

Abstract

Cancer is the most severe disease worldwide. Every year, tens of millions of people are diagnosed with cancer, and over half of those people will ultimately die from the disease. Hence, the discovery of new inhibitors for fighting cancer is necessary. As a result, new indolyl-triazole hybrids were synthesized to target breast and liver cancer cells. The synthetic strategy involves glycosylation of the 4-aryltriazolethiones 3a–b with acetyl-protected α-halosugars in the presence of K2CO3 in acetone to give a mixture of β-S-glycosides 6a–b, 7a–b, and β-N-glycosides 8a–b, 9a–b. Chemo-selective S-glycosylation was achieved using NaHCO3 in ethanol. The migration of glycosyl moiety from sulfur to nitrogen (S → N glycosylmigration) was achieved thermally without any catalyst. Alkylation of the triazole-thiones with 2-bromoethanol and 1-bromopropan-2-ol in the presence of K2CO3 yielded the corresponding S-alkylated products. The synthesized compounds were tested for their cytotoxicity using an MTT assay and for apoptosis induction targeting PARP-1 and EGFR. Compounds 12b, 13a, and 13b exhibited cytotoxic activities with promising IC50 values of 2.67, 6.21, 1.07 μM against MCF-7 cells and 3.21, 8.91, 0.32 μM against HepG2 cells compared to Erlotinib (IC50 = 2.51, 2.91 μM, respectively) as reference drug. Interestingly, compounds 13b induced apoptosis in MCf-7 and HepG2 cells, arresting the cell cycle at the G2/M and S phases, respectively. Additionally, the dual enzyme inhibition seen in compound 13b against EGFR and PARP-1 is encouraging, with IC50 values of 62.4 nM compared to Erlotinib (80 nM) and 1.24 nM compared to Olaparib (1.49 nM), respectively. The anticancer activity was finally validated using an in vivo SEC-cancer model; compound 13b improved both hematological and biochemical analyses inhibiting tumor proliferation by 66.7% compared to Erlotinib’s 65.7%. So, compound 13b may serve as a promising anticancer activity through dual PARP-1/EGFR target inhibition.
Introduction
In 2020, new cancer cases were estimated at 19.3 million, and almost 10.0 million cancer deaths were detected worldwide. Breast and liver cancers are the most diagnosed types of cancers cancer in many countries. Female breast cancer is the most commonly diagnosed cancer, with an estimated 2.3 million new cases (11.7%), while there are 1.6 million new estimated cases of liver cancer (8.3%).1 Research and development in chemotherapy will continue to face the challenge of finding safer and more effective chemotherapeutic drugs, which is a crucial step in transforming cancer medication therapy using nontargeted antitumor agents. DNA damage repair and maintaining genomic stability are two primary functions of poly ADP-ribose polymerase (PARP). It has been found that reducing PARP-1 activity, one of the most prevalent members of the PARP family, is effective in cancer treatment because it prevents tumor cells from repairing DNA damage, prevents tumor cell DNA synthesis, and induces tumor cell apoptosis.2−7
It has been found that EGFRs are the most important molecular targets in cancer treatment. Overexpression of epidermal growth factor receptors in breast cancer is linked to unfavorable tumor characteristics and poor prognosis.8,9 It is a promising target for these new anticancer drugs early in the discovery process. About 50% of triple-negative breast cancer (TNBC) and inflammatory breast cancer (IBC) is EGFR-overexpressing. Consequently, the pursuit of EGFR inhibitors as a therapy for breast cancer has persisted.10−13 Cell proliferation and tumor growth were also inhibited by inhibiting PARP in EGFR mutant tumors, which was achieved by targeting nuclear PKM2.14
Gefitinib, a tyrosine kinase inhibitor that can be reversed, is used to treat breast cancer and has been shown to have potent antiproliferative effects in studies of other cancers, including ovarian, lung, prostate, and colorectal cancers.15,16 It is possible to treat breast cancer and other solid tumors with an orally active medication called Lapatinib (Tykerb or Tyverb). There is an EGFR and Her2 tyrosine kinase inhibitor in it, which blocks their respective signaling pathways. The inhibitor was authorized by FDA for the treatment of advanced breast cancer in March 2007.17−19
A PARP inhibitor called Rubracarib (brand name Rubraca) has shown promise as a cancer treatment. Taken orally, the novel medication Rucaparib inhibits the DNA-repair enzyme poly-ADP ribose polymerase-1 (PARP-1).20 Oliparib, or Lynparza, is an inhibitor of poly ADP ribose polymerase (PARP). PARP is an enzyme involved in DNA repair. Some breast, ovarian, and prostate cancers in families with a BRCA1 or BRCA2 mutation can be prevented by taking this drug21 (Figure 1).
Figure 1.

Approved EGFR and PARP-1 inhibitors.
The indolyl-triazole hybrid systems disclosed promising results in fighting cancer by suppressing the activity of kinase Inhibitors, including EGFR and PARP-1 enzymes.22−25 Besides indoles, 1,2,4-triazole motifs have been widely explored in medicinal chemistry because of their structural features and pharmacophoric activities that could be employed as anticancer agents.26−28
3-Benzylsulfanyl-5-(1H-indol-2-yl)-2H-1,2,4-triazole I was found to have promising antiproliferative activity against HEPG-2 and MCF-7 cancer cell lines with IC50 = 3.58 μg/mL and 4.53 μg/mL, respectively, compared to the standard drug doxorubicin (IC50 = 4.0 μg/mL) this activity was predicted to be through the interaction with tyrosine kinases, namely, Akt, PI3, and EGFR.29
As seen in Figure 2, interesting antiproliferative potential against breast cancer in the low micromolar range was revealed for 3-(allylsulfanyl)-4-phenyl-5-(1H-indol-2-yl)-1,2,4-triazole II and its derivatives. The study’s findings demonstrated that the identified hit compounds effectively impeded cell migration and cell proliferation, most likely interfering with PARP-1 enzymatic activity.30 There was significant suppression of VEGFR-2by indole-triazole hybrid III and its analogs, suggesting that these compounds may have efficacy against kidney cancer.31 In comparison to Sorafenib (IC50 = 2.13 M), the indolyl-triazole hit compound IV showed promising cytotoxic action against the MCF-7 cells with an IC50 of 1.18 M. Remarkably, lead compound IV dramatically enhanced apoptotic breast cancer cell death and showed remarkable VEGFR2 inhibitory activity with an IC50 value of 19.8 nM compared to Sorafenib (IC50 = 30 nM).32
Figure 2.

Indolyl-triazole hybrids showing EGFR and PARP-1 inhibition activity.
Compounds 12b and 13b, two newly synthesized indolyl-triazoles, showed dual PARP-1 and EGFR inhibition and promised anticancer action against breast and liver malignancies.
Results and Discussion
Chemistry
Reaction of indole-2-carbohydrazide 1 with phenyl isothiocyanate and 4-chlorophenyl isothiocyanate in ethanol yielded thethiosemicarbazides 2a–b, which revealed four NH protons around 9.80, 9.90, 10.50, and 11.70 ppm in the 1H NMR and the carbonyl carbon near 161.4 ppm and the thiocarbonyl carbon at 181.3 ppm in the 13C NMR. Cyclization of the thiosemicarbazides 2a–b by reflux in aqueous KOH afforded the indolyl-triazolethiones 3a–b, which showed only two NH protons around 11.90 and 14.20 ppm, and the thiocarbonyl carbon (C=S) at 168.5 ppm. In glycosylation of the indolyltriazolethiones 3a–b with 2,3,4,6-tetra-O-acetyl-α-d-glucopyranosyl bromide 4 and 2-acetamido-3,4,6-tri-O-acetyl-2-deoxy-α-d-glucopyranosyl chloride 5 in acetone and the presence of anhydrous K2CO3, each reaction was found to give a mixture of two products that were separated by alumina column chromatography and characterized as S-glycosides 6a–b and 7a–b and N-glycosides 8a–b and 9a–b. Chemo-selectivity was successfully obtained using NaHCO3 in ethanol to afford S-glycosylated products in good yields (Scheme 1). Rearrangement of the S-glycosides to the corresponding N-glycosides was done by thermal fusion without solvent or catalyst. This glycosyl migration was done in a short time with good to excellent yields, which served as a rapid and economical way. The anomeric protons in the case of S-glycosides appeared at a lower chemical shift (≈ 5.50 ppm) than their respective N-glycosides (≈ 6.30 ppm). The coupling constants of the anomeric protons of S-glycosides were detected to be greater than 10.2 Hz, and its value for N-glycosides was greater than 9.3 Hz, which strongly recommends the β-configuration. The N-glycosides showed the C=S signal near 170.0 ppm.
Scheme 1. Glycosylation of Triazolyl-indoles 3a–b.

Alkylation of the indolyl triazoles 3a–b with 2-bromoethanol 10, and 1-bromopropan-2-ol 11 in the presence of K2CO3 produces S-alkylated products 12a–b and 13a–b (Scheme 2).
Scheme 2. Alkylation of 3a–b with Bromoalkanoles.

The 2-hydroxyethyl-sulfanyl-triazoles 12a–b exhibited the ethylene protons (−S–CH2–CH2−) as triplet at 3.26 ppm and multiplet between 3.65 and 3.70 ppm. The −S–CH2–CH2–OH carbons appeared at 34.96 and 59.62 ppm, respectively. The hydroxy group was found as a triplet at 5.04 ppm. The methyl and methylene protons were shown as a doublet at 1.13 and 3.22 ppm, respectively, in 2-hydroxypropyl-sulfanyl-triazoles 13a–b. Around 3.88–3.92 ppm, the multiplet of the proton in methine was first seen. At 22.44, 40.68, and 65.12 ppm, 2-hydroxypropyl carbons were detected.
Biology
Cytotoxicity against MCF-7 and HepG2 Cells
The MTT assay was used to test the synthesized compounds for cytotoxicity against MCF-7 and HepG2 cells; IC50 values were summarized in Table 1. As seen in the results, compounds 12b, 13a, and 13b exhibited cytotoxic activities with promising IC50 values of 2.67, 6.21, and 1.07 μM against MCF-7 cells and 3.21, 8.91, and 0.32 μM against HepG2 cells compared to Erlotinib as reference drug. However, the cytotoxicity of the remaining compounds was moderate. Compound 12b was found to be the most cytotoxic of the two tested compounds; thus, it was tested on normal cell lines MCF-10A and THLE2. As can be seen in Figure 3, at the highest dose of 100 μM, it caused 80% cell inhibition; this is relatively low in comparison to the other substances tested. In this study, compound 13b was effective against cancer cells while showing no toxicity to healthy cells.
Table 1. Cytotoxicity of the Synthesized Compounds against MCF-7 and HepG2 Cells Using MTT Assay.
| IC50 (μM)a |
||
|---|---|---|
| Compound | MCF-7 | HepG2 |
| 2a | NT | 12.36 |
| 3a | NT | 7.39 |
| 3b | 14.2 | 17.21 |
| 6a | NT | 6.71 |
| 6b | 19.6 | 21.6 |
| 7a | 17.2 | ≥50 |
| 7b | 12.7 | 16.9 |
| 8a | 11.2 | ≥50 |
| 8b | 17.3 | 46.35 |
| 9a | 8.3 | 6.68 |
| 12a | 12.1 | 16.31 |
| 12b | 2.67 | 3.21 |
| 13a | 6.21 | 8.91 |
| 13b | 1.07 | 0.32 |
| Erlotinib | 2.51 | 2.91 |
“IC50values were calculated as the average of three independent trials using a dose-response curve in GraphPad prism”. NT: Not tested.
Figure 3.

Percentage of cell viability compared to concentrations of compound 13b against normal breast (MCF-10A) and normal liver (THLE2) cells using an MTT assay.
As summarized in Figure 4, tested compounds with varying derivatives exhibited varying cytotoxicity against MCF-7 and HepG2 cells. Both compounds 13b and 12b with p-chloro-phenyl at the triazole ring exhibited potent cytotoxicity against MCF-7 and HepG2 cells. Compound 13b (IC50 = 1.07 μM, 0.32 μM) with S-substituted secondary alcohol derivative exhibited cytotoxicity more than compound 12b (IC50 = 2.67 μM, 3.21 μM) with S-substituted primary alcohol derivative. Additionally, 9a and 7b (IC50 range of 6.68–16.9 μM) with sugar and indole substitution exhibited moderate activity, and compounds 13a and 12a (IC50 range of 6.21–16.3 μM) exhibited moderate activity. Other compounds exhibited poor cytotoxicity.
Figure 4.

Effect of derivatization of the synthesized alkylated 1,2,4-triazoles on their cytotoxicity.
Compound 13b Induced Apoptosis in MCF-7 and HepG2 Cells
Compound 13b was tested for its ability to induce apoptosis in MCF-7 and HepG2 cancer cells using cell cycle analysis of the cell population at various stages of the cell cycle after treatment (IC50 = 1.07 and 0.32 M, 48 h).
Figure 5A reveals that compound 13b greatly enhanced total apoptotic breast cancer cell death by 26-fold (27.63% compared to 1.07 for the control). The percentage of cells undergoing early apoptosis was increased by 10.6%, and the percentage of cells undergoing late apoptosis by 16.9% compared to the control group’s 0.8% and 0.2%, respectively. Additionally, it caused a 4.18-fold increase in necrosis-mediated cell death (5.06%, compared to 1.21% for the control).
Figure 5.

Flow cytometry analysis. (A, B) Bar representation of Annexin V/PI staining for apoptosis-necrosis assessment. (C, D) Histograms of DNA content at each phase of untreated and 13b-treated MCF-7 and HepG2 cells with IC50 values of 1.07 and 0.32 μM, 48 h. “*(P ≤ 0.05), and **(P ≤ 0.001) are significantly different using the unpaired test in GraphPad prism”.
Figure 5B shows that the overall apoptotic cell death of liver cancer cells was greatly enhanced by compound 13b, with a 34.68-fold increase (32.95% compared to 0.95 for the control). Compared to the control group’s 0.74 and 0.21%, it produced early apoptosis at 23.61% and late apoptosis at 9.34%. Furthermore, a 6.1-fold increase in necrosis-induced cell death was observed (6.01%, compared to 0.98% for the control).
Cell cycle analysis was performed on both cell lines to determine the percentage of cells in each divisional stage before and after treatment. As seen in Figure 5C and D, compound 13b significantly increased the cell population of MCF-7 at G2/M by 26.4%, compared to 10.32% for control, so it arrested cell division at the G2/M phase, while treatment of HepG2 cells with the compound significantly increased cells at the S phase by 45.21% compared to 32.61%, so it arrested cell division at the S phase.
As a result, compound 13b caused cell cycle arrest at the G2/M and S phases in MCf-7 and HepG2 cells, inducing death in both types of cells.
Enzyme Targeting
Table 2 shows that of the three compounds evaluated, compound 13b showed the most promise as a dual enzyme inhibitor of EGFR (IC50 = 62.4 nM) and PARP-1 (1.24 nM) compared to Erlotinib (IC50 = 80 nM) and Olaparib (IC50 = 1.49 nM). Additionally, compound 12b exhibited dual inhibitory activities in the same manner with IC50 values of 78.1 nM and 2.13 nM, respectively. In contrast, compound 13a showed poor inhibitory activities. As seen in Figure 6, Interestingly, our results of PARP-1 and EGFR inhibition agreed with some 1,2,4-triazole-based derivatives (V)33 showed promising cytotoxic efficacy as an EGFR inhibitor with an IC50 value of 1.5 M, another derivative with the same moiety (VI)34 had intriguing cytotoxic action, inducing apoptosis by blocking PARP-1 at an IC50 of 0.33 μM.
Table 2. EGFR/PARP Target Inhibition of Compounds 12b, 13a, and 13b.
Figure 6.

Highlighted pharmacophoric regions with the previously reported triazole-based PARP-1 and EGFR target inhibition.
In Silico Studies
Both the physical and chemical characteristics of the lead molecule, 13b, and its drug-likeness scores were predicted using ADME pharmacokinetics. Drug candidates that adhere to Lipinski’s “five rules” (Ro5) are seen as having a good chance of being approved.37,38 Compound 9b, according to the calculations, had a molecular weight of 384.89 D, a volume of 322.76 A3, a polar surface area of 66.74 A2, a logP (octanol–water partition coefficient) value of 4.28, 5 H-bond acceptors, and 2 H-bond donors. As a result, it complied with Lipinski’s “five rules” (Ro5) and was found suitable for use as an oral drug Figure 7.
Figure 7.
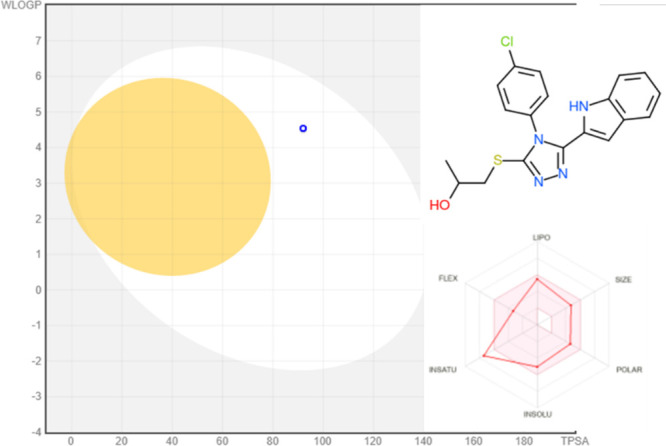
ADME pharmacokinetics of the most active compound 13b using SWIS-ADME: BOILED-Egg model for compound 16 using SwissADME. “Points in the BOILED-yolk Eggs are molecules predicted to passively permeate the blood-brain barrier (BBB), whereas points in the BOILED-white Eggs are molecules predicted to be passively absorbed by the GI tract”.
Based on the promising dual PARP-1 and EGFR inhibitory activities of compound 13b, it was screened for virtual binding toward PARP-1 and EGFR proteins using the molecular docking approach. As summarized in Table 3, compound 13b was docked inside the PARP-1 protein with a binding energy of −24.97 kcal/mol and formed one HB with Gly 863 and arene-cation with His 862, while it was docked inside the EGFR protein with a binding energy of −24.32 kcal/mol and formed one HB with Met 793 and arene-cation with Lys 745 as the key amino acids for enzyme activities. Hence, docking results highlighted the virtual mechanism of the binding of compound 13b toward PARP-1 and EGFR proteins, which agreed with its promising experimental activity.
Table 3. Ligand–Receptor Interactions of the Docked Compound 13b with Binding Energies (kcal/mol) inside the PARP-1 and EGFR Proteinsa.

Binding disposition of compound 13b (green-colored) and co-crystallized ligands (cyan-colored) inside the active sites of PARP-1 and EGFR proteins.
In Vivo
Further validation of the anticancer activity of 13b, solid Ehrlich carcinoma cells were proliferated as a solid tumor model, and the compound was administered IP during the experiment duration. At the end of the experiment, blood and tumor parameters were assayed and summarized in Table 4.
Table 4. Blood and Antitumor Parameters in Normal, Untreated, And Treated SEC-Bearing Micea.
| treatment |
|||||
|---|---|---|---|---|---|
| parameters | normal control | SEC control | SEC + 13b | SEC + Erlotinib | |
| hematological parameters | Hb (g/dL) | 9.03 ± 0.49 | 2.36* ± 0.4 | 7.29# ± 0.67 | 7.9# ± 0.63 |
| RBC count (× 106/μL) | 5.76 ± 0.62 | 2.34* ± 0.48 | 4.99# ± 0.63 | 5.01# ± 0.39 | |
| WBC count (× 103/μL) | 3.04 ± 0.29 | 7.03* ± 0.39 | 4.36# ± 0.49 | 4.02# ± 0.56 | |
| liver parameters | ALT (I/U) | 42.36 ± 1.03 | 69.8* ± 2.03 | 50.6# ± 1.3 | 51.4# ± 1.3 |
| AST (I/U) | 46.6 ± 0. 9 | 89.69* ± 1.23 | 60.3# ± 1.98 | 55.63# ± 1.4 | |
| total protein (g/dL) | 6.7 ± 0.26 | 3.29 ± 0.36 | 5.6 ± 0.39 | 6.2 ± 0.47 | |
| tumor potentiality | tumor weight (mg) | 214.6 ± 3.14 | 74.6 ± 2.79 | 69.3 ± 2.19 | |
| tumor volume (mm3) | 378.6 ± 16.8 | 129.6 ± 13.6 | 114.6 ± 12.5 | ||
| tumor inhibition ratio (TIR%) | 66.7 ± 1.06 | 65.7 ± 1.31 | |||
Mean ± SD values of mice in each group (n = 6). *Values are significantly different (P ≤ 0.05) between SEC control and normal group, while # values are significantly different (P ≤ 0.05) between treated SEC and SEC control mice using the unpaired test in GraphPad prism.
In SEC-bearing mice, all CBC parameters were altered, and Hb content and RBCs were significantly reduced to 2.36 (g/dL) and 2.34 (106/L), respectively, while the WBC count was significantly increased to 7.03 (103/L) compared to normal control levels. Reduced hemoglobin and RBC levels, as well as an increase in WBC counts, are common side effects of tumor progression.39,41 CBC levels were nearly restored to normal after treatment with the compound, which increased Hb (7.29 g/dL), RBC (4.79 × 106/L), and WBC (4.36 × 103/L) levels. Interestingly, our results followed those of refs (40 and 41), which showed improvement in hematological parameters after treatment with the tested compound. The effectiveness of compound 13b was comparable to Erlotinib as standard anticancer medication.
Liver enzymes (ALT and AST) were considerably elevated to 69.8 and 89.69 (U/L), respectively, as compared to normal mice, suggesting hepatocellular injury from tumor inoculation (42.36 and 46.6, respectively). Treatment with 13b, substantially reduced liver enzymes to 50.6, 60.3 U/L, respectively, and increased the reduced total protein content from 3.29 (g/dL) in the SEC-control to 5.6 (g/dL) near the normal control levels. These results indicated a significant improvement in hepatocellular toxicity caused by SEC proliferation.
Accordingly, regarding tumor potentiality, an increase in solid tumor mass of around 214 mg was observed via tumor proliferation. Treatment with 13b and Erlotinib significantly decreased the solid tumor mass to 74.6 mg and 69.3 mg, respectively. Accordingly, treatments with 9b 5-FU significantly inhibited tumor proliferation by 66.7% and 65.7%, respectively, by reducing the tumor volume from 348.6 mm3 in the untreated control to 129 mm3 and 114 mm3.
For validating the anticancer activity of compound 13b, histopathological examinations of liver tissues of normal, untreated, and treated SEC-mice as seen in Figure 8. Compound 13b-treatment exhibited improvement in liver tissues and retained them near normal.
Figure 8.

Histopathological examination of SCE-bearing mice in different groups in normal mice, SEC mice, and SEC-treated with 13b. Mice that are considered to be “normal” have hepatocytes and portal tracts that are both evenly distributed (black arrows). Hydropic degeneration of hepatocytes, a region of lytic necrosis (black arrowhead), and enlargement of the portal tract with chronic inflammatory cells characterize the SEC group (red arrows). Mild hydropic degeneration of hepatocytes in the treated 13b group (red arrows).
Conclusion
New hybrid compounds containing indole, triazole, and alkyl moiety were synthesized via glycosylation and alkylation of 4-aryltriazolethiones. Among the synthesized compounds, the hit compound 13b exhibited cytotoxic activities with promising IC50 values of 1.07 μM against MCF-7 cells and 0.32 μM against HepG2 cells compared to Erlotinib as a reference drug. Interestingly, compounds 13b induced apoptosis in MCf-7 and HepG2 cells, arresting the cell cycle at the G2/M and S phases, respectively. Additionally, compound 13b exhibited promising dual enzyme inhibition EGFR and PARP-1 with IC50 values of 62.4 nM compared to Erlotinib (80 nM) and 1.24 nM compared to Olaparib (1.49 nM), respectively. The anticancer activity was finally validated using an in vivo SEC-cancer model; compound 13b improved both hematological and biochemical analyses inhibiting tumor proliferation by 66.7% compared to Erlotinib by 65.7%. So, compound 13b may serve as a promising anticancer activity through dual PARP-1 and EGFR inhibitory activities.
Experimental Section
Melting points were assigned in open capillaries by a melting-point equipment (SMP10). Thin-layer chromatography (TLC) plates precoated with 0.25 mm thick layers of silica gel 60 F254 were used to monitor the reactions as they unfolded (Merck). UV light illumination and/or treatment with 10% H2SO4 in an aqueous methanol solution were used for detection. The process of flash chromatography utilized commercial alumina. Infrared (IR) spectra were recorded using a Bruker Alpha-I ATR FTIR spectrophotometer and are expressed in v (cm–1). The 1H NMR, 13C NMR, and 2D NMR spectra were acquired using Bruker AC 300–500 spectrometers in CDCl3 and DMSO-d6 with tetramethylsilane as the internal standard. Coupling constants are described in Hz, and chemical changes are described in ppm. The incorporation of D2O verified the OH and NH assignments. Both the Jeol JMS.600H and Finnigan MAT312 mass spectrometers were used to capture the EI mass spectra.The Finnigan MAT 95XP was used to record the mass spectra of HREI experiments. A Jeol JMS HX110 mass spectrometer was used to capture the FAB-MS data. ESI-MS spectra were determined with an Applied Biosystems QStarXL instrument. Using Flash EA-1112 equipment, CHNS microanalysis was carried out.
Synthesis of Thiosemicarbazides 2a–b
A mixture of indole-2-carbohydrazide 1 (1.0 mmol) and phenyl isothiocyanate or its chloro-substituted derivative (1.1 mmol) were refluxed in ethanol for 4 h and then left to cool. The formed precipitate was filtered, washed with ethanol, and dried. The products are pure enough for the next reaction. For more purification, recrystallization from ethanol was done.
1-(1H-Indol-2-yl-carbonyl)-4-Phenyl-thiosemicarbazide (2a)
Yield: 91%, mp 204–205 °C [Lit. 23]; 1H NMR (DMSO-d6, 400 MHz) δ 7.04–7.47 (m, 9H), 7.67 (d, 1H, J = 8.0 Hz), 9.76 (br.s, 1H, NH), 9.88 (br.s, 1H, NH), 10.55 (br.s, 1H, NH), 11.73 (br.s, 1H, NH); 13C NMR (DMSO-d6, 100 MHz) δ 104.02, 112.29, 119.87, 121.66, 123.68, 125.84, 126.90, 127.91, 128.29, 129.65, 136.58, 139.24, 161.07, 181.37; HRMS (EI) calcd for C16H14N4OS (M+): 310.0888. Found: 310.0893.
4-(4-Chlorophenyl)-1-(1H-indol-2-yl-carbonyl)thiosemicarbazide (2b)
Yield: 96%, mp 202–203 °C;1H NMR (DMSO-d6, 300 MHz) δ 7.04 (dd, 1H, J = 7.8, 7.5 Hz), 7.17–7.23 (m, 2H), 7.35–7.63 (m, 5 H), 7.64 (d, 1H, J4,5 7.8 Hz), 9.84 (br.s, 1 H, NH), 9.92 (br.s, 1H, NH), 10.54 (br.s, 1H, NH), 11.72 (br.s, 1H, NH); 13C NMR (DMSO-d6, 75 MHz) δ 104.09, 112.33, 119.93, 121.71, 123.76, 126.92, 127.46, 127.85, 129.00, 129.59, 136.63, 138.27, 161.14, 181.26; HRMS (FAB + ve) calcd for C16H14N4OSCl (M+1): 345.0577. Found: 345.0557.
Synthesis of the Triazole-thiones
Thiosemicarbazides 2a–b (1.0 mmol)were refluxed in 10 mL of 4 N KOH for 6 h. After cooling the solutions were acidified with conc. HCl, the formed precipitates were filtered, washed with distilled water, dried and recrystallized from ethanol.
5-(1H-Indol-2-yl)-4-phenyl-1,2,4-triazole-3(4H)-thione (3a)
Yield: 88%, mp 292–294 °C [Lit. 23]; 1H NMR (DMSO-d6, 300 MHz) δ 5.51 (s, 1H), 6.94 (dd, 1H, J = 8.1, 7.2 Hz,), 7.14 (dd, 1H, J = 8.4, 7.2 Hz), 7.33 (d, 1H, J = 8.1 Hz), 7.39 (d, 1H, J = 8.4 Hz), 7.46–7.49 (m, 2 H), 7.63–7.65 (m, 3H, 2H), 11.87 (br.s, 1H, NH), 14.19 (br.s, 1H, NH); 13C NMR (DMSO-d6, 75 MHz) δ 102.93), 111.78, 119.85, 120.86, 122.75, 123.60, 126.77, 128.89, 129.74, 130.11, 134.60, 136.51, 145.06,168.64; FTIR cm–1: 3360 (NH), 3086 (Aromatic C–H), 2499 (C=S), 1600 (C=C), 1513(C=N); HRMS (EI) calcd for C16H12N4S (M+): 292.0783. Found: 292.0789.
4-(4-Chlorophenyl)-5-(1H-indol-2-yl)-2H-1,2,4-triazole-3(4H)-thione (3b)
Yield: 83%, mp 295–296 °C; 1H NMR (DMSO-d6, 300 MHz) δ 5.67 (s, 1H), 6.95 (dd, 1H, J = 8.1, 6.9 Hz), 7.16 (dd, 1H, J = 6.9, 8.4 Hz), 7.38–7.43 (m, 2H), 7.55 (d, 2H, J = 8.7 Hz), 7.71 (d, 2H, J = 8.4 Hz), 11.88 (br. s, 1H, NH), 14.23 (br. s, 1H, NH); 13C NMR (DMSO-d6, 100 MHz) δ 103.19, 111.83, 119.93, 121.08, 122.62, 123.71, 126.87, 129.90, 130.95, 133.53, 134.79, 136.60, 144.97, 168.70; FTIR cm–1: 3324 (NH), 3061 (Aromatic C–H), 2318 (C=S), 1549 (C=C), 1484 (C=N), 711(C–Cl); HRMS (EI) calcd for C16H11N4SCl (M+): 326.0393. Found: 326.0402.
General Procedures for Glycosylation and Alkylation
Method A
A mixture of the selected 4-aryl-triazole-thione 3a or 3b (1.0 mmol) and K2CO3 (1.1 mmol) in dry acetone (10 mL) was stirred for 1 h, and then the appropriate glycosyl halides 4–5 or alkyl halides 10–11 (1.1 mmol) were added portion wise and stirring was continued overnight. The mixture was filtered off, washed thoroughly with acetone, and evaporated invacuo. The products were purified using alumina column chromatography and ethyl acetate/n-hexane 3:7 as eluent in the case of the glycosides and by recrystallization from ethanol in the remaining alkylated products.
Method B
A mixture of the selected 4-aryl-triazole-thione 3a or 3b (1.0 mmol) and NaHCO3 (1.1 mmol), in abs. ethanol (10 mL) were stirred for 1 h and then the appropriate glycosyl halide 4–5 (1.1 mmol) was added portion wise and stirring was continued for 48 h at room temperature. The solvent was evaporated under a vacuum, the residue was washed thoroughly with water, filtered off, and dried. The products were purified using alumina column chromatography and ethyl acetate/n-hexane 3:7 as eluent.
General Method for Thermal Transformation of S-Glycosides into N-Glycosides
Method C
The selected S-glycoside 6a–b and 7a–b (0.1 mmol) was heated at a temperature higher than its melting point for 5 min; the conversion progress was monitored by TLC. After the glycosyl migration is completed, the fused mass was left to reach ambient temperature, and the resultant solid was grounded and then recrystallized from EtOH to give the corresponding N-glycosides analogs.
5-(1H-Indol-2-yl)-4-phenyl-3-(2,3,4,,6-tetra-O-acetyl-β-d-glucopyranosylsulfanyl)-1,2,4-triazole (6a)
Yields: 67% method A, 62% method B, mp 137–138 °C [Lit. 23]; 1H NMR (600 MHz, CDCl3): δ 1.97, 1.98, 2.00, 2.02 (4s, 12H), 3.75–3.78 (m, 1H), 4.07 (dd, 1H, J = 12.5, 1.9 Hz), 4.22 (dd, 1H, J = 12.5, 4.7 Hz), 5.06–5.11 (m, 2H), 5.27 (dd, 1 H, J = 9.3 Hz), 5.55 (d, 1H, J = 10.4 Hz), 5.71 (d, 1H, J = 1.3 Hz), 7.03 (dd,1 H, J = 8.0, 7.3 Hz), 7.22 (dd, 1H, J = 7.3, 8.3 Hz), 7.36–7.38 (m, 3H), 7.49 (d, 1H, J = 8.3 Hz), 7.60 (dd, 2H, J = 7.6, 8.0 Hz), 7.66 (dd, 1H, J = 7.4, 7.5 Hz), 10.00 (br.s, 1H, NH); 13C NMR (150 MHz, CDCl3): δ 20.59, 20.61, 61.61, 67.85, 69.97, 73.72, 76.196, 84.34, 103.06, 111.68, 120.48, 121.32, 123.35, 124.19, 127.765, 127.985, 130.28, 130.95, 133.40, 136.398, 149.16, 150.22, 169.44, 169.55, 170.02, 170. FTIR cm–1: 3418 (NH), 3092 (aromatic C–H), 2916 (aliphatic C–H), 1740 (C=O ester), 1595 (C=C), 1495 (C=N); HRMS (EI): m/z Calcd for C30H30N4O9S (M+): 622.1733. Found: 622.1731.
4-(4-Chlorophenyl)-5-(1H-indol-2-yl)-3-(2,3,4,,6-tetra-O-acetyl-β-d-glucopyranosylsulfanyl)-1,2,4-triazole (6b)
Yields: 61% method A, 65% method B, mp 155–156 °C; 1H NMR (300 MHz, CDCl3): δ 1.96, 1.98, 1.99, 2.02 (4s, 12H, 3.73–3.77 (m, 1H), 4.06 (dd, 1H, J = 12.6, 2.1 Hz), 4.20 (dd, 1H, J = 12.6, 4.8 Hz), 5.04–5.12 (m, 2H), 5.27 (dd, 1H, J = 9.3 Hz), 5.53 (d, 1H, J = 10.2 Hz), 5.77 (d, 1H, J = 1.5 Hz), 7.05 (dd, 1H, J = 8.1, 7.2 Hz), 7.23 (dd, 1H, J = 7.2, 8.4 Hz), 7.30 (d, 2 H, J 7.8 Hz, 2 HPh), 7.43 (d, 1 H, J4,5 8.1 Hz, H-4Ind.), 7.53 (d, 1 H, J6,7 8.4 Hz, H-7Ind.), 7.57 (d, 2H, J = 9.0 Hz), 10.39 (br.s, 1H, NH); 13C NMR (75 MHz, CDCl3): δ 20.54, 20.61, 61.53, 67.84, 70.02, 73.65, 76.23, 84.49, 103.16, 111.84, 120.56, 121.36, 123.03, 124.29, 127.71, 129.41, 130.57, 131.89, 136.58, 137.12, 148.95, 150.17, 169.38, 169.51, 169.98, 170.47; FTIR cm–1: 3325 (NH), 3065 (aromatic C–H), 2944 (aliphatic C–H), 1749 (C=O), 1596 (C=C), 1493 (C=N), 744 (C–Cl); HRMS (ESI + ve mode): m/z Calcd for C30H30N4O9SCl (M+1): 657.1422. Found: 657.1420.
3-(2-Acetamido-2-deoxy-3,4,6-tri-O-acetyl-2-deoxy-β-d-glucopyranosylsulfanyl)-5-(1H-indol-2-yl)-4-phenyl-1,2,4-triazole (7a)
Yields: 65% method A, 67% method B, mp 133decomp. °C; 1H NMR (500 MHz, DMSO-d6): δ 1.75, 1.89, 1.91, 1.96 (4s, 12H), 3.81–3.85 (m, 1H), 3.88–3.94 (m, 1H), 3.98 (dd, 1H, J = 12.3, 1.9 Hz), 4.16 (dd, 1H, J = 12.3, 5.2 Hz), 4.82 (dd, 1H, J = 9.7, 9.8 Hz), 5.17 (dd, 1H, J = 9.7 Hz), 5.33 (d, 1H, J = 10.5 Hz, H-1), 5.56 (d, 1H, J = 1.55 Hz), 6.93 (dd, 1H, J = 8.0, 7.3 Hz), 7.13 (dd, 1H, J = 7.3, 8.4 Hz), 7.34 (d, 1H, J = 8.0 Hz), 7.42 (d, J = 8.4 Hz), 7.48 (d, 2H, J = 7.3 Hz), 7.63–7.70 (m, 3H), 8.11 (d, 1H, J = 9.2 Hz), 11.92 (br.s, 1H); 13C NMR (125 MHz, DMSO-d6): δ 20.21, 20.25, 20.29, 22.49, 52.26, 61.60, 68.29, 72.88, 74.88, 85.18, 101.53, 111.78, 119.66, 120.66, 123.11, 123.72, 127.05, 128.20, 129.89, 130.48, 133.70, 136.45, 148.23, 149.53, 169.14, 169.38, 169.44, 169.81; FTIR cm–1: 3312 (NH), 3171 (aromatic C–H), 2947 (aliphatic C–H), 1738 (C=O ester), 1660 (C=O amide), 1536 (C=N); HRMS (ESI + ve mode): m/z Calcd for C30H32N5O8S (M+1): 622.1972. Found: 622.1920.
3-(2-Acetamido-2-deoxy-3,4,6-tri-O-acetyl-2-deoxy-β-d-glucopyranosylsulfanyl)-4-(4-chlorophenyl)-5-(1H-indol-2-yl)-1,2,4-triazole (7b)
Yields: 66% method A, 61% method B, mp 210decomp. °C; 1H NMR (500 MHz, DMSO-d6): δ 1.74, 1.89, 1.93, 1.97 (4s, 12 H), 3.80–3.97 (m, 3 H), 4.16 (dd, 1H, J = 12.5, 5.0 Hz), 4.81 (dd, 1H, J = 9.5, 10.0 Hz), 5.15 (dd, 1H, J = 10.0, 9.5 Hz), 5.24 (d, 1H, J = 10.5 Hz), 5.67 (d, 1H, J = 2.0 Hz), 6.95 (dd, 1H, J = 8.5, 7.5 Hz), 7.14 (dd, 1H, J = 7.5, 8.5 Hz), 7.42 (d, 2H, J = 8.5 Hz), 7.56 (d, 2H, J = 8.5 Hz), 7.71 (d, 2H, J = 8.5 Hz), 8.15 (d, 1H, J = 9.5 Hz), 11.99 (br.s, 1H, NH); 13C NMR (125 MHz, DMSO-d6): δ 20.29, 20.31, 20.36, 22.53, 52.32, 61.61, 68.29, 72.88, 74.91, 85.40, 101.80, 111.86, 119.76, 120.90, 123.26, 123.62, 127.15, 130.01, 130.29, 132.72, 135.14, 136.56, 148.12, 149.59, 169.21, 169.46, 169.53, 169.88; FTIR cm–1: 3306 (NH), 3052 (aromatic C–H), 2960 (aliphatic C–H), 1743 (C=O ester), 1661 (C=O amide), 1594 (C=C), 1536 (C=N), 738 (C–Cl); HRMS (ESI + ve mode): m/z Calcd for C30H31N5O8SCl (M+1): 656.1582. Found: 656.1500.
5-(1H-Indol-2-yl)-4-phenyl-2-(2,3,4,,6-tetra-O-acetyl-β-d-glucoopyranosyl)-3-thioxo-1,2,4-triazole (8a)
Yields: 25% method A, 73% method C, mp 280decom. °C; 1H NMR (300 MHz, CDCl3): δ 1.96, 2.04, 2.06, 2.09 (4s, 12H), 4.04–4.06 (m, 1H), 4.17 (dd, 1H, J = 12.6, 2.1 Hz), 4.35 (dd, 1H, J = 12.6, 4.8 Hz), 5.29 (dd, 1H, J = 9.6, 9.9 Hz), 5.45 (dd, 1H, J = 9.3 Hz), 5.69 (d, 1H, J = 1.2 Hz), 5.88 (dd, 1H, J = 9.3 Hz), 6.27 (d, 1H, J = 9.3 Hz), 7.07 (dd, 1H, J = 7.5 Hz), 7.22–7.65 (m, 8 H), 8.96 (br.s, 1H, NH); 13C NMR (100 MHz, CDCl3): δ 20.59, 61.72 (C-6Glc), 67.89, 69.63, 73.61, 74.68, 82.80, 105.83, 111.37, 120.87, 121.22, 121.75, 125.20, 127.32, 128.48, 130.26, 130.86, 136.39, 136.99, 144.49, 169.31, 169.46, 170.04, 170.62, 171.60; FTIR cm–1: 3433 (NH), 3068 (aromatic C–H), 2964 (aliphatic C–H), 1746 (C=O ester), 1607 (C=C), 1426 (C=N); HRMS (EI): m/z Calcd for C30H30N4O9S (M+): 622.1733. Found: 622.1723.
4-(4-Chlorophenyl)-5-(1H-indol-2-yl)-2-(2,3,4,,6-tetra-O-acetyl-β-d-glucopyranosyl)-3-thioxo-1,2,4-triazole (8b)
Yields: 23% method A, 74% method C, mp >300 °C; 1H NMR (300 MHz, CDCl3): δ 1.96, 2.045, 2.065, 2.07, 4.16–4.39 (m, 1H, 4.18 (dd, 1 H, J = 12.6, 2.1 Hz = ), 4.35 (dd, 1H, J = 12.6, 5.1 Hz), 5.30 (dd, 1H, J = 9.6, 9.9 Hz), 5.45 (dd, 1H, J = 9.6 Hz), 5.75 (d, 1H, J = 1.5 Hz), 5.87 (dd, 1H, J = 9.3, 9.6 Hz), 6.25 (d, 1H, J = 9.3 Hz), 7.07 (dd, 1H, J = 8.4, 7.2 Hz), 7.24–7.30 (m 3H), 7.24–7.44 (m, 2H), 7.59 (d, 2H, J = 8.7 Hz), 9.01 (br.s, 1H, NH); 13C NMR (100 MHz, CDCl3): δ 20.60, 20.75, 20.78, 61.75, 67.83, 69.55, 73.54, 74.72, 82.77, 105.79, 111.48, 121.00, 121.24, 121.79, 125.35, 127.32, 129.88, 130.58, 132.77, 136.39, 136.99, 144.49, 169.31, 169.46, 170.04, 170.62, 171.60; FTIR cm–1: 3401 (NH), 3042 (aromatic C–H), 2955 (aliphatic C–H), 1747 (C=O ester), 1612 (C=C), 1494 (C=N); HRMS (ESI + ve mode): m/z Calcd for C30H30N4O9SCl (M+1): 657.1300. Found: 657.1420.
2-(2-Acetamido-2-deoxy-3,4,6-tri-O-acetyl-2-deoxy-β-d-glucopyranosyl)-3-thioxo-5-(1H-indol-2-yl)-4-phenyl-1,2,4-triazole (9a)
Yields: 22% method A, 85% method C, mp >270decomp. °C; 1H NMR (400 MHz, DMSO-d6): δ 1.69, 1.96, 2.01, 2.02 (4s, 12H), 4.11 (dd, 1 H, J = 12.5, 1.2 Hz), 4.14–4.17 (m, 1H), 4.30 (dd, 1H, J = 12.5, 4.5 Hz), 4.56–4.32 (m, 1H), 5.02 (dd, 1H, J = 9.8, 9.9 Hz), 5.53 (d, 1H, J = 1.1 Hz), 5.58 (dd, 1H, J = 9.9, 9.8 Hz), 6.39 (d, 1H, J = 9.5 Hz), 6.96 (dd, 1H, J = 8.0, 7.4 Hz), 7.17 (dd, 1H, J = 7.4, 7.9 Hz), 7.35 (d, 1H, J = 8.0 Hz), 7.45–7.47 (m, 3H), 7.64–7.67 (m, 3H), 8.01 (d, 1H, J = 8.7 Hz), 11.86 (br.s, 1H); 13C NMR (125 MHz, DMSO-d6): δ 20.29, 20.34, 20.53, 22.68, 52.33, 61.70, 68.23, 72.18, 73.17, 82.75, 104.15, 111.99, 120.01, 120.99, 121.82, 123.98, 126.63, 128.66, 129.95, 130.42, 134.65, 136.74, 144.25, 169.40, 169.65, 169.97, 170.26; FTIR cm–1: 3302 (NH), 3055 (aromatic C–H), 2938 (aliphatic C–H), 1744 (C=O ester), 1667 (C=O amide), 1604 (C=C), 1411 (C=N); HRMS (ESI + ve mode): m/z Calcd for C30H32N5O8S (M+1): 622.1972. Found: 622.1920.
2-(2-Acetamido-2-deoxy-3,4,6-tri-O-acetyl-2-deoxy-β-d-glucopyranosyl)-4-(4-chlorophenyl)-5-(1H-indol-2-yl)-3-thioxo-1,2,4-triazole (9b)
Yields: 26% method A, 79% method C, mp 251decomp. °C; 1H NMR (300 MHz, DMSO-d6): δ 1.69, 1.96, 2.01, 2.02 (4s, 12H), 4.09 (dd, 1H, J = 12.6, 1.3 Hz), 4.14–4.19 (m, 1H), 4.30 (dd, 1H, J = 12.6, 4.5 Hz), 4.49–4.62 (m, 1H), 5.02 (dd, 1H, J = 9.6, 9.9 Hz), 5.57 (dd, 1H, J = 9.9, 9.6 Hz), 5.68 (d, 1H, J = 1.8 Hz), 6.37 (d, 1H, J = 9.6 Hz), 6.97 (dd, 1H, J = 8.0, 7.2 Hz), 7.19 (dd, 1H, J = 7.2, J = 8.1 Hz), 7.42–7.53 (m, 4 H), 7.73 (d, 2H, J = 8.7 Hz), 8.03 (d, 1H, J = 8.7 Hz), 11.89 (br.s, 1H); 13C NMR (75 MHz, DMSO-d6): δ 20.37, 20.42, 20.61, 22.77, 52.40, 61.74, 68.24, 72.19, 73.21, 82.87, 104.36, 112.05, 120.10, 121.23, 121.74, 124.11, 126.75, 130.17, 130.77, 133.58, 135.11, 136.84, 144.19, 169.46, 169.75, 170.04, 170.27; HRMS (ESI + ve mode): m/z Calcd for C30H31N5O8SCl (M+1): 656.1582. Found: 656.1490.
3-(2-Hydroxyeth-1-ylsulfanyl)-5-(1H-indol-2-yl)-4-phenyl-1,2,4-triazole (12a)
Yield: 65%, mp 207–209 °C; 1H NMR (DMSO-d6, 500 MHz) δ 3.26 (t, 2H, J = 6.5 Hz), 3.66–3.70 (m, 2H), 5.04 (t, 1H, J = 5.0 Hz), 5.58 (d, 1H, J = 1.5 Hz), 6.93 (dd, 1H, J = 8.0, 7.0 Hz), 7.12 (dd, 1H, J = 7.0, 8.5 Hz), 7.33 (d, 1H, J = 8.0 Hz), 7.41 (d, 1H, J = 8.5 Hz), 7.53–7.55 (m, 2H), 7.64–7.69 (m, 3H), 11.92 (br.s, 1H); 13C NMR (DMSO-d6, 125 MHz) δ 34.78, 59.76, 101.22, 111.78, 119.66, 120.63, 123.02, 123.89, 127.12, 127.99, 130.19, 130.65, 133.71, 136.43, 149.03, 152.01 (C-3Trz.); FTIR cm–1: 3767 (OH), 3076 (aromatic C–H), 2932 (aliphatic C–H), 1589 (C=C), 1494 (C=N); HRMS (EI) calcd for C18H16N4OS (M+): 336.1045. Found: 336.1031.
4-(4-Chlorophenyl)-3-(2-hydroxyeth-1-ylsulfanyl)-5-(1H-indol-2-yl)-1,2,4-triazole (12b)
Yield: 70%, mp 251–252 °C; 1H NMR (DMSO-d6, 400 MHz) δ 3.26 (t, 2H, J = 6.4 Hz,), 3.65–3.70 (m, 2H), 5.03 (t, 1H, J = 5.4 Hz), 5.68 (d, 1H, J = 1.2 Hz), 6.94 (dd, 1H, J = 8.0, 7.2 Hz), 7.13 (dd, 1H, J = 7.2, 8.0 Hz), 7.39–7.43 (m, 2H), 7.62 (d, 2H, J = 8.4 Hz), 7.74 (d, 2H, J = 8.4 Hz), 11.93 (br.s, 1H); 13C NMR (DMSO-d6, 75 MHz) δ 34.96, 59.62, 101.54, 111.78, 119.79, 120.82, 123.21, 123.50, 127.14, 129.97, 130.32, 132.32, 135.34, 136.33, 148.95, 152.03; FTIR cm–1: 3730 (OH), 3365 (NH), 3154 (aromatic C–H), 2858 (aliphatic C–H), 1591 (C=C), 1488 (C=N), 799 (C–Cl); HRMS (EI) calcd for C18H15N4OSCl (M+): 370.0667. Found: 370.0661.
3-(2-Hydroxyprop-1-ylsulfanyl)-5-(1H-indol-2-yl)-4-phenyl-1,2,4-triazole (13a)
Yield: 58%, mp 232–233 °C [Lit. 23]; 1H NMR (DMSO-d6, 400 MHz) δ 1.13 (d, 3H, J = 6.0 Hz), 3.22 (d, 2 H, J = 6.0 Hz), 3.88–3.92 (m, 2H), 5.01 (d, 1H, J = 4.8 Hz), 5.57 (s, 1H), 6.93 (dd, 1H, J = 8.0, 7.2 Hz), 7.12 (dd, 1H, J = 7.2, 8.0 Hz), 7.33 (d, 1H, J = 8.0 Hz), 7.41 (d, 1H, J = 8.0 Hz), 7.53–7.56 (m, 2H), 7.64–7.66 (m, 3H), 11.92 (br.s, 1H); 13C NMR (DMSO-d6, 75 MHz) δ 22.44, 40.68, 65.12, 101.96, 111.96, 119.98, 120.86, 123.35, 123.86, 127.28, 128.13, 130.45, 130.91, 133.85, 136.47, 149.19, 152.61; FTIR cm–1: 3647 (OH), 3159 (NH), 3084 (aromatic C–H), 2922 (aliphatic C–H), 1593 (C=C), 1493 (C=N); HRMS (EI) calcd for C19H18N4OS (M+): 350.1201. Found: 350.1208.
4-(4-Chlorophenyl)-3-(2-hydroxyprop-1-ylsulfanyl)-5-(1H-indol-2-yl)-1,2,4-triazole (13b)
Yield: 64%, mp 243–244 °C; 1H NMR (DMSO-d6, 400 MHz) δ 1.13 (d, 3H, J = 6.4 Hz), 3.16–3.26 (m, 3H), 3.86–3.92 (m, 2H), 5.68 (d, 1H, J = 1.2 Hz), 6.94 (dd, 1H, J = 8.0, 7.2 Hz), 7.12 (dd, 1H, J = 7.2, 8.0 Hz), 7.39–7.42 (m, 2H), 7.62 (d, 2H, J = 8.4 Hz), 7.74 (d, 2H, J = 8.8 Hz), 11.92 (br.s, 1H); 13C NMR (DMSO-d6, 100 MHz) δ 22.44, 40.86, 65.10, 101.73, 111.96, 120.00, 121.00, 123.66, 123.81, 127.31, 130.15, 130.52, 132.75, 135.53, 136.50, 149.09, 152.54; HRMS (EI) calcd for C19H17N4OSCl (M+): 384.0812. Found: 384.0821.
Biology
Cytotoxicity
The MCF-7 and MCF-10A breast cancer cell lines, as well as the HepG2 and THLE2 liver cancer cell lines, were obtained from the National Cancer Institute in Cairo and cultured in RPMI-1640 medium l-glutamine. The cells were cultured in a medium containing 10% fetal bovine serum (FBS) and 1% penicillin-streptomycin. The samples were grown in a CO2 gas atmosphere at 37 °C. Cells were seeded at a density of 5 × 104 per well on day two and incubated with the compounds at concentrations of 0.01, 0.1, 1, 10, and 100 μM. Cell viability was measured using the MTT solution.42 The absorbance was evaluated utilizing an ELISA microplate reader, the viability percentage relative to control was determined, and IC50 values were recorded.
Apoptosis Investigation
MCF-7 and HepG2 cells were incubated overnight in 6-well culture plates (3–5 × 105 cells/well) and then treated with the IC50 values for 48 h with compound 13b. After that, the cells were incubated in a 100 μL solution of Annexin binding buffer 25 mM CaCl2, 1.4 M NaCl, and 0.1 M Hepes/NaOH, pH 7.4 in the dark for 30 min with Annexin V-FITC solution (1:100) and propidium iodide (PI) at a concentration equivalent to 10 g/mL” The labeled cells were then extracted using the Cytoflex FACS machine. CytExpert software was used to analyze the data.43,44
Enzymatic Targeting
To assess the inhibitory potency
of compound 13b against target inhibition of PARP-1 and
EGFR, we used PARP-1 (Bioscience, Cat No. 80580, CA, USA) and EGFR
(ADP-GloTM kinase assay, Cat No. V9261, Promega, USA). The following
equation was used to calculate the percentage of autophosphorylation
inhibition by drugs: Percentage inhibition = 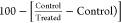 (45,46)
(45,46)
Molecular Docking
Both Chimera-UCSF and AutoDock Vina were used on Linux-based platforms for molecular modeling research. Binding sites within proteins were identified by measuring the dimensions of grid boxes surrounding the cocrystallized ligands; this process was carried out utilizing Maestro to create and optimize the structures of both proteins and compounds. Protein structures were used for docking the tested chemicals PARP-1 (PDB = 5DS3) and EGFR (PDB = 1XKK) using AutoDock Vina software.47 The protein and ligand structures were optimized and energetically favored using Vina. Molecular docking results were interpreted by binding activities in terms of binding energy and ligand–receptor interactions. Chimera was then utilized to make the visualization.
In Vivo
Ethics Statement
The experimental protocol was permitted by the Research Ethics Committee (Approval number REC135/2022), Chemistry Department, Faculty of Science, Suez Canal University.
Animal, Tumor Inoculation Experiment Design
A total of 40 male Swiss albino mice (weighing between 21 and 28 g) were randomly separated into four groups: normal control, SEC control, SEC + 13b, and SEC + Erlotinib. After 10 days of tumor cell inoculation, tumor masses appeared where SEC cells (1 × 106 tumor cells/mouse) had been put subcutaneously into the right thigh of the hind limb. On day seven post-tumor inoculation, the 13b and Erlotinib 5 mg/kg BW, IP” were administered in seven doses.48 Finally, the size of the solid tumor masses was evaluated. The experiment was concluded by sacrificing several groups of animals and collecting blood samples for the hematological parameters Hb, RBC, and WBC levels, serum for the assessment of liver enzyme levels ALT, AST, and total protein, and histopathological examinations.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsomega.2c06531.
Characterization analyses (Figures S1–S47) (PDF)
Author Contributions
M.F.Y., E.E.S., A.T.B., and E.M.G. designed the idea of synthetic organic chemistry and made formal analyses of characterization charts, while M.S.N. designed the idea and carried out all biological analyses with the in vivo study and the molecular docking study. All authors contributed to writing the manuscript with their corresponding parts and agreed to the final manuscript form.
The authors declare no competing financial interest.
Supplementary Material
References
- Sung H.; Ferlay J.; Siegel R. L.; Laversanne M.; Soerjomataram I.; Jemal A.; Bray F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA: A Cancer Journal for Clinicians 2021, 71 (3), 209–249. 10.3322/caac.21660. [DOI] [PubMed] [Google Scholar]
- Almahli H.; Hadchity E.; Jaballah M. Y.; Daher R.; Ghabbour H. A.; Kabil M. M.; Al-shakliah N. S.; Eldehna W. M. Development of Novel Synthesized Phthalazinone-Based PARP-1 Inhibitors with Apoptosis Inducing Mechanism in Lung Cancer. Bioorganic Chemistry 2018, 77, 443–456. 10.1016/j.bioorg.2018.01.034. [DOI] [PubMed] [Google Scholar]
- Bourton E. C.; Ahorner P.-A.; Plowman P. N.; Zahir S. A.; Al-Ali H.; Parris C. N. The PARP-1 Inhibitor Olaparib Suppresses BRCA1 Protein Levels, Increases Apoptosis and Causes Radiation Hypersensitivity in BRCA1± Lymphoblastoid Cells. J. Cancer 2017, 8 (19), 4048–4056. 10.7150/jca.21338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo C.; Wang L.; Li X.; Wang S.; Yu X.; Xu K.; Zhao Y.; Luo J.; Li X.; Jiang B.; Shi D. Discovery of Novel Bromophenol–Thiosemicarbazone Hybrids as Potent Selective Inhibitors of Poly(ADP-Ribose) Polymerase-1 (PARP-1) for Use in Cancer. J. Med. Chem. 2019, 62 (6), 3051–3067. 10.1021/acs.jmedchem.8b01946. [DOI] [PubMed] [Google Scholar]
- Higgins G. S.; Boulton S. J. Beyond PARP—POLθ as an Anticancer Target. Science 2018, 359 (6381), 1217–1218. 10.1126/science.aar5149. [DOI] [PubMed] [Google Scholar]
- Li X.; Li C.; Jin J.; Wang J.; Huang J.; Ma Z.; Huang X.; He X.; Zhou Y.; Xu Y.; Yu M.; Huang S.; Yan X.; Li F.; Pan J.; Wang Y.; Yu Y.; Jin J. High PARP-1 Expression Predicts Poor Survival in Acute Myeloid Leukemia and PARP-1 Inhibitor and SAHA-Bendamustine Hybrid Inhibitor Combination Treatment Synergistically Enhances Anti-Tumor Effects. eBioMedicine 2018, 38, 47–56. 10.1016/j.ebiom.2018.11.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou J.; Ji M.; Zhu Z.; Cao R.; Chen X.; Xu B. Discovery of 2-Substituted 1H-Benzo[d]Immidazole-4-Carboxamide Derivatives as Novel Poly(ADP-Ribose)Polymerase-1 Inhibitors with in Vivo Anti-Tumor Activity. Eur. J. Med. Chem. 2017, 132, 26–41. 10.1016/j.ejmech.2017.03.013. [DOI] [PubMed] [Google Scholar]
- Richard J.; Sainsbury C.; Needham G. K.; Farndon J. R.; Malcolm A. J.; Harris A. L. Epidermal-Growth-Factor Receptor Status as Predictor of Early Recurrence of and Death from Breast Cancer. Lancet 1987, 329 (8547), 1398–1402. 10.1016/S0140-6736(87)90593-9. [DOI] [PubMed] [Google Scholar]
- Salomon D. S.; Brandt R.; Ciardiello F.; Normanno N. Epidermal Growth Factor-Related Peptides and Their Receptors in Human Malignancies. Crit Rev. Oncol Hematol 1995, 19 (3), 183–232. 10.1016/1040-8428(94)00144-I. [DOI] [PubMed] [Google Scholar]
- Burness M. L.; Grushko T. A.; Olopade O. I. Epidermal Growth Factor Receptor in Triple-Negative and Basal-like Breast Cancer: Promising Clinical Target or Only a Marker?. Cancer J. 2010, 16 (1), 23–32. 10.1097/PPO.0b013e3181d24fc1. [DOI] [PubMed] [Google Scholar]
- Rakha E. A.; El-Sayed M. E.; Green A. R.; Lee A. H. S.; Robertson J. F.; Ellis I. O. Prognostic Markers in Triple-Negative Breast Cancer. Cancer 2007, 109 (1), 25–32. 10.1002/cncr.22381. [DOI] [PubMed] [Google Scholar]
- Guérin M.; Gabillot M.; Mathieu M. C.; Travagli J. P.; Spielmann M.; Andrieu N.; Riou G. Structure and Expression of C-ErbB-2 and EGF Receptor Genes in Inflammatory and Non-Inflammatory Breast Cancer: Prognostic Significance. Int. J. Cancer 1989, 43 (2), 201–208. 10.1002/ijc.2910430205. [DOI] [PubMed] [Google Scholar]
- Masuda H.; Zhang D.; Bartholomeusz C.; Doihara H.; Hortobagyi G. N.; Ueno N. T. Role of Epidermal Growth Factor Receptor in Breast Cancer. Breast Cancer Res. Treat 2012, 136 (2), 331–345. 10.1007/s10549-012-2289-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li N.; Feng L.; Liu H.; Wang J.; Kasembeli M.; Tran M. K.; Tweardy D. J.; Lin S. H.; Chen J. PARP Inhibition Suppresses Growth of EGFR-Mutant Cancers by Targeting Nuclear PKM2. Cell Rep 2016, 15 (4), 843–856. 10.1016/j.celrep.2016.03.070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas S. M.; Grandis J. R. Pharmacokinetic and Pharmacodynamic Properties of EGFR Inhibitors under Clinical Investigation. Cancer Treat Rev. 2004, 30 (3), 255–268. 10.1016/j.ctrv.2003.10.003. [DOI] [PubMed] [Google Scholar]
- Woodburn J. R. The Epidermal Growth Factor Receptor and Its Inhibition in Cancer Therapy. Pharmacol Ther 1999, 82 (2–3), 241–250. 10.1016/S0163-7258(98)00045-X. [DOI] [PubMed] [Google Scholar]
- Figueroa-Magalhães M. C.; Jelovac D.; Connolly R.; Wolff A. C. Treatment of HER2-Positive Breast Cancer. Breast 2014, 23 (2), 128–136. 10.1016/j.breast.2013.11.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blackwell K. L.; Burstein H. J.; Storniolo A. M.; Rugo H.; Sledge G.; Koehler M.; Ellis C.; Casey M.; Vukelja S.; Bischoff J.; Baselga J.; O’Shaughnessy J. Randomized Study of Lapatinib Alone or in Combination with Trastuzumab in Women with ErbB2-Positive, Trastuzumab-Refractory Metastatic Breast Cancer. J. Clin Oncol 2010, 28 (7), 1124–1130. 10.1200/JCO.2008.21.4437. [DOI] [PubMed] [Google Scholar]
- Medina P. J.; Goodin S. Lapatinib: A Dual Inhibitor of Human Epidermal Growth Factor Receptor Tyrosine Kinases. Clin Ther 2008, 30 (8), 1426–1447. 10.1016/j.clinthera.2008.08.008. [DOI] [PubMed] [Google Scholar]
- Papeo G.; Posteri H.; Borghi D.; Busel A. A.; Caprera F.; Casale E.; Ciomei M.; Cirla A.; Corti E.; D’Anello M.; Fasolini M.; Forte B.; Galvani A.; Isacchi A.; Khvat A.; Krasavin M. Y.; Lupi R.; Orsini P.; Perego R.; Pesenti E.; Pezzetta D.; Rainoldi S.; Riccardi-Sirtori F.; Scolaro A.; Sola F.; Zuccotto F.; Felder E. R.; Donati D.; Montagnoli A. Discovery of 2-[1-(4,4-Difluorocyclohexyl)Piperidin-4-Yl]-6-Fluoro-3-Oxo-2,3-Dihydro-1H-Isoindole-4-Carboxamide (NMS-P118): A Potent, Orally Available, and Highly Selective PARP-1 Inhibitor for Cancer Therapy. J. Med. Chem. 2015, 58 (17), 6875–6898. 10.1021/acs.jmedchem.5b00680. [DOI] [PubMed] [Google Scholar]
- Fong P. C.; Boss D. S.; Yap T. A.; Tutt A.; Wu P.; Mergui-Roelvink M.; Mortimer P.; Swaisland H.; Lau A.; O’Connor M. J.; Ashworth A.; Carmichael J.; Kaye S. B.; Schellens J. H. M.; de Bono J. S. Inhibition of Poly(ADP-Ribose) Polymerase in Tumors from BRCA Mutation Carriers. New England Journal of Medicine 2009, 361 (2), 123–134. 10.1056/NEJMoa0900212. [DOI] [PubMed] [Google Scholar]
- Sidhu J. S.; Singla R.; Mayank; Jaitak V. Indole Derivatives as Anticancer Agents for Breast Cancer Therapy: A Review. Anticancer Agents Med. Chem. 2015, 16 (2), 160–173. 10.2174/1871520615666150520144217. [DOI] [PubMed] [Google Scholar]
- Sever B.; Altıntop M. D.; Özdemir A.; Akalın Çiftçi G.; Ellakwa D. E.; Tateishi H.; Radwan M. O.; Ibrahim M. A. A.; Otsuka M.; Fujita M.; Ciftci H. I.; Ali T. F. S. In Vitro and In Silico Evaluation of Anticancer Activity of New Indole-Based 1,3,4-Oxadiazoles as EGFR and COX-2 Inhibitors. Molecules 2020, 25 (21), 5190. 10.3390/molecules25215190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sever B.; Altıntop M. D.; Kuş G.; Özkurt M.; Özdemir A.; Kaplancıklı Z. A. Indomethacin Based New Triazolothiadiazine Derivatives: Synthesis, Evaluation of Their Anticancer Effects on T98 Human Glioma Cell Line Related to COX-2 Inhibition and Docking Studies. Eur. J. Med. Chem. 2016, 113, 179–186. 10.1016/j.ejmech.2016.02.036. [DOI] [PubMed] [Google Scholar]
- Rathi A. K.; Syed R.; Singh V.; Shin H.-S.; Patel R. V. Kinase Inhibitor Indole Derivatives as Anticancer Agents: A Patent Review. Recent Pat Anticancer Drug Discov 2017, 12 (1), 55–72. 10.2174/1574892811666161003112119. [DOI] [PubMed] [Google Scholar]
- Özdemir A.; Sever B.; Altıntop M. D.; Temel H. E.; Atlı Ö.; Baysal M.; Demirci F. Synthesis and Evaluation of New Oxadiazole, Thiadiazole, and Triazole Derivatives as Potential Anticancer Agents Targeting MMP-9. Molecules 2017, 22 (7), 1109. 10.3390/molecules22071109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Al Sheikh Ali A.; Khan D.; Naqvi A.; Al-blewi F. F.; Rezki N.; Aouad M. R.; Hagar M. Design, Synthesis, Molecular Modeling, Anticancer Studies, and Density Functional Theory Calculations of 4-(1,2,4-Triazol-3-Ylsulfanylmethyl)-1,2,3-Triazole Derivatives. ACS Omega 2021, 6 (1), 301–316. 10.1021/acsomega.0c04595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sever B.; Altıntop M. D.; Çiftçi G. A.; Özdemir A. A New Series of Triazolothiadiazines as Potential Anticancer Agents for Targeted Therapy of Non-Small Cell Lung and Colorectal Cancers: Design, Synthesis, In Silico and In Vitro Studies Providing Mechanistic Insight into Their Anticancer Potencies. Med. Chem. 2021, 17 (10), 1104–1128. 10.2174/1573406416666201021142832. [DOI] [PubMed] [Google Scholar]
- Boraei A. T. A.; Gomaa M. S.; El Ashry E. S. H.; Duerkop A. Design, Selective Alkylation and X-Ray Crystal Structure Determination of Dihydro-Indolyl-1,2,4-Triazole-3-Thione and Its 3-Benzylsulfanyl Analogue as Potent Anticancer Agents. Eur. J. Med. Chem. 2017, 125, 360–371. 10.1016/j.ejmech.2016.09.046. [DOI] [PubMed] [Google Scholar]
- Boraei A. T. A.; Singh P. K.; Sechi M.; Satta S. Discovery of Novel Functionalized 1,2,4-Triazoles as PARP-1 Inhibitors in Breast Cancer: Design, Synthesis and Antitumor Activity Evaluation. Eur. J. Med. Chem. 2019, 182, 111621. 10.1016/j.ejmech.2019.111621. [DOI] [PubMed] [Google Scholar]
- Mohamady S.; Galal M.; Eldehna W. M.; Gutierrez D. C.; Ibrahim H. S.; Elmazar M. M.; Ali H. I. Dual Targeting of VEGFR2 and C-Met Kinases via the Design and Synthesis of Substituted 3-(Triazolo-Thiadiazin-3-Yl)Indolin-2-One Derivatives as Angiogenesis Inhibitors. ACS Omega 2020, 5 (30), 18872–18886. 10.1021/acsomega.0c02038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nafie M. S.; Boraei A. T. A. Exploration of Novel VEGFR2 Tyrosine Kinase Inhibitors via Design and Synthesis of New Alkylated Indolyl-Triazole Schiff Bases for Targeting Breast Cancer. Bioorganic Chemistry 2022, 122, 105708. 10.1016/j.bioorg.2022.105708. [DOI] [PubMed] [Google Scholar]
- El-Sherief H. A. M.; Youssif B. G. M.; Abbas Bukhari S. N.; Abdelazeem A. H.; Abdel-Aziz M.; Abdel-Rahman H. M. Synthesis, Anticancer Activity and Molecular Modeling Studies of 1,2,4-Triazole Derivatives as EGFR Inhibitors. Eur. J. Med. Chem. 2018, 156, 774–789. 10.1016/j.ejmech.2018.07.024. [DOI] [PubMed] [Google Scholar]
- Boraei A. T. A.; Singh P. K.; Sechi M.; Satta S. Discovery of Novel Functionalized 1,2,4-Triazoles as PARP-1 Inhibitors in Breast Cancer: Design, Synthesis and Antitumor Activity Evaluation. Eur. J. Med. Chem. 2019, 182, 111621. 10.1016/j.ejmech.2019.111621. [DOI] [PubMed] [Google Scholar]
- Schettino C.; Bareschino M. A.; Ricci V.; Ciardiello F. Erlotinib: An EGF Receptor Tyrosine Kinase Inhibitor in Non-Small-Cell Lung Cancer Treatment. Expert Rev. Respir Med. 2008, 2 (2), 167–178. 10.1586/17476348.2.2.167. [DOI] [PubMed] [Google Scholar]
- Gunderson C. C.; Moore K. N. Olaparib: An Oral PARP-1 and PARP-2 Inhibitor with Promising Activity in Ovarian Cancer. Future Oncol 2015, 11 (5), 747–757. 10.2217/fon.14.313. [DOI] [PubMed] [Google Scholar]
- Lipinski C. A. Lead- and Drug-like Compounds: The Rule-of-Five Revolution. Drug Discovery Today: Technologies 2004, 1 (4), 337–341. 10.1016/j.ddtec.2004.11.007. [DOI] [PubMed] [Google Scholar]
- Nafie M. S.; Arafa K.; Sedky N. K.; Alakhdar A. A.; Arafa R. K. Triaryl Dicationic DNA Minor-Groove Binders with Antioxidant Activity Display Cytotoxicity and Induce Apoptosis in Breast Cancer. Chemico-Biological Interactions 2020, 324, 109087. 10.1016/j.cbi.2020.109087. [DOI] [PubMed] [Google Scholar]
- Gad E. M.; Nafie M. S.; Eltamany E. H.; Hammad M. S. A. G.; Barakat A.; Boraei A. T. A. Discovery of New Apoptosis-Inducing Agents for Breast Cancer Based on Ethyl 2-Amino-4,5,6,7-Tetra Hydrobenzo[b]Thiophene-3-Carboxylate: Synthesis, In Vitro, and In Vivo Activity Evaluation. Molecules 2020, 25 (11), 2523. 10.3390/molecules25112523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ElZahabi H. S. A.; Nafie M. S.; Osman D.; Elghazawy N. H.; Soliman D. H.; EL-Helby A. A. H.; Arafa R. K. Design, Synthesis and Evaluation of New Quinazolin-4-One Derivatives as Apoptotic Enhancers and Autophagy Inhibitors with Potent Antitumor Activity. Eur. J. Med. Chem. 2021, 222, 113609. 10.1016/j.ejmech.2021.113609. [DOI] [PubMed] [Google Scholar]
- Nafie M. S.; Arafa K.; Sedky N. K.; Alakhdar A. A.; Arafa R. K. Triaryl Dicationic DNA Minor-Groove Binders with Antioxidant Activity Display Cytotoxicity and Induce Apoptosis in Breast Cancer. Chemico-Biological Interactions 2020, 324, 109087. 10.1016/j.cbi.2020.109087. [DOI] [PubMed] [Google Scholar]
- Mosmann T. Rapid Colorimetric Assay for Cellular Growth and Survival: Application to Proliferation and Cytotoxicity Assays. Journal of immunological methods 1983, 65 (1–2), 55–63. 10.1016/0022-1759(83)90303-4. [DOI] [PubMed] [Google Scholar]
- Boraei A. T. A.; Eltamany E. H.; Ali I. A. I.; Gebriel S. M.; Nafie M. S. Synthesis of New Substituted Pyridine Derivatives as Potent Anti-Liver Cancer Agents through Apoptosis Induction: In Vitro, in Vivo, and in Silico Integrated Approaches. Bioorganic Chemistry 2021, 111, 104877. 10.1016/j.bioorg.2021.104877. [DOI] [PubMed] [Google Scholar]
- Nafie M. S.; Amer A. M.; Mohamed A. K.; Tantawy E. S. Discovery of Novel Pyrazolo[3,4-b]Pyridine Scaffold-Based Derivatives as Potential PIM-1 Kinase Inhibitors in Breast Cancer MCF-7 Cells. Bioorg. Med. Chem. 2020, 28 (24), 115828. 10.1016/j.bmc.2020.115828. [DOI] [PubMed] [Google Scholar]
- Elgawish M. S.; Nafie M. S.; Yassen A. S. A.; Yamada K.; Ghareb N. The Design and Synthesis of Potent Benzimidazole Derivatives via Scaffold Hybridization and Evaluating Their Antiproliferative and Proapoptotic Activity against Breast and Lung Cancer Cell Lines. New J. Chem. 2022, 46 (9), 4239–4256. 10.1039/D1NJ05655G. [DOI] [Google Scholar]
- Nafie M. S.; Khodair A. I.; Hassan H. A. Y.; El-Fadeal N. M. A.; Bogari H. A.; Elhady S. S.; Ahmed S. A. Evaluation of 2-Thioxoimadazolidin-4-One Derivatives as Potent Anti-Cancer Agents through Apoptosis Induction and Antioxidant Activation: In Vitro and In Vivo Approaches. Molecules 2022, 27 (1), 83. 10.3390/molecules27010083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trott O.; Olson A. J. AutoDock Vina: Improving the Speed and Accuracy of Docking with a New Scoring Function, Efficient Optimization, and Multithreading. J. Comput. Chem. 2009, 31 (2), 455–461. 10.1002/jcc.21334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nafie M. S.; Elghazawy N. H.; Owf S. M.; Arafa K.; Abdel-Rahman M. A.; Arafa R. K. Control of ER-Positive Breast Cancer by ERα Expression Inhibition, Apoptosis Induction, Cell Cycle Arrest Using Semisynthetic Isoeugenol Derivatives. Chemico-Biological Interactions 2022, 351, 109753. 10.1016/j.cbi.2021.109753. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.