phylogenetic classification, integration in bacterial genomes, infected hosts, and spread in the environment.
Materials and methods
Growth media
Marine broth (MB) was used both for the liquid cultures and for the plaque and spot assays. This media had the following recipe: 5.0 g/l peptone, 1.0 g/l yeast extract, 0.1 g/l C6H8FeO7, 12.6 g/l MgCl2 × 6H2O, 3.24 g/l Na2SO4, 19.45 g/l NaCl, 2.38 g/l CaCl2 × 2H2O, 0.55 g/l KCl, 0.16 g/l NaHCO3, 0.01 g/l Na2HPO4 × 2H2O, 0.08 g/l KBr, 0.034 g/l SrCl2 × 6H2O, 0.022 g/l H3BO3, 0.004 g/l Na2SiO3 × 3H2O, 0.0024 g/l NaF, and 0.0016 g/l NH4NO3. After autoclavation, the media was completed by adding 1 ml/l of a multivitamin solution (Balch et al. 1979). Artificial sea water (ASW) base medium was used for plaque assays or one-step infection experiments. This medium had the following recipe: 24.32 g/l NaCl, 10 g/l MgCl2 × 6H2O, 1.5 g/l CaCl2 × 6H2O, 0.66 g/l KCl, 4 g/l Na2SO4, 2.38 g/l (4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid) (HEPES), 1 ml KBr (0.84 M), 1 ml H3BO3 (0.4 M), 1 ml SrCl2 (0.15 M), 1 ml NH4Cl (0.4 M), 1 ml KH2PO4 (0.04 M), and 1 ml NaF (0.07 M).
Isolation of the phage ICBM5
Phage ICBM5 was isolated from the coastal North Sea using a phage enrichment procedure, followed by plaque picking and purification. For this purpose, surface seawater was collected in June 2015 from the shoreline near Neuharlingersiel (53°42’09.8”N 7°41’58.9”E) during high tide, transported to the lab on ice, and then filtered through a 0.2-µm filter (Rotilabo-syringe filters, Carl Roth). A phage enrichment was set up by mixing nine parts of freshly filtered seawater with one part of 10× MB and adding an inoculum of exponentially growing S. dubius SH24-1b (Hahnke et al. 2013). After overnight incubation at 20°C and 100 rpm, cells and debris were removed from the enrichment by centrifugation (15 min, 4000 × g, 20°C) and 0.2-µm filtration of the supernatant. To test for the presence of phages, 20 µl of filtrate were spotted on a lawn of S. dubius SH24-1b. The clearing zone was then collected, passed through a 0.2-µm filter to remove cells, and used further in plaque assays, to obtain single plaques. For this purpose, serial dilutions (100, 10–1, etc.) were prepared from the phage fractions by mixing with MB medium. Further, 100 µl of phage dilution were mixed with 280 µl of exponentially growing host culture (optical density (OD) = 0.2–0.3) and incubated for 15 min on ice. The mixture was transferred to 3 ml MB-soft agar (0.6 per cent low melting point Biozym Plaque GeneticPure agarose, Biozym, kept warm at 37°C), mixed by brief vortexing, and poured onto the bottom MB agar layer (1.8 per cent agar). After drying, the plates were incubated at 20°C. Phage plaques were picked and incubated overnight in 500 µl ASW base at 4°C. After centrifugation (10 min, 10,000 × g, 4°C), the supernatant was used for the next round of plaque assays. The procedure of plaque assay, picking of plaques, and re-plating was repeated three times to ensure the purity of the newly isolated phages.
Finally, one plaque was picked and used to infect a liquid culture of S. dubius SH24-1b. After overnight incubation at 20°C and 100 rpm, the phage lysate was obtained by removing cells and debris by centrifugation (15 min, 4000 × g, and 4°C) and 0.2-µm filtration. The phage lysate was stored at 4°C. For long-term storage, two types of glycerol stocks were prepared: (1) stock of free phage particles (one part phage fraction and one part MB media with 50 per cent glycerol) and (2) stock of infected host cells (one part infected cells—375 µl phage fraction added to 375 µl host culture, 15 min on ice for absorption—and one part MB media with 50 per cent glycerol).
Host range of the phage ICBM5
To determine the host range of ICBM5, ninety-four different strains covering the phylogenetic diversity of Rhodobacteraceae (see Table S2) were challenged with the purified ICBM5 phage by spot assay. For the spot assay, 280 µl of exponentially growing host culture (OD = 0.2–0.3) were mixed with 3 ml MB-soft agar and poured onto the bottom MB agar layer (1.8 per cent agar). After drying the top layer, 15 µl of phage fraction, obtained from a liquid infection as described above, were spotted in triplicates onto the top layer. For each strain, three plates were prepared and incubated at 15°C, 20°C, or 28°C. For those hosts showing clearing zones, infection by ICBM5 was further confirmed by plaque assays.
ICBM5 purification via CsCl gradient ultracentrifugation
To generate a high volume of lysate, we prepared sixty double-layer agar plates with confluent ICBM5 lysis. After plaque formation, 5 ml of Sodium chloride Magnesium sulphate (SM) buffer (100 mM NaCl, 8 mM MgSO4, and 50 mM Tris–HCl pH 7.4) were added to each plate, followed by incubation at 4°C for 6 h. The phage containing buffer was then collected and centrifuged for 15 min at 4,000 × g and 4°C, to remove cells and cell debris. Then, phages were precipitated by adding polyethylene glycol (PEG) (Promega) (final concentration 10 per cent) and NaCl (final concentration 0.6 mM) and incubating at 4°C for 2 h. After centrifuging for 2 h at 7197 × g and 4°C, the phage pellet was resuspended in 500 µl SM buffer (100 mM NaCl, 8 mM MgSO4, and 50 mM Tris–HCl pH 7.4), followed by 30-min incubation at 4°C.
Further phage concentration and purification were done by cesium chloride (CsCl) gradient ultracentrifugation. A density gradient was set up by layering from bottom up: 1.5 ml of 1.65 g/ml CsCl, 2 ml of 1.5 g/ml CsCl, 2 ml of 1.4 g/ml CsCl, and 1 ml of 1.2 g/ml CsCl. The PEG concentrated phage fraction was added on top, followed by ultracentrifugation for 4 h at 20°C and 25,000 rpm (Beckman, SW 41 Ti). Afterward, the visible band corresponding to the phages was collected with a syringe and needle through the side wall of the ultracentrifuge tube (∼500 µl). Removal of CsCl was done by dialysis in Slide-A-Lyzer G2 Dialysis Cassettes 10K MWCO (Thermo Fisher Scientific) against ASW base, for a total of 21 h, with buffer exchange after 3 h and 18 h. The selected phage fraction was tested for lysis by spot assay.
Transmission electron microscopy of phage ICBM5
To prepare for transmission electron microscopy (TEM), 30 µl of CsCl-purified ICBM5 stock were pipetted on top of a carbon-coated grid (Formvar 162, 200 mesh) and phages were allowed to absorb for 3 min. This was followed by staining with 30 µl 2 per cent uranyl acetate for 45 s and gentle removal of the liquid with filter paper. After air-drying for 15 min, the grids were visualized with the transmission electron microscope Zeiss EM902A. Images were documented with the Proscan High-Speed Slow Scan Charge Coupled Device (SSCCD) camera and analyzed using the software ImageSP viewer (Version 1.2.5.16). Negatively stained phages were used for capsid size measurements.
Testing the ssDNA nature of the ICBM5 phage genome
Phage genomic DNA was extracted from a CsCl-concentrated phage stock by mixing with the same amount of phenol:chloroform:isoamyl (Roth) solution and then gently inverting and centrifuging for 15 min, at 12,000 × g and 4°C. The aqueous phase was then mixed with an equal amount of ice-cold absolute ethanol (Th.Geyer) and the DNA was precipitated at −80°C for 30 min. The DNA was pelleted by centrifugation (20 min, 12,000 × g and 4°C) and resuspended in nuclease-free water (Thermo Fisher Scientific). Afterward, the DNA was purified with the NucAway spin column kit (Thermo Fisher Scientific) and quantified using the Nanodrop 2000 spectrophotometer.
To determine the genomic architecture of ICBM5, the phage DNA was exposed to four different enzymes: S1 nuclease (Thermo Fisher Scientific), TURBO DNase (Thermo Fisher Scientific), Exonuclease VII (New England Biolabs), and Hind III (New England Biolabs). Exonuclease VII and S1 strictly target ssDNA, while TURBO DNase digests both ssDNA and dsDNA. Hind III targets only dsDNA. For each enzyme, a 50-µl reaction was set up, by adding 1 µl of enzyme, 1 µg of extracted phage DNA, corresponding reaction buffers, and water. The four reactions were incubated for 30 min at 37°C, followed by 10 min at 95°C, for enzyme inactivation. For visualization of the digestion products, 2 µl of digested DNA were mixed with 5 µl loading buffer (BlueJuice Gel Loading Buffer, Thermo Fisher Scientific) and loaded on a 0.9 per cent agarose gel. The gel was run for 30 min at 80 V and pre-stained with SYBR Gold (Thermo Fisher Scientific). The gel was analyzed with the FAS Digi Gel Documentation System (NIPPON Genetics Europe) and evaluated using the BioDocAnalyze software (Biometra GmbH).
Sequencing of Ascunsovirus oldenburgi ICBM5 via Illumina sequencing
The phage lysate from plates with confluent plaques was first concentrated using 15-ml Amicon ultracentrifugal filter columns (Merck Millipore), then 0.2 µm filtered to remove bacteria and cell debris, and finally purified on an OptiPrep density gradient (Sigma Aldrich). The gradient was set up by layering OptiPrep solutions in a concentration range from 10 per cent to 50 per cent, with an incremental step of 5 per cent. After allowing the gradient to settle for 2 h at room temperature, 1 ml of phage solution was added, followed by ultracentrifugation for 12 h, at 40,000 × g and 20°C (Beckman, SW 41 Ti). The gradient was divided into 1-ml fractions, which were then tested for the presence of phages by spot assays. The fraction with the highest concentration of ICBM5 was then washed and concentrated using 0.5-ml Amicon columns, during which the OptiPrep was replaced by SM buffer.
Extracellular DNA was removed by incubating the phage concentrate with 0.043 units/µl of TURBO DNase (Thermo Fisher Scientific) for 30 min at 37°C, followed by enzyme inactivation for 10 min at 75°C with 15 mM ethylenediaminetetraacetic acid (EDTA). Further, the phage DNA was extracted using the ChargeSwitch gDNA Mini Bacteria Kit (Thermo Fisher Scientific), according to the instructions manual, but without using lysozyme in the first step. The ICBM5 ssDNA genome was converted to dsDNA by using the REPLI-g Mini kit (Qiagen), following the manufacturer’s instructions. Throughout these procedures, the concentration and quality of the DNA were checked fluorometrically with Qubit 2.0 and the Qubit dsDNA HS Assay, spectrophotometrically with Nanodrop 2000 spectrophotometer, and visually by regular gel electrophoresis (0.7 per cent agarose gel, 50 V, SYBR Gold staining).
An Illumina shotgun library was prepared using the Nextera XT DNA Sample Preparation Kit (Illumina). To assess the quality and size of the library, the samples were run on an Agilent Bioanalyzer 2100 using an Agilent High Sensitivity DNA Kit as recommended by the manufacturer (Agilent Technologies). Library DNA concentration was determined using the Qubit dsDNA HS Assay Kit as recommended by the manufacturer (Life Technologies GmbH). Sequencing was performed on an MiSeq system with the reagent kit v3 with 600 cycles (Illumina) as recommended by the manufacturer, resulting in 785.119 paired-end reads.
Assembly and annotation of the ICBM5 phage genome
The Illumina raw reads were cleaned with BBDuk in two steps. In the first step, the adaptors were removed, using the following parameters for BBDuk: ‘ktrim=r k=21 mink=8 tbo tpe ftm=5 rcomp=t ordered t=8’. In the second step, any contaminating reads (from the host or from phiX174), as well as low-quality ends, were removed, using the following parameters for BBDuk: ‘k=31 rcomp=t hdist=1 qtrim=rl trimq=20, maq=20 minlen=30 ordered t=8’. Afterward, the cleaned reads were assembled with Tadpole (parameters ‘k=50 t=8’). Both BBDuk and Tadpole are part of the BBTools package (https://jgi.doe.gov/data-and-tools/bbtools/). After assembly, direct terminal repeats were detected at the end of the contig, indicating that the contig can be circularized and that the genome is complete. For further analyses, the genome was linearized and one of the repeats was removed. Open reading frames (ORFs) were predicted using the MetaGeneAnnotator (Noguchi, Taniguchi, and Itoh 2008) implemented in VirClust (Moraru 2021). A first ORF annotation was done by using Domain Enhanced Lookup Time Accelerated Basic Local Alignment Search Tool (DELTA-BLAST) to search for homologous proteins in the non-redundant (NR) database (http://ncbi.nlm.nih.gov/). The ICBM5 phage genome is available in the National Center for Biotechnology Information (NCBI) GenBank database under the following accession number: OM782324. The sequences of the complete genome and the encoded proteins can also be found at the end of the supplementary information (SI) file 1.
Obtaining a Sulfitobacter dubius SH24-1b strain resistant to ICBM5
Turbid plaques were detected after ∼48 h of incubation in plaque assays of S. dubius SH24-1b and phage ICBM5. Several turbid plaques were picked, resuspended in 50 µl of MB medium, serially diluted to 10−2–10−5, and then plated on MB agar. After incubation at 20°C, single bacterial colonies were picked and transferred to new MB agar plates. The presence of ICBM5 in cultures derived from these single colonies was tested by polymerase chain reaction (PCR) with ICBM5-specific primers (Supplementary Table S1). One such ICBM5-positive culture was selected for further experiments and sequenced using two long-read technologies: PacBio and Nanopore.
Bacterial genome sequencing and assembly via PacBio sequencing
The original S. dubius SH24-1b strain and the ICBM5 carrier strain were genome sequenced using PacBio. Bacterial genomic DNA was extracted with the Genomic-tip 100/G kit (Qiagen). A SMRTbell™ (PacificBiosciences) template library was prepared according to the manufacturer’s ‘Procedure & Checklist - 20 kb Template Preparation Using BluePippin™ Size-Selection System’ protocol. Shortly, sheared genomic DNA was end-repaired and ligated to hairpin adapters applying components from the DNA/Polymerase Binding Kit P6 (Pacific Biosciences). BluePippin™ Size-Selection to 7 kb was performed as recommended by the manufacturer (Sage Science). single-molecule real-time sequencing (SMRT) sequencing was carried out on the PacBio RSII (Pacific Biosciences) taking 240-min movies, which resulted in 166,457 and 90,339 post-filtered reads with a mean read length of 14,035 bp and 14,014 bp, respectively. Illumina libraries were prepared with the Nextera XT DNA Sample Preparation Kit (Illumina) modified after (Baym et al. 2015), and paired-end sequencing was performed on the NextSeq 500 (PE75).
Long-read genome assembly was performed with the ‘RS_HGAP_Assembly.3’ protocol in SMRTPortal version 2.3.0. The assembled contigs were error-corrected by mapping of Illumina short reads using the Burrows–Wheeler Aligner (BWA 0.6.2) (Li and Durbin 2009) and subsequent variant and consensus calling using VarScan 2.3.6 (Koboldt et al. 2012). The final assembly was trimmed, circularized, and adjusted to the replication system as a start point (https://github.com/boykebunk/genomefinish) and checked via the mapping of Illumina reads (BWA) and PacBio reads (Bridgemapper). The genome was annotated with Prokka 1.13 (Seemann 2014) with subsequent manual curation of the replication systems. For the infected strain, PacBio reads were not only assembled but also mapped on the original genome including the phage ICBM5 genome. PacBio reads were also compared with BLASTN against the genome of phage ICBM5 but no hit was detected.
Genome sequencing and assembly of the ICBM5 carrier host strain via Nanopore
Phenol–chloroform-extracted DNA was used to prepare a sequencing library using the Rapid Barcoding Kit, according to the manufacturer’s instructions (Oxford Nanopore Technologies). The sequencing was performed on a MINION Flow Cell (R9.4.1), controlled by the MinIon software.
After sequencing, the quality control was performed using FastQC (https://www.bioinformatics.babraham.ac.uk/projects/fastqc/) (fastqc -t 16 $input‘01_reads.fastq’ -o $Step2_out—nano). Adapters were removed using PoreChop (https://github.com/rrwick/Porechop) (porechop -i $input‘01_reads.fastq’ -t 16 -v 2 -o $output‘03_reads_trimmed.fastq’ > $output‘03_porechop.log’). Assembly was performed using a set of tools from the pomoxis suite (https://github.com/nanoporetech/pomoxis). Minimap2 (minimap2 -x ava-ont -t $threads $output‘01_reads.fastq’ $output‘01_reads.fastq’ | gzip −1 > $mapping‘08_mapping.paf.gz’) and miniasm (miniasm -f $output‘01_reads.fastq’ $mapping‘08_mapping.paf.gz’ > $mapping‘08_miniasm_reads.gfa’) were used to map the .fastq files onto each other to find overlaps. Afterward, the .gfa file was converted to .fasta (awk ‘$1 ∼/S/ {print “>” $2”\ n”$3}’ $mapping‘08_miniasm_reads.gfa’ > $assembly‘09_miniasm_reads.fasta’). Assembly was done by minimap2 (minimap2 $assembly‘09_miniasm_reads.fasta’ $output‘01_reads.fastq’ > $assembly‘09_minimap_reads.paf’) and polishing was done by racon (racon -t $threads $output‘01_reads.fastq’ $assembly‘09_minimap_reads.paf’ $assembly‘09_miniasm_reads.fasta’ > $polished‘10_racon.fasta’). A last quality control was performed by quast (quast.py -t $threads -o $Step10_QC $polished‘10_racon.fasta’ $assembly‘09_miniasm_reads.fasta’) (Gurevich et al. 2013).
One-step growth experiment with phage ICBM5
Sulfitobacter dubius SH24-1b was grown in MB media for three consecutive generations, to ensure consistent growth. In the last generation, when the bacteria reached an OD of 0.3, an ICBM5 phage stock prepared via CsCl gradient ultracentrifugation was added to a final multiplicity of infection (MOI) of 6.5. Previously, the concentration of the phage stock was determined by performing plaque assays. The concentration of the bacterial cells was determined just before setting up the one-step infection experiment, by calculating the cell concentration from the OD600 measurements (see Supplementary Fig. S1). In parallel, a second flask was prepared where ASW base was added instead of phage stock, to serve as a negative control throughout the infection. After 20 min of incubation at 20°C, to allow phage adsorption, free phages were removed by centrifugation. The cells were resuspended in fresh media and incubated at 20°C and 100 rpm for 200 min. Through the infection experiment, samples were taken every 15 min. Plaque assays were used to quantify both the free and total (free and cell-bound) phages. The free fraction was obtained by filtering the infected culture through a 0.22-µm syringe filter. The samples for phage-targeted direct-geneFISH were fixed by adding paraformaldehyde (Electron Microscopy Sciences) to a final concentration of 4 per cent and incubating at room temperature (RT) for 1 h. Afterward, the cells were pelleted by centrifugation at 5,000 × g for 10 min, resuspended in 1× phosphate-buffered saline (PBS) (137 mM NaCl, 2.7 mM KCl, 8 mM Na2HPO4, and 2 mM KH2PO4) (Invitrogen) and again pelleted by centrifugation at 5,000 × g for 10 min. The supernatant was discarded and the cell pellet was resuspended in a 1:1 ratio in 1× PBS and absolute ethanol (Th. Geyer). The fixed cells were then stored at −20°C until further processing.
The plaque-forming units (PFU)-based burst size was determined from the one-step growth infection cycle experiment, using the following formula: (average free phages after lysis − average free phages before lysis)/average free phages average at T1.
ICBM5-targeted direct-geneFISH
To detect intracellular phages, we have used a modified version of the direct-geneFISH protocol (Barrero-Canosa et al. 2017), in which we used ICBM5 genome-specific probes. This protocol was applied both on the ICBM5-infected culture and on the control, not infected culture.
Design and synthesis of ICBM5-specific genome probes
To target the ICBM5 phage genome, eight dsDNA polynucleotides (Supplementary Fig. S2 and Supplementary Table S3) were designed using geneProber (gene-prober.icbm.de/). The corresponding dsDNA polynucleotides were chemically synthesized (Integrated DNA Technologies) and further labeled with Alexa 594, as described previously (Barrero-Canosa and Moraru 2021).
Immobilization on solid support
The cells from the one-step growth experiment were immobilized by spotting 20 µl of fixed cell suspension on SuperFrost Plus glass slides (Electron Microscopy Sciences, USA), on which silicone isolators (Grace Bio-Lab, USA) were placed to create wells. The cells were dried at 37°C, then washed for 1 min in 0.22-µm-filtered, deionized, and autoclaved water, and 10–30 s in absolute ethanol (Th. Geyer, Germany).
Permeabilization and RNA removal
The samples were overlaid with a solution containing 0.5 mg/ml lysozyme (Sigma Aldrich, USA), RNase Cocktail (500 U/ml RNase A, 20.000 U/ml RNase T1) (Thermo Fisher, USA), 0.05 M EDTA pH 8.0, and 0.1 M Tris–HCl pH 8.0. Then, the samples were incubated for 30 min at 37°C, followed by washing twice for 5 min in 1× PBS, 1 min in MQ water, and 10–30 s in absolute ethanol, and finally, by air-drying.
Denaturation and hybridization
A hybridization buffer containing 45 per cent formamide, 5× SSC (750 mM NaCl and 0.075 mM sodium citrate), 20 per cent dextran sulfate, 0.1 per cent sodium dodecyl sulfate (SDS), 20 mM EDTA, 0.25 mg/ml sheared salmon sperm DNA, 0.25 mg/ml yeast RNA, and 1 per cent blocking reagent for nucleic acids (Roche, Switzerland) was used. To this buffer, the ICBM5 genome probes were added at a final concentration of 30 pg/µl for each polynucleotide probe. The samples were covered with the probe-hybridization buffer mixture and denatured for 40 min at 85°C, followed by 2 h of hybridization at 46°C. After hybridization, the samples were quickly rinsed in washing buffer I (2× SSC, 0.1 per cent SDS) at room temperature and washed in washing buffer II (0.1× SSC, 0.1 per cent SDS) for 30 min at 48°C. Finally, the samples were washed for 15 min in 1× PBS and 1 min in water and air-dried.
Counterstaining and embedding for microscopy
The samples were counterstained using 5 ng/ml 4′,6-diamidino-2-phenylindole (DAPI) dissolved in SlowFade Gold (Invitrogen, USA) and covered with a #1.5 high-precision coverslip (Marienfeld, Germany).
Microscopy
Samples generated from phageFISH were visualized using an Axio Imager 0.72 m fluorescent microscope (Carl Zeiss, Germany), with the help of its associated software AxioVision (version 4.8.2.0) (Carl Zeiss, Microlmaging GmbH, 2006-2010). For each field of view, a set of images was created using different exposure times for the two fluorescent channels, DAPI and Alexa594. For DAPI, the following filter set was used: 365 excitation, 445/50 emission, and 395 Beam Splitter. For Alexa594, the following filter set was used: 562/40 excitation, 624/40 emission, and 593 Beam Splitter. The exposure times were 40, 80, and 150 ms for DAPI and 80, 200, 600, 1200, 3000, and 5000 ms for Alexa594. Each exposure time was saved individually as TIFF for further image analysis.
Image analysis
Image analysis was performed using CellProfiler v. 3.1.9 (McQuin et al. 2018) and had two purposes: (1) to quantify the fraction of infected cells and (2) to quantify the number of phage genomes per cell. Cell Profiler was used to define a semi-automatic workflow for image analysis. First, for every microscopic field of view, images from two different fluorescent channels (DAPI for cell counterstaining and Alexa594 for phage detection) were imported. The respective images were previously acquired with exposure times, allowing the detection of single, weak phage signals but avoiding the overexposure of strong phage signals. Second, the contours of bacterial cells were automatically identified from the DAPI channel and manually curated. Cell clumps were avoided, due to the difficulties in identifying cell borders. In parallel, the contours of 100 small, dot-like phage signals were identified in the first sampling time point after infection. These were considered to approximate a single ICBM5 genome. Third, for each corresponding Alexa594 image a mean background level (gray value/pixel) was measured in cell-free image areas and further subtracted from the entire Alexa 594 image. Fourth, the mean Alexa 594 intensity (gray value/pixel) and area were determined for each cell or phage contour and exported. Fifth, an additional background correction was performed by averaging the Alexa 594 gray values of the cells in the negative control images (samples without an infection) and substracting these values from the average intensity of the infected cells. Finally, the total Alexa 594 intensity (gray value) was calculated for each cell, by multiplying the average corrected intensity value by the cell area. The amount of per-cell phage genome copies was calculated by dividing the per-cell total Alexa 594 intensity by the total intensity of a single phage. For each time point, a total of 100 phage-positive cells were quantified.
The fraction of infected cells was calculated by manually counting from the images the cells with visible Alexa 594 signals and then reporting this number to the total cell number, as defined from the DAPI images. For each time point, a total of 500 cells were investigated.
Detection and curation of ICBM5-like regions in bacterial genomes
Proteins from phage ICBM5 were used to query the NR database from NCBI, using DELTA-BLAST, with two iterations. Proteins detected as similar were downloaded in GenBank format, imported into Geneious v 9.1.5 (http://www.geneious.com (Kearse et al. 2012)), and identified as part of a viral or bacterial genome based on their organism name and taxonomy (see the next section for the analysis of viral sequences). Bacterial strains having hits with at least two different ICBM5 phage proteins were considered to potentially harbor ICBM5-like prophages and were selected for further analysis.
To determine the prophage borders, close relatives (same species) of each bacterial strains were searched in the GenBank sequence database, in order to have very similar bacterial genomes both with and without the prophage. Similar bacterial genomes were aligned using MAUVE (Darling et al. 2004), and prophage regions were precisely identified from these alignments. We refer to these prophages as ‘sure border prophages’ (SBPs). For the remaining bacterial strains, referred as ‘unsure border prophages’ (UBPs), a larger genomic region surrounding the phage-like genes was selected and prophage regions were further refined. First, proteins of these UBPs were predicted using MetaGeneAnnotator and clustered with proteins of ICBM5, of other publicly available ssDNA phages and also of the SBPs. To this end, an all against all BLASTp (e-value < 1e − 5 and bitscore > 50) was performed and proteins were clustered using the mcl program, with the parameters ‘-I 2 --abc’. Using the defined protein clusters (PCs), the following steps were performed to better identify the borders of UBPs: (1) the UBP genes were judged as phage genes if they were annotated as MCP, replication initiation protein, lysis, or pilot protein, or if they grouped in PCs with proteins from the SBPs or the reference ssDNA phages; (2) if, on a UBP, genes encoding hypothetical proteins were located between the previously determined phage genes, they were kept and labeled as phage genes; and (3) if, on a UPB, genes not classified in any of the above categories were located at the periphery of a region encoding phage genes, they were considered of bacterial origin and the UBPs were shortened correspondingly. Those genes that occurred in the opposite direction on the contig, in comparison to the rest of the genes, were removed from the dataset as well as those genes at the border that were unique or could not be identified. If a ribosomal binding site could be determined for the first gene, this has been used as the start point for the prophage region. Otherwise, the start of the prophage region was determined at the start codon of the first gene.
Detection of ICBM5-like regions in viral metagenomes
A set of 2,944 publicly available viromes (see SI file 2) were downloaded as raw reads that were first cleaned by removing potential adapters with cutadapt (Martin 2011) and trimmed using Trimmomatic (Bolger, Lohse, and Usadel 2014). Each dataset was then individually assembled into contigs using Newbler 2.6 (454; Life Sciences, Branford, CT, USA), IDBA_UD (Peng et al. 2012), or megahit (Li et al. 2015) with default parameters, depending on the sequencing technology. Details about these viromes (database source, sequencing technology, publication, etc.) can be found in SI file 2. In order to detect Microviridae similar to ICBM5, circular contigs between 3 and 8 kb were extracted and their proteins were predicted using prodigal (Hyatt et al. 2012). Using MMseqs (Steinegger and Soedin 2017) with a threshold of 50 on the bitscore, these proteins were compared to the MCP sequence of ICBM5 and sixteen circular contigs were found to have a protein similar to ICBM5 MCP (Steinegger et al. 2019). In addition to these contigs found in the above viromes, we also added the environmental viral genomes (EVGs) retrieved from NCBI using DELTA-BLAST, during our search for ICBM5-like regions in the NR database described in the previous section.
Clustering Microviridae proteins and genomes
The dataset used to classify phage ICBM5 was assembled by combining reference Microviridae genomes from publicly available sequence databases (NCBI), the ICBM5 phage genome, the newly detected ICBM5-like regions in bacterial genomes, as well as ICBM5-related EVGs. Here, we define as reference those microvirus genomes, be it from phage isolates or EVGs, that have been previously assigned to different Microviridae subfamilies. In addition, we have included in the analysis phage genomes from the Obscuriviridae family (Bartlau et al. 2021).
The genomes from all ssDNA phages in the above dataset were clustered hierarchically with VirClust, based on their protein super-supercluster content (PSSC; Moraru 2021). Shortly, the genomes were translated into proteins, and the proteins were subjected to three clustering steps. In the first step, which grouped the proteins into PCs, the following parameters were used: BLASTp-based similarity (e-value > 0.00001, bitscore ≥ 30 and coverage > 70) and clustering based on log e-values. In the second step, which grouped PCs into protein super-clusters (PSCs), the following parameters were used: hidden Markov model (HMM)-based similarity (probability ≥ 90, coverage ≥ 60, no threshold on alignment length) and clustering based on log e-values. In the third step, which grouped PSCs into PSSCs, the following parameters were used: HMM-based similarity (probability ≥ 90, no threshold on coverage and alignment length). We refer from here on to PSSCs as ‘protein clusters’. Afterward, the phage genomes were clustered hierarchically with a clustering distance of 0.9.
To annotate the resulting PCs, we compared the individual protein sequences to various databases using VirClust. The NR database from NCBI was queried using BLASTp 2.6.0+, and the InterPro database v 66.0 (Finn et al. 2017) was queried using Inter-ProScan 5.27-66.0 tool (Jones et al. 2014). The prokaryotic Viruses Orthologous Groups database (Grazziotin, Koonin, and Kristensen 2017), the Virus Orthologous Group database (Kiening et al. 2019), and the Prokaryotic virus Remote Homologous Groups database were searched using hhsearch (Steinegger et al. 2019). Finally, the efam database (Zayed et al. 2021) was searched using hmmscan (Eddy 2011). At the end, the annotations of each protein were manually curated and compared with the annotations of other proteins in the same PC. The annotation of each PC represented a consensus of the annotations of all proteins in the cluster (see SI file 3).
Phylogenetic analysis of the ssDNA phages based on their MCP and Rep proteins
The recognizable MCP and Rep proteins from all phages in the dataset were aligned using Clustal Omega (Sievers et al. 2011) and then concatenated. A maximum-likelihood phylogeny was computed based on RAxML v8.2.12 (Stamatakis 2014), using an automatic determination of the best protein model (option -m PROTGAMMAAUTO) and 100 bootstrap replicates. The resulting phylogenetic tree was further visualized and refined with interactive tree of life (iTOL) (Letunic and Bork 2021). This analysis did not include Obscuriviridae phages, because they have no recognizable MCP genes.
Phylogenetic analysis of all host 16S rRNA genes and species assignment for Sulfitobacter dubius SH24-1b
A neighbor-joining tree of the 16S ribosomal RNA (rRNA) gene sequences from all phage hosts for which we could find the 16S rRNA gene (see SI file 4 Table 3) in this study was constructed with the ARB software package (Ludwig et al. 2004). Tree calculation was performed using the reference dataset SSU Ref NR 111, with Jukes–Cantor correction, termini filter, and 1,000 bootstrap replicates. Members of the genus Acidobacterium served as an outgroup. For species assignment of S. dubius SH24-1b, the average nucleotide identity (ANI) value between SH24-1b and the S. dubius type strain DSM 16472T was calculated with FastANI (Jain et al. 2018) and the digital DNA–DNA hybridization (dDDH) value was calculated with genome-to-genome distance calculation (GGDC) (applying Formula 2) (Meier-Kolthoff et al. 2022).
Results
‘Ascunsovirus oldenburgi’ ICBM5—a novel Microviridae isolate
Phage ICBM5 was isolated from surface seawater, collected from the shoreline of the North Sea (53°42’09.8”N 7°41’58.9”E) during high tide in June 2015. The host, S. dubius SH24-1b, was isolated from a seawater sample taken on 12 May 2007 in the southern North Sea (54°42’N, 06°48’E) during a phytoplankton bloom (Hahnke et al. 2013). Its 16S rRNA has 99.8 per cent identity with S. dubius type strain DSM 16472T. According to its corresponding dDDH value of 70 per cent and ANI value of 96.9 per cent, the strain SH24-1b belongs to the species S. dubius. A host range assessment performed on almost 100 bacterial strains showed that ICBM5 has a narrow host range, infecting only its original host S. dubius SH24-1b and S. dubius DSM 16472T (SI file 1 Supplementary Table S2). On S. dubius SH24-1b, ICBM5 formed turbid plaques.
ICBM5 has an icosahedral capsid and no tail, as revealed by TEM of uranyl-acetate-stained samples (Fig. 1A). The capsid diameter measured 28.68 ± 1.95 nm (100 phages measured and three measurements per phage). Enzymatic digestion revealed that ICBM5 has an ssDNA genome (Fig. 1B). Sequencing and assembly resulted in a 5,581-base-long contig, circularly closed. Six protein-encoding genes were detected. Using BLASTp, only one ICBM5 protein was similar to proteins from previously classified Microviridae, namely the replication initiation protein (Rep). However, DELTA-BLAST, a remote homology tool, showed three more proteins distantly related to reference Microviridae: the MCP, a pilot protein, and a lysis protein (see Fig. 1C). The presence of these Microviridae core genes, alongside their genome characteristics and virion morphology, clearly indicates that ICBM5 is a new member of this family. However, a first hint that ICBM5 is distantly related to known microviruses comes from the detection of MCP similarity to reference Microviridae only by a remote homology tool, although this protein is generally well conserved and often used to build Microviridae phylogenies. We consider thus that ICBM5 is the representative of a new phage species, which we tentatively named here ‘Ascunsovirus oldenburgi’ ICBM5 (from the Romanian word ‘ascuns’, meaning ‘hidden’, and the town Oldenburg), using the binomial nomenclature recently adopted by ICTV.
Figure 1.
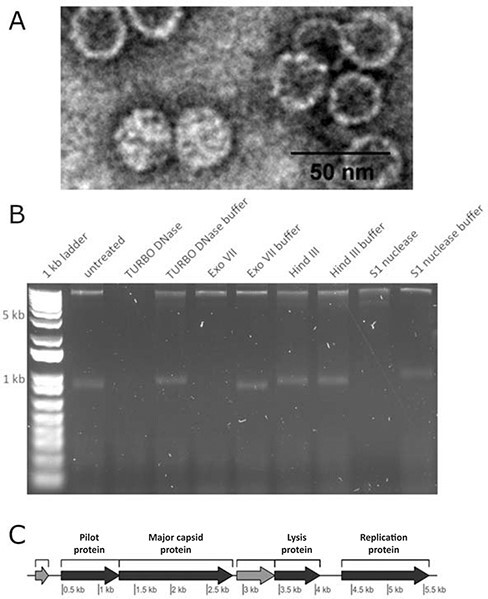
(A) ICBM5 morphology determined by TEM of uranyl-acetate-stained virions. (B) Agarose gel shows enzymatic digestion of ICBM5 ssDNA phage. The DNA was digested by TURBO DNase, Exo VII, and S1 nuclease, but was not affected by treating it via restriction enzyme Hind III, which only targets dsDNA, or the exclusion of nucleases (usage of only buffer). The 1 kb plus ladder was used to track the DNA migration. However, it was not used to infer the size of the ICBM5 genome, because the ladder comprises from linear dsDNA molecules, in contrast to the ICBM5 genome, which comprises a circular, ssDNA molecule. (C) Genome map of ICBM5. In dark gray—identified proteins, with labels on top of each gene. In light gray—hypothetical proteins.
‘Ascunsovirus oldenburgi’ ICBM5 has both a lytic and a carrier-state infection strategy on its Sulfitobacter dubius SH24-1b host
To characterize the infection cycle of the phage ICBM5, we performed one-step infection curves, in two separate experiments. The MOI was 6.5 in both experiments. Through the infections, samples were collected at different time points, in ∼15-min increments. For each time point, we quantified: (1) the free and total phages using plaque assays (see Fig. 2A), (2) the percentage of infected cells (see Fig. 2A), and (3) the variation of the amount of ICBM5 genomes per infected cell (see Fig. 2B for Experiment 1, Fig. 3 and SI file 1 Supplementary Fig. S3 for Experiment 2). The latter two measurements were obtained by using ICBM5-targeted direct-geneFISH, a single-cell method. We noticed a progressive increase in the per-cell ICBM5 genome numbers from ∼50 min post-infection (p.i.) until ∼110 min p.i. At 50 min p.i., the median number of per-cell ICBM5 genomes was 6.5× and 2× higher than at 35 min p.i., for Experiment 1 and Experiment 2, respectively. At 110 min p.i., it was 61× and 81× higher, for Experiment 1 and Experiment 2, respectively. Therefore, ICBM5 was replicating its genome at least as early as 50 min p.i. Cell lysis events were visually observed between 110 min and 140 min p.i. (see Fig. 3) in both experiments. In agreement, the free phage particles increased in numbers starting with ∼110 min p.i. This corresponded with the decline of the cell population with a high amount of ICBM5 genomes and the emergence of a cell population with a low amount of ICBM5 genomes, as indicated by the progressive drop in the median number of ICBM5 genomes per cell (see Fig. 2B). The population with a low amount of ICBM5 genomes was stably maintained from 155 min to 185 min p.i., when the experiments ended. Therefore, the major lysis event took place starting at 110 min p.i. and newly released virions infected new cells, leading toward a second wave of infection. Overall, these results show that ICBM5 can undergo a complete lytic cycle on S. dubius SH24-1b.
Figure 2.
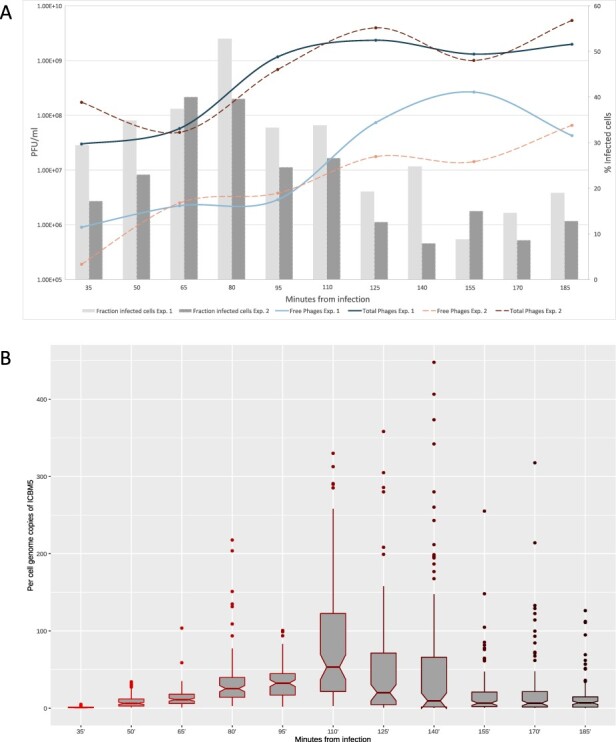
ICBM5—S. dubius SH24-1b infection dynamics. A. The fraction of ICBM5-infected cells (bars) and the abundance of free (extracellular) and total (intra- and extracellular) phages, given in PFU/ml (lines). The fraction of infected cells was calculated from the proportion of cells showing a phage signal, after subtraction of the false-positive signals detected in the negative control cultures. B. The variation of the per-cell ICBM5 genome copies through the infection, as calculated from measuring the phage signal intensities using direct-geneFISH. The box plot borders represent the first and third quartiles, and the middle line represents the second quartile. The whiskers extend from the first or third quartiles with 1.5 × IQR (distance between the first and third quartiles). The data beyond the whiskers are plotted individually. The plot was generated using the ggplot2 R package (Hadley 2016). The different shades of red for the box plots are just indicating the progress of the infection time, from bright red at the beginning of the experiment, to dark red at the end of the experiment.
Figure 3.
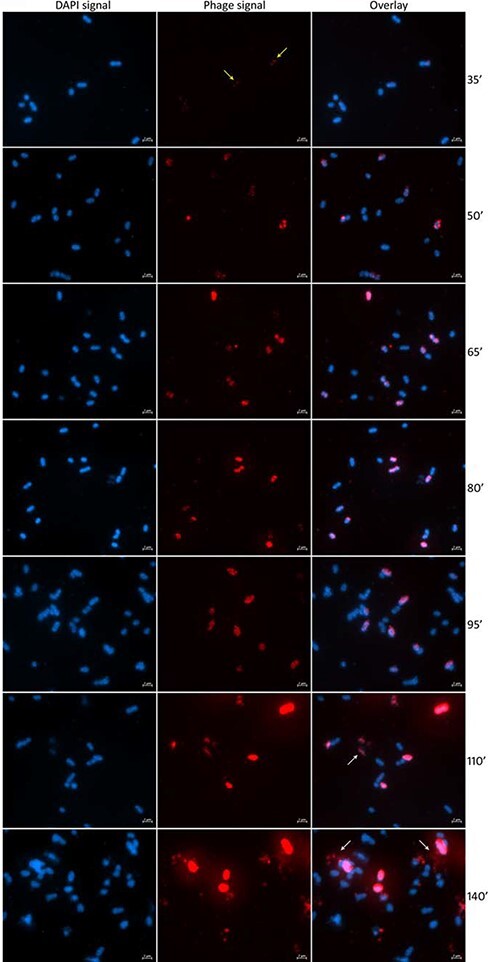
Visualization of the infection of S. dubius SH24-1b by ICBM5 using direct-geneFISH. First column: cells as visualized by DAPI staining. Second column: intracellular phages, as visualized by ICBM5-genome-targeted probes. Third column: overlay of DAPI and phage signal. The phage signal increases progressively through the infection. The very small dots in the first time point (yellow arrows) represent early infections, with just a few copies per cell. Larger, cell-wide signals in the following time points represent phages in the replication and maturation phases. Finally, at 110ʹ and 140ʹ, cell lysis events can be noticed (white arrows).
The fraction of ICBM5-infected cells reached a maximum at 80 min p.i., after which it progressively decreased (see Fig. 2A). This, together with the ability of ICBM5 to form turbid plaques, suggested the emergence of a resistant S. dubius SH24-1b sub-population. To test if the resistance was conferred by the integration of ICBM5 as prophage in the host cells, we collected surviving bacterial cells from turbid plaques and plated them to obtain single colonies that we screened by PCR for the presence of the phage ICBM5. When ICBM5-positive cultures derived from the phage-positive colonies were challenged with ICBM5 in a spot assay, no clearing zones were formed. Therefore, the new cultures were resistant to ICBM5. Using the Nanopore long-read sequencing technology on the entire genome, without any size-exclusion during library preparation, the phage ICBM5 was detected as an independent, circular contig. Using PacBio long-read technology, using a 7 kb size selection threshold for library preparation, ICBM5-specific sequences were not detected. Therefore, no evidence of integration in the bacterial chromosome was found; that is, no hybrid ICBM5–S. dubius SH24-1b reads were present. ICBM5-targeted direct-geneFISH on this resistant culture showed that ICBM5 was present in ∼4.5 per cent of the cells. The geneFISH signal varied among cells. Some cells had small, dot-like signals, a characteristic of a low number of ICBM5 genome copies. Other cells had larger, diffuse signals, indicating the presence of a higher number of ICBM5 genomes. No cell lysis events were noticed. Together, these results showed that ICBM5 does not undergo lysogeny as integrated prophage in the tested conditions. However, ICBM5 can survive and replicate in a sub-population of sensitive S. dubius SH24-1b cells, which co-exists in parallel with a dominant ICBM5 resistant sub-population. This is indicative of a carrier-state infection strategy.
ICBM5-related genomes are widespread within bacterial genomes, both as prophages and episomes, and in environmental viromes
To better understand the spread of ICBM5-related phages in bacterial hosts and environmental samples, as well as their phylogenetic classification, we searched for similar genomes in two publicly available data sources: bacterial genomic data and viral metagenomes.
First, ICBM5 proteins were used to find potential prophages within prokaryotic genomes. Out of the seventy-two Microviridae-like genomic regions found in bacterial genomes, seven have been previously described (Krupovic and Forterre 2011; Quaiser et al. 2015; Zheng et al. 2018) and sixty-five are new (see Fig. 4 and see Supplementary SI file 4 Table 1 and SI file 5). For thirty-nine of them, we were able to determine the borders, by comparing them with related bacterial strains free from these regions. For the rest, we narrowed down the borders by keeping only proteins with a clear phage origin (see Materials and methods). The length of these regions varied between 3.3 and 6.6 kb for clear border regions and between 3.5 and 8.2 kb for unclear border regions. The majority of Microviridae-like genomic regions occurred in bacteria from Alphaproteobacteria (53.5 per cent), followed by Bacteroidia (29.5 per cent) and Gammaproteobacteria (5.6 per cent). The rest was distributed among Bacilli, Clostridia, Erysipelotrichia, Negativicutes, Cyanophycea, and Flavobacteriia (see Fig. 5). Most of the new Microviridae-like genomic regions had a chromosomal localization and three were localized on large plasmids (0.1–1.6 Mb, see SI file 4 Table 1), indicating that they are bona fide prophages. Seven genomic regions consisted of small contigs similar in size to Microviridae genomes, most likely representing episomes (see SI file 4 Table 3 and Fig. 4) from a carrier-state life strategy.
Figure 4.

Hierarchical clustering of the ssDNA (pro)-phage genomes, based on their PSSC. The annotations show the following: (1) hierarchical clustering tree and (2) distribution of the PCs (PSSCs) in each viral genome. PCs not shared with any other phage genomes in this dataset are not shown. The color encodes different protein annotations; (3) phage genome category; (4) the label given to each phage genome, consisting of accession numbers and names of the phage isolate or the environmental contig or of the bacterial host in which a (pro)-phage was predicted. For a high-resolution image, see SI extended Fig. 4.
Figure 5.
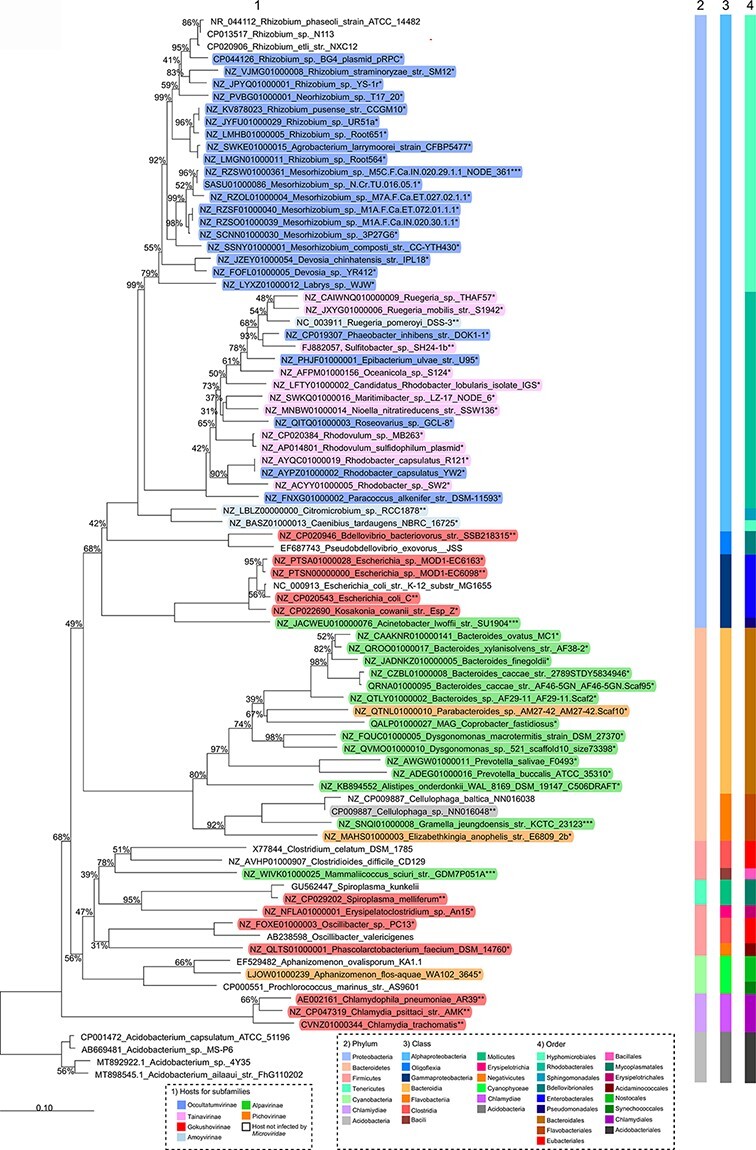
16S rRNA phylogenetic analysis of the (pro)-phage hosts. Neighbor-joining tree based on the 16S rRNA gene sequence similarity showing the position of S. dubius SH24-1b and other bacterial hosts for Microviridae-like (pro)-phages. The 16S rRNA gene was not detected for four bacterial strains in Fig. 4 and, therefore, not included in this tree. Bootstrap values are derived from 1,000 replicates. GenBank accession numbers are given as prefixes, followed by species and strain names. The bar represents ten substitutions per nucleotide position. The stars encode the following: * hosts of predicted prophages, integrated into chromosomes or plasmids; ** hosts of isolated phages; *** hosts of predicted episomes, represented by short contigs.
In addition to these potential prophages, we also searched for ICBM5-related genomes among EVGs sequenced from virions from environmental samples, which can be found in NR and public viromes. We have retrieved thirty-one environmental phage genomes from our search of the NR NCBI database, alongside twenty-three EVGs already affiliated to know microvirus subfamilies. Their size ranged between 4.2 and 6.6 kb. These NR sequences were generated from ten viromes associated with humans, animals, or plants (see Fig. 4 and SI file 6). In addition, we used the MCP from ICBM5 to screen 2,944 previously published viral metagenomes yet only available as raw reads and had to be newly assembled in this study. A total of fifteen circular contigs representing potential full-length genomes were retrieved in eight different viromes from fresh- or reclaimed water and soil (see Fig. 4 and SI file 2).
All newly found prophages, episomes, and EVGs were then pooled with ICBM5 and reference Microviridae genomes and compared in terms of gene content using VirClust. Their proteins are of course related and formed clusters with proteins from cultivated and uncultivated reference Microviridae. They shared no PCs with the Obscuriviridae, which are also ssDNA phages with icosahedral capsids. Most genomes newly detected here had the usual Microviridae proteins: pilot, MCP, lysis, and Rep proteins, with interspersed hypothetical proteins. Hierarchical clustering of the genomes based on their PC content resulted in thirteen major viral genome clusters (VGCs) (see Fig. 4). Each VGC had its own set of PCs, with few PCs being shared between the VGC. The phylogenetic analysis of the MCP and Rep proteins was generated to have a more precise idea of the relationships between these viruses and was coherent with the VirClust analysis, as the thirteen genome clusters mostly corresponded to major phylogenetic clades (see Figs 4 and 6).
Figure 6.
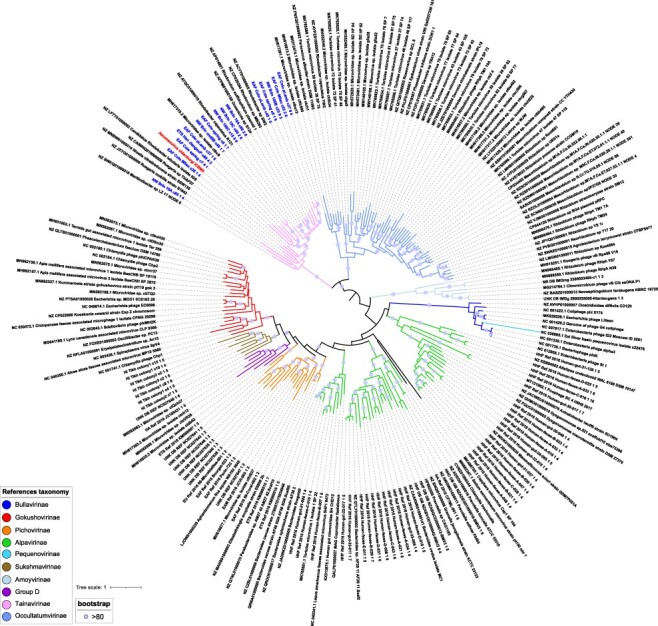
MCP- and Rep-based phylogeny. Phylogeny on the concatenated major capsid and replication proteins of the ssDNA (pro)-phage genomes. Branches are colored according to the taxonomic affiliation into subfamilies. Clear blue circles are placed on internal branches when bootstrap values are over 80. ICBM5 is highlighted in red, while closely related virome contigs are in blue (BLASTp on major capsid proteins, bitscore > 300). For a high-resolution image, see SI file extended Fig. 6.
About half of the newly predicted prophages, episomes, and EVGs, as well as phage ICBM5, were gathered in a group separated from previously defined Microviridae subfamilies (Fig. 6). Viruses from this large group were clustered in two clades in the phylogenetic tree (Fig. 6) that correspond to VGC1 and VGC2 in the VirClust analysis (Fig. 4, SI file 7). The first cluster comprised ICBM5 as the only cultivated phage, alongside sequences newly identified. Indeed, VGC1 encompassed ten newly found prophages and all the fifteen EVGs newly assembled in this study from eight viromes generated in three different previous studies (Colombo et al. 2017; Han et al. 2017; Brinkman et al. 2018). Only the contig MH617319.2_Microviridae_sp._isolate_ctbc9 was previously identified as Microviridae, but not further classified (Tisza et al. 2020). The second cluster was larger, with twenty-one new prophages and two episomes, thirty EVGs, and three recent isolated phages infecting Rhizobium (Jannick et al. 2021). All the EVGs here were previously identified as Microviridae in different viromics studies (see SI file 6) (Creasy et al. 2018; Orton et al. 2020; Tisza et al. 2020; Collins et al. 2021). In two of the studies, the respective EVGs were recognized already to represent a separate group from the known Microviridae subfamilies (Creasy et al. 2018; Orton et al. 2020). However, no further classification of these phages was performed.
Considering that two VGCs were generated, clearly separated from each other and even more distantly related to known Microviridae subfamilies in the phylogeny, here we tentatively propose two new subfamilies: (1) ‘Tainavirinae’ (from the Romanian word ‘taina’, which means secret), representing the clade with phage ICBM5, and (2) ‘Occultatumvirinae’ (from the Latin word ‘occultatum’, which means hidden), representing the clade with the Rhizobium phages. The genomic diversity within the two new subfamilies was high, with the nucleotide-based intergenomic identity ranging between 0.0 and 99.9 per cent. Most of the phage pairs had an intergenomic identity lower than 40 per cent. Few phages had an intergenomic identity above 95 per cent, which would place them into the same species: five EVGs into two species in the ‘Tainavirinae’ and two Rhizobium phages into one species in the ‘Occultatumvirinae’ (see SI file 8 and SI file 9).
The two subfamilies comprise phages present in different environments (Fig. 4) and spread worldwide (Fig. 7), with only Alphaproteobacteria as known hosts (Fig. 5). Tainavirus prophages and ICBM5 were only found in Rhodobacterales hosts. Most occultatumvirus prophages, episomes, and cultivated phages infected Hyphomicrobiales, the rest infecting the related bacterial order Rhodobacterales. A habitat overview showed that these phages, including the EVGs and their hosts are usually found in the terrestrial and marine environment, often in association with plants and animals (see Fig. 4, SI file 4, Table 2, and SI file 6). We found occultatumvirus EVGs in viromes from the lizard Heloderma suspectum (Collins et al. 2021); the fishes Carassius carassius, Lutjanus campechanus, and Pimephales sp.; the snail Haliotis sp. (Tisza et al. 2020); the tortoise Gopherus morafkai (Orton et al. 2020); and the sea squirt Ciona robusta (Creasy et al. 2018). All Hyphomicrobiales infected by Occultatumvirinae were isolated from soil or plant root nodules. The Rhodobacteraceae infected by occultatumviruses were isolated either from the soil or marine algae and seawater (see SI file 4, Table 2). We found tainavirus EVGs in viromes from paddy soils (Han et al. 2017), freshwater rivers (Colombo et al. 2017), and wastewater (Brinkman et al. 2018). The Rhodobacteraceae hosts infected by tainaviruses were isolated from terrestrial environments, including ponds, sediments, and soil, and marine environments, including sediments, water column, sponges, copepods, and dinoflagellates (see SI file 4, Table 2).
Figure 7.
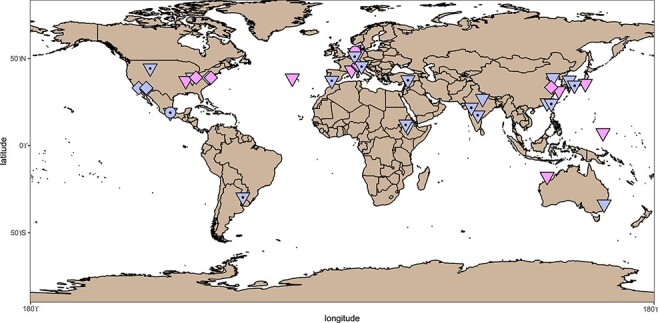
Biogeographical distribution of tainaviruses (pink) and occultatumviruses (lavender). The points on the map represent the sampling place for phage isolates (circles), for bacterial hosts harboring prophages and episomes (triangles), and for the viromes from which the EVGs have been assembled (squares). The triangles with a central circle represent locations for Rhizobium and Mesorhizobium infected by occultatumviruses.
Discussion
Knowledge of the diversity of tailless, icosahedral ssDNA phages is still in progress, as evidenced by the constant sequencing of new viral genomes. As a result, their classification is under regular revision, as it is clear by now that there are several major clusters of icosahedral ssDNA viruses. These are defined so far as subfamilies—Bullavirinae, Gokushovirinae, ‘Pichovirinae’, ‘Aravirinae’, ‘Alpavirinae’, ‘Stockavirinae’, ‘Pequenovirinae’, ‘Sukshmavirinae’, and ‘Amoyvirinae’, and grouped under the umbrella of the Microviridae family (Creasy et al. 2018; Zheng et al. 2018). Only the first two subfamilies are currently accepted in ICTV. This study illustrates such a process, because (1) the new phage isolated here is far from references and (2) the use of this phage as a stepping stone lifted the veil on two new major groups of Microviridae—‘Tainavirinae’ and ‘Occultatumvirinae’. These proposed new subfamilies represent a major contribution to the known Microviridae diversity, as indicated by the MCP–Rep phylogeny, where the two groups are distant from all current subfamilies (see Fig. 6).
In addition, the data collected here extends our understanding of Microviridae lifestyle. So far, only a handful of microvirus-like prophages are predicted bioinformatically, contrarily to inoviruses, the other major group of ssDNA phages. Indeed, inoviruses have been for a long time known to integrate into their host genomes, as part of their chronic life cycle (Mai-Prochnow et al. 2015), and a recent bioinformatics study found a huge diversity of inovirus-like prophages, spread throughout many bacterial and archaeal taxa (Roux et al. 2019). Here, the cultivation and sequencing of ICBM5 enabled the prediction of many more Microviridae-like prophages and episomes. Furthermore, the known host clades, so far restricted to the Bacteroidia class, Enterobacteriaceae family (Gammaproteobacteria class), and one Hyphomicrobiales family (Alphaproteobacteria class) (Krupovic and Forterre 2011; Roux et al. 2012; Quaiser et al. 2015; Zheng et al. 2018), were here expanded to include several Firmicutes classes, Flavobacteriia, and new Alphaproteobacteria and Gammaproteobacteria. Within the Alphaproteobacteria, this is the first report of prophages in genomes of the Rhodobacteraceae family, an environmentally significant clade in the marine environment. Microviridae prophages have been recently reported in a few genomes from Rhodobacteraceae (Forcone et al. 2021); however, a closer look revealed that they are phiX174, most likely an unremoved addition from the sequencing process. The many (pro)-phages we found both in Hyphomicrobiales and in Rhodobacterales are unrelated with the Amoyvirinae prophage previously found in C. tardaugens (Hyphomicrobiales; Zheng et al. 2018). Similarly, they are not related to the previously isolated R. pomeroyi phages, which according to our analysis belong to Amoyvirinae.
Occultatumviruses and tainaviruses with known hosts infect Hyphomicrobiales and Rhodobacterales, but of course, hosts are not known for the EVGs. Yet, their phylogenetic relatedness to phage isolates and prophages and the homogeneity of the known bacterial hosts suggest that these EVGs also infect Rhodobacterales or Hyphomicrobiales. Indeed, representatives of these two bacterial orders have been found in habitats similar to those in which the EVGs were found, either in diverse aquatic systems (sewage, freshwater, or marine), in soil, or to be associated with eukaryotes (animals, plants, or microalgae). For example, Rhodobacteraceae were found in the gut of a Ciona species (Dishaw et al. 2014), suggesting that the occultatumvirus EVGs found in C. robusta could also infect bacteria in this family. Similarly, Rhodobacteraceae and Hyphomicrobiales were found in the gut or skin of similar fish species from which occultatumvirus EVGs were retrieved (Nielsen et al. 2018; DeBofsky et al. 2020; Tarnecki et al. 2022). In the same vein, the tortoise G. morafkai harbors in its nasal microbiome both Hyphomicrobiales and Rhodobacterales (Weitzman, Sandmeier, and Richard 2018), making members of the two orders very likely the hosts of the occultatumviruses found in the fecal viromes from similar tortoises. Furthermore, Rhodobacteraceae are common in wastewater (Numberger et al. 2019), freshwater rivers (Liu et al. 2019), paddy soils (Wang et al. 2018), and marine sediments and water column (Hahnke et al. 2013). For example, here, Rhodovulumsp. MB263 and Rhodovulum sulfidophilum, which belong to the same species (Fig. 5), are respectively found in soil and marine systems. Accordingly, their tainaviruses appear as closely related (Figs 4 and 6). Rhodobacteraceae are also known to have a free or associated lifestyle. For example, the tainavirus host Epibacterium ulvae(Breider et al. 2019) was isolated from macroalgae. It is thus very likely that tainavirus EVGs also infect Rhodobacteraceae living in diverse aquatic systems (sewage, freshwater, and marine) and soil. Concerning occultatumviruses, which infect two bacterial orders, Rhodobacterales and Hyphomicrobiales, it has to be noted that these orders are closely related in the bacterial tree. Furthermore, all occultatumviruses infecting Hyphomicrobiales form a monophyletic group (Fig. 6). The Hyphomicrobiales order, containing Rhizobium and Mesorhizobium strains, is particularly interesting. These bacteria are commonly found in soil and, in conditions of nitrogen starvation, they migrate in the root hairs of legumes, where they transform into nitrogen-fixing endosymbionts (Poole et al. 2018; Clúa et al. 2018). Considering the worldwide spread of both wild and cultivable, economically important legumes (Sprent, Ardley, and James 2017), it can well be that occultatumviruses are cosmopolitan. Certainly, the varied geographical locations in which occultatumvirus harboring Rhizobium, Neorhizobium and Mesorhizobium were found (see Fig. 7) support this hypothesis. To summarize, the two related groups ‘Tainavirinae’ and ‘Occultatumvirinae’ infect, respectively, only Rhodobacterales or Hyphomicrobiales and Rhodobacterales, two related orders of Alphaproteobacteria, reinforcing our belief that these viral groups are evolutionary linked. Either the ancestor of these viruses was already infecting the ancestor of this branch of Alphaproteobacteria or the viral ancestor infected only Rhodobacterales and during the evolution it infected Hyphomicrobiales. Concerning the rest of the Microviridae, some subfamilies proposed earlier, like Pichovirinae and Alpavirinae, appear to be polyphyletic when adding new prophages and EVGs. In addition, these two subfamilies infect, respectively, two and three bacterial phyla, suggesting that these subfamilies might be too large and need to be revised.
Phage infections are usually described in terms of lytic, lysogenic, or chronic lifestyles. Alternative lifestyles such as pseudolysogeny and carrier state have been observed among different phage groups, although their definitions are not always consistent and the two terms have been used interchangeably (reviewed in (Mäntynen et al. 2021)). The same phage can exhibit different lifestyles on the same host. For example, the P22 phage displayed the following lifestyles when infecting its host, Salmonella Typhimurium: (1) a lytic strategy resulting in cell lysis, (2) a pseudolysogenic strategy characterized by the existence of a P22 episome, which, after cell division, was transmitted only to one of the daughter cells, and (3) a lysogenic strategy, arising from the cell which inherited the episomal phage (Cenens et al. 2015). The discovery of prophages in other Rhodobacteraceae has prompted us to ask if ICBM5, in addition to its lytic strategy, also has a lysogenic lifestyle. When investigating an ICBM5-resistant strain isolated from turbid plaques, we found no evidence that ICBM5 integrates into the genome of S. dubius SH24-1b. However, we found that ICBM5 can undergo a carrier-state life strategy. A sub-population of resistant strain cells carried the ICBM5 genome as an episome present in variable numbers. A second, numerically dominant sub-population carried no ICBM5, likely being resistant to this phage and conferring to the host strain the resistance to ICBM5 observed in spot assays. Considering that this strain was isolated from a single colony, there are two possible mechanisms by which the two sub-populations were produced. In the first scenario, the single cell from which the colony arose harbored the ICBM5 genome intracellularly. Asymmetrical cell division would have resulted in the transmission of ICBM5 only to one of the daughter cells. In a second scenario, the ICBM5 phage particle somehow became attached extracellularly to the initial colony-forming cell. Upon subsequent cell divisions, sensitive cells would have arisen and would have become infected by ICBM5. What factors confer resistance to the ICBM5 is for now unknown. During P22 infection of Salmonella Typhimurium, P22-free daughter cells resulting from the asymmetric division of pseudolysogenic cells were transiently immune to P22. The resistance was conferred by immunity factors cytoplasmically transmitted from the mother cell and, thus, inevitably diluted by subsequent cell divisions (Cenens et al. 2015). Considering the high proportion of noninfected cells in our ICBM5-resistant strain, it is unlikely that a similar mechanism is responsible for conferring resistance. Further experiments are required to characterize the ICBM5 carrier-state life strategy and to elucidate the host resistance mechanism. The carrier state does not seem to be confined to the infection of S. dubius SH24-1b by ICBM5. We predicted Microviridae-like episomes in Mesorhizobium, Neorhizobium, Prevotella, Aphanizomenon, Gramella, Mammalicoccus, and Acinetobacter, hosts belonging to various phyla. Likely, these phages used a carrier-state life strategy to survive in their host cultures, without having a dramatic effect on the culture’s growth. Furthermore, a similar carrier state was recently shown for a gokushovirus ‘revived’ from its prophage state in the host genome by molecular cloning (Kirchberger and Ochman 2020). Together, this indicates that such a carrier state is spread among Microviridae phages, very likely enabling coexistence with their hosts in environmental samples.
Fluorescence in situ hybridization targeting phage genes was previously used to characterize the lytic life cycle of PSA-HP1, a Pseudoalteromonas infecting dsDNA phage (Allers et al. 2013). The method used multiple probes labeled with digoxigenin, followed by a signal amplification step mediated by antibody binding and enzymatic tyramide deposition. Here, we have applied the relatively new direct-geneFISH method (Barrero-Canosa et al. 2017; Barrero-Canosa and Moraru 2021), which uses multiple probes directly labeled with fluorochromes and thus avoids signal amplification steps. This method was used recently for intracellular virus detection of dsDNA archaeal and picoeukaryotic viruses in environmental samples and pure cultures (Castillo et al. 2020, 2021; Rahlff et al. 2021). In our study, using phage-targeted genome-wide probes, we were able to characterize the lytic cycle of an ssDNA phage. The time until lysis for ICBM5, in the tested conditions, was 110 min. This is shorter than the ∼3 h reported for the vB_Cib_ssDNA_P1 phage infecting Citromicrobium sp. (Zheng et al. 2018) and the vB_RpoMi_Mini infecting R. pomeroyi DSS-3 (Zhan and Chen 2019b). In contrast, the duration of the lytic cycle for phiX174, the best characterized to date Microviridae, was only 20 min on Escherichia coli (Hutchison and Sinsheimer 1963). The difference comes most likely not only from the genetic differences between the phages, but also from the differences in the physiology of the two hosts. Furthermore, by measuring the total phage signal intensity values per cell and normalizing them to the intensity of a single phage, we were able to quantify the per-cell genome numbers of ICBM5. In the last phases of the lytic cycle, most of the cells had up to 125 ICBM5 genome copies. However, some cells reached >300 ICBM5 genome copies. Our measurements do not show how many of these genomes were packed into mature virions and released from the cells. However, these values are similar to the burst size of 250 phages per cell calculated from the PFU measurements. In comparison, phiX174 and vB_Cib_ssDNA_P1 have a burst size of ∼170 phages per cell (Hutchison and Sinsheimer 1966), and vB_RpoMi_Mini of only ∼8 phages/cell.
The discovery of so many new Microviridae-like prophages and episomes raises the question of whether they are more widespread in bacterial genomes than previously recognized. Our prophage prediction approach using ICBM5 proteins to search the NCBI-Blast database with DELTA-BLAST, followed by several rounds of PSI Blast, is a relatively unsophisticated procedure. It was able to find not only ICBM5-related prophages but also distant relatives, for example, prophages of Bacteroidetes which group with Alpavirinae or Gokushovirinae. This suggests that most of the findable prophage and episome diversity at the date of our search has been recovered. The addition of new bacterial genomes or the discovery of new Microviridae-like sequence diversity, either by phage cultivation or metagenomics, could reveal further microvirus diversity in bacterial genomes.
Supplementary Material
Acknowledgements
This work was supported by the Deutsche Forschungsgemeinschaft within the Transregional Collaborative Research Centre Roseobacter (TRR51). We thank Cathrin Spröer for sequencing support. The work conducted by the U.S. Department of Energy Joint Genome Institute (S.R.), a Department of Energy (DOE) Office of Science User Facility, is supported by the Office of Science of the U.S. Department of Energy under contract no. DE-AC02-05CH11231.
Contributor Information
Falk Zucker, Institute for Chemistry and Biology of the Marine Environment, University of Oldenburg, Carl-von-Ossietzky-Str. 9−11, Oldenburg D-26111, Germany.
Vera Bischoff, Institute for Chemistry and Biology of the Marine Environment, University of Oldenburg, Carl-von-Ossietzky-Str. 9−11, Oldenburg D-26111, Germany.
Eric Olo Ndela, Laboratoire Microorganismes: Genome Environment (LMGE), Université Clermont Auvergne, CNRS, 1 Imp. Amélie Murat, Aubière 63170, Frankreich.
Benedikt Heyerhoff, Institute for Chemistry and Biology of the Marine Environment, University of Oldenburg, Carl-von-Ossietzky-Str. 9−11, Oldenburg D-26111, Germany.
Anja Poehlein, Department of Genomic and Applied Microbiology & Göttingen Genomics Laboratory, Georg-August-University Göttingen, Institute of Microbiology and Genetics, Grisebachstr. 8, Göttingen D-37077, Germany.
Heike M Freese, Leibniz-Institut DSMZ, Deutsche Sammlung von Mikroorganismen und Zellkulturen GmbH, Inhoffenstraße 7 B, Braunschweig D-38124, Germany.
Simon Roux, Lawrence Berkeley National Laboratory, DOE Joint Genome Institute, Berkeley, CA 94720, USA.
Meinhard Simon, Institute for Chemistry and Biology of the Marine Environment, University of Oldenburg, Carl-von-Ossietzky-Str. 9−11, Oldenburg D-26111, Germany.
Francois Enault, Laboratoire Microorganismes: Genome Environment (LMGE), Université Clermont Auvergne, CNRS, 1 Imp. Amélie Murat, Aubière 63170, Frankreich.
Cristina Moraru, Institute for Chemistry and Biology of the Marine Environment, University of Oldenburg, Carl-von-Ossietzky-Str. 9−11, Oldenburg D-26111, Germany.
Supplementary data
Supplementary data are available at Virus Evolution online.
Conflict of interest:
The authors declare no conflict of interest.
Author contribution
C.M. designed the study, isolated the phage ICBM5, carried out part of the prophage prediction and phage classification using VirClust, analyzed the data, and wrote the manuscript. F.Z. performed the direct-geneFISH, the experiments proving the single-stranded DNA nature of ICBM5 and the Nanopore sequencing, carried out part of the prophage prediction and phage classification using VirClust, analyzed the data, and wrote the manuscript. V.B. performed TEM and host range assays for ICBM5, calculated the 16S tree, and wrote the manuscript. Genomes of Sulfitobacter dubius SH24-1b strains based on PacBio sequencing were generated by H.F. B.H. isolated the ICBM5-resistant strain. A.P. sequenced the ICBM5 phage. S.R., E.O.N., and F.E. constructed the major capsid protein - replication initiation protein (MCP-REP) tree and participated in the phage classification. All authors revised the manuscript.
References
- Allers E. et al. (2013) ‘Single-cell and Population Level Viral Infection Dynamics Revealed by phageFISH, a Method to Visualize Intracellular and Free Viruses’, Environmental Microbiology, 15: 2306–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Angly F. E. et al. (2006) ‘The Marine Viromes of Four Oceanic Regions’, PLoS Biology, 4: e368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balch W. E. et al. (1979) ‘Methanogens: Reevaluation of a Unique Biological Group’, Microbiological Reviews, 43: 260–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barrero-Canosa J., and Moraru C. (2021) ‘Linking Microbes to Their Genes at Single Cell Level with direct–geneFISH’. In: Azevedo, N. F., and Almeida, C. (eds) Fluorescence In-Situ Hybridization (FISH) for Microbial Cells, pp. 169–205. Methods and Concepts: Springer US (Methods in Molecular Biology), Vol. 2246. Humana: New York, NY. [DOI] [PubMed] [Google Scholar]
- Barrero-Canosa J. et al. (2017) ‘Direct-geneFISH. A Simplified Protocol for the Simultaneous Detection and Quantification of Genes and rRNA in Microorganisms’, Environmental Microbiology, 19: 70–82. [DOI] [PubMed] [Google Scholar]
- Bartlau N. et al. (2021) ‘highly diverse flavobacterial phages isolated from north sea spring blooms’, The ISME Journal: 555–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baym M. et al. (2015) ‘Inexpensive Multiplexed Library Preparation for Megabase-sized Genomes’, PloS One, 10: e0128036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bergh Ø. et al. (1989) ‘High Abundance of Viruses Found in Aquatic Environments’, Nature, 340: 467–8. [DOI] [PubMed] [Google Scholar]
- Bolger A. M., Lohse M., and Usadel B. (2014) ‘Trimmomatic: A Flexible Trimmer for Illumina Sequence Data’, Bioinformatics (Oxford, England), 30: 2114–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Breider S. et al. (2019) ‘Genome Sequence of Epibacterium Ulvae Strain DSM 24752T, an Indigoidine-producing, Macroalga-associated Member of the Marine Roseobacter Group’, Environmental Microbiome, 14: 4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brentlinger K. L. et al. (2002) ‘Microviridae, a Family Divided: Isolation, Characterization, and Genome Sequence of phiMH2K, a Bacteriophage of the Obligate Intracellular Parasitic Bacterium Bdellovibrio bacteriovorus’, Journal of Bacteriology, 184: 1089–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brinkman N. E. et al. (2018) ‘Reducing Inherent Biases Introduced during DNA Viral Metagenome Analyses of Municipal Wastewater’, PloS One, 13: e0195350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bryson S. J. et al. (2015) ‘A Novel Sister Clade to the Enterobacteria Microviruses (Family Microviridae) Identified in Methane Seep Sediments’, Environmental Microbiology, 17: 3708–21. [DOI] [PubMed] [Google Scholar]
- Castillo Y. M. et al. (2021) ‘Seasonal Dynamics of Natural Ostreococcus Viral Infection at the Single Cell Level Using VirusFISH’, Environmental Microbiology, 23: 3009–19. [DOI] [PubMed] [Google Scholar]
- ——— et al. (2020) ‘Visualization of Viral Infection Dynamics in a Unicellular Eukaryote and Quantification of Viral Production Using Virus Fluorescence in Situ Hybridization’, Frontiers in Microbiology, 11: 2306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cenens W. et al. (2015) ‘Viral Transmission Dynamics at Single-Cell Resolution Reveal Transiently Immune Subpopulations Caused by a Carrier State Association’, PLoS Genetics, 11: e1005770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chipman P. R. et al. (1998) ‘Structural Analysis of the Spiroplasma Virus, SpV4: Implications for Evolutionary Variation to Obtain Host Diversity among the Microviridae’, Structure, 6: 135–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clúa J. et al. (2018) ‘Compatibility between Legumes and Rhizobia for the Establishment of a Successful Nitrogen-Fixing Symbiosis’, Genes, 9.doi: 10.3390/genes9030125 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collins C. L. et al. (2021) ‘Genome Sequences of Microviruses Identified in Gila Monster Feces’, Microbiology Resource Announcements, 10: e00163–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colombo S. et al. (2017) ‘Viromes as Genetic Reservoir for the Microbial Communities in Aquatic Environments: A Focus on Antimicrobial-Resistance Genes’, Frontiers in Microbiology, 8: 1095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Creasy A. et al. (2018) ‘Unprecedented Diversity of ssDNA Phages from the Family Microviridae Detected within the Gut of a Protochordate Model Organism (Ciona Robusta)’, Viruses, 10: 404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Darling A. C. E. et al. (2004) ‘Mauve: Multiple Alignment of Conserved Genomic Sequence with Rearrangements’, Genome Research, 14: 1394–403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeBofsky A. et al. (2020) ‘Differential Responses of Gut Microbiota of Male and Female Fathead Minnow (Pimephales promelas) to a Short-term Environmentally-relevant, Aqueous Exposure to Benzoapyrene’, Chemosphere, 252: 126461. [DOI] [PubMed] [Google Scholar]
- Desnues C. et al. (2008) ‘Biodiversity and Biogeography of Phages in Modern Stromatolites and Thrombolites’, Nature, 452: 340–3. [DOI] [PubMed] [Google Scholar]
- Dishaw L. J. et al. (2014) ‘The Gut of Geographically Disparate Ciona intestinalis Harbors a Core Microbiota’, PloS One, 9: e93386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doore S. M., and Fane B. A. (2016) ‘The Microviridae: Diversity, Assembly, and Experimental Evolution’, Virology, 491: 45–55. [DOI] [PubMed] [Google Scholar]
- Eddy S. R. (2011) ‘Accelerated Profile HMM Searches’, PLoS Computational Biology, 7: e1002195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Everson J. S. et al. (2002) ‘Biological Properties and Cell Tropism of Chp2, a Bacteriophage of the Obligate Intracellular Bacterium Chlamydophila abortus’, Journal of Bacteriology, 184: 2748–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fane B. et al. (2006) ‘ØX174 Et Al., The Microviridae’. In: The Bacteriophages, 2nd edn. pp. 129–45. Oxford University Press: New York, NY. [Google Scholar]
- Finn R. D. et al. (2017) ‘InterPro in 2017-beyond Protein Family and Domain Annotations’, Nucleic Acids Research, 45: D190–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Forcone K. et al. (2021) ‘Prophage Genomics and Ecology in the Family Rhodobacteraceae’, Microorganisms, 9: 1115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grazziotin A. L., Koonin E. V., and Kristensen D. M. (2017) ‘Prokaryotic Virus Orthologous Groups (Pvogs). A Resource for Comparative Genomics and Protein Family Annotation’, Nucleic Acids Research, 45: D491–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gurevich A. et al. (2013) ‘QUAST: Quality Assessment Tool for Genome Assemblies’, Bioinformatics (Oxford, England), 29: 1072–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hadley W. (2016) Ggplot2. Elegrant Graphics for Data Analysis, 2nd edn. Springer (Use R!): Switzerland. [Google Scholar]
- Hahnke S. et al. (2013) ‘Physiological Diversity of Roseobacter Clade Bacteria Co-occurring during a Phytoplankton Bloom in the North Sea’, Systematic and Applied Microbiology, 36: 39–48. [DOI] [PubMed] [Google Scholar]
- Han -L.-L. et al. (2017) ‘Genetic and Functional Diversity of Ubiquitous DNA Viruses in Selected Chinese Agricultural Soils’, Scientific Reports, 7: 45142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hay I. D., and Lithgow T. (2019) ‘Filamentous Phages: Masters of a Microbial Sharing Economy’, EMBO Reports, 20: e47427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holmfeldt K. et al. (2013) ‘Twelve Previously Unknown Phage Genera are Ubiquitous in Global Oceans’, Proceedings of the National Academy of Sciences of the United States of America, 110: 12798–803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hopkins M. et al. (2014) ‘Diversity of Environmental Single-stranded DNA Phages Revealed by PCR Amplification of the Partial Major Capsid Protein’, The ISME Journal, 8: 2093–103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hutchison C. A., and Sinsheimer R. L. (1963) ‘Kinetics of Bacteriophage Release by Single Cells of φX174-infected E. Coli’, Journal of Molecular Biology, 7: 206–8. [DOI] [PubMed] [Google Scholar]
- ——— (1966) ‘The Process of Infection with Bacteriophage ΦX174. X. Mutations in a phiX Lysis Gene’, Journal of Molecular Biology, 18: 429–IN2. [DOI] [PubMed] [Google Scholar]
- Hyatt D. et al. (2012) ‘Gene and Translation Initiation Site Prediction in Metagenomic Sequences’, Bioinformatics (Oxford, England), 28: 2223–30. [DOI] [PubMed] [Google Scholar]
- Jain C. et al. (2018) ‘High Throughput ANI Analysis of 90K Prokaryotic Genomes Reveals Clear Species Boundaries’, Nature Communications, 9: 5114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jannick V. C. et al. (2021) ‘Spatial Patterns in Phage-Rhizobium Coevolutionary Interactions across Regions of Common Bean Domestication’, The ISME Journal, 15: 2092–106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones P. et al. (2014) ‘InterProScan 5: Genome-scale Protein Function Classification’, Bioinformatics (Oxford, England), 30: 1236–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kearse M. et al. (2012) ‘Geneious Basic: An Integrated and Extendable Desktop Software Platform for the Organization and Analysis of Sequence Data’, Bioinformatics (Oxford, England), 28: 1647–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kiening M. et al. (2019) ‘Conserved Secondary Structures in Viral mRNAs’, Viruses, 11: 401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirchberger P. C., and Ochman H. (2020) ‘Resurrection of a Global, Metagenomically Defined Gokushovirus’, eLife, 9: e51599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koboldt D. C. et al. (2012) ‘VarScan 2: Somatic Mutation and Copy Number Alteration Discovery in Cancer by Exome Sequencing’, Genome Research, 22: 568–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koonin E. V. et al. (2020) ‘Global Organization and Proposed Megatax-onomy of the Virus World’, Microbiology and Molecular Biology Reviews, 84: e00061–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krupovic M., and Forterre P. (2011) ‘Microviridae Goes Temperate. Microvirus-related Proviruses Reside in the Genomes of Bacteroidetes’, PloS One, 6: e19893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Labonté J. M., and Suttle C. A. (2013) ‘Metagenomic and Whole-genome Analysis Reveals New Lineages of Gokushoviruses and Biogeographic Separation in the Sea’, Frontiers in Microbiology, 4: 404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Letunic I., and Bork P. (2021) ‘Interactive Tree of Life (Itol) V5: An Online Tool for Phylogenetic Tree Display and Annotation’, Nucleic Acids Research, 49: W293–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li D. et al. (2015) ‘MEGAHIT: An Ultra-fast Single-node Solution for Large and Complex Metagenomics Assembly via Succinct de Bruijn Graph’, Bioinformatics (Oxford, England), 31: 1674–6. [DOI] [PubMed] [Google Scholar]
- Li H., and Durbin R. (2009) ‘Fast and Accurate Short Read Alignment with Burrows-Wheeler Transform’, Bioinformatics (Oxford, England), 25: 1754–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu J. et al. (2019) ‘Biogeography and Diversity of Freshwater Bacteria on a River Catchment Scale’, Microbial Ecology, 78: 324–35. [DOI] [PubMed] [Google Scholar]
- Loeb T. (1960) ‘Isolation of a Bacteriophage Specific for the F+ and Hfr Mating Types of Escherichia coli K-12’, Science, 131: 932–3. [DOI] [PubMed] [Google Scholar]
- López-Bueno A. et al. (2009) ‘High Diversity of the Viral Community from an Antarctic Lake’, Science (New York, N.Y.), 326: 858–61. [DOI] [PubMed] [Google Scholar]
- Ludwig W. et al. (2004) ‘ARB: A Software Environment for Sequence Data’, Nucleic Acids Research, 32: 1363–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mai-Prochnow A. et al. (2015) ‘Big Things in Small Packages: The Genetics of Filamentous Phage and Effects on Fitness of Their Host’, FEMS Microbiology Reviews, 39: 465–87. [DOI] [PubMed] [Google Scholar]
- Mäntynen S. et al. (2021) ‘Black Box of Phage-bacterium Interactions: Exploring Alternative Phage Infection Strategies’, Open Biology, 11: 210188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin M. (2011) ‘Cutadapt Removes Adapter Sequences from High-throughput Sequencing Reads’, EMBnet. Journal, 17: 10–2. [Google Scholar]
- McQuin C. et al. (2018) ‘CellProfiler 3.0: Next-generation Image Processing for Biology’, PLoS Biology, 16: e2005970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meier-Kolthoff J. P. et al. (2022) ‘TYGS and LPSN: A Database Tandem for Fast and Reliable Genome-based Classification and Nomenclature of Prokaryotes’, Nucleic Acids Research, 50: D801–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moraru C. (2021) ‘VirClust, a Tool for Hierarchical Clustering, Core Gene Detection and Annotation of (Prokaryotic) Viruses’, BioRXiv. 10.1101/2021.06.14.448304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nielsen K. M. et al. (2018) ‘Alterations to the Intestinal Microbiome and Metabolome of Pimephales promelas and Mus musculus following Exposure to Dietary Methylmercury’, Environmental Science & Technology, 52: 8774–84. [DOI] [PubMed] [Google Scholar]
- Noguchi H., Taniguchi T., and Itoh T. (2008) ‘MetaGeneAnnotator: Detecting Species-specific Patterns of Ribosomal Binding Site for Precise Gene Prediction in Anonymous Prokaryotic and Phage Genomes’, DNA Research: An International Journal for Rapid Publication of Reports on Genes and Genomes, 15: 387–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Numberger D. et al. (2019) ‘Characterization of Bacterial Communities in Wastewater with Enhanced Taxonomic Resolution by Full-length 16S rRNA Sequencing’, Scientific Reports, 9: 9673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orton J. P. et al. (2020) ‘Virus Discovery in Desert Tortoise Fecal Samples: Novel Circular Single-Stranded DNA Viruses’, Viruses, 12: 143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peng Y. et al. (2012) ‘IDBA-UD: A de Novo Assembler for Single-cell and Metagenomic Sequencing Data with Highly Uneven Depth’, Bioinformatics (Oxford, England), 28: 1420–8. [DOI] [PubMed] [Google Scholar]
- Poole P. et al. (2018) ‘Rhizobia: From Saprophytes to Endosymbionts’, Nature Reviews. Microbiology, 16: 291–303. [DOI] [PubMed] [Google Scholar]
- Quaiser A. et al. (2015) ‘Diversity and Comparative Genomics of Microviridae in Sphagnum-dominated Peatlands’, Frontiers in Microbiology, 6: 375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rahlff J. et al. (2021) ‘Lytic Archaeal Viruses Infect Abundant Primary Producers in Earth’s Crust’, Nature Communications, 12: 4642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rakonjac J., Bennet N. J., and Spagnuolo J. (2011) ‘Filamentous Bacteriophage: Biology, Phage Display and Nanotechnology Applications’, Current Issues in Molecular Biology, 13: 51–76. [PubMed] [Google Scholar]
- Rosario K. et al. (2012) ‘Diverse Circular ssDNA Viruses Discovered in Dragonflies (Odonata: Epiprocta)’, The Journal of General Virology, 93: 2668–81. [DOI] [PubMed] [Google Scholar]
- Roux S. et al. (2016) ‘Ecogenomics and Potential Biogeochemical Impacts of Globally Abundant Ocean Viruses’, Nature, 537: 689–93. [DOI] [PubMed] [Google Scholar]
- ——— et al. (2019) ‘Cryptic Inoviruses Revealed as Pervasive in Bacteria and Archaea across Earth’s Biomes’, Nature Microbiology, 4: 1895–906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ——— et al. (2012) ‘Evolution and Diversity of the Microviridae Viral Family through a Collection of 81 New Complete Genomes Assembled from Virome Reads’, PloS One, 7: e40418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanger F., Nicklen S., and Coulson A. R. (1977) ‘DNA Sequencing with Chain-terminating Inhibitors’, Proceedings of the National Academy of Sciences, 74: 5463–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seemann T. (2014) ‘Prokka: Rapid Prokaryotic Genome Annotation’, Bioinformatics (Oxford, England), 30: 2068–9. [DOI] [PubMed] [Google Scholar]
- Sertic V., and Boulgakov N. (1935) ‘Classification Et Identication Des Typhi-Phages’, C R Soc Bio, 119: 1270–2. [Google Scholar]
- Sievers F. et al. (2011) ‘Fast, Scalable Generation of High-quality Protein Multiple Sequence Alignments Using Clustal Omega’, Molecular Systems Biology, 7: 539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sprent J. I., Ardley J., and James E. K. (2017) ‘Biogeography of Nodulated Legumes and Their Nitrogen-fixing Symbionts’, The New Phytologist, 215: 40–56. [DOI] [PubMed] [Google Scholar]
- Stamatakis A. (2014) ‘RAxML Version 8: A Tool for Phylogenetic Analysis and Post-analysis of Large Phylogenies’, Bioinformatics (Oxford, England), 30: 1312–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steinegger M., and Soedin J. (2017) ‘MMseqs2 Enables Sensitive Protein Sequence Searching for the Analysis of Massive Data Sets: Nat’, Biotechnology, 35: 1026–8. [DOI] [PubMed] [Google Scholar]
- Steinegger M. et al. (2019) ‘HH-suite3 for Fast Remote Homology Detection and Deep Protein Annotation’, BMC Bioinformatics, 20: 473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suttle C. A. (2005) ‘Viruses in the Sea’, Nature, 437: 356–61. [DOI] [PubMed] [Google Scholar]
- Sźekely A. J., and Breitbart M. (2016) ‘Single-stranded DNA Phages: From Early Molecular Biology Tools to Recent Revolutions in Environmental Microbiology’, FEMS Microbiology Letters, 363: fnw027. [DOI] [PubMed] [Google Scholar]
- Tarnecki A. M. et al. (2022) ‘Dispersed Crude Oil Induces Dysbiosis in the Red Snapper Lutjanus Campechanus External Microbiota’, Microbiology Spectrum, 10: e0058721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tisza M. J. et al. (2020) ‘Discovery of Several Thousand Highly Diverse Circular DNA Viruses’, eLife, 9: e51971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Touchon M., Bernheim A., and Rocha E. P. (2016) ‘Genetic and Life-history Traits Associated with the Distribution of Prophages in Bacteria’, The ISME Journal, 10: 2744–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tucker K. P. et al. (2011) ‘Diversity and Distribution of Single-stranded DNA Phages in the North Atlantic Ocean’, The ISME Journal, 5: 822–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y.-Q. et al. (2018) ‘Differentiated Mechanisms of Biochar Mitigating Straw-Induced Greenhouse Gas Emissions in Two Contrasting Paddy Soils’, Frontiers in Microbiology, 9: 2566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weitzman C. L., Sandmeier F. C., and Richard T. C. (2018) ‘Host Species, Pathogens and Disease Associated with Divergent Nasal Microbial Communities in Tortoises’, Royal Society Open Science, 5: 181068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wommack K. E., and Colwell R. R. (2000) ‘Virioplankton: Viruses in Aquatic Ecosystems’, Microbiology and Molecular Biology Reviews, 64: 69–114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zayed A. A. et al. (2021) ‘Efam: An Expanded, Metaproteome-supported HMM Profile Database of Viral Protein Families’, Bioin-formatics, 16: btab451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhan Y., and Chen F. (2019a) ‘Bacteriophages that Infect Marine Roseobacters: Genomics and Ecology’, Environmental Microbiology, 21: 1885–95. [DOI] [PubMed] [Google Scholar]
- ——— (2019b) ‘The Smallest ssDNA Phage Infecting a Marine Bacterium’, Environmental Microbiology, 21: 1916–28. [DOI] [PubMed] [Google Scholar]
- Zheng Q. et al. (2018) ‘A Virus Infecting Marine Photoheterotrophic Alphaproteobacteria (Citromicrobium Spp.) Defines A New Lineage of ssDNA Viruses’, Frontiers in Microbiology, 9: 403. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.