

Abstract
One‐third of the human proteome is comprised of membrane proteins, which are particularly vulnerable to misfolding and often require folding assistance by molecular chaperones. Calnexin (CNX), which engages client proteins via its sugar‐binding lectin domain, is one of the most abundant ER chaperones, and plays an important role in membrane protein biogenesis. Based on mass spectrometric analyses, we here show that calnexin interacts with a large number of nonglycosylated membrane proteins, indicative of additional nonlectin binding modes. We find that calnexin preferentially bind misfolded membrane proteins and that it uses its single transmembrane domain (TMD) for client recognition. Combining experimental and computational approaches, we systematically dissect signatures for intramembrane client recognition by calnexin, and identify sequence motifs within the calnexin TMD region that mediate client binding. Building on this, we show that intramembrane client binding potentiates the chaperone functions of calnexin. Together, these data reveal a widespread role of calnexin client recognition in the lipid bilayer, which synergizes with its established lectin‐based substrate binding. Molecular chaperones thus can combine different interaction modes to support the biogenesis of the diverse eukaryotic membrane proteome.
Keywords: calnexin, chaperone, membrane protein
Subject Categories: Membranes & Trafficking, Translation & Protein Quality
Chaperoning of misfolded membrane proteins by calnexin in the ER involves not only lectin‐based glycan binding, but also direct transmembrane domain interactions within the lipid bilayer.
Introduction
Integral membrane proteins (IMPs) play essential roles in biology, including transporting molecules and signals across lipid bilayers, functioning as metabolic enzymes, and mediating cell–cell interactions. Around one‐third of all human genes code for membrane proteins (Fagerberg et al, 2010). In eukaryotic cells, the biosynthesis of both IMPs and soluble secretory pathway proteins occurs at the endoplasmic reticulum (ER) (Shao & Hegde, 2011). IMPs, however, often pose more complex challenges to the ER folding and quality control system than soluble proteins since the formation of their native state requires multiple topologically distinct folding events (Marinko et al, 2019). These include correct structuring of soluble domains on both the ER lumenal and cytosolic side and the proper lipid bilayer integration often including intra‐ and intermolecular assembly of transmembrane domain (TMDs) (Hegde & Keenan, 2021; O'Keefe et al, 2021). Further complicating these processes is the fact that TMDs of multipass membrane proteins are highly diverse in nature. They often contain polar residues, breaks or kinks and may exhibit flexibility in the membrane which gives rise to complex membrane integration and folding pathways (Ota et al, 1998; Lu et al, 2000; Hessa et al, 2005; Sadlish et al, 2005; Kauko et al, 2010; Feige & Hendershot, 2013). Notwithstanding, these deviations from ideal hydrophobic membrane anchors allow membrane proteins to fulfill their wide functional repertoire including transport of hydrophilic molecules through the lipid bilayer and specific recognition events in an apolar environment (Illergard et al, 2011). At the same time, they render IMPs vulnerable to incorrect folding and assembly in the lipid bilayer. This is demonstrated by the many severe human pathologies that are caused by membrane protein misfolding (Partridge et al, 2004; Guerriero & Brodsky, 2012; Marinko et al, 2019). Intramembrane chaperones and quality control factors that can efficiently recognize and monitor the folding status of IMPs directly within the lipid bilayer to support folding and detect misfolding are thus a prerequisite for protein homeostasis of any eukayotic cell.
One of the first chaperones identified to be involved in membrane protein folding was calnexin (CNX; Anderson & Cresswell, 1994; Hammond & Helenius, 1994; Jackson et al, 1994). CNX, which is integrated into to the ER membrane via its single TMD, plays a crucial role in glycoprotein folding and transiently interacts with a wide range of newly synthesized proteins that transit the ER. As a part of the ER quality control system, CNX prevents incompletely folded substrates from leaving the ER and recruits cochaperones that accelerate slow folding reactions including disulfide bond formation (Hebert & Molinari, 2012). The marked preference of CNX for monoglucosylated oligosaccharides on its clients results from its ER‐lumenal lectin domain (Hebert et al, 1995). A highly similar lectin domain is found within calreticulin (CRT), the soluble homolog of CNX in the ER (Kozlov et al, 2010b). Despite their almost identical lectin domains, genetic deletions of CNX and CRT have different effects. Whereas knockout cell lines of either protein are viable, organism develelopment is compromised. CRT deletion in mice leads to failures in heart development and prenatal lethality (Mesaeli et al, 1999). By contrast, deletion of CNX strongly affects nerve fibers and causes early postnatal death (Denzel et al, 2002). A major discriminating feature between CRT and CNX is the TMD of CNX. Previous work has found that anchoring CRT in the membrane through fusion with the TMD of CNX can alter the spectrum of proteins associated with CRT to be similar to that of CNX (Wada et al, 1995; Danilczyk et al, 2000). Although a simple explanation for functional differences may thus be the different localization of CNX and CRT, in the membrane versus ER‐lumenal, this cannot account for many observations concerning the client binding of CNX. CNX directly interacts with the TM region of MHCI (Margolese et al, 1993). Furthermore, CNX recognizes and binds to truncated IMP substrates that lack one or more TM segments—presumably because they are recognized as misfolded and/or unassembled. Interestingly, in several of these studies, CNX:substrate binding occurred in a glycan‐independent manner (Cannon & Cresswell, 2001; Swanton et al, 2003; Fontanini et al, 2005; Wanamaker & Green, 2005; Li et al, 2010; Coelho et al, 2019). These and numerous other studies indicate that the CNX TMD is more than just a simple membrane anchor and potentially adds an essential function for intramembrane client recognition. Despite the key role of CNX in ER protein folding, only limited insights are available into a potential intramembrane client recognition by CNX and its biological impact.
Here, we show that the substrate spectrum of CNX contains a large number of nonglycosylated membrane proteins and provide an in‐depth analysis of intramembrane client recognition by CNX. Our study defines features within the TMD of CNX, as well as within its clients, that allow CNX to bind its substrates in the lipid bilayer. We identify a motif within the CNX TMD that is required for efficient client binding. This allows us to reveal a protective role of intramembrane client binding for CNX substrates. Together, these structural, mechanistic, and systematic analyses provide a comprehensive understanding of intramembrane substrate recognition by CNX. Molecular chaperones can thus combine different binding modes to safeguard the biosynthesis of membrane proteins in the ER.
Results
Calnexin interacts with nonglycosylated membrane proteins in a chaperone‐like manner
Although several studies indicate glycan‐independent membrane protein client recognition by CNX, no systematic analysis on this exists yet. We thus decided to perform a global analysis of the CNX interactome. Toward this end, we established CNX knockout cells and complemented these with a CNX construct with a C‐terminal ALFA‐tag (Fig 1A). The ALFA‐tag is lysine‐free (Götzke et al, 2019) and thus allows for the uncompromised use of lysine‐crosslinkers, like DSSO, in co‐immunoprecipitation experiments to stabilize transient CNX:client complexes. CNX itself has a large number of well‐dispersed lysines, which should allow for in situ DSSO‐crosslinking (Fig 1B). Indeed, DSSO crosslinking led to many CNX crosslinks that could be immunoprecipitated using ALFA‐tag nanobodies (Appendix Fig S1A). Mass spectrometry on the DSSO‐crosslinked interactome of CNX revealed known ER‐lumenal interactors like EDEM1, ERp29, and PDIA3, supporting the validity of our approach (Fig 1C and Dataset EV1). It furthermore revealed that approximately half of the interacting proteins were membrane proteins. Of note, among those, 44% were predicted to be not glycosylated (Fig 1C and Appendix Fig S1B). These include a small number of known functional interactors of CNX (e.g., the oxidoreductase TMX1 and the translocon subunit Sec63, Appendix Fig S1B). Although it should be noted that our approach does not distinguish between direct and indirect interactors, most of the interactors are likely to be CNX clients.
Figure 1. Calnexin interacts with nonglycosylated membrane proteins in a chaperone‐like manner.
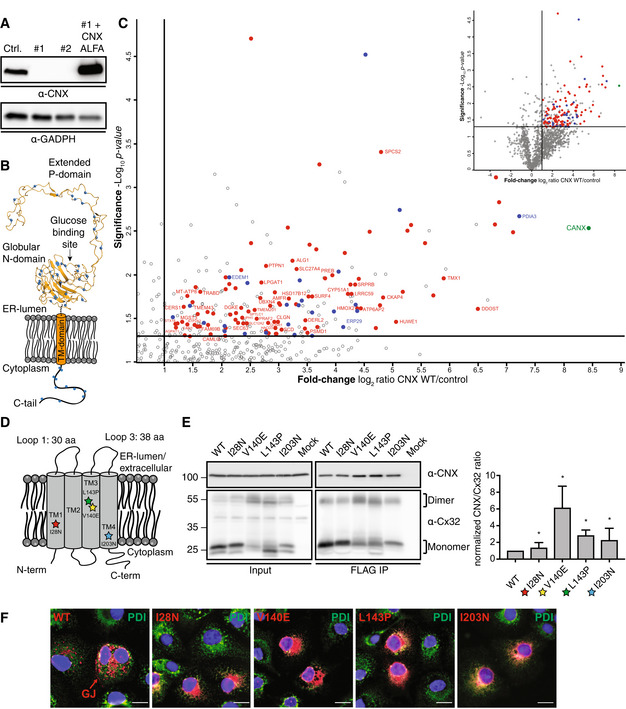
- Immunoblot analysis of overexpressed and ALFA‐tagged CNX in CNX knockout cell lines clones #1 and #2, which were used for mass spectrometry. Endogenous CNX level of wildtype HEK293T cells is shown as a control.
- Schematic of CNX showing its lysine residues as blue dots. Lysines can be crosslinked by DSSO. The exact location of lysine residues is pictured in the crystal structure of the lumenal domain of CNX whereas the position of lysines in the cytoplasmic tail (C‐tail) is an approximation.
- Volcano plots derived from LC–MS/MS analyses of ALFA‐tagged CNX, immunoprecipitated in 1% NP‐40 buffer from transfected CNX‐deficient HEK293T cells after DSSO crosslinking and compared to empty vector (EV) control co‐IPs. Depicted is an enlarged section of the associated complete volcano plot in the upper right. Among the significantly enriched proteins, annotations are Uniprot gene names. CNX is shown in green. ER proteins with the GO‐term annotation “endoplasmic reticulum”, which are localized to the ER lumen, are highlighted in blue and among those several known CNX interaction partners are indicated with their names. Membrane proteins are shown in red and those which are nonglycosylated are additionally labeled with their gene names. Further details on the classification of the identified membrane proteins can be found in Appendix Fig S1B. Cut‐off values (solid lines) in the volcano plot were defined as log2 = > 1 (2‐fold enrichment) and −log10 = (P‐value) > 1.3 (P < 0.05).
- Schematic of Cx32 structure and its orientation within the membrane. Positions of mutations analyzed in this study are indicated with stars. The overall topology and loop lengths were obtained from the UniProt server (human GJB1 gene).
- Co‐immunoprecipitations of endogenous CNX with FLAG‐tagged Cx32 WT and its mutants including quantification and statistical analysis. Constructs were expressed in HEK293T cells. One representative immunoblot is shown. Monomers and dimers of Cx32 are indicated with brackets. All samples were normalized to WT (mean ± SEM, N ≥ 4, *P‐value < 0.05, two‐tailed Student's t tests).
- Immunofluorescence images of Cx32 and its mutants. COS‐7 cells were transiently transfected with the indicated FLAG‐tagged Cx32 constructs and immunofluorescence microscopy was performed using anti‐PDI (green) as an ER marker. Detection of Cx32 was performed using anti FLAG antibodies and suitable labeled secondary antibodies (red). Nuclei were stained with DAPI (blue). All three channels are overlayed. Images are representative of cells from at least three different biological replicates. GJ denotes gap junctions observed at cell–cell junctions of cells transfected with WT Cx32. Scale bars correspond to 20 μm.
Source data are available online for this figure.
The relatively large number of membrane proteins that interact with CNX, which are predicted to be nonglycosylated, suggests that this kind of interaction is very common. Furthermore, proteome‐wide analyses in CNX k/o cells revealed reduced levels for several nonglycosylated membrane proteins (Appendix Fig S1C and Dataset EV2), implying a chaperone function of CNX for this protein class. Together, the interaction of CNX with nonglycosylated membrane proteins thus warrants further investigation.
To provide mechanistic insights into possible intramembrane, glycan‐independent client recognition and chaperoning processes by CNX, we first focused on a model protein, which a recent study from us has revealed to interact with CNX: Connexin 32 (Cx32; Coelho et al, 2019). Cx32 is a tetra‐spanning integral membrane protein (Fig 1D) that forms homo‐hexameric connexons. Two of these connexons embedded in different membranes can dock onto each other to form a gap junction channel (Pantano et al, 2008; Maeda et al, 2009). Mutations in Cx32 cause X‐linked Charcot–Marie–Tooth disease (CMTX), a common genetic disorder of the peripheral nervous system (Scherer & Kleopa, 2012). Cx32 is not glycosylated (Appendix Fig S1D) and possesses only short ER‐lumenal loops (Fig 1D). The observed CNX binding can thus neither rely on recognition of sugar moieties nor on binding of large ER‐lumenal domains by the CNX lectin domain. Taken together, we considered Cx32 a relevant starting point toward defining possible intramembrane client binding by CNX.
To investigate the nature of the Cx32:CNX interaction in more detail, we expressed FLAG‐tagged Cx32 in human HEK293T cells and analyzed its interactions with endogenous CNX in co‐immunoprecipitation experiments. These confirmed interaction of Cx32 with CNX (Fig 1E) and revealed interaction of CNX with monomeric and dimeric Cx32 (Appendix Fig S1E). If CNX indeed acted as a chaperone on Cx32, one would expect a preferential interaction with misfolded variants of the client. We thus proceeded to study four disease‐causing mutants of Cx32 (Bone et al, 1997; Rouger et al, 1997; Kleopa et al, 2006). All of these contain mutations in their TMDs (Fig 1D) but are properly integrated into the lipid bilayer (Appendix Fig S1D). In contrast to Cx32 wild‐type (Cx32 WT), all mutants were retained in the ER, arguing for misfolding and recognition by the ER quality control system (Fig 1F). Strikingly, the mutants showed an up to ~6‐fold stronger signal in CNX co‐immunopecipitation experiments, arguing for increased interaction with CNX compared with Cx32 WT (Fig 1E). Taken together, our data show that CNX interacts with a large number of nonglycosylated membrane proteins, is important for normal cellular levels of this protein class and that interactions occur in a chaperone‐like manner.
The transmembrane domain of Calnexin binds clients in the membrane
Our data suggest that CNX can recognize misfolded Cx32 in the membrane where the mutations are located. To further test this hypothesis, we individually fused each of the four Cx32 TMDs to an antibody light chain constant domain (CL; Fig 2A). A related system was established previously to assess chaperone:client interactions for soluble proteins in a systematic manner (Feige & Hendershot, 2013; Behnke et al, 2016). Using a glycosylation reporter site downstream of the TMD, we could verify that the majority of each TMD segment was integrated into the ER membrane. The only exception was TMD2 (Appendix Fig S2A), which did not integrate properly, in agreement with a recent study (Coelho et al, 2019) and the predicted membrane integration potentials (Fig 2A). TMD2 was thus excluded from further analyses. As a small portion of the TMD3 and TMD4 constructs could also enter the ER, we replaced the sugar‐accepting Asn residue downstream of each TMD by a Gln residue, to exclusively monitor glycan‐independet interaction with CNX. Using this system, we probed whether CNX could bind individual TM segments and if so, which of the TMDs from Cx32 was bound by CNX. Interaction was observed for all three TM segments and among those, by far the strongest interaction was observed with TMD1 (Fig 2B). Introducing mutations into the individual TMDs compromised their proper membrane integration (Appendix Fig S2B and C), precluding further direct analyses on the effects of mutations on binding. Collectively, our data show that CNX can differentially bind individual TM segments of a nonglycosylated client.
Figure 2. The transmembrane domain of Calnexin binds clients in the membrane.
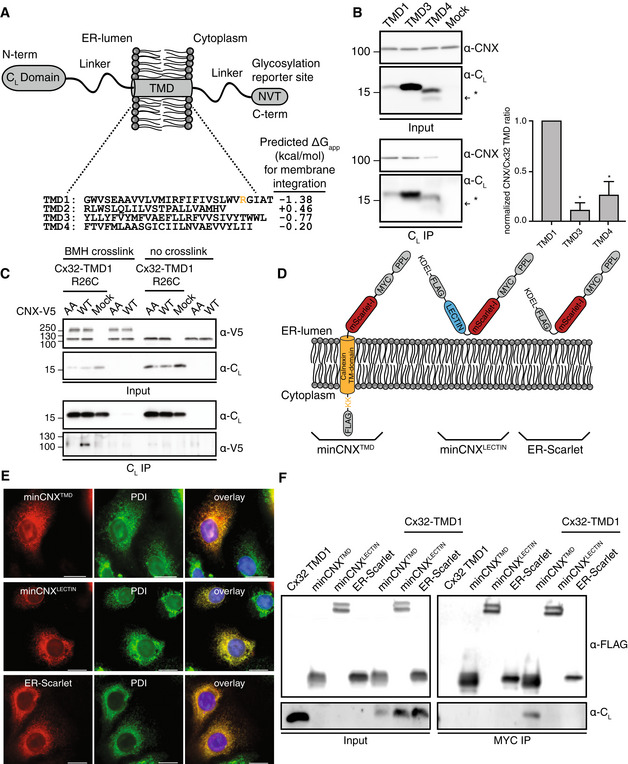
- Schematic of the reporter construct, where an antibody CL domain was fused to sequences of interest, to assess CNX interactions with individual Cx32 TMD segments. The four WT sequences of the Cx32‐TMDs are shown. The Arg residue exchanged against Cys for BMH crosslinking is highlighted in TMD1. Predicted membrane integration energies are given using DGPred (Hessa et al, 2007). Sequences of TMD1 and TMD3 were inverted in the respective constructs to have the same amino acid composition and the same amino acids exposed to the lipid bilayer as in the corresponding Cx32 TMDs.
- Co‐immunoprecipitations of endogenous CNX with the different CL‐TMD constructs (NVT glycosylation site mutated to QVT) including quantification and statistical analysis. One representative immunoblot is shown. The asterisk (*) denotes a fraction of cleaved species as observed previously for similar constructs (Behnke et al, 2016). All samples were normalized to the construct containing TMD1 (mean ± SEM, N ≥ 3, *P‐value < 0.05, two‐tailed Student's t tests).
- Interaction analysis of Calnexin with the first transmembrane domain of Cx32. Intracellular BMH crosslinking of transiently transfected V5‐tagged CNX with the transiently transfected R26C mutant of Cx32‐TMD1. In the AA mutant of CNX, the two cysteines within the CNX TMD were replaced by alanines. The species at approx. 250 kDa are possibly CNX dimers.
- Schematic of the minimal CNX construct (minCNX tmd ). This consists of a preprolactin (PPL) ER import sequence, followed by an N‐terminal MYC tag, the monomeric red fluorescent protein mScarlet‐I, the TM segment of CNX including an endogenous double lysine motif downstream of the TM region and a C‐terminal FLAG‐tag. Flexible linker regions connect the individual components. Parts derived from CNX are shown in orange. A control construct, minCNXLECTIN contains the entire lumenal lectin domain of CNX whereas another control, ER‐Scarlet, is lacking any CNX segments. A C‐terminal KDEL sequence was included in both control constructs for ER retention.
- COS‐7 cells were transiently transfected with the indicated constructs and immunofluorescence microscopy was performed using anti‐PDI (green) as an ER marker. Detection of minCNX tmd and both control constructs was carried out by mScarlet‐I fluorescence (red). Nuclei were stained with DAPI (blue). Images are representative of cells from at least three different biological replicates. Scale bars correspond to 20 μm.
- Interaction of Cx32‐TMD1 with the minCNX system. MinCNX tmd constructs and controls (see (D)) were co‐transfected with Cx32‐TMD1 into HEK293T cells. Interaction between the proteins was analyzed by co‐immunoprecipitation experiments followed by immunoblotting.
Source data are available online for this figure.
To assess whether binding was direct, we made use of cysteine crosslinking in the membrane. CNX possesses two intrinsic Cys residues at the cytoplasmic side of its TMD (Appendix Fig S2D). We reasoned that these should be in proximity to the cytoplasmic side of a substrate TMD if interactions were direct. Thus, within the first TMD of Cx32, we replaced an Arg at a suitable position with Cys (Fig 2A). Using the membrane‐permeable crosslinker bismaleimidohexane (BMH), we could observe a crosslink between CNX and the TMD1 of Cx32, which was dependent on the CNX‐intrinsic cysteines (Fig 2C). Together, this argues for direct binding of the CNX TMD to a substrate TMD.
Based on these findings, we next aimed to define which structural elements of CNX were necessary and sufficient for this interaction. Toward this end, we designed a minimal CNX construct that only contained the CNX TMD and a few additional C‐terminal residues (minCNX tmd ). The ER‐lumenal domain of CNX was replaced by the fluorescent protein mScarlet‐I (Fig 2D). To maintain ER retention of the construct, an endogenous cytosolic di‐lysine motif was left in place (Fig 2D and Appendix Fig S2D; Jackson et al, 1990). ER‐localization was confirmed by fluorescence microscopy (Fig 2E). Using this construct, we found that the CNX TMD was necessary and sufficient for binding to full‐length Cx32 (Appendix Fig S2E). Further extending this finding, we could show that the first TMD of Cx32 was sufficient to bind to the minCNX tmd construct (Fig 2F). In either case, a control construct containing only the lumenal lectin domain of CNX (minCNXLECTIN, Fig 2D and E, and Appendix Fig S2F) as well a second ER‐retained control construct lacking any CNX regions (ER‐Scarlet, Fig 2D and E, and Appendix Fig S2G) did not bind to the different clients (Fig 2F and Appendig Fig S2E). Thus, binding to nonglycosylated IMP clients can occur via the CNX TMD and does not require the CNX lectin domain or its C‐terminal tail. Accordingly, the CNX TMD contains the relevant features for client recognition in the membrane, and this can be recapitulated with a single TMD derived from a client protein.
Development of a tool to systematically assess intramembrane recognition processes
Our data show that CNX directly binds to individual TM segments of its clients and that the CNX TMD is sufficient for binding to occur. This established a minimal system for client recognition by a chaperone in the membrane. Based on these findings, we next aimed to define which intramembrane features of its clients CNX recognizes. This would be a major step forward in our understanding of intramembrane chaperones but is a difficult task to accomplish using (parts of) natural proteins, as these generally will possess complex sequences and structures which preclude unbiased analyses. We thus designed a client protein that allows for the systematic analysis of intramembrane recognition processes. Toward this end, we performed a multiple sequence alignment of 200 randomly selected human single pass plasma membrane proteins from the Membranome database (Lomize et al, 2017, 2018). This gave rise to an average TMD of a transport‐competent protein, which can thus be expected to lack major chaperone recognition sites. We termed this protein minimal consensus membrane protein (ConMem). The design of ConMem included a superfolder GFP (sfGFP) moiety for microscopic localization studies, a C‐terminal HA‐epitope tag, and two glycosylation sites to analyze the membrane topology and intracellular transport of the construct (Fig 3A). In agreement with our assumption that, on average, TM segments from single‐pass cell surface TMD proteins are close to optimal, the most frequent amino acid at most positions turned out to be Leu (Fig 3A). TM Leu‐zippers, however, have a strong self‐assembly propensity that could compromise our analyses (Gurezka et al, 1999). We thus proceeded with the second most frequently occuring amino acids, which were also forming a benign TM sequence (Hessa et al, 2007). The multiple sequence alignment resulted in a TMD of 26 amino acid in length in ConMem, which is in very good agreement with studies on average TMD lengths in the plasma membrane (Sharpe et al, 2010; Singh & Mittal, 2016). Interestingly, it was flanked by an N‐terminal Pro‐residue, breaking the helical TMD structure (Cordes et al, 2002), and a C‐terminal Lys residue (Fig 3A and Appendix Fig S3). A C‐terminal Lys will induce a type I orientation (von Heijne & Gavel, 1988), placing the Lys residue in the cytoplasm, which reflects the nature of our sequence set (76% of the proteins were type I). Individually mutating the first or second consensus glycosylation site in ConMem showed that it was exclusively modified at the first site, which confirms the predicted topology (Fig 3B). Enzymatic deglycosylation with EndoH (which only removes N‐linked sugars not further modified in the Golgi) or with PNGaseF (which also removes Golgi‐modifed N‐linked sugars) revealed that ConMem glycosylation was EndoH‐resistant, arguing that it was able to traverse the Golgi as expected (Fig 3B). Localization to the plasma membrane was confirmed by microscopic studies (Fig 3C). Taken together, ConMem provided us with an ideal tool to systematically dissect intramembrane substrate recognition by CNX.
Figure 3. Design and validation of ConMem, a tool to query TM‐domain recognition.
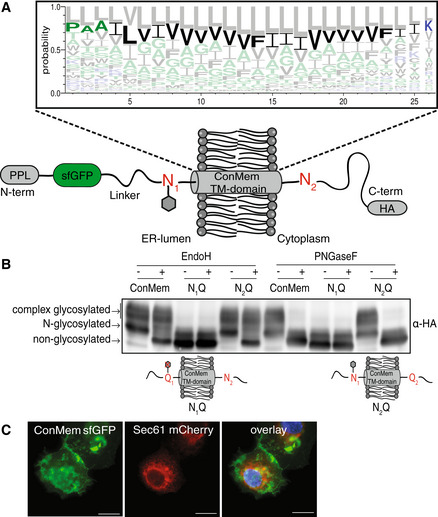
- Schematic of the the minimal consensus membrane protein ConMem which contains a preprolactin (PPL) ER import sequence, a superfolder GFP (sfGFP), a TMD flanked by two individual NVT glycosylation motifs (N1 and N2) and a C‐terminal HA‐tag. Individual construct components are connected by flexible linker regions. As illustrated, in the predicted topology of ConMem, only the first NVT glycosylation site (N1) is accessible to the ER glycosylation machinery (gray hexagon). The TMD of ConMem was designed on the basis of a multiple sequence alignment of 200 human single pass TMD sequences as illustrated in the sequence logo. Hydrophilic amino acid residues are depicted in blue, neutral ones in green and hydrophobic residues in black. The predicted amino acid sequence with the second highest score was selected as the TMD consensus sequence for ConMem (bold). Note that the orientation of the TMD was not included in this analysis, thus giving rise to a consensus sequence purely based on amino acid composition.
- HEK293T cells were transiently transfected with plasmids expressing ConMem or the indicated variants, where either the first (N1) or the second (N2) NVT glycosylation motif was ablated (N to Q mutation). Lysates were treated with or without EndoH or PNGaseF as indicated, and analyzed by immunoblotting. N‐glycosylation occurs in the ER, complex glycosylation in the Golgi.
- COS‐7 cells were transfected with the indicated constructs and fluorescence microscopy was performed using Sec61 mCherry (red) as an ER marker. ConMem, detected by sfGFP fluorescence (green), localizes to the plasma membrane. Nuclei were stained with DAPI (blue). Images are representative of cells from at least three different biological replicates. Scale bars correspond to 20 μm.
Source data are available online for this figure.
Defining intramembrane recognition motifs for Calnexin
To dissect features recognized by CNX in the membrane, we first individually replaced the central Val residue at position 13 in ConMem with all other 19 amino acids. Of note, for all of these substitutions, ConMem was still predicted to be stably integrated into the membrane (Fig 4A), which was experimentally confirmed for several constructs and different locations of polar amino acids (Appendix Fig S4A). Using this ConMem panel, we first established suitable conditions for co‐immunoprecipitation experiments (Appendix Fig S4B) and then investigated interactions with CNX. Although ConMem containing an ER‐lumenal glycosylation site bound stronger to CNX, significant binding was also observed without this site. This shows that glycosylation of ConMem increases binding to CNX but is not required for it to occur (Fig 4B), in agreement with our mass spectrometric analyses and the binding to Cx32 (Fig 1). No binding of sfGFP (ConMemΔ tmd ) to CNX was observed (Appendix Fig S4C), neither did Cx32 bind to ConMem (Appendix Fig S4D), showing the specificity of the ConMem:CNX interactions. Together, this further corroborated binding of CNX to TM regions in the membrane. For all subsequent experiments, ConMem lacking its ER‐lumenal glycosylation site was used to specifically investigate glycan‐independent binding to CNX. Since CNX also bound to ConMem with an unaltered TM segment, this allowed to assess features increasing and decreasing binding to CNX. Using this approach revealed a distinct binding pattern for CNX to the 20 ConMem variants with the membrane‐central amino acid exchanged (Fig 4A). For some, for example, Arg, binding was significantly increased, whereas for others, for example, Pro, binding was decreased (Fig 4C and Appendix Fig S4E). Arg introduces a polar residue into the membrane, whereas Pro acts as a TMD helix breaker. Based on these findings, we selected one amino acid where increased binding to CNX was observed if it was present in a central location in the ConMem TM segment (Arg), and one that decreased binding (Pro; Fig 4C). For these, we moved the mutation site through the entire ConMem TMD to analyze a possible positional dependency on CNX binding (Fig 4D). In each case, the central position 13 (of 26 amino acids) showed the strongest effects, but replacements at other positions also influenced binding, with slightly different positional dependencies for the two selected amino acids (Fig 4D). In summary, these data show that CNX can differently recognize clients with changes of single amino acids in the membrane.
Figure 4. Defining intramembrane recognition preferences for Calnexin.
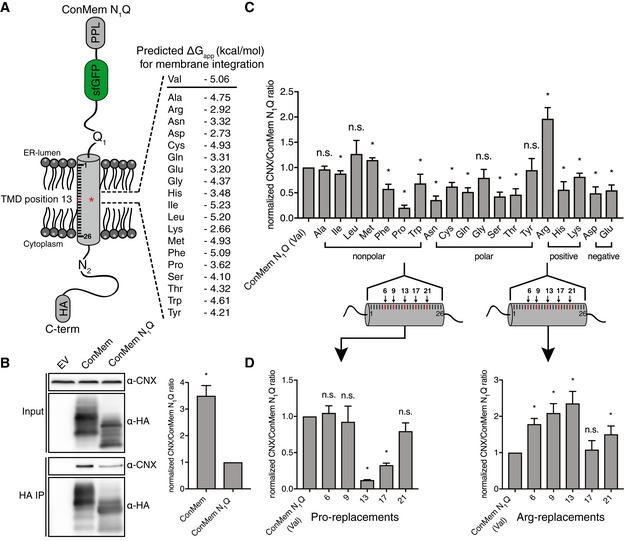
- Schematic of ConMem N1Q where a central Val residue at position 13 within its TMD was replaced by all other 19 amino acids. Free energies for TMD insertion were predicted according to Hessa et al (2007).
- Representative blots from co‐immunoprecipitation experiments from HEK293T cells transfected with the indicated ConMem constructs with or without ER‐lumenal glycosylation site (N1; mean ± SEM, N = 3, *P‐value < 0.05, two‐tailed Student's t tests). Individual values were normalized to ConMem N1Q (Val) values that were set to 1.
- Interaction of ConMem N1Q variants as described in (A) with endogenous CNX (mean ± SEM, N ≥ 5, *P‐value < 0.05, two‐tailed Student's t tests). Individual values were normalized to ConMem N1Q (Val) values that were set to 1.
- Same as in (C) only that selected amino acids (proline and arginine) where shifted through the ConMem N1Q TMD segment to five different positions as shown in the illustration, revealing positional binding dependencies (mean ± SEM, N ≥ 4, *P‐value < 0.05, two‐tailed Student's t tests). Individual values were normalized to ConMem N1Q (Val) values that were set to 1.
Source data are available online for this figure.
A structural understanding of intramembrane Calnexin:client recognition
Having defined client‐intrinsic binding patterns for CNX in the membrane, we next proceeded to analyze the features within the CNX TM segment that allow client binding to occur. Toward this end, we performed molecular dynamics simulations on either the CNX TM region together with the ConMem TMD or with the first TMD of Cx32 (Cx32‐TMD1), which our data have shown to be interacting systems in cells (Figs 2 and 4). In agreement with these experimental findings, molecular dynamics simulations also showed interactions for both systems (Fig 5A). Of note, for ConMem, as well as for Cx32‐TMD1, similar regions in the CNX TMD region were involved in the binding process. These regions involved a Tyr, a Thr and a Leu on the same face of the CNX TMD region (termed YTL‐motif), which we found in the simulations always to be oriented toward the substrate TMDs if a complex was formed (Fig 5A–C). For ConMem, apolar interactions seemed to dominate complex formation (Fig 5B). For Cx32‐TMD1, polar interactions including hydrogen bonds between the CNX Tyr and the Cx32 backbone but also between an Arg in Cx32‐TMD1 and a Ser in the CNX TMD as well as possible cation‐π interactions with a Phe in the CNX TMD were observed (Fig 5C). Supporting the relevance of the YTL‐motif, in silico free energy calculations revealed that exchanging this motif to Val residues caused significant destabilization of the CNX‐ConMem complex by approx. 50% in comparison with the wt complex (details in the Materials and Methods section) although our simulations also suggested flexibility in the molecular details of binding. To assess our computational predictions experimentally, we mutated these predicted interaction sites also to Val residues in a CNX construct that we expressed in mammalian cells. This construct was furnished with a V5 epitope tag for specific immunoprecipitation and overexpression was only slightly higher than the endogenous CNX level (Appendix Fig S5A). Strikingly, whereas ConMem and Cx32‐TMD1 co‐immunoprecipitated with V5‐tagged CNX as expected, mutation of the YTL‐motif, and even of only the Y and T residues to Val, significantly reduced interactions between CNX and ConMem (Fig 5D). This was even more pronounced for Cx32‐TMD1 (Fig 5E), and when the YTL‐motif was replaced by Ala instead of Val, a similar reduction in binding was observed (Appendix Fig S5B). This argues that mutating the interaction site per se, but not the choice of the residues mutated to, accounts for the effects. Furthermore, when we assessed two of the nonglycosylated TM protein interactors of CNX our mass spectrometry analysis had revealed (Fig 1C and Appendix Fig S1B) we also observed significant reduction in binding to CNX when the YTL‐motif was mutated (Appendix Fig S5C). Together, these findings supported our simulation data and pointed toward a conserved interaction motif in CNX, necessary for client binding in the membrane. Of note, when ConMem with its ER‐lumenal glycosylation site was used, mutations in the CNX TMD region did not significantly affect binding, arguing that for this glycosylated client with a well‐behaving TMD, binding via the lectin domain is dominant (Fig 5F). The importance of the intramembrane YTL‐motif obtained with these model clients was corroborated when we investigated CNX interactions with full‐length Cx32 (Fig 5G) as well as the Cx32 V140E mutant (Appendix Fig S5D).
Figure 5. A structural analysis of intramembrane Calnexin:client recognition.
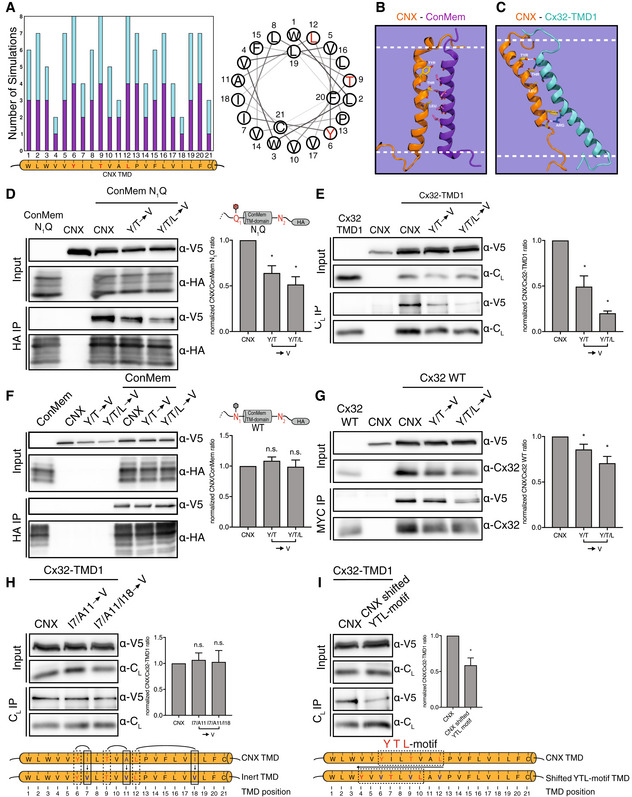
- Meta analysis of MD simulations (N = 8) where complex formation between CNX and Cx32‐TMD1 or CNX and ConMem was observed. On the x‐axis the CNX TMD residues obtained from UniProt are shown. The height of the bars corresponds to the number of simulations in which a specific CNX TMD residue interacted with the substrate helix CX32‐TMD1 (turquoise) or ConMem (violet). Only CNX TMD residues Y6, T9 and L12 (highlighted in red in the helical wheel) interacted with CX32‐TMD1 or ConMem in all sampled complexes.
- The YTL‐motif of CNX (orange) interacts with ConMem (violet) mainly via hydrophobic interactions in MD simulations.
- Interactions between the CNX (orange) YTL‐motif and Cx32‐TMD1 (turquoise) include general hydrophobic interactions and hydrogen bonding between Y6 and the CX32‐TMD1 backbone. The intramembrane arginine of Cx32 is involved in CNX interactions via cation‐π stacking as well as polar interactions.
- Interaction of nonglycosylated ConMem N1Q constructs with V5‐tagged CNX. In CNX variants, amino acids important for interaction as revealed in (A) were replaced by valine residues. HEK293T cells were transiently transfected with the indicated constructs and cell lysates and HA‐immunoprecipitates were analyzed for HA‐tagged ConMem N1Q and co‐immunoprecipitating CNX mutants. One representative blot and quantifications are shown (mean ± SEM, N ≥ 9, *P‐value < 0.05, two‐tailed Student's t tests).
- The same as in (D) for Cx32‐TMD1 (mean ± SEM, N ≥ 6, *P‐value < 0.05, two‐tailed Student's t tests).
- Analysis of mutations in the CNX TMD region and their effect on binding glycosylated ConMem. One representative blot and quantifications are shown (mean ± SEM, N ≥ 3, n.s.: not significant, two‐tailed Student's t tests).
- The same as in (D) for Cx32 full‐length (mean ± SEM, N ≥ 4, *P‐value < 0.05, two‐tailed Student's t tests).
- Interaction of Cx32‐TMD1 with CNX mutants, where amino acids not predicted to be important for interaction as shown in (A) were exchanged by valine residues. HEK293T cells were transiently transfected with the indicated constructs. One representative blot and quantifications are shown (mean ± SEM, N ≥ 9, n.s.: not significant, two‐tailed Student's t tests).
- Shifting of the residues Y, T and L, most important vor CNX substrate interaction, and replacing these against valine residues, impacts interactions with Cx32‐TMD1. HEK293T cells were transiently transfected with the indicated constructs. One representative blot and quantifications are shown (mean ± SEM, N ≥ 9, *P‐value < 0.05, two‐tailed Student's t tests).
Source data are available online for this figure.
To further verify the specificity of the binding motif we have identified within the CNX TMD, we selected three non‐Val residues that had the lowest overall predicted client interactions from our simulations (an Ile (TMD position 7), an Ala (TMD position 11) and another Ile (TMD position 18), Fig 5A). Replacing either the first two or all three of these three residues by Val did not affect binding of CNX to its client Cx32‐TMD1 (Fig 5H), further supporting our molecular interpretation of CNX client binding in the membrane. By contrast, when we shifted the identified YTL‐motif N‐terminally within the CNX TMD, this again significantly reduced binding to Cx32‐TMD1 (Fig 5I), revealing positional specificity in the recognition process. Our combined computational and experimental approach thus points toward a molecular recognition motif within the CNX TMD that is involved in CNX client binding in the lipid bilayer.
Biological functions of intramembrane client recognition by Calnexin
Our comprehensive analyses provided us with detailed molecular insights into CNX:client recognition in the membrane. Using these insights, we were able to generate a CNX variant that was compromised in client binding within the membrane while still having a functional lectin domain (Fig 5). To define functional consequences of weakening intramembrane client binding, we used rhodopsin (Rho) as a model protein, as its biogenesis is highly dependent on CNX (Rosenbaum et al, 2006). To avoid confounding effects by endogenous CNX, we used our CNX knockout cell line (Fig 1A). CNX knockout did not cause detectable ER stress and neither did the re‐expression of CNX or its mutant compromised in intramembrane client binding (Appendix Fig S6A). Furthermore, both CNX variants showed normal ER localization in the knockout cells (Appendix Fig S6B), which together are prerequisites for unbiased analyses. We first analyzed interaction of Rho with either reconstituted wt CNX or the mutant lacking the intramembrane YTL‐motif. In agreement with our data on a panel of other model proteins (Fig 5 and Appendix Fig S5), Rho binding to the CNX mutant was significantly reduced (Fig 6A), qualifying Rho as another candidate of the intramembrane client binding by CNX. In agreement with this finding, Rho as a strong CNX client was hardly detectable if CNX was completely absent (Fig 6A). Building on these insights, we assessed the effects of CNX on Rho stability in cells. Strikingly, Rho degradation was significantly accelerated in CNX knockout cells complemented with the YTL‐motif mutant in comparison to wt CNX (Fig 6B), revealing a protective role of intramembrane substrate binding by CNX for a labile client. This protective role of intramembrane client binding appeared to work synergistically with lectin‐based client binding as the complete absence of CNX had an even stronger effect on Rho degradation, which was again hardly detectable in the complete absence of CNX (Fig 6B). Thus, CNX intramembrane client binding synergizes with lectin‐based binding to chaperone labile clients.
Figure 6. Biological functions of intramembrane client recognition by Calnexin.
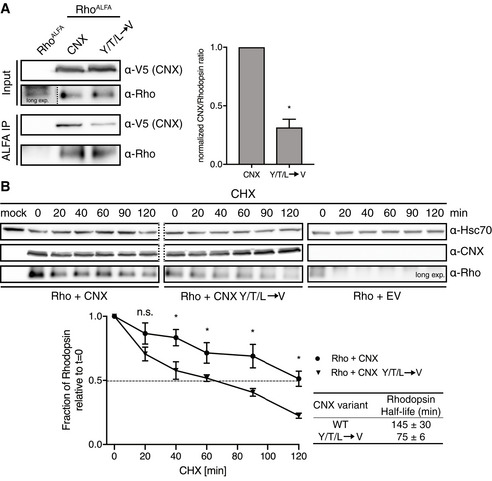
- Analysis of the interaction of the binding deficient CNX YTL TMD variant with Rhodopsin. CNX knockout HEK293T cells were transiently transfected with either N‐terminal ALFA‐tagged Rhodopsin alone or in combination with the indicated V5‐tagged CNX constructs. In the absence of any CNX, Rho is hardly detectable (long exposure shown). Cell lysates and ALFA‐immunoprecipitates were analyzed for Rhodopsin and co‐immunoprecipitating CNX. One representative blot is shown on the left, quantifications on the right (mean ± SEM, N = 4, *P‐value < 0.05, two‐tailed Student's t tests).
- Analysis of Rhodopsin degradation and its dependency on CNX. CNX knockout HEK293T cells that were transfected with the indicated constructs were incubated with cycloheximide and lysates were collected at different timepoints. CNX WT and CNX YTL immunoblots are from the same blot (dashed line), showing similar expression levels. Hsc70 was used as a loading control (also from the same blot, dashed line). Quantifications are shown below representative immunoblots (mean ± SEM, N ≥ 8, *P‐value < 0.05, two‐tailed Student's t tests).
Source data are available online for this figure.
Discussion
Focusing on CNX, one of the key chaperones of the ER, this study advances our understanding of the molecular mechanisms that underlie membrane protein chaperoning. We show that CNX recognizes membrane protein clients in the lipid bilayer, even if nonglycosylated, which reconciles a long debate about this chaperone. Furthermore, we identify chaperone‐ and substrate‐intrinsic motifs that allow client recognition in the membrane, establishing a systematic bilateral analysis on this important event in cell biology. And lastly, our data show that a large variety of CNX clients are nonglycosylated and some depend on CNX for their physiological levels, thus providing a new perspective on the client repertoire of this key chaperone.
Among the nonglycosylated membrane protein clients of CNX, we find roughly equal numbers of single‐pass but also multipass TM proteins. It has been suggested that the TM helix of CNX may act as a placeholder until assembly of multipass TM proteins in the lipid bilayer is complete (Cannon & Cresswell, 2001). This is consistent with our findings that CNX binds to several multipass membrane proteins. It is also consistent with our data that CNX has a preference for intramembrane mutants of TM proteins, where helix assembly in the bilayer may fail. The large number of nonglycosylated single‐pass TM protein clients of CNX we identify, however, suggests additional functions, in particular since not all of those are (known to) be part of multiprotein complexes. A likely additional function is to link substrate recognition in the membrane to ER lumenal chaperone action: whenever CNX binds to TM helices of clients in the membrane, its ER lumenal domain can recruit further folding factors like the PDI ERp57 (Oliver et al, 1997) or the PPIase CypB (Kozlov et al, 2010a). Our study suggests that this recruitment is not necessarily dependent on client glycosylation, which significantly extends the substrate repertoire of the CNX‐centered folding machinery. It is noteworthy that ~50% (1,400 of 2,790 proteins) of all human multipass membrane proteins do not contain any domains outside the membrane (defined as continuous stretchers longer than 100 amino acids, TM domains predicted with Phobius (Käll et al, 2004) using the UniProt databank (UniProt Consortium, 2021)). By contrast, among the human proteins with only one TM domain, 90% do contain domains outside the membrane. This highlights the need for intramembrane chaperoning for multipass membrane proteins—as well as the need of recruiting folding machineries for soluble domains to single pass membrane proteins.
For CNX itself, we find a surprisingly simple principle of client engagement in the membrane: its single TMD can apparently directly bind TM clients, involving a YxxTxxL motif in the CNX TM region (with x being any amino acid, although we did not analyze this further; Fig 7). The amino acids in this motif point toward the same face of the CNX TMD, and our data show that the position of the motif within the membrane is important for client binding. It should be noted that CNX can also form (ERp29‐mediated) dimers (Nakao et al, 2021), which may affect intramembrane client binding and CNX may, for example, in fact be a dimer when it recognizes TMDs. Our data can also not rule out that further factors are involved in CNX client binding, as CNX has a large repertoire of interacting proteins in the ER that among others involve the membrane protein chaperone EMC (Christianson et al, 2011). This may suggest that CNX can also act synergistically with other TM protein chaperones in the ER. Taken together, however, our data favor the interpretation of direct client engagement by the CNX TMD. We thus performed a bioinformatic analysis on the occurrence of the YxxT motif in CNX from various species but also in other membrane proteins in yeast and humans (leaving out the very common L residue for this analysis) of the ER and inner nuclear membrane. In CNX from different species, the YxxT motif is strictly conserved (Appendix Fig S7A). Interestingly, similar motifs are found in the TMDs of yeast Spf1, Asi2, Hrd1 and Dfm1 (Appendix Fig S7B), factors that are all involved in protein quality control and may recognize clients in the lipid bilayer (Sato et al, 2009; Neal et al, 2018; McKenna et al, 2020; Natarajan et al, 2020). In humans, among others, this motif is found in Bap31 and RHBL4 (Appendix Fig S7C), which are also known to be involved in membrane protein quality control (Geiger et al, 2011; Fleig et al, 2012). This could potentially suggest a more general role of a YxxT or similar motif in this process—and now allows to test this experimentally.
Figure 7. Intramembrane client recognition potentiates the chaperone functions of Calnexin.
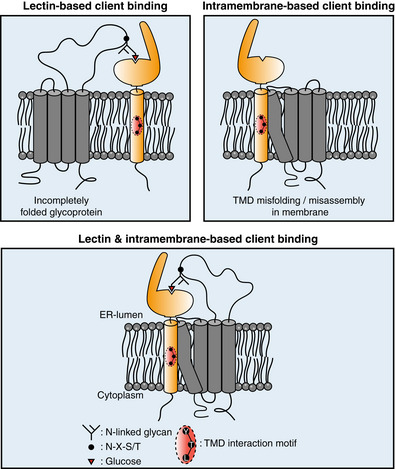
A model for the recognition and quality control of membrane proteins by CNX. Calnexin can recognize clients via mono‐glucosylated glycans (lectin‐based binding mode). Additionally, membrane proteins containing a misfolded/mis‐assembled TMD are recognized and bound by calnexin via a substrate interaction motif located in its TMD (intramembrane‐based client binding). For glycosylated membrane proteins, both modes can work synergistically to chaperone clients.
The identification of a client recognition motif within the CNX TM region allowed us to assess the biological impact of lectin‐versus TM‐based binding of CNX. We find that for the single‐pass client ConMem, binding can occur via the CNX TMD or, if ConMem was glycosylated, via the CNX lectin domain. Consistent with this, for Rhodopsin, a multipass glycoprotein, both recognition modes work synergistically: ablating binding in the membrane reduces Rhodopsin stability, but to lesser extent than complete CNX deletion from cells. This highlights the functional crosstalk between both binding modes, which, characteristic for chaperones, each need to be dynamic and of rather low affinity (Fig 7).
Recent work has revealed that several previously ill‐characterized proteins act as membrane protein chaperones, including the EMC (Jonikas et al, 2009; Satoh et al, 2015; Chitwood et al, 2018; Shurtleff et al, 2018; Coelho et al, 2019), the PAT complex (Meacock et al, 2002; Chitwood & Hegde, 2020) and the Asi complex in yeast, which appears to control assembly of membrane proteins (Natarajan et al, 2020). Many of these factors likely work synergistically to support membrane protein biogenesis (McGilvray et al, 2020). Our systematic analysis revealed TM‐intrinsic preferences for client binding by CNX. The strongest binding is observed for a centrally located Arg residue. Arg, among all amino acids, has a high propensity to be protonated even within the lipid bilayer (Yoo & Cui, 2008). This may also affect its integration into the membrane, for example, TMD tilting, involving snorkeling of the Arg residue toward the negatively charged headgroups of the lipid bilayer, can occur and thus influence binding. For the PAT complex, polar residues within the TMD of Rhodopsin were found to be important for binding, again in particular Arg (Chitwood & Hegde, 2020). Of note, in addition to the PAT complex, CNX was among the most highly enriched interactors of early intermediates in Rhodposin biogenesis (Chitwood & Hegde, 2020). This suggests a concerted action of CNX and the PAT complex on membrane proteins in the process of their biogenesis, which our study now reveals not to be restricted to the lectin functions of CNX. Arg residues seem to be a hotspot of TM chaperone recognition, including CNX and the PAT complex. Of note, Arg‐introducing mutations are often functionally detrimental for a membrane protein (Fink et al, 2012). For the EMC, chaperoning functions have also been proposed for polar TMDs (Shurtleff et al, 2018; Coelho et al, 2019; Tian et al, 2019) but also generally on first Nexo transmembrane helices to allow for their correct integration (Chitwood et al, 2018). It is noteworthy, that for none of these three factors, EMC (when acting as a chaperone), PAT or CNX, it is currently known how clients are released. It may be simply a competition between folding and binding, as suggested for the PAT complex (Chitwood & Hegde, 2020), but for soluble chaperones, generally energy‐dependent processes regulate client release. In this light, the dual binding mode for CNX, via its TMD and lectin domains, is an interesting finding as it allows for synergistic binding of clients that expose two chaperone recognition sites at the same time, yet will likely lead to reduced affinity if only one site is engaged, for example, due to modification of glycans within the client or TMD folding/assembly.
Taken together, from functional (Conti et al, 2015) and structural studies (McGilvray et al, 2020), the picture of a complex molecular machinery supporting membrane protein biogenesis in proximity to the translocon emerges. Recent work together with this study now allow us to begin to systematically, mechanistically and structurally understand how individual membrane protein chaperones in this complex system recognize their clients and synergistically function to allow cells to correctly produce one third of their proteome.
Materials and Methods
DNA constructs
ConMem WT constructs, CNX, minCNX tmd and relevant control constructs were obtained from GeneArt Gene Synthesis (ThermoFisher) in a pcDNA3.4 TOPO expression vector optimized for mammalian expression. The TMD sequence of ConMem WT was designed by multiple sequence alignment of 200, randomly selected, human single pass plasma membrane proteins (152 type I and 48 type II orientation) whose TMD sequences were obtained from the Membranome 2.0 Database using Unipro U Gene software. The human CNX sequence was obtained from UniProt (P27824) and complemented with a C‐terminal epitope tag. For construction of the minCNX tmd construct, the human CNX TMD sequence was obtained from UniProt, its TMD helix verified according (Hessa et al, 2007) to avoid artificially shortening of the TMD sequence and finally complemented by endogenous amino acids flanking the TMD region N‐ and C‐terminally (Appendix Fig S2D). Human Cx32 and Rhodopsin cDNA was obtained from Origene and cloned into a pSVL vector (Amersham). CL‐Cx32‐TMD1 reporter constructs were synthesized by GeneArt Gene Synthesis (Thermo Fisher) and cloned into a pSVL vector. Individual construct components of the designed proteins and N‐ as well as C‐terminal epitope tags were separated by (GGGS)2, (GSGS)2 or (GGGA)2 linkers. The full sequence of the CL‐Cx32‐TMD1 construct, which was mainly used in this study, is: MAWISLILSLLALSSGAISQAGQPKSSPSVTLFPPSSEELETNKATLVCTITDFYPGVVTVDWKVDGTPVTQGMETTQPSKQSNNKYMASSYLTLTARAWERHSSYSCQVTHEGHTVEKSLSRADSRGSGSGSGSGSGWVSEAAVVLVMIRFIFIVSLWVRGIATGSGSGSGSGSQVTSS. For all other CL‐CX32‐TMD constructs, the TMD sequence shown in italic letters was replaced by the TMD sequence of TMD2, TMD3 or TMD4 of Cx32, which are shown in Fig 2A. Cloning into the mammalian pSVL expression vector was performed using restriction enzymes BamHI and XhoI followed by T4 DNA ligase (Promega) ligation. Introduction of epitope tags and point mutations was carried out via site‐directed mutagenesis PCR using overlapping, complementary mutagenesis primers, Pfu polymerase (Promega) and subsequent DpnI (NEB) digestion. All constructs were verified by sequencing prior to use (Eurofins Genomics).
Cell culture and transient transfections
HEK293T (ECACC) cells were cultivated in Dulbecco's modified eagle's medium (DMEM), high glucose (Sigma‐Aldrich), supplemented with 10% (v/v) fetal bovine serum (Biochrom or Gibco) and 1% (v/v) antibiotic‐antimycotic solution (25 μg/ml amphotericin B, 10 mg/ml streptomycin, 10,000 units of penicillin (Sigma‐Aldrich); complete DMEM) at 37°C and 5% CO2. Twenty‐four hours prior to transfection, 250,000 HEK293T cells were seeded per p35 plate (precoated Corning BioCoat Poly‐D‐Lysine 35 mm #354467 or uncoated Nunclon multidish six‐well plates, Thermo Scientific #140685 coated with 50 μg/ml Poly‐D‐Lysine solution (Gibco A38904‐01) per well according to manufacturer's instructions) or 300,000 per p60 plate (Tissue Culture Dish 60, TPP). Transient transfection was performed using GeneCellin (Eurobio) according to the manufacturer's protocol. For transient transfections using pcDNA (strong CMV promotor), the amount of DNA was reduced to half the amount suggested by the manufacturer.
Cycloheximide chase assays
Cells were transfected with the indicated constructs. Inhibition of further protein biosynthesis and determination of protein half‐lives was performed by the replacement of complete medium with complete medium containing 50 μg/ml Cycloheximide (Sigma‐Aldrich) 24 h after transfection and the collection of lysates, as described below, at different time points. Controls were prepared in the same manner without the addition of CHX (t = 0 sample).
Induction of ER stress
The induction of the unfolded protein response was carried out by the addition of 5 μg/ml tunicamycin (Sigma‐Aldrich) in medium and incubation for 6 h prior cell lysis or the supplementation of pre‐warmed medium with 10 mM DTT (Sigma‐Aldrich) for 1 h. Cleavage analysis of the activating transcription factor 6 (ATF6) based on immunoblotting was performed following immunoprecipitation to increase the amount of detectable protein. Determination of endogenous phosphorylation by immunoblotting (total amount and phosphorylation at Ser51 only) of the eukaryotic initiation factor 2α (eIF2α) was performed following cell lysis using lysis buffer additionally supplemented with 1 × phosphatase inhibitor mix (Serva). To assess XBP1 splicing, RNA was extracted using RNeazy Mini Kit (Qiagen) in an RNase‐free environment. Subsequently RT–PCR of the purified RNA was performed using OligodT20 Primer (18418020, Thermo Fisher) and SuperScript III Reverse Transcriptase (18080044, Thermo Fisher). Following amplification of the XBP1 transcript of the resulting cDNA, XBP1 splicing events were analyzed on 2% agarose gels.
DSSO crosslinking
Transiently transfected cells were washed twice with ice‐cold PBS prior the addition of 2 mM DSSO (Thermo Fisher) in PBS per sample, diluted from 100 mM stock DSSO in anhydrous DMSO (Thermo Fisher). Crosslinking was performed on ice for 1 h including regular agitation. Inhibition of the crosslinking reaction was achieved by washing of the samples with quenching solution (50 mM Tris–HCl pH 8.0) for 15 min. In the following, cell lysis, immunoprecipitation, and sample preparation for mass spectrometry was performed as described in the relevant sections.
BMH crosslinking
Transiently transfected cells were washed twice with ice‐cold PBS prior the addition of 500 μM BMH (Thermo Fisher) in PBS per sample, diluted from a 20 mM stock of BMH in anhydrous DMSO (Thermo Fisher). Crosslinking was performed at room temperature in the dark for 1 h including regular agitation. The crosslinking reaction was ended by washing of the samples with PBS supplemented with 5 mM 2‐mercaptoethanol for 15 min. Subsequently, the cells were washed twice with PBS and cell lysis, and immunoprecipitation were performed as described in the relevant sections.
Cell lysis
Cells were harvested 24 h after transfection (if not described otherwise). All steps were performed on ice or at 4°C using ice‐cold solutions. Cells were washed using PBS and then lysed for 20 min by adding 1 ml (p60) or 500 μl (p35) lysis buffer (50 mM Tris–HCl pH 7.5, 150 mM NaCl complemented with either 1% Digitonin (Sigma‐Aldrich) or 0.5% NP‐40 (Sigma‐Aldrich) along with 0.5% DOC) for ConMem, Cx32, CNX and minCNX tmd or minCNX tmd control constructs. For MS experiments, lysis buffer containing 1% NP‐40, 1 mM MgCl2 and 5% Glycerol was applied to the cells. CL Cx32‐TMD constructs were lysed in NP‐40 buffer only when conducting co‐immunoprecipitation experiments otherwise Digitonin based buffer was used. All Digitionin and NP‐40 based buffers were supplemented with cOmplete protease inhibitor cocktail (Roche) prior to lysis. The resulting lysate was centrifuged for 15 min at 15,000 × g and the supernatant was used for subsequent experiments.
Deglycosylation experiments
Samples were digested for 1 h at 37°C with either EndoHf, or PNGase F (NEB) according to the manufacturer's instructions. Negative controls were prepared in the same manner without the addition of enzymes. The digested proteins were thereafter supplemented with 5× Laemmli buffer and 2% (v/v) β‐mercaptoethanol and boiled for 10 min at 37°C for samples containing full‐length Cx32 or at 95°C for samples containing CL Cx32‐TMD constructs.
Immunoprecipitation
Before co‐immunoprecipitation (co‐IP), 2% of cell lysate were supplemented with 5× Laemmli buffer containing 2% (v/v) β‐mercaptoethanol as input samples. The remaining lysate was incubated for 2 h with 1.5 μg antibody and subsequently for 1 h with 30 μl protein A/G agarose beads (sc‐2003, Santa Cruz), or for 3 h either with 25 μl of M2 anti‐FLAG affinity gel (A2220, Sigma‐Aldrich) or ALFA Selector ST Agaraose (N1511, NanoTag) under rotation at 4°C. Beads were thereafter washed three times with 1 ml wash buffer (50 mM Tris/HCl, pH 7.5, 150 mM NaCl complemented with either 0.5% Digitonin (Sigma‐Aldrich) or 0.5% NP‐40 (Sigma‐Aldrich) along with 0.5% DOC) for ConMem, Cx32, CNX or minCNX tmd and minCNX tmd control constructs. For CL‐Cx32‐TMD constructs 0.5% GDN (Anatrace) was used and centrifugation steps for 1 min at 8,000 × g and 4°C. Proteins were then eluted by addition of 33 μl 2× Laemmli supplemented with β‐mercaptoethanol and subsequent incubation at 95°C for 10 min, or 50°C for all samples containing ConMem and 37°C for 30 min for all samples containing full‐length Cx32.
SDS–PAGE and immunoblotting
Proteins were separated by SDS–polyacrylamide gel electrophoresis (PAGE) in 10% to 14% SDS–PAGE gels. The polyacrylamide gels were then either imaged directly using a Typhoon 9200 Variable Mode gel scanner (GE Healthcare; Cy5 filter setting, excitation 633 nm, emission 670 nm, bandwidth 30 nm or GFP excitation 526 nm, emission 532 nm, short pass) or subsequently used for western blots. Proteins were blotted overnight at 4°C onto polyvinylidene difluoride (PVDF) membranes (Biorad) and subsequently, the membranes were blocked for 6 h at RT (or overnight at 4°C) with Tris‐buffered saline supplemented with skim milk powder and Tween‐20 (M‐TBST; 25 mM Tris/HCl, pH 7.5, 150 mM NaCl, 5% (w/v) skim milk powder, 0.05% (v/v) Tween‐20). Primary antibodies in M‐TBST were applied at 4°C overnight. After washing (1 × 5 min TBS, 2 × 5 min TBST, 3 × 5 min TBS), the blots were decorated for 1 h at RT with the respective HRP‐conjugated secondary antibody diluted in M‐TBST (at 10‐fold lower dilution than the respective primary antibody). Blots were imaged using Amersham ECL prime solution (GE Life Sciences) and a Fusion Pulse 6 imager (Vilber Lourmat). Quantifications were conducted using Bio‐1D software (Vilber Lourmat).
Antibodies
For Western blots, the following primary antibodies were used at the dilutions listed: rabbit monoclonal anti‐EMC4 (ab 184544, Abcam) at 1:5,000; mouse monoclonal (M2) anti‐FLAG (F1804, Sigma‐Aldrich) at 1:1,000; mouse monoclonal (16B12) anti‐HA (901513, Biolegend) at 1:500; mouse monoclonal (C8.B6) anti‐calnexin (MAB3126, Chemicon) at 1:1,000; monoclonal rat (W17077C) anti‐calnexin (699402, Biolegend) at 1:1,000; mouse monoclonal (B‐6) anti‐Hsc70 (sc‐7298, Santa Cruz) at 1:1,000; polyclonal rabbit anti‐connexin32 (C3595, Sigma‐Aldrich) at 1:500; polyclonal goat anti‐mouse lambda (1060‐01, Southern Biotech) at 1:250; monoclonal mouse anti‐Rhodopsin (MA1‐722, Invitrogen) at 1:250; mouse monoclonal anti‐ATF6 (ab122897, Abcam) at 1:500; polyclonal rabbit anti‐(Ser51)elF2alpha (9721, Cell Signaling) at 1:500; polyclonal rabbit anti‐eIF2alpha (9722, Cell Signaling) at 1:1,000; polyclonal rabbit anti‐BiP (C50B12, Cell Signaling) at 1:500; polyclonal rabbit anti‐SPCS2 (14872‐1‐AP, Proteintech) at 1:1,000; polyclonal rabbit anti‐ATP6AP2 (HPA003156, Sigma‐Aldrich) at 1:250. The following HRP‐conjugated secondary antibodies or proteins were used for development of western blots at 1:5,000–1:10,000: mouse IgGκ‐binding protein (m‐IgGκ BP‐HRP; sc‐516102, Santa Cruz); mouse monoclonal anti‐rabbit IgG‐HRP (sc‐2357, Santa Cruz); mouse monoclonal anti‐goat IgG‐HRP (sc‐2354, Santa Cruz); goat polyclonal anti‐rat IgG (Poly4054, Biolegend). For IP, the following antibodies were employed: polyclonal goat anti‐mouse lambda (1060‐01, Southern Biotech); mouse monoclonal (C8.B6) anti‐calnexin (MAB3126, Chemicon); polyclonal rabbit anti‐HA.11 (Poly9023, Biolegend).
Immunofluorescence
Seeding and transfection
Thirty‐six microliter DMEM containing 3.6 μg DNA was mixed with 1.2 μl TorpedoDNA transfection reagent (ibidi) and incubated for 15 min at RT. Two hundred microliter of COS‐7 (ECACC) cell suspension with 4 × 105 cells/ml was added and mixed gently. Thirty microliter of the resulting suspension was applied per inlet of a μ‐Slide IV 0.4 (ibidi) and the μ‐Slide was incubated for 3 h at 37°C, 5% CO2. According to the manufacturer's instructions, medium was replaced 3 h after seeding. For this, 60 μl complete DMEM was added per reservoir, and the μ‐Slides were incubated for additional 21 h at 37°C, 5% CO2.
Staining
For fixation, liquid was removed from all reservoirs and channels and 60 μl glyoxal fixation solution (20% EtOH, 7.825% glyoxal, 0.75% acetic acid; Richter et al, 2018) was added. Samples were then incubated for 30 min on ice and thereafter further 30 min at RT. The reaction was quenched by aspirating the fixation solution and adding 60 μl of 100 mM NH4Cl (Sigma‐Aldrich) and subsequent incubation at RT for 20 min. Afterwards, samples were washed twice for 5 min with 100 μl 4°C PBS. Then, 60 μl of blocking solution (2.5% BSA (Sigma‐Aldrich), 0.1% Triton X‐100 in PBS) was added and samples were incubated for 5 min at RT following three further washing steps at RT. For FLAG‐tagged constructs, 30 μl of anti‐FLAG mouse monoclonal antibody (F1804, Sigma‐Aldrich) at 1:500 dilution in blocking solution was added and incubated for 2 h at RT and subsequently washed three times for 5 min with 100 μl PBS. As an ER‐marker, 30 μl of anti‐PDI antibody conjugated to Alexa Fluor® 488 (#5051, Cell Signaling Technology) at 1:50 dilution in blocking solution was added and incubated for 1 h at RT in the dark. For FLAG‐tagged constructs, donkey polyclonal anti‐mouse IgG antibody conjugated with TexasRed (PA1‐28626, Thermo‐Fisher) at 1:300 dilution and for ALFA tagged CNX constructs ALFA‐FluoTag‐X2 nanobodies (NanoTag Biotechnologies) at 1:250 dilution were added to the PDI‐staining solution. The antibody solution was washed out using 100 μl PBS and the samples were thereafter washed three times. All liquid was removed, and 25 μl of DAPI solution (Sigma‐Aldrich; 0.01% in PBS) was added to stain Nuclei and incubated for 2 min. The samples were then washed three times with PBS, the liquid was aspirated, and mounting medium (ibidi) was added to cover the inlets of the slides.
Microscopy
Imaging was performed on a DMi8 CS Bino inverted widefield fluorescence microscope (Leica) using a 100× (NA = 1.4) or 63× (NA = 1.4) oil immersion objective. The employed dichroic filters were chosen to image Alexa 488 and sfGFP (GFP channel; excitation/bandpass: 470/40 nm; emission/bandpass: 525/50 nm), mScarlet‐I, Sec61mCherry and TexasRed (TXR channel; excitation/bandpass: 560/40 nm; emission/bandpass: 630/75 nm), or DAPI (excitation/bandpass: 350/50 nm; emission/bandpass: 460/50 nm). For image analysis and processing, the LAS X (Leica) analysis software and ImageJ (NIH) where used. Adjustments of acquired images were restricted to homogenous changes in brightness and contrast over the whole image.
Quantification and statistics
Immunoblots were quantified using the Bio‐1D software (Vilber Lourmat). Binding of CNX to full‐length Cx32 WT and mutants, individual Cx32‐TMD domains, Rhodopsin or ConMem TMD helices was calculated as the ratio of intensities of the co‐immunoprecipitated protein (CNX) to the overall intensity of immunoprecipitated protein. In case of ConMem, the overall intensity of immunoprecipitated protein was determined as the sum of both glycosylated and nonglycosylated species—beginning at the molecular weight of HA‐tagged nonglycosylated ConMem until the upper protein smear represented by the complex glycosylated species. In case of ConMem N1Q, the area around the nonglycosylated species was included into quantification. Subsequently, to facilitate comparability of individual experiments, formed ratios were each normalized to the ConMem N1Q (ConMem lacking the lumenal glycosylation site) dataset of each experiment or CNX when examining binding to the binding deficient variants. The decay following CHX treatment was calculated by normalizing the quantified intensity at each time point to the intensity of the control sample (t = 0 min) not treated with CHX. Determination of half‐life was performed by logarithmic linearization of the CHX decays and determining the point of intersection of this line to ln (0.5). Statistical analyses were performed using Prism (GraphPad Software). Where indicated, data were analyzed with two‐tailed, unpaired Student's t tests, and differences were considered to be statistically significant when P < 0.05.
Sequence analysis and structural modeling
TMD regions of Cx32 and ConMem WT as well as mutant variants were annotated by using the ΔG prediction server v1.0 (Stockholm University). The same tool was used to predict the ΔGapp for helix insertions using the full protein scan mode. Structural analysis of and identification of lysine residues as potential DSSO crosslinking targets within the lumenal domain of CNX was performed using PyMol and mammalian (Canis lupus familiaris, Schrag et al, 2001) CNX as template. Helical wheel projection of the CNX TMD sequence was performed using the web based NetWheels application (http://lbqp.unb.br/NetWheels/).
Bioinformatic analysis
Reference proteomes of Homo sapiens (UP000005640) and Saccharomyces cerevisiae (UP00000231) were downloaded from the UniProt database (Consortium, 2021). Subcellular locations were assigned to the proteins according to their UniProt annotation. Only proteins that have been found to be located in the endoplasmic reticulum or nucleus inner membrane were taken into the analysis. Sequence positions of transmembrane (TM) regions were predicted using Phobius (Käll et al, 2004). The YxxT motifs were detected in the TM sequences by matching the corresponding regular expression (script in python). The first one and last three residues of TM regions were excluded from the search for the motif. Alignment of CNX TM domains was performed based on the extraction of orthologous sequences of human CNX from selected species (see Appendix Fig S7A) obtained from OMA database (Altenhoff et al, 2021). Subsequently, multiple sequence alignment (MSA) of the selected sequences was built using MUSCLE v3.8.1551 (Edgar, 2004) and TM domains of human CNX were predicted by Phobius (Käll et al, 2004). Ultimately, the region of the MSA that corresponds to the TM domain of human CNX was visualized in UGENE (Okonechnikov et al, 2012).
Gene knockouts
CNX‐deficient cells were generated using the CRISPR/Cas9 system. The vector pSpCas9(BB)‐2A‐Puro (PX459) V2.0 was provided from Feng Zhang (plasmid 62988; Addgene, Cambridge, MA; Ran et al, 2013). The design of guide RNA was performed using the German Cancer Research Center (DKFZ) E‐CRISP design tool. In order to prevent the occurrence of off‐target effects two different guide RNA sequences for the CNX target gene were designed (gRNA #1: 5′‐CACCGCTTGGAACTGCTATTGTTG‐3′; gRNA #2: 5′‐CACCGTGGTTGCTGTGTATGTTAC‐3′) and subsequently cloned into PX459 according to published protocols (Ran et al, 2013). In the following, HEK293T cells were transfected using GeneCellin (Eurobio) according to the manufacturer's protocol and cultured for 2 days. Selection of cells was carried through to the addition of 1.5 μg/ml puromycin for 72 h. Subsequently, single colonies were isolated and CNX protein levels were determined by immunoblotting. To ensure a complete knockout of the target gene and loss of the respective protein this process was repeated during several passages. Furthermore, following the amplification of the genomic CRISPR/Cas9 target area by the usage of specific primers, the obtained amplicons were sequenced and the successful CNX knockout verified by NGS CRISPR amplicon sequencing (CCIB DNA Core, Massachusetts General Hospital, Boston, MA).
MS sample preparation and measurement of CNX enrichment
HEK293T CNX KO cells were seeded in P100 plates and cultivated for 24 h prior transfection with 10 μg of EV or ALFA‐tagged CNX variants with a total of three replicates for each construct. DSSO crosslinking and cell lysis was performed as described above using NP‐40 lysis buffer additionally supplemented with 1 mM MgCl2 and 5% Glycerol. Immunoprecipitations were performed in the same buffer using anti‐ALFA Selector ST agarose beads as described above followed by two additional washing steps in detergent free buffer. During further proceedings, immunoprecipitated proteins destined for MS analysis were digested and eluted from beads prior to desalting and purification of the samples as otherwise described (Keilhauer et al, 2015). Nanoflow liquid chromatography‐mass spectrometry (MS)/MS analyses were performed using a combination of an UltiMate 3000 Nano HPLC system (Thermo Fisher) together with an Orbitrap Fusion mass spectrometer (Thermo Fisher). Peptides were first loaded on an Acclaim C18 PepMap100 75 μm ID × 2 cm trap column together with 0.1% FA before transfer to an Aurora reversed phase UHPLC analytical column, 75 μm ID × 25 cm, 120 Å pore size (Ionopticks). Columns were constantly heated to 40°C. Subsequent separation was performed using a first gradient ranging from 5 to 22% acetonitrile in 0.1% FA for 105 min followed by a second gradient ranging from 22 to 32% acetonitrile in 0.1% FA for 10 min at an overall flow rate of 400 nl/min. Peptides were ionized via electrospray ionization. Orbitrap Fusion was carried out in a top speed data dependent mode using a cycle time of 3 s. Full scan (MS1) acquisition (scan range of 300–1,500 m/z) was performed in the orbitrap at a defined resolution of 120,000 as well as with an automatic gain control (AGC) ion target value of 2e5 whereby dynamic exclusion was set to 60 s. For fragmentation, precursors with a charge state of 2–7 and a minimum intensity of 5e3 were selected and isolated in the quadrupole using a window of 1.6 m/z. Subsequent fragment generation was achieved using higher‐energy collisional dissociation (HCD, collision energy: 30%). The MS2 AGC was adjusted to 1e4 and 50 ms were selected as the maximum injection time for the ion trap (with inject ions for all available parallelized time enabled). Scanning of fragments was performed by applying the rapid scan rate.
MS search and bioinformatics for CNX enrichment
Thermo raw files were analyzed with MaxQuant software (version 1.6.17.0) with most default settings and a protein database containing human sequences (downloaded July 2019 from Uniprot, taxonomy ID: 9606, 74,349 entries). The ALFA tagged version of Calnexin was not implemented. The following parameter settings were used: PSM and protein FDR 1%; enzyme specificity trypsin/P; minimal peptide length: 7; variable modifications: methionine oxidation, N‐terminal acetylation; fixed modification: carbamidomethylation. The minimal number of unique peptides for protein identification was set to 2. For label‐free protein quantification, the MaxLFQ algorithm was used as part of the MaxQuant environment: (LFQ) minimum ratio count: 2; peptides for quantification: unique. Statistical analysis was performed in Perseus (version 1.6.14.0). Proteins identified only by site, reverse hits or potential contaminants were removed. LFQ intensities were log2 transformed, and data were then filtered for at least two valid values in at least one group (wt). Then, missing values were imputed from normal distribution (width: 0.3, down shift: 1.8 standard deviations, mode: over whole matrix). The replicate groups were compared via a two‐ sided, two‐sample Student's t test (S0 = 0, permutation‐based FDR method with FDR = 0.05 and 250 randomizations). Enrichment values and corresponding −log10 P‐values were plotted. Cutoffs in the volcano plot were set to twofold enrichment and a P‐value of 0.05.
MS sample preparation and measurement parameters: proteome
HEK293T CNX KO cells were seeded in P100 plates and transfected with empty vector or reconstituted with vector coding for CNX WT. Four plates of either condition were used for the four independent replicates. Lysis was performed 2 days after transfection by removing the supernatant and adding 500 μl of lysis buffer (as described above) to the cells followed by 10‐min incubation on ice. Cells were removed from plates by scraping and precleared by centrifugation at 15,000 g for 10 min at 4°C. Protein concentration was determined using a BCA assay (Life Technologies), and 300 μg of each sample was precipitated according to Wessel and Flügge (1984) and proteins afterward resuspended in 8 M urea, 0.1 M Tris/HCl pH 8.5 for denaturation. Samples were loaded on molecular cutoff spin columns (Microcon 30 kDa, Merck Millipore) and subsequently reduced, alkylated, and digested with trypsin on the filters following the FASP protocol described by Wiśniewski et al (2009). The obtained peptide mixtures were lyophilized, desalted on 50 mg SepPac C18 columns (Waters) and afterward fractionated on self‐made SCX stage tips (3 disks of Cation extraction material, Empore, in 200 μl pipette tips). Peptides bound to the SCX material were stepwise eluted with five different concentrations of AcONH4 (20, 50, 100, 250 and 500 mM), 0.1% TFA, 15% acetonitrile. Overall 40 samples (5 fractions for each of the two conditions and four replicates) were further evaporated in a Speed Vac, desalted on self‐made C18 stage tips (2 layers of C18 disks, Empore) and after final evaporation resuspended in 25 μl 0.1% formic acid (FA) and filtered through equlibrated 0.2 μm Millipore filters before MS measurement.
Nanoflow liquid chromatography‐mass spectrometry (MS)/MS analysis for the proteome analysis was performed on a TimsTOF Pro mass spectrometer equipped with an CaptiveSpray nano‐electrospray ion source (Bruker Daltonics) coupled to an UltiMate 3000 Nano HPLC system (Thermo Fisher). The column setup for peptide separation was as described above. Gradient length on the TimsTOF Pro was 73 min, while acetonitrile in 0.1% FA was step wise increased from 5 to 28% in 60 min and from 28 to 40% in 13 min, followed by a washing and equilibration step of the column.
The timsTOF Pro was operated in PASEF mode (Meier et al, 2015, 2018).
Mass spectra for MS and MS/MS scans were recorded between 100 and 1,700 m/z. Ion mobility resolution was set to 0.85–1.40 V s/cm over a ramp time of 100 ms. Data‐dependent acquisition was performed using 10 PASEF MS/MS scans per cycle with a near 100% duty cycle. A polygon filter was applied in the m/z and ion mobility space to exclude low m/z, singly charged ions from PASEF precursor selection. An active exclusion time of 0.4 min was applied to precursors that reached 20,000 intensity units. Collisional energy was ramped stepwise as a function of ion mobility.
MS search and bioinformatics for proteome analysis
TimsTOF raw files were loaded into MaxQuant (software version 2.0.3.0) and the default settings for TimsTOF files were applied except that the TOF MS/MS match tolerance was set to 0.05 Da. For the Andromeda search, the human protein database downloaded from Uniprot in September 2021 was used. Other search parameters were set as described for the CNX enrichment dataset and also statistical analysis, performed in Perseus (version 1.6.15.0) followed the same steps. Data filtration was chosen for at least three valid values in at least one group, followed by imputation of missing values and comparison of replicate groups of CNX knock out cells versus CNX reconstituted cells via a two‐sided, two‐sample Student's t‐test (S0 = 0, permutation‐based FDR method with FDR = 0.05 and 250 randomizations). Cutoffs were set for the P‐value < 0.05 and a difference in fold change of 20%.
Molecular dynamics/computational modeling
MD simulations
All MD simulations were performed with the GPU accelerated version of PMEMD (Salomon‐Ferrer et al, 2013), part of the AMBER18 package (www.ambermd.org). Amino acids were described with ff14SB (Maier et al, 2015), lipids with LIPID17, a successor of LIPID14 (Dickson et al, 2014) and water molecules with TIP3P (Jorgensen et al, 1983). The sequences of the TM helices used for the simulations are given below. All simulations were prepared with the membrane builder (Wu et al, 2014) of CHARMM‐GUI (Jo et al, 2008), utilizing the AMBER‐FF compatibility (Lee et al, 2020). The simulation boxes consisted of approx. 200 POPC molecules, 13,500 water molecules with box dimensions (after equilibration and pressurization) of 84 × 84 × 95Å. Simulations were performed in a 0.15 M KCl solution. Before sampling, the systems were treated with the equilibration protocol, suggested by CHARMM‐GUI (see Table below).
| Steps | Time step | K protein | K lipid | K dihed. | Temperature | Pressure | |
|---|---|---|---|---|---|---|---|
| Min | 2,000 | Minimization | 10.0 | 2.5 | 250 | — | — |
| Eq1 | 125,000 | 1 fs | 10.0 | 2.5 | 100 | 303.15 K | NVT |
| Eq2 | 125,000 | 1 fs | 5.0 | 2.5 | 50 | 303.15 K | NVT |
| Eq3 | 125,000 | 1 fs | 2.5 | 1.0 | 50 | 303.15 K | 1 bar |
| Eq4 | 250,000 | 2 fs | 1.0 | 0.5 | 50 | 303.15 K | 1 bar |
| Eq5 | 250,000 | 2 fs | 0.5 | 0.1 | 25 | 303.15 K | 1 bar |
| Eq6 | 250,000 | 2 fs | 0.1 | 0.0 | 0 | 303.15 K | 1 bar |
Overview over the seven equilibration steps performed for all simulations. The initial minimization is followed by six equilibration simulations with decreasing force constants (K, in kcal/mol/Å2) on positional restraints of amino acids (Protein, all atoms), positional restraints on lipid headgroups (Lipid, phosphorus atom) as well as restraints on the lipid dihedrals (Dihed. K in kcal/mol).
In all cases, periodic boundary conditions were used, and the temperature was set to 303.15 K using the Langevin thermostat (Goga et al, 2012) with a friction coefficient of 1.0 ps−1. To sample NpT ensembles, the Berendsen manostat (Berendsen et al, 1984) was utillized and set to 1.0 bar with a relaxation time of 1 ps. All nonbonded interactions were calculated directly up to a distance of 9 Å, after which electrostatic interactions were treated with the particle mesh Ewald method (Darden et al, 1993), while long range van‐der‐Waals interactions were estimated by a dispersion correction model. To achieve time‐steps of 4 fs, the Shake algorithm (Anderson, 1983) as well as hydrogen mass repartitioning (Hopkins et al, 2015) were used.
Prior to building the dimeric simulation systems, the CNX, Cx32 and ConMem helices were simulated in isolation for 1 μs in a POPC bilayer in order to equilibrate the structure and to predict their orientation in the membrane. All simulation snapshots were rendered using VMD (Humphrey et al, 1996).
CNX‐ConMem dimerization simulations
Initially, five simulations of one CNX helix and one ConMem helix in the same simulation box, were performed. Each of the five simulations was started with different relative orientations of the two helices which were placed at random positions. After preparing the simulation systems using CHARMM‐GUI, the systems were equilibrated using the aforementioned protocol. Subsequently, unrestricted sampling simulations of at least 2.5 μs were performed. Only one of the starting structures led to the formation of a stable complex. In order to inprove statistics, two more simulations were started from this initial conformation. Altogether, two successful dimerization events were observed in these simulations. All simulations were sampled for at least 4 μs.
To obtain binding statistics on the amino acid level that are more generally valid, we also performed simulations with CNX and two ConMem mutants: V13P and V13R. Several at least 4 μs long simulations were performed for both mutants; however, only two resulted in the formation of CNX‐ConMem interactions along the entire TMD: One simulation featuring V13R and one with the V13P mutant. Of interest: While the V13P mutant binds to CNX in a fashion very similar to WT ConMem, the V13R mutant uses an alternative interface to bind to CNX. CNX, on the contrary, bound all ConMem variants utilizing basically the same binding interface. Altogether, 22 μs time‐scale simulations of this system were performed. However, for analysis, we only selected the four simulations that exhibited broad TMD‐TMD interactions between CNX and ConMem. Data analysis was performed on 4 μs long trajectories in each case.
CNX‐Cx32 dimerization simulations
For this system five, at least 4 μs long simulations were performed. The simulation setup was identical to the one used for the CNX‐ConMem system. In four of the five cases, we were able to sample at least transient interactions between the helices, involving the entire TMD of CNX. These four simulations were then selected for data analysis on 4 μs of the sampled trajectories.
CNX SEQUENCE in Simulations: [ACE GLN MET ILE GLU ALA ALA GLU GLU ARG PRO TRP LEU TRP VAL VAL TYR ILE LEU THR VAL ALA LEU PRO VAL PHE LEU VAL ILE LEU PHE CYS CYS SER GLY LYS LYS GLN THR SER GLY MET GLU TYR NME].
Cx32‐TMD1 SEQUENCE in Simulations: [ACE SER GLY SER GLY SER GLY SER GLY SER GLY THR ALA ILE GLY ARG VAL TRP LEU SER VAL ILE PHE ILE PHE ARG ILE MET VAL LEU VAL VAL ALA ALA GLU SER VAL TRP GLY SER GLY SER GLY SER GLY SER GLY SER GLY NME].
ConMem (WT) SEQUENCE in Simulations: [ACE GLY GLY GLY GLY SER PRO ALA ALA ILE LEU VAL ILE VAL VAL VAL VAL VAL VAL PHE ILE ILE ILE VAL VAL VAL VAL VAL PHE ILE ILE LYS GLY GLY GLY GLY SER NME].
Free energy calculations
In order to analyze the significance of the YTL‐motif in‐silico, we conducted additional simulations of the CNX‐ConMem complex: First, a CNX mutant where the YTL‐motif was replaced by valines, was created. This structure was then used to perform three independent 1 μs long simulations of the complex between mutant‐CNX and ConMem. The starting structure for this was taken from one of the previously sampled wt‐CNX‐ConMem complexes. As a control, we performed three additional 1 μs simulations of the wt‐CNX‐ConMem complex starting from the same initial conformation as the mutant. Complex stabilites were then estimated using the MM‐PB/SA method as implemented in the mmpbsa.py routine (Miller III et al, 2012) in AMBER18. For every system, 1,500 simulation snapshots were analyzed with ε = 1 for protein and ε = 2 for the lipid bilayer. The calculations were performed assuming a salt concentration of 150 mM. Contributions of conformational entropy were neglected; therefore, the resulting free energies cannot be directly compared with experimental ΔG values. However, since the compared systems are highly similar to each other, the resulting affinity estimates allow for comparison between the mutants. For the wt‐CNX‐ConMem complex a ΔGbind of −53.56 ± 0.37 kcal/mol (s.e.m., n = 1,500) was found. ΔGbind of the mutant‐CNX‐ConMem complex was predicted to be −27.72 ± 0.57 kcal/mol (s.e.m., n = 1,500).
Author contributions
Nicolas Bloemeke: Investigation; visualization; writing – original draft; writing – review and editing. Kevin Meighen‐Berger: Investigation; writing – review and editing. Manuel Hitzenberger: Investigation; writing – review and editing. Nina C Bach: Investigation; writing – review and editing. Marina Parr: Investigation; writing – review and editing. Joao PL Coelho: Investigation; writing – review and editing. Dmitrij Frishman: Resources; funding acquisition; writing – review and editing. Martin Zacharias: Resources; funding acquisition; writing – review and editing. Stephan A Sieber: Resources; funding acquisition; writing – review and editing. Matthias J Feige: Conceptualization; resources; funding acquisition; writing – original draft; project administration; writing – review and editing.
Disclosure and competing interests statement
The authors declare that they have no conflict of interest.
Supporting information
Appendix
Dataset EV1
Dataset EV2
Source Data for Appendix
Source Data for Figure 1
Source Data for Figure 2
Source Data for Figure 3
Source Data for Figure 4
Source Data for Figure 5
Source Data for Figure 6
Acknowledgements
We are grateful to Carolin Klose (TUM), Bastian Bräuning (MPI of Biochemistry) and Marius Lemberg (University of Cologne) for helpful comments on the manuscript. The Sec61 mCherry contruct was a kind gift of Gia Voeltz, University of Boulder, Colorado. SAS gratefully acknowledges funding of the European Research Council (ERC) and the European Union's Horizon 2020 research and innovation program (grant agreement no. 725085, CHEMMINE, ERC consolidator grant). MJF gratefully acknowledges funding by a German Research Foundation (DFG) research grant, number FE1581/1‐2. Open Access funding enabled and organized by Projekt DEAL.
See also: KP Guay et al (December 2022)
Data availability
The mass spectrometry data can be found in Datasets EV1 and EV2 and have been deposited to the ProteomeXchange Consortium via the PRIDE (Perez‐Riverol et al, 2022) partner repository https://www.ebi.ac.uk/pride/ with the dataset identifier PXD031140.
References
- Altenhoff AM, Train CM, Gilbert KJ, Mediratta I, Mendes de Farias T, Moi D, Nevers Y, Radoykova HS, Rossier V, Warwick Vesztrocy A et al (2021) OMA orthology in 2021: website overhaul, conserved isoforms, ancestral gene order and more. Nucleic Acids Res 49: D373–D379 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson HC (1983) Rattle: a “velocity” version of the shake algorithm for molecular dynamics calculations. J Comput Phys 52: 24–34 [Google Scholar]
- Anderson KS, Cresswell P (1994) A role for calnexin (IP90) in the assembly of class II MHC molecules. EMBO J 13: 675–682 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Behnke J, Mann MJ, Scruggs FL, Feige MJ, Hendershot LM (2016) Members of the Hsp70 family recognize distinct types of sequences to execute ER quality control. Mol Cell 63: 739–752 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berendsen HJC, Postma JPM, van Gunsteren WF, DiNola A, Haak JR (1984) Molecular dynamics with coupling to an external bath. J Chem Phys 81: 3684–3690 [Google Scholar]
- Bone LJ, Deschenes SM, Balice‐Gordon RJ, Fischbeck KH, Scherer SS (1997) Connexin32 and X‐linked Charcot‐Marie‐Tooth disease. Neurobiol Dis 4: 221–230 [DOI] [PubMed] [Google Scholar]
- Cannon KS, Cresswell P (2001) Quality control of transmembrane domain assembly in the tetraspanin CD82. EMBO J 20: 2443–2453 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chitwood PJ, Hegde RS (2020) An intramembrane chaperone complex facilitates membrane protein biogenesis. Nature 584: 630–634 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chitwood PJ, Juszkiewicz S, Guna A, Shao S, Hegde RS (2018) EMC is required to initiate accurate membrane protein topogenesis. Cell 175: 1507–1519 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christianson JC, Olzmann JA, Shaler TA, Sowa ME, Bennett EJ, Richter CM, Tyler RE, Greenblatt EJ, Harper JW, Kopito RR (2011) Defining human ERAD networks through an integrative mapping strategy. Nat Cell Biol 14: 93–105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coelho JPL, Stahl M, Bloemeke N, Meighen‐Berger K, Alvira CP, Zhang ZR, Sieber SA, Feige MJ (2019) A network of chaperones prevents and detects failures in membrane protein lipid bilayer integration. Nat Commun 10: 672 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conti BJ, Devaraneni PK, Yang Z, David LL, Skach WR (2015) Cotranslational stabilization of Sec62/63 within the ER Sec61 translocon is controlled by distinct substrate‐driven translocation events. Mol Cell 58: 269–283 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cordes FS, Bright JN, Sansom MSP (2002) Proline‐induced distortions of transmembrane helices. J Mol Biol 323: 951–960 [DOI] [PubMed] [Google Scholar]
- Danilczyk UG, Cohen‐Doyle MF, Williams DB (2000) Functional relationship between calreticulin, calnexin, and the endoplasmic reticulum luminal domain of calnexin. J Biol Chem 275: 13089–13097 [DOI] [PubMed] [Google Scholar]
- Darden T, York D, Pedersen L (1993) Particle mesh Ewald: An N·log(N) method for Ewald sums in large systems. J Chem Phys 98: 10089–10092 [Google Scholar]
- Denzel A, Molinari M, Trigueros C, Martin JE, Velmurgan S, Brown S, Stamp G, Owen MJ (2002) Early postnatal death and motor disorders in mice congenitally deficient in calnexin expression. Mol Cell Biol 22: 7398–7404 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dickson CJ, Madej BD, Skjevik AA, Betz RM, Teigen K, Gould IR, Walker RC (2014) Lipid14: the Amber lipid force field. J Chem Theory Comput 10: 865–879 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edgar RC (2004) MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res 32: 1792–1797 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fagerberg L, Jonasson K, von Heijne G, Uhlén M, Berglund L (2010) Prediction of the human membrane proteome. Proteomics 10: 1141–1149 [DOI] [PubMed] [Google Scholar]
- Feige MJ, Hendershot LM (2013) Quality control of integral membrane proteins by assembly‐dependent membrane integration. Mol Cell 51: 297–309 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fink A, Sal‐Man N, Gerber D, Shai Y (2012) Transmembrane domains interactions within the membrane milieu: principles, advances and challenges. Biochim Biophys Acta 1818: 974–983 [DOI] [PubMed] [Google Scholar]
- Fleig L, Bergbold N, Sahasrabudhe P, Geiger B, Kaltak L, Lemberg MK (2012) Ubiquitin‐dependent intramembrane rhomboid protease promotes ERAD of membrane proteins. Mol Cell 47: 558–569 [DOI] [PubMed] [Google Scholar]
- Fontanini A, Chies R, Snapp EL, Ferrarini M, Fabrizi GM, Brancolini C (2005) Glycan‐independent role of calnexin in the intracellular retention of Charcot‐Marie‐tooth 1A Gas3/PMP22 mutants. J Biol Chem 280: 2378–2387 [DOI] [PubMed] [Google Scholar]
- Geiger R, Andritschke D, Friebe S, Herzog F, Luisoni S, Heger T, Helenius A (2011) BAP31 and BiP are essential for dislocation of SV40 from the endoplasmic reticulum to the cytosol. Nat Cell Biol 13: 1305–1314 [DOI] [PubMed] [Google Scholar]
- Goga N, Rzepiela AJ, de Vries AH, Marrink SJ, Berendsen HJ (2012) Efficient algorithms for Langevin and DPD dynamics. J Chem Theory Comput 8: 3637–3649 [DOI] [PubMed] [Google Scholar]
- Götzke H, Kilisch M, Martínez‐Carranza M, Sograte‐Idrissi S, Rajavel A, Schlichthaerle T, Engels N, Jungmann R, Stenmark P, Opazo F et al (2019) The ALFA‐tag is a highly versatile tool for nanobody‐based bioscience applications. Nat Commun 10: 4403 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guerriero CJ, Brodsky JL (2012) The delicate balance between secreted protein folding and endoplasmic reticulum‐associated degradation in human physiology. Physiol Rev 92: 537–576 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gurezka R, Laage R, Brosig B, Langosch D (1999) A heptad motif of leucine residues found in membrane proteins can drive self‐assembly of artificial transmembrane segments. J Biol Chem 274: 9265–9270 [DOI] [PubMed] [Google Scholar]
- Hammond C, Helenius A (1994) Folding of VSV G protein: sequential interaction with BiP and calnexin. Science 266: 456–458 [DOI] [PubMed] [Google Scholar]
- Hebert DN, Molinari M (2012) Flagging and docking: dual roles for N‐glycans in protein quality control and cellular proteostasis. Trends Biochem Sci 37: 404–410 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hebert DN, Foellmer B, Helenius A (1995) Glucose trimming and reglucosylation determine glycoprotein association with calnexin in the endoplasmic reticulum. Cell 81: 425–433 [DOI] [PubMed] [Google Scholar]
- Hegde RS, Keenan RJ (2021) The mechanisms of integral membrane protein biogenesis. Nat Rev Mol Cell Biol 23: 107–124 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hessa T, White SH, von Heijne G (2005) Membrane insertion of a potassium‐channel voltage sensor. Science 307: 1427 [DOI] [PubMed] [Google Scholar]
- Hessa T, Meindl‐Beinker NM, Bernsel A, Kim H, Sato Y, Lerch‐Bader M, Nilsson I, White SH, von Heijne G (2007) Molecular code for transmembrane‐helix recognition by the Sec61 translocon. Nature 450: 1026–1030 [DOI] [PubMed] [Google Scholar]
- Hopkins CW, Le Grand S, Walker RC, Roitberg AE (2015) Long‐time‐step molecular dynamics through hydrogen mass repartitioning. J Chem Theory Comput 11: 1864–1874 [DOI] [PubMed] [Google Scholar]
- Humphrey W, Dalke A, Schulten K (1996) VMD: visual molecular dynamics. J Mol Graph 14: 27–38 [DOI] [PubMed] [Google Scholar]
- Illergard K, Kauko A, Elofsson A (2011) Why are polar residues within the membrane core evolutionary conserved? Proteins 79: 79–91 [DOI] [PubMed] [Google Scholar]
- Jackson MR, Nilsson T, Peterson PA (1990) Identification of a consensus motif for retention of transmembrane proteins in the endoplasmic reticulum. EMBO J 9: 3153–3162 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jackson MR, Cohen‐Doyle MF, Peterson PA, Williams DB (1994) Regulation of MHC class I transport by the molecular chaperone, calnexin (p88, IP90). Science 263: 384–387 [DOI] [PubMed] [Google Scholar]
- Jo S, Kim T, Iyer VG, Im W (2008) CHARMM‐GUI: a web‐based graphical user interface for CHARMM. J Comput Chem 29: 1859–1865 [DOI] [PubMed] [Google Scholar]
- Jonikas MC, Collins SR, Denic V, Oh E, Quan EM, Schmid V, Weibezahn J, Schwappach B, Walter P, Weissman JS et al (2009) Comprehensive characterization of genes required for protein folding in the endoplasmic reticulum. Science 323: 1693–1697 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jorgensen WL, Chandrasekhar J, Madura JD, Impey RW, Klein ML (1983) Comparison of simple potential functions for simulating liquid water. J Chem Phys 79: 926–935 [Google Scholar]
- Käll L, Krogh A, Sonnhammer EL (2004) A combined transmembrane topology and signal peptide prediction method. J Mol Biol 338: 1027–1036 [DOI] [PubMed] [Google Scholar]
- Kauko A, Hedin LE, Thebaud E, Cristobal S, Elofsson A, von Heijne G (2010) Repositioning of transmembrane alpha‐helices during membrane protein folding. J Mol Biol 397: 190–201 [DOI] [PubMed] [Google Scholar]
- Keilhauer EC, Hein MY, Mann M (2015) Accurate protein complex retrieval by affinity enrichment mass spectrometry (AE‐MS) rather than affinity purification mass spectrometry (AP‐MS). Mol Cell Proteomics 14: 120–135 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kleopa KA, Zamba‐Papanicolaou E, Alevra X, Nicolaou P, Georgiou DM, Hadjisavvas A, Kyriakides T, Christodoulou K (2006) Phenotypic and cellular expression of two novel connexin32 mutations causing CMT1X. Neurology 66: 396–402 [DOI] [PubMed] [Google Scholar]
- Kozlov G, Bastos‐Aristizabal S, Määttänen P, Rosenauer A, Zheng F, Killikelly A, Trempe JF, Thomas DY, Gehring K (2010a) Structural basis of cyclophilin B binding by the calnexin/calreticulin P‐domain. J Biol Chem 285: 35551–35557 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kozlov G, Pocanschi CL, Rosenauer A, Bastos‐Aristizabal S, Gorelik A, Williams DB, Gehring K (2010b) Structural basis of carbohydrate recognition by calreticulin. J Biol Chem 285: 38612–38620 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee J, Hitzenberger M, Rieger M, Kern NR, Zacharias M, Im W (2020) CHARMM‐GUI supports the Amber force fields. J Chem Phys 153: 035103 [DOI] [PubMed] [Google Scholar]
- Li Q, Su YY, Wang H, Li L, Wang Q, Bao L (2010) Transmembrane segments prevent surface expression of sodium channel Nav1.8 and promote calnexin‐dependent channel degradation. J Biol Chem 285: 32977–32987 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lomize AL, Lomize MA, Krolicki SR, Pogozheva ID (2017) Membranome: a database for proteome‐wide analysis of single‐pass membrane proteins. Nucleic Acids Res 45: D250–D255 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lomize AL, Hage JM, Pogozheva ID (2018) Membranome 2.0: database for proteome‐wide profiling of bitopic proteins and their dimers. Bioinformatics 34: 1061–1062 [DOI] [PubMed] [Google Scholar]
- Lu Y, Turnbull IR, Bragin A, Carveth K, Verkman AS, Skach WR (2000) Reorientation of aquaporin‐1 topology during maturation in the endoplasmic reticulum. Mol Biol Cell 11: 2973–2985 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maeda S, Nakagawa S, Suga M, Yamashita E, Oshima A, Fujiyoshi Y, Tsukihara T (2009) Structure of the connexin 26 gap junction channel at 3.5 A resolution. Nature 458: 597–602 [DOI] [PubMed] [Google Scholar]
- Maier JA, Martinez C, Kasavajhala K, Wickstrom L, Hauser KE, Simmerling C (2015) ff14SB: improving the accuracy of protein side chain and backbone parameters from ff99SB. J Chem Theory Comput 11: 3696–3713 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Margolese L, Waneck GL, Suzuki CK, Degen E, Flavell RA, Williams DB (1993) Identification of the region on the class I histocompatibility molecule that interacts with the molecular chaperone, p88 (calnexin, IP90). J Biol Chem 268: 17959–17966 [PubMed] [Google Scholar]
- Marinko JT, Huang H, Penn WD, Capra JA, Schlebach JP, Sanders CR (2019) Folding and misfolding of human membrane proteins in health and disease: from single molecules to cellular proteostasis. Chem Rev 119: 5537–5606 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGilvray PT, Anghel SA, Sundaram A, Zhong F, Trnka MJ, Fuller JR, Hu H, Burlingame AL, Keenan RJ (2020) An ER translocon for multi‐pass membrane protein biogenesis. Elife 9: e56889 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKenna MJ, Sim SI, Ordureau A, Wei L, Harper JW, Shao S, Park E (2020) The endoplasmic reticulum P5A‐ATPase is a transmembrane helix dislocase. Science 369: eabc5809 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meacock SL, Lecomte FJ, Crawshaw SG, High S (2002) Different transmembrane domains associate with distinct endoplasmic reticulum components during membrane integration of a polytopic protein. Mol Biol Cell 13: 4114–4129 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meier F, Beck S, Grassl N, Lubeck M, Park MA, Raether O, Mann M (2015) Parallel accumulation‐serial fragmentation (PASEF): multiplying sequencing speed and sensitivity by synchronized scans in a trapped ion mobility device. J Proteome Res 14: 5378–5387 [DOI] [PubMed] [Google Scholar]
- Meier F, Brunner AD, Koch S, Koch H, Lubeck M, Krause M, Goedecke N, Decker J, Kosinski T, Park MA et al (2018) Online parallel accumulation‐serial fragmentation (PASEF) with a novel trapped ion mobility mass spectrometer. Mol Cell Proteomics 17: 2534–2545 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mesaeli N, Nakamura K, Zvaritch E, Dickie P, Dziak E, Krause KH, Opas M, MacLennan DH, Michalak M (1999) Calreticulin is essential for cardiac development. J Cell Biol 144: 857–868 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller BR III, McGee TD Jr, Swails JM, Homeyer N, Gohlke H, Roitberg AE (2012) MMPBSA.py: an efficient program for end‐state free energy calculations. J Chem Theory Comput 8: 3314–3321 [DOI] [PubMed] [Google Scholar]
- Nakao H, Seko A, Ito Y, Sakono M (2021) Dimerization of ER‐resident molecular chaperones mediated by ERp29. Biochem Biophys Res Commun 536: 52–58 [DOI] [PubMed] [Google Scholar]
- Natarajan N, Foresti O, Wendrich K, Stein A, Carvalho P (2020) Quality control of protein complex assembly by a transmembrane recognition factor. Mol Cell 77: 108–119 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neal S, Jaeger PA, Duttke SH, Benner C, Glass CK, Ideker T, Hampton RY (2018) The Dfm1 Derlin is required for ERAD retrotranslocation of integral membrane proteins. Mol Cell 69: 306–320 [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Keefe S, Pool MR, High S (2021) Membrane protein biogenesis at the ER: the highways and byways. FEBS J 10.1111/febs.15905 [DOI] [PubMed] [Google Scholar]
- Okonechnikov K, Golosova O, Fursov M (2012) Unipro UGENE: a unified bioinformatics toolkit. Bioinformatics 28: 1166–1167 [DOI] [PubMed] [Google Scholar]
- Oliver JD, van der Wal FJ, Bulleid NJ, High S (1997) Interaction of the thiol‐dependent reductase ERp57 with nascent glycoproteins. Science 275: 86–88 [DOI] [PubMed] [Google Scholar]
- Ota K, Sakaguchi M, von Heijne G, Hamasaki N, Mihara K (1998) Forced transmembrane orientation of hydrophilic polypeptide segments in multispanning membrane proteins. Mol Cell 2: 495–503 [DOI] [PubMed] [Google Scholar]
- Pantano S, Zonta F, Mammano F (2008) A fully atomistic model of the Cx32 connexon. PLoS ONE 3: e2614 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Partridge AW, Therien AG, Deber CM (2004) Missense mutations in transmembrane domains of proteins: phenotypic propensity of polar residues for human disease. Proteins 54: 648–656 [DOI] [PubMed] [Google Scholar]
- Perez‐Riverol Y, Bai J, Bandla C, García‐Seisdedos D, Hewapathirana S, Kamatchinathan S, Kundu DJ, Prakash A, Frericks‐Zipper A, Eisenacher M et al (2022) The PRIDE database resources in 2022: a hub for mass spectrometry‐based proteomics evidences. Nucleic Acids Res 50: D543–D552 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ran FA, Hsu PD, Wright J, Agarwala V, Scott DA, Zhang F (2013) Genome engineering using the CRISPR‐Cas9 system. Nat Protoc 8: 2281–2308 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richter KN, Revelo NH, Seitz KJ, Helm MS, Sarkar D, Saleeb RS, D'Este E, Eberle J, Wagner E, Vogl C et al (2018) Glyoxal as an alternative fixative to formaldehyde in immunostaining and super‐resolution microscopy. EMBO J 37: 139–159 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenbaum EE, Hardie RC, Colley NJ (2006) Calnexin is essential for rhodopsin maturation, Ca2+ regulation, and photoreceptor cell survival. Neuron 49: 229–241 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rouger H, LeGuern E, Birouk N, Gouider R, Tardieu S, Plassart E, Gugenheim M, Vallat JM, Louboutin JP, Bouche P et al (1997) Charcot‐Marie‐Tooth disease with intermediate motor nerve conduction velocities: characterization of 14 Cx32 mutations in 35 families. Hum Mutat 10: 443–452 [DOI] [PubMed] [Google Scholar]
- Sadlish H, Pitonzo D, Johnson AE, Skach WR (2005) Sequential triage of transmembrane segments by Sec61alpha during biogenesis of a native multispanning membrane protein. Nat Struct Mol Biol 12: 870–878 [DOI] [PubMed] [Google Scholar]
- Salomon‐Ferrer R, Götz AW, Poole D, Le Grand S, Walker RC (2013) Routine microsecond molecular dynamics simulations with AMBER on GPUs. 2. Explicit solvent particle mesh Ewald. J Chem Theory Comput 9: 3878–3888 [DOI] [PubMed] [Google Scholar]
- Sato BK, Schulz D, Do PH, Hampton RY (2009) Misfolded membrane proteins are specifically recognized by the transmembrane domain of the Hrd1p ubiquitin ligase. Mol Cell 34: 212–222 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Satoh T, Ohba A, Liu Z, Inagaki T, Satoh AK (2015) dPob/EMC is essential for biosynthesis of rhodopsin and other multi‐pass membrane proteins in Drosophila photoreceptors. Elife 4: e06306 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scherer SS, Kleopa KA (2012) X‐linked Charcot‐Marie‐Tooth disease. J Peripher Nerv Syst 17: 9–13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schrag JD, Bergeron JJ, Li Y, Borisova S, Hahn M, Thomas DY, Cygler M (2001) The Structure of calnexin, an ER chaperone involved in quality control of protein folding. Mol Cell 8: 633–644 [DOI] [PubMed] [Google Scholar]
- Shao S, Hegde RS (2011) Membrane protein insertion at the endoplasmic reticulum. Annu Rev Cell Dev Biol 27: 25–56 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharpe HJ, Stevens TJ, Munro S (2010) A comprehensive comparison of transmembrane domains reveals organelle‐specific properties. Cell 142: 158–169 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shurtleff MJ, Itzhak DN, Hussmann JA, Schirle Oakdale NT, Costa EA, Jonikas M, Weibezahn J, Popova KD, Jan CH, Sinitcyn P et al (2018) The ER membrane protein complex interacts cotranslationally to enable biogenesis of multipass membrane proteins. Elife 7: e37018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh S, Mittal A (2016) Transmembrane domain lengths serve as signatures of organismal complexity and viral transport mechanisms. Sci Rep 6: 22352 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swanton E, High S, Woodman P (2003) Role of calnexin in the glycan‐independent quality control of proteolipid protein. EMBO J 22: 2948–2958 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tian S, Wu Q, Zhou B, Choi MY, Ding B, Yang W, Dong M (2019) Proteomic analysis identifies membrane proteins dependent on the ER membrane protein complex. Cell Rep 28: 2517–2526 [DOI] [PMC free article] [PubMed] [Google Scholar]
- UniProt Consortium (2021) UniProt: the universal protein knowledgebase in 2021. Nucleic Acids Res 49: D480–D489 [DOI] [PMC free article] [PubMed] [Google Scholar]
- von Heijne G, Gavel Y (1988) Topogenic signals in integral membrane proteins. Eur J Biochem 174: 671–678 [DOI] [PubMed] [Google Scholar]
- Wada I, Imai S, Kai M, Sakane F, Kanoh H (1995) Chaperone function of calreticulin when expressed in the endoplasmic reticulum as the membrane‐anchored and soluble forms. J Biol Chem 270: 20298–20304 [DOI] [PubMed] [Google Scholar]
- Wanamaker CP, Green WN (2005) N‐linked glycosylation is required for nicotinic receptor assembly but not for subunit associations with calnexin. J Biol Chem 280: 33800–33810 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wessel D, Flügge UI (1984) A method for the quantitative recovery of protein in dilute solution in the presence of detergents and lipids. Anal Biochem 138: 141–143 [DOI] [PubMed] [Google Scholar]
- Wiśniewski JR, Zougman A, Nagaraj N, Mann M (2009) Universal sample preparation method for proteome analysis. Nat Methods 6: 359–362 [DOI] [PubMed] [Google Scholar]
- Wu EL, Cheng X, Jo S, Rui H, Song KC, Dávila‐Contreras EM, Qi Y, Lee J, Monje‐Galvan V, Venable RM et al (2014) CHARMM‐GUI Membrane Builder toward realistic biological membrane simulations. J Comput Chem 35: 1997–2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoo J, Cui Q (2008) Does arginine remain protonated in the lipid membrane? Insights from microscopic pKa calculations. Biophys J 94: L61–L63 [DOI] [PMC free article] [PubMed] [Google Scholar]