

Abstract
Introduction
Limbic predominant age related TDP‐43 encephalopathy neuropathological change (LATE‐NC) is a recently characterized brain disease that mimics Alzheimer's disease (AD) clinically. To date, LATE‐NC is difficult to diagnose antemortem using clinical information or biomarkers. Recent studies suggest concentrations of extracellular vesicle (EVs) protein cargo derived from neuronal and glial cells may serve as useful diagnostic biomarkers for AD and other neurodegenerative diseases.
Methods
TDP‐43 was evaluated in neuronal (NDEVs), astrocyte (ADEVs), and microglial derived extracellular vesicles (MDEVs). EV preparations were isolated from the plasma of research subjects with autopsy‐confirmed diagnoses, including many with LATE (n = 22). Quantified TDP‐43 concentrations were compared to the cohort that included healthy controls, mild cognitively impairment (MCI), and AD dementia with diagnoses other than LATE‐NC (n = 42).
Results
TDP‐43 was significantly elevated in plasma ADEVs derived from autopsy confirmed LATE‐NC subjects, with or without comorbid AD pathology. Measurable levels of TDP‐43 were also detected in EV‐depleted plasma; however, TDP‐43 levels were not significantly different between persons with and without eventual autopsy confirmed LATE‐NC. No correlation was observed between EV TDP‐43 levels with cognition‐based variables, sex, and APOE carrier status.
Discussion
Blood‐based EVs, specifically measuring TDP‐43 accumulation in ADEVs, may serve as a potential diagnostic tool to rapidly identify subjects who are currently living with LATE‐NC.
Keywords: Alzheimer's Disease, biomarkers; TDP‐43, extracellular vesicles, Limbic predominant age related TDP‐43 encephalopathy (LATE)
1. INTRODUCTION
Abnormal accumulation of TAR DNA binding protein43 (TDP‐43) in its phosphorylated state has been associated with cognitive dysfunction and neurodegeneration in subjects who suffer from frontotemporal lobar degeneration (FTLD‐TDP), 1 , 2 amyotrophic lateral sclerosis (ALS), 1 , 3 and Alzheimer's disease (AD). 4 Recently, limbic predominant age‐related TDP43 encephalopathy (LATE) neuropathological change (NC) was characterized as a distinct condition that is primarily marked by the accumulation of TDP‐43 proteinopathy in the hippocampus and entorhinal cortex of older adults. 5 Subjects with LATE‐NC often have comorbid brain pathologies, including amyloid‐β plaques and tau pathology, and display similar clinical manifestations to those who suffer from AD. 5 To date, LATE can only be diagnosed retrospectively (essentially impossible to diagnose with confidence in living subjects).
The need to identify neurodegenerative disease in the clinical setting, and at early and more treatable timepoints, has fueled research into blood‐based biomarkers. Increased plasma concentrations of TDP‐43 have been observed in subjects with FTLD 2 ; however, its correlation with cognitive dysfunction and disease progression in AD is not well established. 6 Moreover, elevated plasma concentrations of TDP‐43 in subjects with LATE have yet to be described.
RESEARCH IN CONTEXT
Systematic Review: The authors reviewed the literature using traditional (e.g., PubMed, medical literature, abstracts, presentations) sources. A consensus working group report recently described limbic predominant age‐related TDP43 encephalopathy (LATE) neuropathological change (NC) as a distinct condition, separate from those diagnosed with pathologically confirmed Alzheimer's disease (AD). However, elevated plasma concentrations of TDP‐43 in subjects with LATE have yet to be described. The relevant citations describing the cognitive manifestations, public health impact, and genetics LATE and AD are properly cited.
Interpretation: Measurable levels of TDP‐43 can be quantified in the plasma of healthy controls and demented patients. Our findings describe, for the first time, that levels of TDP‐43 quantified in plasma astrocyte derived extracellular vesicles (ADEVs) can accurately identify subjects with LATE‐NC.
Future Directions: LATE is an under‐recognized condition with a large impact on public health, for which the lack of a clinical biomarker, is a glaring unmet need. Continued work is urgently required to identify those suffering from the neurodegenerative disease at earlier and more treatable timepoints. Ultimately, our study must be validated in a larger and more ethnically and socioeconomically diverse patient cohort.
Extracellular vesicles (EVs) of endocytic origin are released from various cell types, including neurons and glial cells. 7 Their diverse contents, including DNA, mRNA, and proteins, are thought to be reflective of the intracellular environment of the parent cell. 7 Secreted EVs from neuronal cells are thought to mediate the propagation of TDP‐43 in ALS and AD brains. Neuronal (NDEVs) and astrocyte (ADEVs) derived extracellular vesicles have been successfully isolated from the plasma of demented subjects and healthy controls. 8 , 9 , 10 , 11 , 12 While numerous studies suggest that protein cargo extracted from NDEVs and ADEVs may serve as useful diagnostic biomarkers for AD and other neurodegenerative diseases, 8 , 9 , 10 , 11 , 12 very few studies have characterized the biomarker potential of protein cargo extracted from microglial derived EVs (MDEVs). 13 , 14
In the current study, TDP‐43 accumulation was quantified in NDEV, ADEV, and MDEV preparations isolated from the plasma of research subjects with autopsy‐confirmed diagnoses, including many with LATE neuropathologic changes (LATE‐ NC(+)). Quantified TDP‐43 concentrations were compared in a cohort that included healthy controls, mild cognitively impairment (MCI), and AD dementia with diagnoses other than LATE‐NC (‐).
2. METHODS
2.1. Blood collection and baseline characteristics of subjects
Plasma samples from autopsy‐validated LATE‐NC positive and negative cases were acquired through the University of Kentucky Alzheimer's Disease Research Center Biobank. Fasting plasma samples were collected and processed within 2 hours following collection into EDTA tubes. Samples were spun at 2000×g for 10 minutes at 4°C, and aliquoted into 250 μl aliquots. Aliquots were immediately frozen on dry ice and moved into –80°C for long‐term storage within 8 hours.
Patient characterization details are listed in Table 1 . LATE positive subjects had neuropathological changes associated with LATE‐NC (+) and LATE‐NC (–) subjects had no TDP‐43 proteinopathy. 5 , 15 Final clinical diagnoses were rendered at a consensus conference and varied between cognitively normal controls (CNC); subjects with an established diagnosis of mild cognitive impairment (MCI), possible AD (AD), and subjects with probable AD.
TABLE 1.
Patient demographics for the 64 patients’ blood samples that were included in the study. EV preparations were enriched against neuronal (NDEVs), astrocyte (ADEVs), and microglial sources (MDEVs). EV cargo protein, TDP‐43 was measured by human ELISA. Categorical variables are summarized using counts and percentages (%) with continuous variables summarized using means and standard deviation (SD). Two‐tailed t‐test; LATE‐NC (–) versus LATE‐ NC(+)
| Patient demographic data (n = 64) | ||||
|---|---|---|---|---|
| Characteristic, P value |
LATE – NC (–) (N = 42) |
LATE – NC (+) (N = 22) |
Combined (N = 64) |
|
| Age of death, mean (SD) P = .936 | 80 (9.6) | 84 (9.7) | 81 (9.8) | |
| MMSE, mean (SD) P < .05 | 22 (8.0) | 17 (11) | 21 (9.2) | |
| Yrs of Ed mean (SD), P = .281 | 16 (3.6) | 18 (2.9) | 17 (3.4) | |
| Last Clinical Index | Control | 13 (31%) | 3 (13.6%) | 16 (25%) |
| MCI or Impaired | 9 (21.4%) | 3 (13.6%) | 12 (18.75%) | |
| Demented (AD) | 20 (47.6%) | 16 (72.7%) | 36 (56.25%) | |
| Sex | Female | 21 (50.0%) | 9 (40.9%) | 30 (46.9%) |
| Male | 21 (50.0%) | 13 (59.1%) | 34 (53.1%) | |
| APOEε4 | Negative | 24 (57.1%) | 10 (45.5%) | 35 (54.7%) |
| Positive | 17 (40.5%) | 11 (50%) | 28 (43.8%) | |
| (Missing) | 1 (2.4%) | 1 (4.5%) | 2 (1.5%) | |
| Race | White | 40 (95.2%) | 21 (95.5%) | 61 (95.3%) |
|
Black or African American |
2 (4.8%) | 1 (4.5%) | 3 (4.7%) | |
| Hispanics | 0 (0.0%) | 0 (0.0%) | 0 (0.0%) | |
| Asian | 0 (0.0%) | 0 (0.0%) | 0 (0.0%) | |
| American Indian or Alaska Native | 0 (0.0%) | 0 (0.0%) | 0 (0.0%) | |
2.2. Isolation and neuronal enrichment of blood‐based EVs derived neuronal (NDEVs), astrocyte (ADEVs), and microglial (MDEVs) sources
EVs isolation was conducted as previously described. 1 Briefly, 500 μl of human plasma were incubated with 5 μl purified thrombin (System Biosciences, Inc.; Catalog # TMEXO‐1) at room temperature (RT) for 5 minutes. After centrifugation at 10,000 rmp for 5 minutes, supernatants were incubated with 120 μl ExoQuick exosome precipitation solution (System Biosciences, Inc.; Catalog # EXOQ5TM‐1) for 30 minutes at 4°C. Resultant suspensions were centrifuged at 1500×g for 1 hour at 4°C. The supernatant was collected and the resultant pellet was suspended in 350 μl of 1× phosphate buffer saline (PBS) (diluted from 10× PBS; Thermo Fisher Scientific; Catalog# AM9625) with Halt protease and phosphatase inhibitor cocktail EDTA‐free (Thermo Fisher Scientific; Catalog # 78443) and stored at −80°C until immunochemical enrichment of EVs from both neuronal and glial sources.
Neuronal, astrocyte, and microglial enrichment was conducted per the manufacturer's instructions (System Biosciences, Inc.; Catalog # CSFLOWBASICA‐1). Briefly, 45 μl of 9.1 μm, streptavidin magnetic Exo‐Flow beads (System Biosciences, Inc.; Catalog # CSFLOWBASICA‐1) were incubated with 100 ng/μl of mouse anti‐human CD171 (L1‐CAM, neural adhesion protein) biotinylated antibody (clone 5G3, eBioscience/Thermo Fisher Scientific; Catalog # 13‐1719‐82); mouse anti‐human GLAST (ACSA‐1) biotinylated antibody (Miltenyi Biotec, Inc., Auburn, CA, USA; Catalog # 130‐118‐984; or Purified anti‐TMEM119 (Extracellular) Antibody (Biolegend, Catalog # 853302) for 2 hours on the ice, with gently flicking every 30 minutes to mix. Anti‐TMEM119 was biotinylated prior to EV enrichment (biotinylation Kit / biotin conjugation kit (Fast, Type A)—Lightning‐LinK, Catalog # ab201795). Bead‐antibody (Ab) complex was washed three times in bead wash buffer (Systems Biosciences, Inc.; CSFLOWBASICA‐1) using a magnetic stand. Bead‐Ab complex was suspended with 400 μl of bead wash buffer and 100 μl of total EV suspensions rotating overnight at 4°C. Bead‐Ab‐EV (BAE) complex was washed three times with bead wash buffer and then suspended in 240 μl of Exosome stain buffer and 10 μl of Exo‐FITC Exosome FACS stain (Systems Biosciences, Inc.; Catalog # CSFLOWBASICA‐1) for 2 hours on ice, with gently flicking to mix. BAE‐FITC complex was washed three times in bead wash buffer and then suspended in 300 μl of bead wash buffer prior to loading into BD FACS Aria II for sorting. Flow‐sorted, BAE‐FITC complexes were incubated with 350 μl of exosome elution buffer (System Biosciences, Inc.; Catalog # CSFLOWBASICA‐1) at 25°C for 30 minutes. Finally, the supernatant containing eluted EVs were incubated with 1 μl of Exo‐FlowIP clearing reagent (System Biosciences, Inc.; Catalog # EXOFLOW32A) at 37°C for 30 minutes then stored at −80°C.
Nanoparticle Tracking Analysis (NTA) was used to characterize L1‐CAM‐positive (NDEVs), GLAST‐positive (ADEVs), and TMEM119‐positive (MDEVs) based on size distribution. EV preparations were confirmed by tetraspanin EV marker CD81 (Cusabio, American Research Products, Waltham, MA, USA; Catalog # CSB‐EL004960HU).
2.2.1. TDP‐43 assay
Protein concentrations for all EV preparations were determined using a bicinchoninic acid (BCA) Protein Assay kit (Pierce Biotechnology). EV cargo proteins were quantified by commercially available human‐specific ELISAs kits for Human TAR DNA‐binding protein 43 (TARDBP/TDP‐43) (CSB‐E17007h; Cusabio; Waltham, MA); Human TDP‐43 DuoSET ELISA kit DuoSet Ancillary Reagent Kit 2 (#DY008, R&D Systems, Minneapolis, MN, USA), and tetraspanin EV marker CD81 (CSB‐EL004960HU; Cusabio; Waltham, MA) according to suppliers' directions.
Briefly, 96 well plates were coated overnight with capture Ab Human Recombinant TDP‐43 (mAb 5044.48.42(7‐12‐16), IgG1, aa 261 – 393, gift from Dr. John Q. Trojanowski, UPENN), washed 3× in 1× wash buffer (895003, R&D Systems), blocked in 1% BSA for 1 hour at RT. Coated plates were washed 3× in 1× wash buffer, air dried overnight at 37°C and then stored at 4°C in sealed plastic covers prior to use. Coated plates are incubated with 100 μl EV preparations and standards overnight at 4°C followed by Human TDP‐43 ‐ biotin detection antibody (Recombinant Anti‐TDP43 antibody (ab255922) + biotinylation kit/biotin conjugation kit (Type A) (ab201795)) for 2 hours at RT. Signal detection of TDP‐43 protein concentration was quantified by Streptavidin‐HRP and then measured at an optical density of 450 nm using a microplate reader (Bio‐Rad, Hercules, California, USA). Absorbance values were converted into concentrations using a standard curve of TDP‐43 in the range of 0–20 ng/ml with a detection range from 0.198 to 20 ng/ml. Any value that fell below this detectable range was marked as N.D. and excluded. For the ADEV assay, over 95% of the readings fell below the cutoff of 0.198, there a value of 0 was added in order to complete statistical analyses. The mean value for all determinations of CD81 in each assay group was set at 1.00, and the relative values for each sample were used to normalize their recovery.
2.3. Statistical analyses
The statistical significance of differences between means for cross‐sectional patient groups and their respective control group was determined by an unpaired, non‐parametric Mann–Whitney t‐test or a one‐way ANOVA with Newman‐Keuls Multiple Comparison post hoc test (Prism 9; GraphPad Software, La Jolla, CA, USA).
3. RESULTS
Sixty‐four subjects were included in the study: 22 autopsy confirmed LATE‐NC (+) (mean age at death, 84 ± 9.7 years) and 42, age‐matched subjects without LATE‐NC (‐) diagnosis (mean age at death, 80 ± 9.6 years, P = .281). The percentage of APOE ε4 carriers (50% vs. 40.5%) and the percentage of males versus females (males, 46.9%, females, 53.1%) were similar across both groups. The LATE‐NC (+) group contained a significantly higher number of autopsy‐confirmed AD neuropathologic changes (ADNC) 16 (72.7% vs. 47.6%, P < .05) with a lower MMSE score (17 vs. 22, P < .05) as compared to the LATE‐NC (‐) group.
Plasma NDEVs, ADEVs, and MDEVs were characterized by FACs sorting, 8 nanoparticle tracking analysis (NTA) 9 and human specific ELISAs. The significant degree of flow separation from the non‐EV negative control suggested a successful enrichment of EV preparations against neuronal, astrocyte, and microglial sources (Figure 1A ). Extracted plasma EVs were similar in size to previously published studies with an average distribution size of 94.33 ± 4.86 nm for NDEVs, 95.67 ± 3.03 nm for ADEVs, and 83.50 ± 1.4 nm for MDEVs (Figure 1B , P < ns) 9 . Assessments of the EV membrane marker CD81 confirmed resultant EV preparations that were derived from LATE‐NC (+) and LATE‐NC (–) samples. Concentrations of CD81 were not statistically different between NDEV (9.045 ± 0.299 ng/ml), ADEV (10.52 ± 0.4635 ng/ml), and MDEV (11.63 ± 0.533 ng/ml) preparations (Figure 1C , One‐way Anova, P = ns).
FIGURE 1.
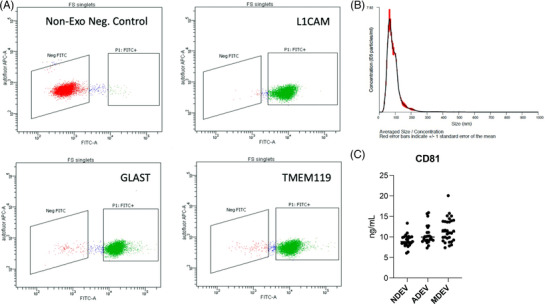
Fluorescent activated cell sorting (FACS) enrichment of plasma NDEVs, ADEVs, and MDEVs from LATE‐NC subjects. Representative FACS plot for non‐EV, negative control (red) and BAE—FITC complexes generated from EVs (green) isolated a LATE‐NC subject and enriched against anti‐human CD171 biotin (L1‐CAM), anti‐GLAST, and anti‐TMEM119 antibody. (A) Representative plot of size/concentration determined by nanoparticle tracking analysis (NTA) for extracted plasma EVs from a LATE‐NC subject (B). Plasma concentrations of EV marker, CD81 as measured by hu‐specific ELISA. CD81 was not statistically different between the three groups (C).
A direct comparison between an in‐house, sandwich‐based ELISA and the commercially available Cusabio kit was conducted in this study. TDP‐43 protein concentration was measured in NDEV, ADEV, and MDEV preparations from all samples. Across both ELISA platforms, NDEV concentrations of TDP‐43 were not significantly different between LATE‐NC (+) and LATE‐NC (–) subjects (Figure 2A , in house, TDP‐ 1.734 ± 0.705 ng/ml vs. TDP+ 2.425 ± 0.847 ng/ml, Two‐tailed t‐test, mean ± SEM). (Figure 2D , Cusabio, TDP‐ 0.5325 ± 0.050 ng/ml vs. TDP+ 0.4528 ± 0.062 ng/ml, Two‐tailed t‐test, mean ± SEM).
FIGURE 2.
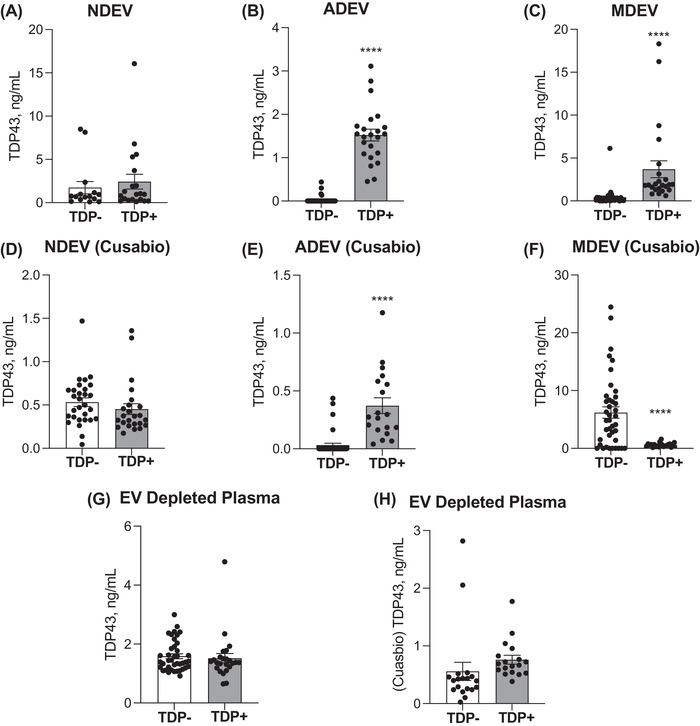
TDP‐43 is elevated in ADEVs extracted from subjects with an autopsy‐confirmed diagnosis of LATE. Plasma concentrations of TDP‐43 were detected in NDEVs, ADEVs, and MDEVs (A‐F) and in EV depleted plasma (G,H) as measured by our in‐house ELISA and Cusabio kit. NDEV concentrations TDP‐43 was not significantly different between LATE‐NC (+) and LATE‐NC (–) subjects (A,D). ADE concentrations of TDP‐43 were significantly increased in LATE‐NC (+) as compared to LATE‐NC (–) subjects (B,E). MDEV concentrations of TDP‐43 were significantly increased in LATE ‐ NC (+), as measured by our in‐house ELISA (C), while MDE concentrations of TDP‐43 were significantly decreased in LATE‐NC (+), as measured by the Cusabio kit (F). EV depleted plasma concentrations of TDP‐43 were not significantly different between LATE‐NC (+) (n = 22) and LATE‐NC (–) subjects (n = 42).
Across both platforms, ADEV concentrations of TDP‐43 were significantly increased in LATE‐NC (TDP+) as compared to LATE‐NC (TDP‐) subjects (Figure 2B , in house, TDP ‐ 0.026 ± 0.013 ng/ml vs. TDP+ 1.522 ± 0.136 ng/ml, Two‐tailed t‐test, mean ± SEM, P < .0001). (Figure 2E , Cusabio, TDP‐ 0.0322 ± 0.016 ng/ml vs. TDP+ 0.372 0.068 ng/ml, Two‐tailed t‐test, mean ± SEM, P < .0001).
MDEV concentrations of TDP‐43 were significantly increased in LATE ‐ NC (TDP+), as measured by our in‐house ELISA (Figure 2C , TDP‐ 0.4217 ± 0.159 ng/ml vs. TDP+ 3.686 ± 0.985 ng/ml, Two‐tailed t‐test, mean ± SEM, P < .0001), while MDEV concentrations of TDP‐43 were significantly decreased in LATE‐NC (TDP+), as measured by the Cusabio kit (Figure 2F , TDP‐ 6.193 ± 0.983 ng/ml vs. TDP+ 0.5850 ± 0.075 ng/ml, Two‐tailed t‐test, mean ± SEM, P < .0001).
In EV depleted plasma, measurable levels of TDP‐43 concentrations were not significantly different between LATE‐NC (TDP+) and LATE‐NC (TDP‐) subjects. These findings were consistent across both ELISA platforms. (Figure 2G , H ). In sum, significantly higher levels of TDP‐43 protein were detected by our in‐house ELISA kit as compared to the Cusabio kit in the NDEV, ADEV, and MDEV preparations and in the EV depleted plasma (Figure 2G , in house, 1.56 ± 0.078 ng/ml vs. Figure 2H , Cusabio, 0.66 ± 0.089 ng/ml, mean ± SEM, P < .001).
Correlations between TDP‐43 accumulation in NDEVs, ADEVs, and MDEVs against cognition‐based variables including MMSE score and Braak NFT staging, sex, and APOEε4 carrier status were established to compare results between LATE‐NC (+) and LATE‐NC (‐) subjects. As determined by our in‐house platform, no correlation was observed with TDP‐43 accumulation in NDEVs, ADEVs, and MDEVs with a lower MMSE score (Figure 3A – C ) or Braak NFT stage (Figure 3D – F ). Moreover, there was no significant difference in NDEV, ADEV, and MDEV concentrations of TDP‐43 between females vs. males (Figure 3G – I ) or between APOEε4 carriers and non‐carriers (Figure 3J – L ). Similarly, no correlation was observed with TDP‐43 accumulation in NDEVs, ADEVs, and MDEVs as measured by the Cusabio kit (Figure 4A – L ).
FIGURE 3.
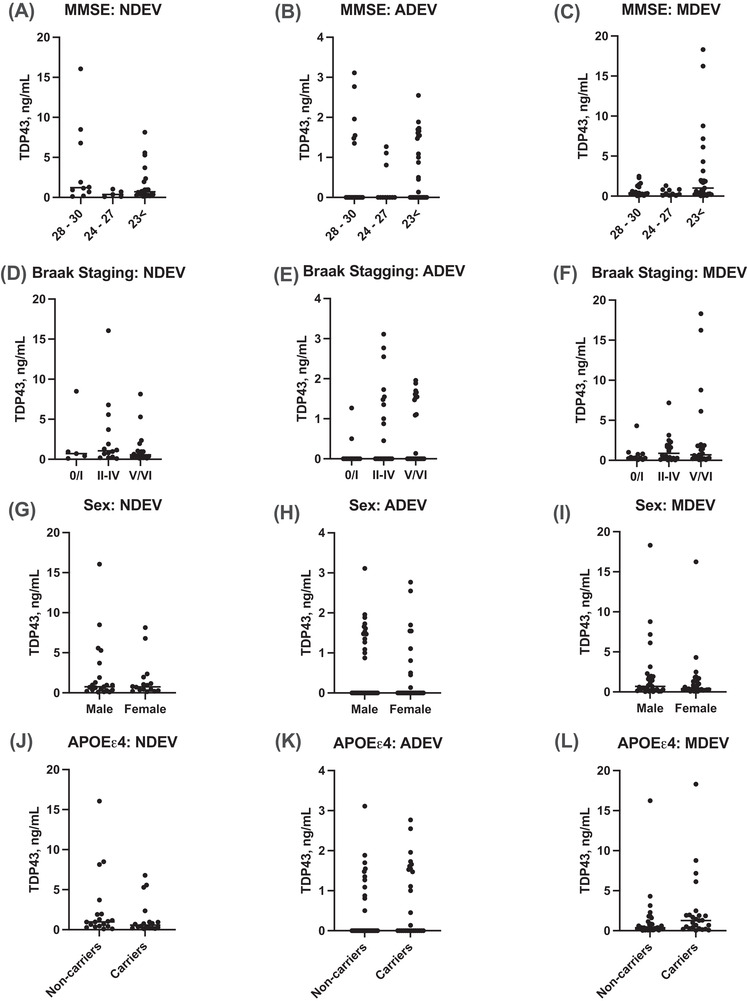
Correlations between TDP‐43 accumulation in NDEVs, ADVEs, and MDVEs against cognition‐based variables including MMSE score and Braak NFT staging; sex; and APOEε4 carrier status as measured by our in‐house ELISA. No correlation was observed with TDP‐43 accumulation in NDEVs, ADEVs, and MDEVs with a lower MMSE score (A–C) or Braak NFT stage (D–F). NDEV, ADEV, and MDEV concentrations of TDP‐43 were not significantly different between females versus males (G–I) or between APOEε4 carriers and non‐carriers (J–L). (n = 22, LATE‐NC (+); n = 42, LATE‐NC (–) subjects).
FIGURE 4.
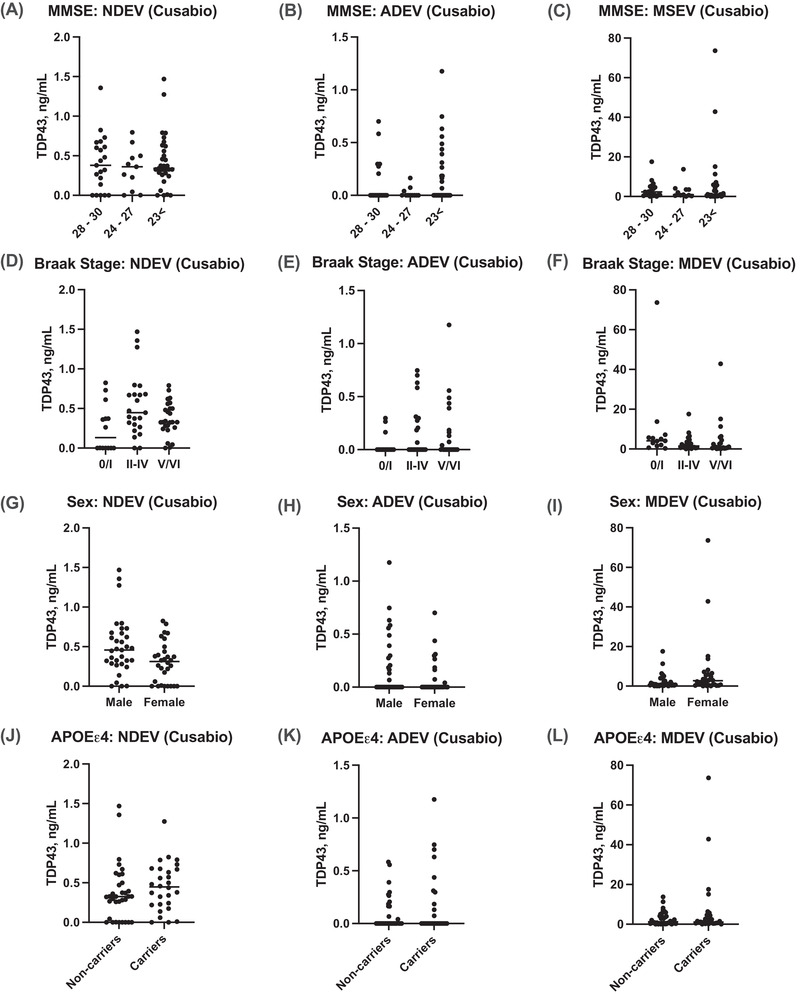
Correlations between TDP‐43 accumulation in NDEVs, ADEVs, and MDEVs against cognition‐based variables including MMSE score and Braak NFT staging; sex; and APOEε4 carrier status as measured by our Cusabio kit. No correlation was observed with TDP‐43 accumulation in NDEVs, ADEs, and MDEs with a lower MMSE score (A–C) or Braak NFT stage (D–F). NDEV, ADE, and MDE concentrations of TDP‐43 were not significantly different in females versus males (G–I) or between APOEε4 carriers and non‐carriers (J–L). (n = 22, LATE‐NC (+); n = 42, LATE‐NC (–) subjects).
4. DISCUSSION
In the current study, we generated a sensitive, sandwich‐based ELISA that can detect TDP‐43 in NDEV, ADEV, and MDEV preparations isolated from human plasma, and we compared between LATE‐NC (+) and LATE‐ NC (–) subjects. We determined that ADEV cargo levels contain significantly elevated levels of TDP‐43 in subjects with an autopsy‐confirmed diagnosis of LATE‐NC. These data suggest that plasma ADEVs may serve as useful diagnostic biomarker to identify subjects with LATE‐NC. To our knowledge, this is the first study to report these findings.
Measurable levels of TDP‐43 were also detected in EV depleted plasma; however, TDP‐43 levels were not significantly different between persons with and without eventual autopsy confirmed LATE‐NC. Our results indicate that there is a diagnostic advantage to measuring EV levels of TDP‐43. There is accumulating evidence suggesting TDP‐43 induces proinflammatory cytokines release from astrocytes and dysregulates astroglial metabolic support of neurons in ALS 17 and FTD. 18 However, the biological rationale for these molecular underpinnings is currently not fully understood and await future studies. In the meantime, without understanding the underlying biology, the ADEV TDP‐43 may still provide the basis for a useful clinical biomarker. We note that our “in‐house” ELISA detected higher EV levels of TDP‐43 as compared to the Cusabio ELISA, and consider it likely that differences are due to the specificity of the TDP‐43 detection antibodies that were used in both platforms. 19 Antibody sensitivity and specificity across the two platforms is likely explains our controversial results for the EV accumulation of TDP‐43 in our MDEV preparations. Here, we used a TDP‐43 specific monoclonal antibody (MAb) that recognized a specific epitope (aa 261—393 TDP‐43) 20 while the Cusabio antibody is speculated to recognize full length TDP‐43. Protein accumulation within EVs is hypothesized to vary across disease state and cellular origin. Methods such unbiased proteomic profiling be instrumental in assessing how the exosome proteome changes in relation to these cellular states. Currently, we are characterizing several novel TDP‐43 specific MAbs targeted against epitopes distributed over the entire length of TDP‐43 protein to further address this discrepancy.
A limitation of the current study is the sample size. The small sample size may partly explain our inability to identify a correlation between EV TDP‐43 levels with cognition‐based variables, sex, and APOE carrier status. However, we do note that there was very robust association between ADEV TDP‐43 levels and LATE‐NC status even with the sample size that was used, and despite the inclusion of a broad spectrum of dementia diagnosis controls. Prior studies have found an association between APOE genotype with AD 21 LATE‐NC. 22 Zhang et al. recently reported that TDP‐43 is elevated in plasma NDEVs of subjects with AD, yet they did not observe a correlation with TDP‐43, APOE genotype and a collection of neuropsychiatric symptoms. 19 Similarly, we did not observe an association between NDEV levels and TDP‐43 in the present study. Here, we found a dramatic difference in ADEV levels of TDP‐43 in LATE‐NC, suggesting that there is a unique interaction with TDP‐43 and astrocytes that also requires further investigation.
The lack of ethnoracial diversity in the current study is another major limitation. In our sample, 95% of subjects were identified as non‐Hispanic Caucasian. Hispanics/Latinos and African Americans/blacks have a higher prevalence of AD as compared to non‐Hispanic whites. 23 Early studies indicate that LATE‐NC occurs with similar prevalence in African Americans/blacks and whites. 24 Recent observational studies have reported the impact of race on AD biomarker performance 25 , 26 , 27 but more work is urgently required in the clinical, pathological, genetic, and biomarker domains with the goals of better serving broad populations. 24 Ultimately, our study must be validated in a larger and more diverse patient cohort.
In conclusion, LATE is an under‐recognized condition with a large impact on public health for which, to date, the lack of a clinical biomarker is a glaring unmet need. 5 Blood‐based EVs, specifically measuring TDP‐43 accumulation in ADEVs, may serve as a potential diagnostic tool to rapidly identify subjects who are currently living with LATE‐NC. Our findings are limited due to our inability to apply these methods to longitudinal examinations. It is possible that baseline, or longitudinal change in these markers may reflect a different diagnostic outcome as well as expanded potential context of uses (COUs) beyond patient identification and/or selection.
CONFLICT OF INTEREST
The authors declare no competing financial interests and have nothing to disclosure regarding the content of this manuscript. Author disclosures are available in the supporting information.
Supporting information
Supporting Information
ACKNOWLEDGMENTS
This work was supported by NIH grants to R.R. (AG0518440, AG051848, AG058533, AG062429), C.W. (AG070390) and P.N. (AG072946, AG061111, NS118584) and V.L (AG062418).
Winston CN, Sukreet S, Lynch H, et al. Evaluation of blood‐based, extracellular vesicles as biomarkers for aging‐related TDP‐43 pathology. Alzheimer's Dement. 2022;14:e12365. 10.1002/dad2.12365
REFERENCES
- 1. Geser F, Lee VM, Trojanowski JQ. Amyotrophic lateral sclerosis and frontotemporal lobar degeneration: a spectrum of TDP‐43 proteinopathies. Neuropathology. 2010;30(2):103‐112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Foulds PG, Davidson Y, Mishra M, et al. Plasma phosphorylated‐TDP‐43 protein levels correlate with brain pathology in frontotemporal lobar degeneration. Acta Neuropathol. 2009;118(5):647‐658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Jo M, Lee S, Jeon Y‐M, Kim S, Kwon Y, Kim H‐J. The role of TDP‐43 propagation in neurodegenerative diseases: integrating insights from clinical and experimental studies. Exp Mol Med. 2020;52(10):1652‐1662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Meneses A, Koga S, O'Leary J, Dickson DW, Bu G, Zhao N. TDP‐43 pathology in Alzheimer's disease. Mol Neurodegener. 2021;16(1):84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Nelson PT, Dickson DW, Trojanowski JQ, et al. Limbic‐predominant age‐related TDP‐43 encephalopathy (LATE): consensus working group report. Brain. 2019;142(6):1503‐1527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Foulds P, Mcauley E, Gibbons L, et al. TDP‐43 protein in plasma may index TDP‐43 brain pathology in Alzheimer's disease and frontotemporal lobar degeneration. Acta Neuropathol. 2008;116(2):141‐146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Colombo M, Raposo G, Théry C. Biogenesis, secretion, and intercellular interactions of exosomes and other extracellular vesicles. Annu Rev Cell Dev Biol. 2014;30:255‐289. [DOI] [PubMed] [Google Scholar]
- 8. Winston CN, Romero HK, Ellisman M, et al. Assessing neuronal and astrocyte derived exosomes from individuals with mild traumatic brain injury for markers of neurodegeneration and cytotoxic activity. Front Neurosci. 2019;13:1005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Winston CN, Goetzl EJ, Akers JC, et al. Prediction of conversion from mild cognitive impairment to dementia with neuronally derived blood exosome protein profile. Alzheimers Dement (Amst). 2016;3:63‐72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Fiandaca MS, Kapogiannis D, Mapstone M, et al. Identification of preclinical Alzheimer's disease by a profile of pathogenic proteins in neurally derived blood exosomes: a case‐control study. Alzheimers Dement. 2015;11(6):600‐607 e1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Goetzl EJ, Mustapic M, Kapogiannis D, et al. Cargo proteins of plasma astrocyte‐derived exosomes in Alzheimer's disease. FASEB J. 2016;30(11):3853‐3859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Winston CN, Goetzl EJ, Schwartz JB, Elahi FM, Rissman RA. Complement protein levels in plasma astrocyte‐derived exosomes are abnormal in conversion from mild cognitive impairment to Alzheimer's disease dementia. Alzheimers Dement (Amst). 2019;11:61‐66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Kumar A, Kim S, Su Y, et al. Brain cell‐derived exosomes in plasma serve as neurodegeneration biomarkers in male cynomolgus monkeys self‐administrating oxycodone. EBioMedicine. 2021;63:103192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Guo M, Hao Y, Feng Y, et al. Microglial exosomes in neurodegenerative disease. Front Mol Neurosci. 2021;14:630808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Nelson PT. LATE neuropathologic changes with little or no Alzheimer disease is common and is associated with cognitive impairment but not Frontotemporal Dementia. J Neuropathol Exp Neurol. 2021;80(7):649‐651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Kapasi A, Yu L, Boyle PA, Barnes LL, Bennett DA, Schneider JA. Limbic‐predominant age‐related TDP‐43 encephalopathy, ADNC pathology, and cognitive decline in aging. Neurology. 2020;95(14):e1951‐e1962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Smethurst P, Risse E, Tyzack GE, et al. Distinct responses of neurons and astrocytes to TDP‐43 proteinopathy in amyotrophic lateral sclerosis. Brain. 2020;143(2):430‐440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Velebit J, Horvat A, Smolič T, et al. Astrocytes with TDP‐43 inclusions exhibit reduced noradrenergic cAMP and Ca. Sci Rep. 2020;10:6003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Zhang N, Gu D, Meng M, Gordon ML. TDP‐43 is elevated in plasma neuronal‐derived exosomes of patients with Alzheimer's disease. Front Aging Neurosci. 2020;12:166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Kwong LK, Irwin DJ, Walker AK, et al. Novel monoclonal antibodies to normal and pathologically altered human TDP‐43 proteins. Acta Neuropathol Commun. 2014;2:33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Josephs KA, Whitwell JL, Weigand SD, et al. TDP‐43 is a key player in the clinical features associated with Alzheimer's disease. Acta Neuropathol. 2014;127(6):811‐824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Robinson JL, Lee EB, Xie SX, et al. Neurodegenerative disease concomitant proteinopathies are prevalent, age‐related and APOE4‐associated. Brain. 2018;141(7):2181‐2193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Raman R, Quiroz YT, Langford O, et al. Disparities by race and ethnicity among adults recruited for a preclinical Alzheimer disease trial. JAMA Netw Open. 2021;4(7):e2114364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Nag S, Barnes LL, Yu L, Wilson RS, Bennett DA, Schneider JA. Limbic‐predominant age‐related TDP‐43 encephalopathy in Black and White decedents. Neurology. 2020;95(15):e2056‐e2064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Howell JC, Watts KD, Parker MW, et al. Race modifies the relationship between cognition and Alzheimer's disease cerebrospinal fluid biomarkers. Alzheimers Res Ther. 2017;9(1):88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Morris JC, Schindler SE, Mccue LM, et al. Assessment of racial disparities in biomarkers for Alzheimer disease. JAMA Neurol. 2019;76(3):264‐273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Garrett SL, Mcdaniel D, Obideen M, et al. Racial disparity in cerebrospinal fluid amyloid and tau biomarkers and associated cutoffs for mild cognitive impairment. JAMA Netw Open. 2019;2(12):e1917363. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting Information