

Abstract
Myelin oligodendrocyte glycoprotein antibody–associated disease (MOGAD) is a recently described CNS inflammatory disorder that may manifest with optic neuritis, myelitis, seizures, and/or acute disseminated encephalomyelitis. While MOG-specific antibodies in patients with MOGAD are IgG1, a T-cell–dependent antibody isotype, immunologic mechanisms of this disease are not fully understood. Thymic hyperplasia can be associated with certain autoimmune diseases. In this report we describe a case of MOGAD associated with thymic hyperplasia in a young adult.
Case Presentation
An 18-year-old first-year college student with no significant medical history presented to an outside hospital with an 8-day course of worsening blurred vision in the left eye and pain with eye movements. She was of non-Hispanic European and East Asian ancestry and had no family history of neurologic or autoimmune disorders. On examination, she was found to have poor visual acuity in the left eye (<20/400), left color desaturation, an afferent pupillary defect, and moderate optic disc swelling, without any hemorrhages observed. The remainder of her neurologic examination was normal. An MRI of the brain and orbits with and without contrast demonstrated a gadolinium-enhancing, longitudinally extensive left optic nerve lesion involving the optic sheath, extending to, but not involving, the optic chiasm (Figure 1A). Serum testing (eTable 1, links.lww.com/NXI/A781) was significant for an erythrocyte sedimentation rate (ESR) of 21 mm/hr (reference range 0–21 mm/hr) and C-reactive protein (CRP) of 17.3 mg/L (reference range 0.0–9.0 mg/L), a negative aquaporin-4 (AQP4) antibody (Ab), and a positive anti–myelin oligodendrocyte glycoprotein (MOG) immunoglobulin G (IgG) with a titer of 1:80 (reference range <1:10), both cell-based assays.
Figure 1. MRI Images of Orbit and Brain.

(A) MRI orbits with and without contrast showing longitudinal hyperintensity and enhancement along the left optic nerve and perineural sheath. (B) MRI brain with and without contrast showing meningeal contrast enhancement and T2 fluid‐attenuated inversion recovery (FLAIR) hyperintensities seen over frontal and parietal lobes, including extension in the right parietal parenchyma, consistent with meningeal inflammation.
She was treated with 1 gram (g) of IV methylprednisolone (IV-MP) for 3 days. On the last day of infusion, she reported an estimated 85% improvement in vision and was discharged without a steroid taper. Three days after completion of steroids, she developed recurrent left eye blurred vision. She was admitted to the outside hospital and received an additional 5 days of 1 g of IV-MP, followed by an 11-day oral prednisone taper. On the ninth day of the taper, she developed a headache with associated nausea and vomiting, followed by a generalized tonic-clonic seizure. She was seen in the emergency department (ED) and was discharged on levetiracetam. She returned to the ED the next day because of worsening headache, nausea, and vomiting. She was afebrile, somnolent but easily arousable to voice, and was able to converse without signs of impaired cognition. Her neurologic examination was otherwise normal. An MRI examination of the brain with and without contrast was notable for meningeal enhancement with 1 focus of parenchymal involvement (Figure 1B). CSF analysis (eTable 1, links.lww.com/NXI/A781) showed a mild, lymphocytic-predominant pleocytosis consistent with aseptic meningoencephalitis. Infectious and inflammatory workup (CSF and serum) and cytology (CSF) showed negative results.
She was treated with another course of IV-MP 1 g daily for 5 days followed by a 5-month prednisone taper. Given her clinical presentation and positive MOG Ab, a diagnosis of MOG antibody–associated disease (MOGAD) was made. Owing to the recurrence of symptoms, a decision to proceed with immunosuppression was made, so she was initiated with rituximab (1 g on day 1 and 1 g on day 15), before discharge from her second hospitalization.
She was seen as second opinion at a tertiary academic center 3 months later. Her repeat MOG-Ab titer at 3 months after her first course of rituximab was found to be persistently elevated at 1:80. Because of the elevation in CRP and ESR observed in this young adult, a systemic workup for additional autoimmune and malignant entities was performed. No significant additional laboratory abnormalities were noted. CT scans of her abdomen and pelvis showed negative results. However, CT of the chest revealed trace residual thymus. Follow-up imaging revealed increased thymic enlargement, 3.2 by 2.3 cm 6 months later, and 3.7 by 2.8 cm at 12-month follow-up. MRI of the chest with contrast and PET with CT, performed approximately 12 months later (Figure 2), demonstrated homogenous enhancement and moderate homogenous fluorodeoxyglucose uptake, respectively. She was evaluated by thoracic surgery, and it was felt that the findings were most supportive of thymic hyperplasia, but did not completely exclude a thymoma.
Figure 2. Thymic Imaging.

PET with CT scan showing a 37 × 28-mm thymic bed mass with homogenous fluorodeoxyglucose uptake, circled.
The patient's condition and probable thymic hyperplasia are currently under clinical and radiologic surveillance. Owing to clinical relapse after the first event and persistent MOG seropositivity (at 3 months and 18 months of follow-up), the patient has continued rituximab. She has been clinically stable since, with visual acuity of 20/20 in each eye. Given this stability and potential surgical complications, the patient decided not to pursue further tissue biopsy or resection.
Differential Diagnosis
This patient presented with an optic neuritis (ON) and subsequently developed subacute meningoencephalitis. Her workup revealed positive MOG-Ab, abnormal systemic inflammatory markers, and thymic hyperplasia.
Optic Neuritis
ON is inflammation of the optic nerve, which may be due to a myriad of causes (Table 1). The characteristics of our patient's ON—severe, anterior with moderate optic disc edema, longitudinally extensive, and perineural involvement—is most suggestive of MOGAD1,2 (Table 1). Aquaporin-4 IgG or seronegative neuromyelitis optica spectrum disorders (NMOSD) may present with longitudinally extensive lesions, but are more likely to be posterior–even involving the optic chiasm and tracts–and less commonly involve the perineural structures of the optic nerve.2 Multiple sclerosis–associated ON is often unilateral in adults, is usually clinically mild-moderate in vision loss, and presents with short-segment anterior involvement of the optic nerve and has not been reported to involve the optic nerve sheath. The steroid responsiveness and dependence (i.e., recurrence of ON after withdrawal of steroids) is also typical of MOGAD and atypical, though possible, for NMOSD.2-4 Chronic relapsing inflammatory ON (CRION) may have similar presentation as our patient; however, it would be a diagnosis only if MOGAD and NMOSD testing results were negative and if she did not go on to have extraoptic neurologic manifestations. Notably, many, though not all, cases of “CRION” have been shown to be MOG-Ab or aquaporin-4 Ab positive. Granulomatous disease and tuberculosis are also potential causes for such a presentation.1 However, the CSF profile with mild lymphocytosis and normal protein and glucose and negative infectious studies in the CSF and serum (eTable 1, links.lww.com/NXI/A781) make tuberculosis or other granulomatous disease unlikely etiologies. Moreover, there were no systemic manifestations of these diseases. Finally, although anti-MOG IgG was only modestly elevated, its persistence further supports MOGAD as the etiology.4 Nevertheless, transient MOG-Ab in MOGAD is well described, particularly in the pediatric population.
Table 1.
Selected Differential Diagnosis of Optic Neuritis

Meningoencephalitis
Aseptic meningitis is meningeal inflammation with negative routine bacterial studies. Meningitis has a wide range of presentations including headache, neck pain, nuchal rigidity, photosensitivity, and fever. When there is also a change in cognition, from psychiatric symptoms to frank coma, seizures, or evidence of parenchymal inflammation by imaging, meningoencephalitis is diagnosed. Our patient was diagnosed with aseptic meningoencephalitis after typical presentation and exclusion of infectious etiologies (eTable 1, links.lww.com/NXI/A781). This patient's presentation of aseptic meningoencephalitis, elevated CRP and ESR, and relatively low MOG-Ab titer5 prompted further investigation of possible systemic diseases, such as sarcoidosis, granulomatosis with angiitis, and autoimmune encephalitis.6 Granulomatosis with angiitis was considered unlikely with negative anti-neutrophil cytoplasm antibodies (eTable 1, links.lww.com/NXI/A781) and lack of other organ involvement. Full body PET with CT scan did not reveal findings typical of sarcoid, and her CSF profile with normal protein and glucose would also be atypical for neurosarcoidosis. Another consideration for this presentation is anti-glial fibrillary acidic protein astrocytopathy, whose primary presentation is a meningoencephalomyelitis7 and has rarely been reported to overlap with MOGAD.8 Unfortunately, her CSF was not tested for glial fibrillary acidic protein antibodies.
Thymic Enlargement
The thymus is a primarily immunologic and endocrine organ that serves to train T cells to distinguish self from nonself (Figure 3). The thymus normally begins to atrophy beginning at puberty, with an average diameter of 1.1 cm (SD 0.4 cm) from age 6–19 years and 0.5 cm (SD 0.27 cm) from age 20–50 years, as measured on conventional CT (the thymus may appear larger on MRI).9 As the thymus involutes, it is replaced with adipose tissue, and as such may appear as lymphoid aggregates in the midst of fat.10 Our patient's enlarged thymus was discovered on chest CT and was then noted to enlarge over time, initially from trace residual thymus to frank enlargement at 3.7 by 2.8 cm.
Figure 3. Thymic Selection of T Cells and Their Potential Role in Promoting MOGAD Pathogenesis.

Precursor (pre)-T cells emerge from the bone marrow and migrate to the thymus. Pre-T cells (CD4 and CD8 double negative) enter the cortex, undergo T-cell receptor gene rearrangement and become double positive for CD4 and CD8 molecules. There, CD4+CD8+ thymocytes are exposed to cortical epithelial cells (cTECs) that express major histocompatibility complex (MHC) I and II molecules that bind self-antigen peptides. CD4+CD8+ thymocytes that recognize MHC/peptides on cTECs with low affinity undergo apoptosis. CD4+CD8+ thymocytes that exhibit intermediate or high affinity for MHC I/peptide or MHC II/peptide on cTECs are believed to be positively selected to become single positive CD8+ T cells or CD4+ T cells, respectively, and then migrate to the medulla. Medullary thymic epithelial cells (mTECS), which also express MHC I and MHC II, regulate negative selection (i.e., deletion) of CD8+ and CD4+ T cells that exhibit high affinity to self (tissue-specific) antigens. This process of thymic education first ensures adequate binding to the MHC molecules on antigen-presenting cells (APCs) (positive selection, cortex) and subsequent deletion of those that have a high affinity for self-antigen (negative selection, medulla). Approximately 2% of T cells survive thymic education and exit as mature T cells. A small number of T cells that recognize self-antigen may escape deletion, even in the normal thymus of healthy individuals. In MOGAD, as in certain other autoimmune diseases such as myasthenia gravis, the self-reactive T cells are believed to be more active and lead to disease pathogenesis. This may reflect failure of deletion of high-affinity MOG-reactive T cells or failure to regulate MOG-reactive T cells peripherally. MOG-reactive T cells may have distinct roles in MOGAD. First, it is known from animal models that proinflammatory MOG-reactive T cells (Teff) can induce encephalomyelitis and optic neuritis independent of MOG-specific antibodies.21 Second, MOG-specific antibodies in MOGAD are IgG1, a T-cell–dependent Ig subtype.22 As T follicular helper (Tfh) cells are required for maturation of B cells into Ab-secreting plasmablasts and plasma cells, it is believed that MOG-specific Tfh promote maturation of MOG-specific B cells into IgG1-secreting plasma cells. Therefore, MOG-specific Teff along with MOG-specific antibodies may enter the CNS causing damage to myelin and oligodendrocytes. The extent to which complement participates in this inflammatory process is not clear. Figure art created by Xavier Studio. MOGAD = myelin oligodendrocyte glycoprotein antibody–associated disease.
Abnormal enlargement of the thymus may be due to thymic hyperplasia–true thymic hyperplasia or thymic lymphocytic hyperplasia (TLH)–or neoplasm, most commonly thymoma or, rarely, thymic carcinoma or other thymic tumors (Figure 4).10 True thymic hyperplasia is an enlarged but normally organized thymus, which may be caused by chemotherapy, corticosteroid use, irradiation, or thermal burns, and is not associated with autoimmune conditions.9 TLH is defined by an increased number of lymphoid follicles with germinal centers within the thymus and is associated with numerous autoimmune conditions, notably myasthenia gravis and Graves disease.9 Thymoma is a usually benign or low-grade epithelioid neoplasm, which is also associated with similar autoimmune conditions.9
Figure 4. Thymic Pathology.
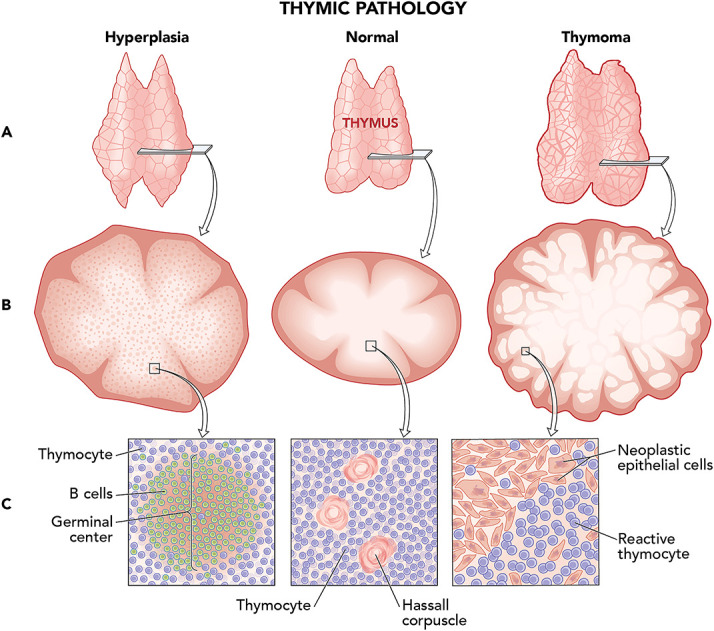
Here demonstrated are the 2 most likely thymic pathologies to exist in our patient and normal thymic tissue. (A) Gross: Triangular shape, as seen in our patient, is more frequently observed with thymic hyperplasia (left), whereas more rounded enlargement favors thymoma (right). (B) Low magnification: Lobular architecture is preserved in thymic hyperplasia (left), whereas thymoma has increased numbers of lobules (right). (C) High magnification: (Left) B-cell germinal centers with surrounding thymocytes characterize thymic hyperplasia. (Center) Hassall corpuscles are groups of concentrically arranged eosinophilic epithelial cells in the normal thymic medulla that are believed to support thymocyte development; they mark thymic tissue apart from other lymphoid tissues and may be seen in the normal as well as pathologic thymus. (Right) Neoplastic thymic epithelial cells with increased reactive thymocytes are seen in thymoma. Figure art created by Xavier Studio.
Imaging characteristics may be helpful in distinguishing underlying etiologies of an enlarged thymus. Diffuse enlargement favors hyperplasia, whereas focal enlargement is concerning for neoplasia; a round shape indicates thymoma, and, by contrast, triangularity supports hyperplasia. Fatty infiltration is also helpful with identifying hyperplasia. MRI chemical phase change (CSR) is a technique that detects even microscopic fat.10 A high CSR is indicative of fatty infiltration and favors hyperplasia over thymic neoplasms.10 Our patient had a diffusely enlarged, triangular thymus, favoring hyperplasia; however, her thymus demonstrated low CSR at 1.02 (i.e., essential absence of fat). Hence, this was felt to favor thymic hyperplasia with poor fatty infiltration, but thymoma could not be excluded.
Differentiating between TLH and physiologic (true) thymic hyperplasia is important when there is coexisting autoimmune disease. Depending on the autoimmune disease, surgical resection of TLH may be indicated (e.g., myasthenia gravis). As previously discussed, true thymic hyperplasia results from physiologic stress and then returns to normal size. For example, thymic hyperplasia has been reported after chemotherapy and noted to grow up to a mean of approximately 4 months before returning to normal size.9 Histologic examination is required to distinguish TLH from true thymic hyperplasia.9 In our patient, the continued enlargement 8 months after steroid cessation makes true thymic hyperplasia less likely.
Final Diagnosis
The final diagnosis was relapsing MOGAD presenting as ON and meningoencephalitis, associated with abnormal thymus and imaging characteristics that favor TLH.
Discussion
MOGAD is a recently described, rare CNS inflammatory disorder mostly reported in White individuals11 and one-third of pediatric cases with first case of demyelination.12 Multiple associated syndromes have been described including ON, myelitis, encephalitis, seizures, acute disseminated encephalomyelitis, and aseptic meningitis.1,2 Approximately 30%–35% patients with NMOSD do not have AQP4-specific antibodies. Of those, it has been reported that 50% have MOGAD.13
Several features support the diagnosis of MOGAD: (1) ON clinicoradiological presentation was characteristic of MOGAD, (2) persistent MOG-Ab positivity, (3) sensitivity to steroid treatment, and (4) lack of findings to support other etiologies. Furthermore, the concomitant presentation of ON and meningoencephalitis is increasingly recognized in MOGAD.14 Finally, seizures are a well-described phenomenon occurring in association with MOGAD, estimated to occur in approximately 20% of patients with MOGAD,15 compared with approximately 3% of patients with MS16,17 and less than 1% of patients with NMOSD.6 Seizures in MOGAD have also been reported to cluster around other demyelinating events, such as ON,18 as seen in our patient.
The pathophysiology of MOGAD has not been fully elucidated (Figure 3). Human MOGAD lesions have been shown to have perivenous demyelination dominated by macrophages and CD4+ T cells, with macrophages ingesting MOG-dominant debris.19 It is, however, unclear whether MOG IgG is directly pathogenic. Introduction of human MOG-IgG alone into rodents has failed to induce demyelination,20 yet anti-MOG antibodies can promote CNS damage in the setting of inflammation.21 Anti-MOG antibodies are IgG1, a T-cell–dependent isotype, requiring T follicular helper cells to class switch from IgM to IgG.21 Some data suggest that MOG IgG1 is able to activate complement and induce complement-mediated cytotoxicity,22 yet pathologic studies have shown relatively rare complement within active lesions.19 Therefore, MOG-Abs may or may not be pathogenic, depending on the presence of T-cell inflammation.20,21
It is well recognized that thymic hyperplasia and thymoma are associated with autoimmunity.23 The “sick” thymus is believed to be causative in its association with autoimmunity, in many but not all (i.e., Graves) cases. The thymus provides a complex environment that promotes the maturation of a diverse pool of T cells that go through positive and negative selection (Figure 3). In this context, medullary thymic epithelial cells express certain genes (e.g., Aire and Fezf2) that control negative selection of CD4+ and CD8+ T cells targeting tissue-specific antigens, and defects in Aire expression have been associated with certain humoral autoimmune diseases.24
Several autoimmune diseases are associated with TLH and thymoma. MG with acetylcholine receptor antibodies (85% of cases of MG) is the prototypical autoimmune disease where thymic pathogenesis is well described. In seropositive MG, especially early-onset cases, 50%–60% of patients exhibit TLH or thymoma3 and 85% have some thymic abnormality.23 Furthermore, thymoma is associated with 30%–40% chance of eventually developing MG10 and removal of TLH or thymoma in those with MG has been proven to be therapeutic.25 Besides MG, thymic hyperplasia has not been consistently described in other autoimmune neurologic diseases, although there are isolated case reports of thymic hyperplasia and N‐methyl‐D aspartate receptor encephalitis or relapsing-remitting longitudinally extensinve transverse myelitis (LETM), which have been cured by thymectomy.26,27 Similarly, paraneoplastic AQP4 + NMO has been well reported, including some cases of thymoma, for which treatment of the underlying malignancy could also be therapeutic.28 MG and AQP4 + NMO have also been noted to coexist more than would be expected.29 Of the published cases, MG almost always preceded NMO, and most of those had thymectomy before developing NMO,29 perhaps reflecting loss of thymic-mediated T-cell regulation. It is of interest that the chest and thymus are rarely imaged in CNS demyelinating diseases. Therefore, it is unknown whether the potential association between MOGAD and thymic hyperplasia is truly rare.
Conclusion
MOGAD has not been previously associated with thymic abnormality, either hyperplasia or thymoma. We now report a case of MOGAD in association with thymic abnormality. The potential implications of thymic pathology associated with MOGAD raise potential insights into the immunology of this disease, at least in our patient. Further studies are needed to investigate the pathophysiologic link between thymus and MOGAD and to assess whether removal of thymic tissue would have therapeutic benefit.
Acknowledgment
The authors thank Dr. Cooper Kumarasen, MD, MBChB, DPhil, from the Department of Pathology at the University of Pennsylvania for his help regarding thymic histology and pathology.
Glossary
- Ab
antibody
- AQP4
aquaporin 4 antibody
- CRION
chronic relapsing inflammatory ON
- CRP
C-reactive protein
- ED
emergency department
- ESR
erythrocyte sedimentation rate
- FLAIR
fluid-attenuated inversion recovery
- IgG
immunoglobulin G
- IV-MP
IV methylprednisolone
- MOG
myelin oligodendrocyte glycoprotein
- MOGAD
MOG antibody–associated disease
- NMOSD
neuromyelitis optica spectrum disorders
- NMO
neuromyelitis optica
- ON
optic neuritis
- TLH
thymic lymphocytic hyperplasia
Appendix. Authors
Study Funding
The authors report no targeted funding.
Disclosure
B. Hurtubise, E.M. Frohman, S. Galetta, L.J. Balcer, T.C. Frohman, L.P. Lisak, S.D. Newsome, J. Graves, S.S. Zamvil, and L. Amezcua report no disclosures relevant to the manuscript. Go to Neurology.org/NN for full disclosure.
References
- 1.Bennett JL. Optic neuritis. Continuum (Minneap Minn). 2019;25(5):1236-1264. doi: 10.1212/con.0000000000000768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Gospe SM, Chen JJ, Bhatti MT. Neuromyelitis optica spectrum disorder and myelin oligodendrocyte glycoprotein associated disorder-optic neuritis: a comprehensive review of diagnosis and treatment. Eye. 2021;35(3):753-768. doi: 10.1038/s41433-020-01334-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Jarius S, Paul F, Aktas O, et al. MOG encephalomyelitis: international recommendations on diagnosis and antibody testing. J Neuroinflammation. 2018;15(1):134. doi: 10.1186/s12974-018-1144-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Gombolay GY, Gadde JA. Aseptic meningitis and leptomeningeal enhancement associated with anti-MOG antibodies: a review. J Neuroimmunol. 2021;358:577653. doi: 10.1016/j.jneuroim.2021.577653. [DOI] [PubMed] [Google Scholar]
- 5.Sechi E, Buciuc M, Pittock SJ, et al. Positive predictive value of myelin oligodendrocyte glycoprotein autoantibody testing. JAMA Neurol. 2021;78(6):741-746. doi: 10.1001/jamaneurol.2021.0912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Etemadifar M, Abhari AP, Sabeti F, et al. “NMDA receptor spectrum disorder” in the differential diagnosis of demyelinating disorders of the CNS: optic neuritis and myelitis. Neurol Sci. 2021;42(1):151-157. doi: 10.1007/s10072-020-04518-y. [DOI] [PubMed] [Google Scholar]
- 7.Flanagan EP, Hinson SR, Lennon VA, et al. Glial fibrillary acidic protein immunoglobulin G as biomarker of autoimmune astrocytopathy: analysis of 102 patients. Ann Neurol. 2017;81(2):298-309. doi: 10.1002/ana.24881. [DOI] [PubMed] [Google Scholar]
- 8.Martin AJ, Strathdee J, Wolfe N. Coexistent anti-GFAP and anti-MOG antibodies presenting with isolated meningitis and papillitis: more support for overlapping pathophysiology. BMJ Neurol Open. 2022;4(1):e000236. doi: 10.1136/bmjno-2021-000236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Nishino M, Ashiku SK, Kocher ON, Thurer RL, Boiselle PM, Hatabu H. The thymus: a comprehensive review. RadioGraphics. 2006;26(2):335-348. doi: 10.1148/rg.262045213. [DOI] [PubMed] [Google Scholar]
- 10.Priola AM, Priola SM. Imaging of thymus in myasthenia gravis: from thymic hyperplasia to thymic tumor. Clin Radiol. 2014;69(5):e230-e245. doi: 10.1016/j.crad.2014.01.005. [DOI] [PubMed] [Google Scholar]
- 11.Cobo-Calvo A, Ruiz A, Maillart E, et al. Clinical spectrum and prognostic value of CNS MOG autoimmunity in adults: the MOGADOR study. Neurology. 2018;90(21):e1858-e1869. doi: 10.1212/wnl.0000000000005560. [DOI] [PubMed] [Google Scholar]
- 12.Waters P, Fadda G, Woodhall M, et al. Serial anti-myelin oligodendrocyte glycoprotein antibody analyses and outcomes in children with demyelinating syndromes. JAMA Neurol. 2020;77(1):82-93. doi: 10.1001/jamaneurol.2019.2940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hamid SHM, Whittam D, Mutch K, et al. What proportion of AQP4-IgG-negative NMO spectrum disorder patients are MOG-IgG positive? A cross sectional study of 132 patients. J Neurol. 2017;264(10):2088-2094. doi: 10.1007/s00415-017-8596-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gutman JM, Kupersmith M, Galetta S, Kister I. Anti-myelin oligodendrocyte glycoprotein (MOG) antibodies in patients with optic neuritis and seizures. J Neurol Sci. 2018;387:170-173. doi: 10.1016/j.jns.2018.01.042. [DOI] [PubMed] [Google Scholar]
- 15.Shen C-H, Zheng Y, Cai MT, et al. Seizure occurrence in myelin oligodendrocyte glycoprotein antibody-associated disease: a systematic review and meta-analysis. Mult Scler Relat Disord. 2020;42:102057. doi: 10.1016/j.msard.2020.102057. [DOI] [PubMed] [Google Scholar]
- 16.Marrie RA, Reider N, Cohen J, et al. A systematic review of the incidence and prevalence of sleep disorders and seizure disorders in multiple sclerosis. Mult Scler. 2015;21(3):342-349. doi: 10.1177/1352458514564486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Burman J, Zelano J. Epilepsy in multiple sclerosis: a nationwide population-based register study. Neurology. 2017;89(24):2462-2468. doi: 10.1212/wnl.0000000000004740. [DOI] [PubMed] [Google Scholar]
- 18.Kleerekooper I, Trip SA, Plant GT, Petzold A. Expanding the phenotype of MOG antibody-associated disease (MOGAD): half a century of epilepsy and relapsing optic neuritis. J Neurol Neurosurg Psychiatry. 2021;92(3):340-342. doi: 10.1136/jnnp-2020-324323. [DOI] [PubMed] [Google Scholar]
- 19.Takai Y, Misu T, Kaneko K, et al. Myelin oligodendrocyte glycoprotein antibody-associated disease: an immunopathological study. Brain. 2020;143(5):1431-1446. doi: 10.1093/brain/awaa102. [DOI] [PubMed] [Google Scholar]
- 20.Reindl M, Waters P. Myelin oligodendrocyte glycoprotein antibodies in neurological disease. Nat Rev Neurol. 2019;15(2):89-102. doi: 10.1038/s41582-018-0112-x. [DOI] [PubMed] [Google Scholar]
- 21.Molnarfi N, Schulze-Topphoff U, Weber MS, et al. MHC class II–dependent B cell APC function is required for induction of CNS autoimmunity independent of myelin-specific antibodies. J Exp Med. 2013;210(13):2921-2937. doi: 10.1084/jem.20130699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zamvil S.S., Slavin A.J. Does MOG Ig-positive AQP4-seronegative opticospinal inflammatory disease justify a diagnosis of NMO spectrum disorder?. Neurol Neuroimmunol Neuroinflamm 2015;2:e62 doi: 10.1212/NXI.0000000000000062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Shelly S, Agmon-Levin N, Altman A, Shoenfeld Y. Thymoma and autoimmunity. Cell Mol Immunol. 2011;8(3):199-202. doi: 10.1038/cmi.2010.74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Cheng M, Anderson MS. Thymic tolerance as a key brake on autoimmunity. Nat Immunol. 2018;19(7):659-664. doi: 10.1038/s41590-018-0128-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wolfe GI, Kaminski HJ, Aban IB, et al. Randomized trial of thymectomy in myasthenia gravis. N Engl J Med. 2016;375:511-522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Palleiko BA, Salamatbad G, Bludevich BM, Maxfield MW. Thymectomy for treatment of anti-NMDA receptor encephalitis. Eur J Cardiothorac Surg. 2022;62(4):ezac329. doi: 10.1093/ejcts/ezac329. [DOI] [PubMed] [Google Scholar]
- 27.Hammond ER, Pardo CA, Kerr DA. Thymic hyperplasia in a patient with recurrent transverse myelitis with clinical resolution after thymectomy. J Neurol Neurosurg Psychiatry. 2008;79(3):334-335. doi: 10.1136/jnnp.2007.127423. [DOI] [PubMed] [Google Scholar]
- 28.Beauchemin P, Iorio R, Traboulsee AL, Field T, Tinker AV, Carruthers RL. Paraneoplastic Neuromyelitis Optica Spectrum Disorder: a single center cohort description with two cases of histological validation. Mult Scler Relat Disord. 2018;20:37-42. doi: 10.1016/j.msard.2017.12.012. [DOI] [PubMed] [Google Scholar]
- 29.Jarius S, Paul F, Franciotta D, et al. Neuromyelitis optica spectrum disorders in patients with myasthenia gravis: ten new aquaporin-4 antibody positive cases and a review of the literature. Mult Scler J. 2012;18(8):1135-1143. doi: 10.1177/1352458511431728. [DOI] [PubMed] [Google Scholar]
- 30.Chen JJ, Flanagan EP, Jitprapaikulsan J, et al. Myelin oligodendrocyte glycoprotein antibody-positive optic neuritis: clinical characteristics, radiologic clues, and outcome. Am J Ophthalmol. 2018;195:8-15. doi: 10.1016/j.ajo.2018.07.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Jurynczyk M, Messina S, Woodhall MR, et al. Clinical presentation and prognosis in MOG-antibody disease: a UK study. Brain. 2017;140(12):3128-3138. doi: 10.1093/brain/awx276. [DOI] [PubMed] [Google Scholar]
- 32.Zhao Y, Tan S, Chan TCY, et al. Clinical features of demyelinating optic neuritis with seropositive myelin oligodendrocyte glycoprotein antibody in Chinese patients. Br J Ophthalmol. 2018;102(10):1372-1377. doi: 10.1136/bjophthalmol-2017-311177. [DOI] [PubMed] [Google Scholar]
- 33.de Mol CL, Wong Y, van Pelt E, et al. The clinical spectrum and incidence of anti-MOG-associated acquired demyelinating syndromes in children and adults. Mult Scler. 2020;26(7):806-814. doi: 10.1177/1352458519845112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Akaishi T, Himori N, Takeshita T, et al. Five-year visual outcomes after optic neuritis in anti-MOG antibody-associated disease. Mult Scler Relat Disord. 2021;56:103222. doi: 10.1016/j.msard.2021.103222. [DOI] [PubMed] [Google Scholar]
- 35.Burman J, Raininko R, Fagius J. Bilateral and recurrent optic neuritis in multiple sclerosis. Acta Neurol Scand. 2011;123(3):207-210. doi: 10.1111/j.1600-0404.2010.01388.x. [DOI] [PubMed] [Google Scholar]
- 36.Beck RW, Cleary PA, Anderson MM, et al. A randomized, controlled trial of corticosteroids in the treatment of acute optic neuritis. N Engl J Med. 1992;326(9):581-588. doi: 10.1056/nejm199202273260901. [DOI] [PubMed] [Google Scholar]
- 37.Malik MT, Healy BC, Benson LA, et al. Factors associated with recovery from acute optic neuritis in patients with multiple sclerosis. Neurology. 2014;82(24):2173-2179. doi: 10.1212/wnl.0000000000000524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wingerchuk DM, Hogancamp WF, O'Brien PC, Weinshenker BG. The clinical course of neuromyelitis optica (Devic's syndrome). Neurology. 1999;53(5):1107-1114. doi: 10.1212/wnl.53.5.1107. [DOI] [PubMed] [Google Scholar]
- 39.Papais-Alvarenga RM, Carellos SC, Alvarenga MP, et al. Clinical course of optic neuritis in patients with relapsing neuromyelitis optica. Arch Ophthalmol. 2008;126(1):12-16. doi: 10.1001/archophthalmol.2007.26. [DOI] [PubMed] [Google Scholar]
- 40.Merle H, Olindo S, Jeannin S, et al. Treatment of optic neuritis by plasma exchange (add-on) in neuromyelitis optica. Arch Ophthalmol. 2012;130(7):858-862. doi: 10.1001/archophthalmol.2012.1126. [DOI] [PubMed] [Google Scholar]