

Abstract
Lysosome integrity is essential for cell viability, and lesions in lysosome membranes are repaired by the ESCRT machinery. Here, we describe an additional mechanism for lysosome repair that is activated independently of ESCRT recruitment. Lipidomic analyses showed increases in lysosomal phosphatidylserine and cholesterol after damage. Electron microscopy demonstrated that lysosomal membrane damage is rapidly followed by the formation of contacts with the endoplasmic reticulum (ER), which depends on the ER proteins VAPA/B. The cholesterol‐binding protein ORP1L was recruited to damaged lysosomes, accompanied by cholesterol accumulation by a mechanism that required VAP–ORP1L interactions. The PtdIns 4‐kinase PI4K2A rapidly produced PtdIns4P on lysosomes upon damage, and knockout of PI4K2A inhibited damage‐induced accumulation of ORP1L and cholesterol and led to the failure of lysosomal membrane repair. The cholesterol–PtdIns4P transporter OSBP was also recruited upon damage, and its depletion caused lysosomal accumulation of PtdIns4P and resulted in cell death. We conclude that ER contacts are activated on damaged lysosomes in parallel to ESCRTs to provide lipids for membrane repair, and that PtdIns4P generation and removal are central in this response.
Keywords: cholesterol, lysosome, membrane contact site, membrane repair, phosphoinositide
Subject Categories: Membranes & Trafficking
The cholesterol‐binding protein ORP1L mediates ESCRT‐independent lysosome damage repair via contact sites between lysosomes and the endoplasmic reticulum.
Introduction
Lysosomes are the main degradative organelles of the cell and play crucial roles in metabolism, signaling, homeostasis, and immunity (Ballabio & Bonifacino, 2020; Holland et al, 2020). Because of their high luminal concentrations of protons, Ca2+ and hydrolases, lysosome rupture can cause cell lethality through several pathways (Papadopoulos & Meyer, 2017). To prevent untimely death, cells are therefore equipped with mechanisms that repair damaged lysosomes or degrade them if the damage is beyond repair. The endosomal sorting complex required for transport (ESCRT) machinery is rapidly recruited to damaged lysosome membranes to mediate repair (Lopez‐Jimenez et al, 2018; Radulovic et al, 2018; Skowyra et al, 2018), whereas the autophagy machinery is recruited with slower kinetics to sequester and degrade severely damaged lysosomes (Hung et al, 2013; Maejima et al, 2013).
The observation that ESCRT depletion does not fully prevent lysosome repair has suggested that additional repair mechanisms may exist (Radulovic et al, 2018; Skowyra et al, 2018). Indeed, an ESCRT‐independent mechanism that involves lysosomal sphingomyelin scrambling and consequent sphingomyelinase‐mediated generation of ceramide has recently been described (Niekamp et al, 2022).
Here, we have performed lipidomic analyses of isolated lysosomes at different time points after membrane damage to obtain a detailed view of lysosomal lipid composition upon damage. Motivated by our discoveries that lysosome damage causes increases in lysosomal phosphatidylserine (PS), cholesterol, and a phosphorylated PtdIns species, accompanied by rapid mobilization of endoplasmic reticulum (ER) around damaged lysosomes, we have here uncovered a third mechanism for lysosome repair, executed via ER–lysosome contacts.
Results
Changes in lysosome lipid composition upon membrane damage
To elucidate the effects of membrane damage on the lipid compositions of lysosomes, we quantitatively profiled lipids in HeLa cells treated with a lysosomotropic compound that causes rapid and reversible damage to lysosome membranes, L‐leucyl‐L‐leucine methyl ester (LLOMe) (Thiele & Lipsky, 1990) for 0, 10, or 45 min. We immuno‐affinity purified lysosomes from the prepared post‐nuclear fractions using a primary antibody against LAMP1 and a secondary antibody conjugated to magnetic microbeads (Stahl‐Meyer et al, 2022; Fig EV1A).
Figure EV1. LLOMe treatment alters lysosomal lipid composition.
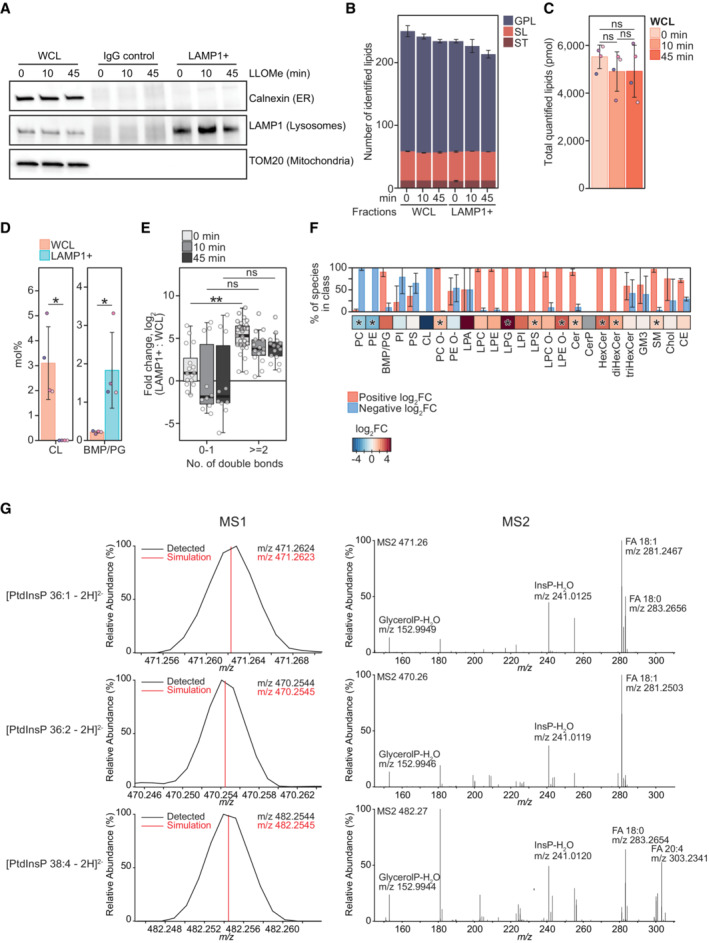
- Representative western blot showing purity of immuno‐affinity purified lysosomal (LAMP1+) and negative control (IgG Control) treated with 250‐μM LLOMe for 0, 10 min, and 45 min. Membrane was probed using anti‐TOM20 and anti‐Calnexin antibodies to verify the absence of mitochondrial and ER membrane, respectively. Enrichment of lysosomes was judged by using an anti‐LAMP1 antibody.
- Numbers of lipid species identified in the produced WCLs and LAMP1+ fraction, belonging to the lipid categories of glycerophospholipids (GPL), sphingolipids (SL), and sterol lipids (ST).
- Total molar quantities of lipids identified in aliquots of the whole‐cell lysates (WCL) produced during lysosome purification.
- Mol% values of the cardiolipin (CL) and bis(monoacylglycero)phosphate/phosphatidylglycerol (BMP/PG) classes in the LAMP1+ fraction and WCL of untreated HeLa cells. BMP and PG classes are isobaric and reported together as BMP/PG.
- Enrichment of individual BMP/PG species after purification (log2‐transformed fold change, mol% in LAMP1+: mol% in WCL) presented as box plots with individual species as circles, the central band representing the median, the box the upper and lower quartiles, and the whiskers the highest and lowest values. Species are grouped according to the total number of acyl double bonds.
- Enrichment of monitored lipid classes (log2‐transformed fold change, mol% in LAMP1+: mol% in WCL) after purification of lysosomes from untreated cells, presented in a heatmap. The bar plot depicts the percentage of species in the individual classes having positive or negative values of log2 fold changes.
- Representative mass spectra of three PtdInsP species. Precursor ions detected in the MS1 and their theoretical m/z values (MS1) and MS2 spectra obtained after their fragmentations, displaying the relative intensities of the ions.
Data information: Bars and error bars denote mean ± SD from n = 3–4 independently performed purifications. Multiple t‐tests were performed to assess changes between means of pmol (C) and mol% values and log2‐transformed mol% values (D–F). Resultant P‐values were corrected for multiple testing using Benjamini–Hochberg correction and differences were considered statistically significantly changed with adjusted P‐values (q‐values) of 0.05 or less. *q < 0.05; **q < 0.01. Abbreviations: FA, fatty acid; glycerolP, glycerol phosphate; InsP, phosphorylinositol headgroup. Additional abbreviations are as in Fig 1.
Quantitative mass spectrometry‐based shotgun lipidomic analysis of the purified lysosomes (LAMP1+ fraction) identified more than 200 species in 25 lipid classes belonging to the categories of glycerophospholipids (GPLs), sphingolipids (SLs), and sterol lipids (STs), and roughly the same numbers in the corresponding whole‐cell lysates (WCLs) (Figs 1B and EV1B). The purification yielded in total ~1,500 picomoles (pmol) of these lipids from untreated (0 min) cells, in comparison with only ~150 pmol in the negative control, in which the anti‐LAMP1 antibody was replaced with a control IgG antibody (Figs 1A and EV1C). Further supporting its purity, the LAMP1+ fraction had on average ~30 fold higher molar percentage (mol%, molar quantity relative to the total pmol of all lipids) values of bismonophosphatidic acid (BMP) species, the primarily polyunsaturated GPLs residing in the intraluminal vesicles of lysosomes (van Meer et al, 2008; Nielsen et al, 2020; Stahl‐Meyer et al, 2022) than the WCL (Bilgin et al, 2017; Nielsen et al, 2020; Fig EV1D and E). The LAMP1+ fraction was abundant in STs and SLs (Fig 1B) but depleted of most other diacyl‐GPL classes than BMP, including the major lipid class of HeLa ER membrane phosphatidylcholine (PC) (Scrima et al, 2022) and the fully depleted mitochondrial cardiolipin (CL) (Figs EV1D and EV2).
Figure 1. Lysosome lipid composition is altered upon membrane damage.
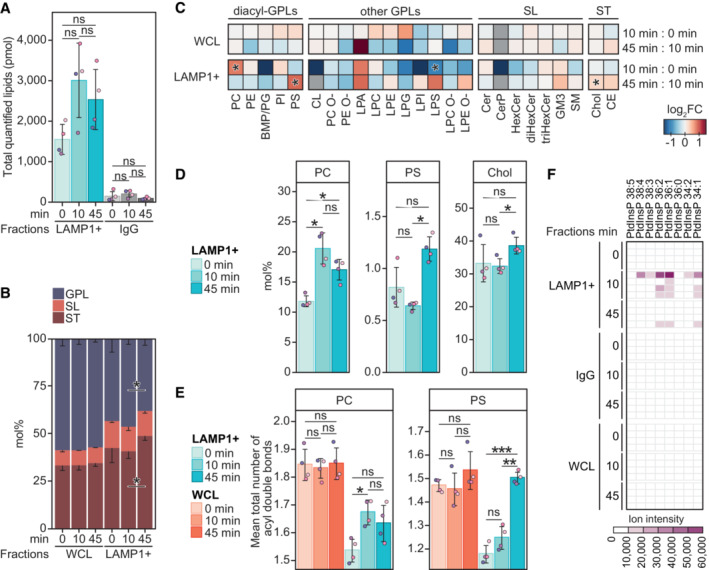
- Total molar quantity of lipids identified in the produced LAMP1‐positive (LAMP1+) and the corresponding IgG negative control fractions, produced from HeLa cells treated with LLOMe for 0, 10, or 45 min.
- Molar percentages (mol%) values of lipid categories of glycerophospholipids (GPL), sphingolipids (SL), and sterol lipids (ST) in the LAMP1+ fractions and the corresponding whole‐cell lysate (WCL).
- LLOMe‐induced fold change (log2‐transformed) in the mol% values of lipid classes in the WCLs and LAMP1+ fractions, in the first 10 min (mol% at 10 min: mol% at 0 min) and the following 45 min (mol% at 45 min: mol% at 10 min) of treatment.
- Mol% values of diacyl‐GPL lipid classes phosphatidylcholine (PC) and phosphatidylserine (PS), and cholesterol (Chol) in the LAMP1+ fractions.
- Mean total numbers of acyl double bonds in the PC and PS classes in the WCLs and LAMP1+ fractions.
- Ion intensities of dehydrated phosphorylinositol headgroup recorded in MS2 after fragmentation of [M‐2H]2− ions of phosphatidylinositol phosphate (PtdInsP) species detected in the negative ion mode MS1. Intensities measured in the four replicates are displayed in individual rows.
Data information: Bars and error bars denote mean ± SD from n = 3–4 independently performed purifications. Multiple t‐tests were performed to assess changes between means of pmol (A), lipid quantities in mol% (B–D), or the mean total numbers of double bonds in diacyl‐GPL classes (E). Resultant P‐values were corrected for multiple testing using Benjamini–Hochberg correction and differences were considered statistically significantly changed with adjusted P‐values (q‐values) of 0.05 or less. *q < 0.05; **q < 0.01; ***q < 0.001. Heatmaps are for simplicity only fitted with a single asterisk regardless of the magnitude of the q‐value. Additional abbreviations: BMP, bismonophosphatidic acid; CE, cholesteryl ester; Cer, ceramide; CerP, ceramide‐1‐phosphate; CL, cardiolipin; diHexCer, dihexosylceramide; GM3, ganglioside GM3; HexCer, hexosylceramide; LPA, lyso‐phosphatidic acid; LPC, lysoPC; LPC O‐, alkyl‐LPC; LPE, lysoPE; LPE O‐, alkyl‐LPE; LPG, lysoPG; LPI, lysoPI; LPS, lysoPS; PC O‐, acyl‐alkyl‐PC; PE, phosphatidylethanolamine, PE O‐, acyl‐alkyl‐PE; PG, phosphatidylglycerol; PI, phosphatidylinositol; SM, sphingomyelin; triHexCer, trihexosylceramide.
Figure EV2. Mol% of lipid classes in whole‐cell lysates (WCL) and purified lysosomes (LAMP1+ fractions).
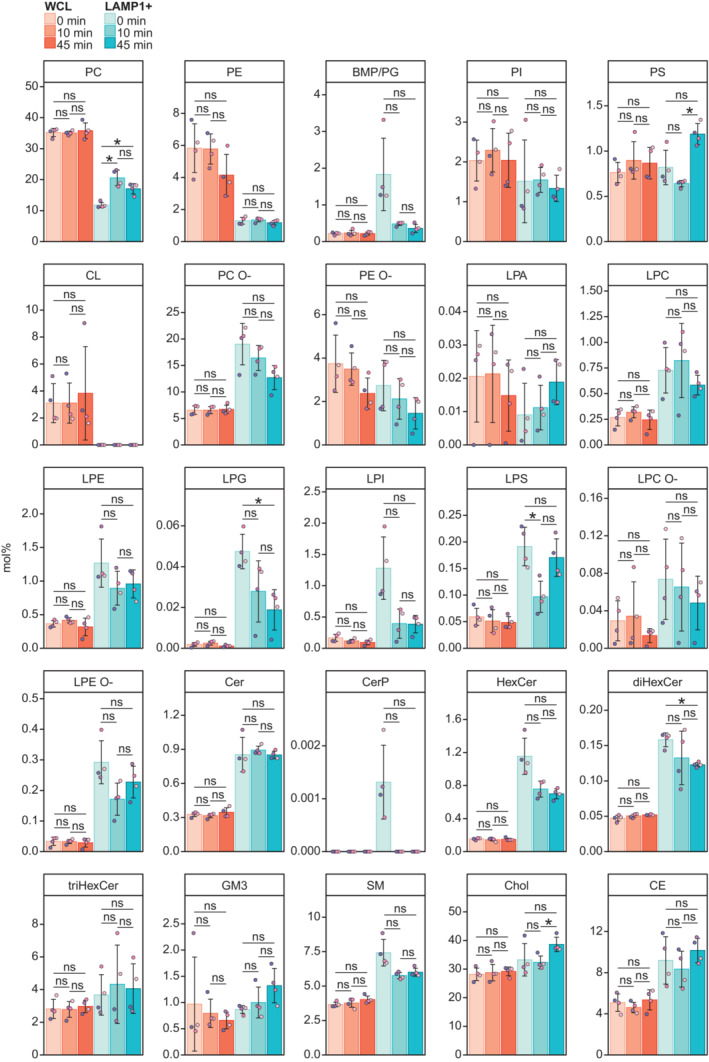
Mol% of all monitored lipid classes in the WCLs and LAMP1+ fractions from cells untreated or treated with LLOMe for 10 or 45 min.
Data information: Bars and error bars denote mean ± SD from n = 3–4 independently performed purifications. Multiple t‐tests were performed to assess changes between means. Resultant P‐values were corrected for multiple testing using Benjamini–Hochberg correction and differences were considered statistically significantly changed with adjusted P‐values (q‐values) of 0.05 or less. *q < 0.05. Abbreviations are as in Fig 1.
L‐leucyl‐L‐leucine methyl ester treatment for 10 min did not significantly alter the mol% values of the 25 monitored lipid classes in the WCL (Figs 1C and EV2). In contrast, this treatment increased the total lipid amount yielded into the LAMP1+ fraction (to ~3,000 pmol, not statistically significant) (Fig 1A). Moreover, it increased the PC class from 11.8 to 20.5 mol% (Fig 1C and D) and rendered it less saturated (Figs 1E and EV3) to better resemble the PC class in the ER (van Meer et al, 2008; Scrima et al, 2022). LLOMe at 10 min did not significantly affect the mol% of any other lipid classes except the minor lysophosphatidylserine (LPS) (Fig 1C). Extending LLOMe treatment to 45 min did not enhance these trends observed after 10 min (Fig 1A and C), but instead increased the mol% values of cholesterol and the PS class (Fig 1C and D), of which the latter similar to the PC class increased the mean total number of acyl double bonds (Figs 1E and EV3).
Figure EV3. Mean total numbers of double bonds in whole‐cell lysates (WCLs) and purified lysosomal fractions (LAMP1+ fractions).
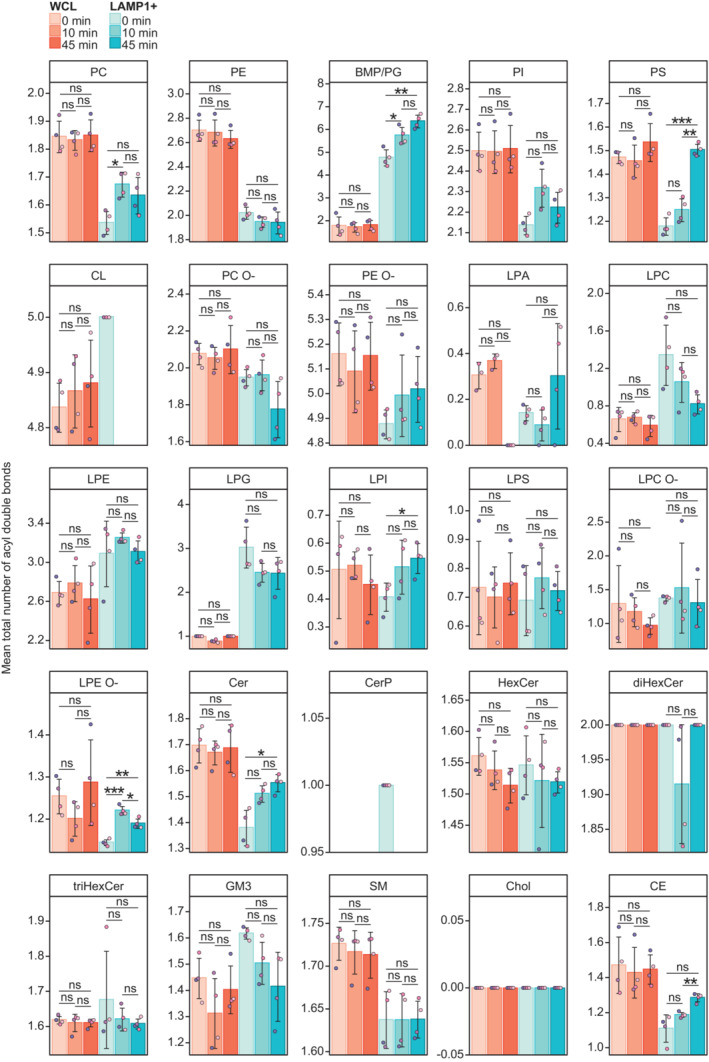
Mean total numbers of double bonds in the monitored lipid classes in the WCLs and LAMP1+ fractions from cells untreated or treated with LLOMe for 10 or 45 min. The double bonds are in the acyl (alkyl) groups of glycerophospholipids and sterol lipids, and in the acyl group and long‐chain base groups of sphingolipids.
Data information: Bars and error bars denote mean ± SD from n = 3–4 independently performed purifications. Multiple t‐tests were performed to assess changes between means. Resultant P‐values were corrected for multiple testing using Benjamini–Hochberg correction and differences were considered statistically significantly changed with adjusted P‐values (q‐values) of 0.05 or less. *q < 0.05; **q < 0.01; ***q < 0.001. Abbreviations are as in Fig 1.
Cholesterol and the PS class are two specific ligands of OSBP‐related proteins (ORPs) exporting these lipids out of the ER to late secretory compartments (Maeda et al, 2013; Moser von Filseck et al, 2015). The observation that membrane damage increased the lysosomal contents of these two lipids (Fig 1C and D) encouraged us to re‐examine the shotgun lipidomic data for phosphatidylinositol 4‐phosphate (PtdIns4P), yet another ligand of ORPs (Moser von Filseck et al, 2015). Even though we were unable to quantify them due to the lack of a PtdIns4P standard in our lipidomic pipeline, we detected precursor ions and associated fragment ions matching multiple PtdIns4P (or other PtdInsPs) species (Figs 1F and EV1G). These PtdInsP species were nearly exclusively detected in the LAMP1+ fraction after LLOMe treatment for 10 min, and not in any WCLs. Overall, the lipidomic data revealed that membrane damage rapidly increases PC and PtdInsP associated with lysosomes, followed by later elevations in cholesterol and the PS class.
Lysosome damage induces VAP‐dependent ER–lysosome contacts
The increase in lipid yield observed in the LAMP1+ fraction after LLOMe treatment suggests that lipid transfer is involved in lysosome repair. Because ER membranes are involved in lipid transfer (Lagace & Ridgway, 2013; Ikonen & Zhou, 2021), we investigated whether lysosome damage increases the extent of ER–lysosome contacts. Indeed, incubation of HeLa cells with LLOMe for only 5 min led to a strong increase in contact sites between ER and lysosomes, which was not further increased by more prolonged incubation (Fig 2A and B). Many of the known contact sites between ER and other organelles contain ER proteins of the VAP family, and we therefore monitored LLOMe‐induced contacts in HeLa cells with double knockout of VAP‐A and VAP‐B (Dong et al, 2016). Interestingly, these cells showed no increase in ER–lysosome contacts upon LLOMe treatment (Fig 2C), indicating that the damage‐induced ER–lysosome contacts are VAP dependent.
Figure 2. Lysosome damage induces VAP‐dependent ER–Lysosome contacts.
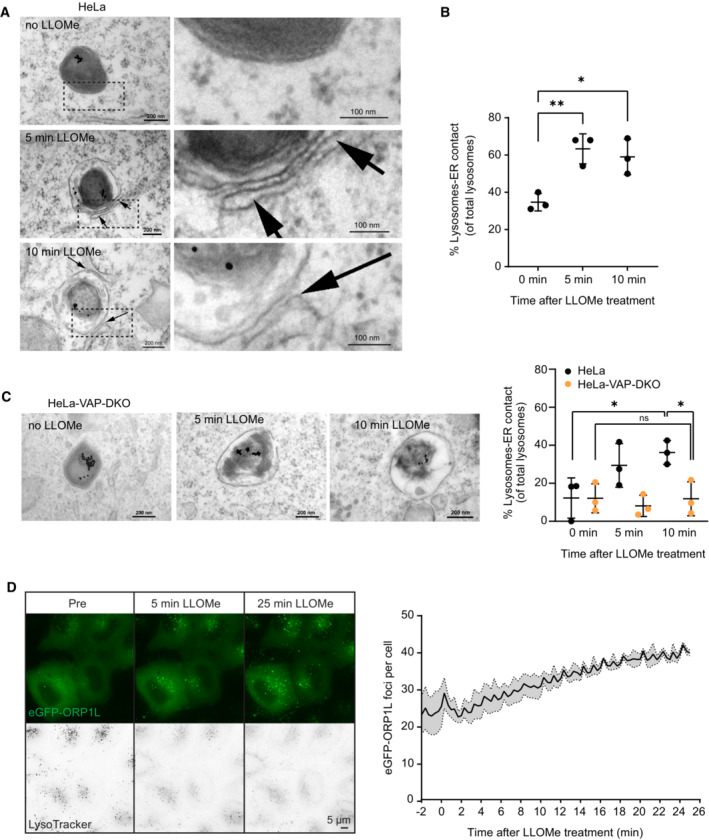
- Representative electron micrographs of HeLa cells treated or not with 250 μM LLOMe for 5 or 10 min before chemical fixation. Cells were incubated for 4 h following washing steps and overnight incubation with 15‐nm gold nanoparticles conjugated to bovine serum albumin to visualize lysosomes (black spots inside lysosomes are internalized gold nanoparticles) before treatment with LLOMe and processing of samples for electron microscopy. Arrows indicate areas with increased lysosome–ER contacts.
- Quantification of electron micrographs showing an increase in membrane contact sites between ER and lysosomes following 250‐μM LLOMe treatment for 5 or 10 min. Error bars denote ± SD from n = 3 independent experiments, > 48 lysosomes were quantified per experiment for each condition. Statistical significance was determined using one‐way ANOVA with Dunnett's multiple comparisons test, *P < 0.05, **P < 0.01.
- Representative electron micrographs of VAP double knockout HeLa cells that were either left untreated or incubated with 250 μM LLOMe for 5 or 10 min before chemical fixation. Cells were incubated with 15‐nm gold nanoparticles conjugated to bovine serum albumin for 4 h to visualize lysosomes. After 4 h, gold nanoparticles were washed off and cells were incubated overnight before treatment with LLOMe and processing samples for electron microscopy (same as in A). Quantification of electron micrographs showing no increase in membrane contact sites between ER and lysosomes following 250‐μM LLOMe treatment for 5 or 10 min in VAP double knockout cells. Error bars denote ± SD from n = 3 independent experiments, > 40 lysosomes were quantified per experiment for each condition. Statistical significance was determined using one‐way ANOVA with Tukey's multiple comparisons test, *P < 0.05, ns = not statistically significant.
- Representative movie stills of a live‐cell imaging experiment illustrating the recruitment of transiently expressed eGFP‐ORP1L to lysosomes following 250‐μM LLOMe treatment. Cells were pre‐treated for 30 min with 75 nM Lysotracker Deep Red to monitor lysosome damage (judged by the decreased number of Lysotracker spots after LLOMe treatment). The graph represents the quantification of eGFP‐ORP1L foci per cell after LLOMe treatment. Error bars denote ± SEM from n = 3 independent live‐cell imaging experiments, > 25 cells were analyzed per experiment.
ORP1L is recruited to lysosomes upon damage
OSBP‐related protein family proteins constitute a group of proteins that form VAP‐dependent contact sites between ER and other organelles (Olkkonen & Ikonen, 2022). Whereas several ORP proteins are involved in PS transport, others mediate cholesterol transport. Since the cholesterol transport protein ORP1L has been implicated in contacts between the ER and late endosomes/lysosomes (Rocha et al, 2009), we investigated whether this protein is recruited to lysosomes upon damage. Interestingly, live microscopy of HeLa cells expressing eGFP‐tagged ORP1L showed that this protein is recruited to lysosomes already at 5 min after LLOMe addition, and recruitment was further increased up to 30 min of LLOMe treatment (Fig 2D, Movie EV1). This suggests that ORP1L‐containing ER–lysosome contacts are formed upon lysosome damage.
ORP1L is recruited by PI4K2A‐dependent accumulation of PtdIns4P on damaged lysosomes
The damage‐induced recruitment of ORP1L to lysosomes raised the question of how such recruitment is triggered. Since ORP1L contains a phosphoinositide‐binding PH domain (Dong et al, 2019; Boutry & Kim, 2021), and since the lipidomic data indicated a rapid increase in a PtdInsP species after LLOMe addition, we examined cells expressing fluorescently tagged probes for various phosphoinositides and monitored their possible recruitment to lysosomes upon LLOMe treatment. Strikingly, PtdIns4P, visualized with a tandem PtdIns4P‐binding domain from the bacterial protein SidM (Hammond et al, 2014), was rapidly recruited to lysosomes after LLOMe treatment (Fig 3A, Movie EV2), whereas probes for PtdIns3P (Ellson et al, 2001), PtdIns(3,5)P2 (Li et al, 2013), and PtdIns(4,5)P2 (Watt et al, 2002) showed no increase (Fig EV4). This indicates that PtdIns4P is acutely produced on lysosome membranes upon their damage.
Figure 3. ORP1L is recruited by PI4K2A‐dependent accumulation of PtdIns4P on damaged lysosomes.
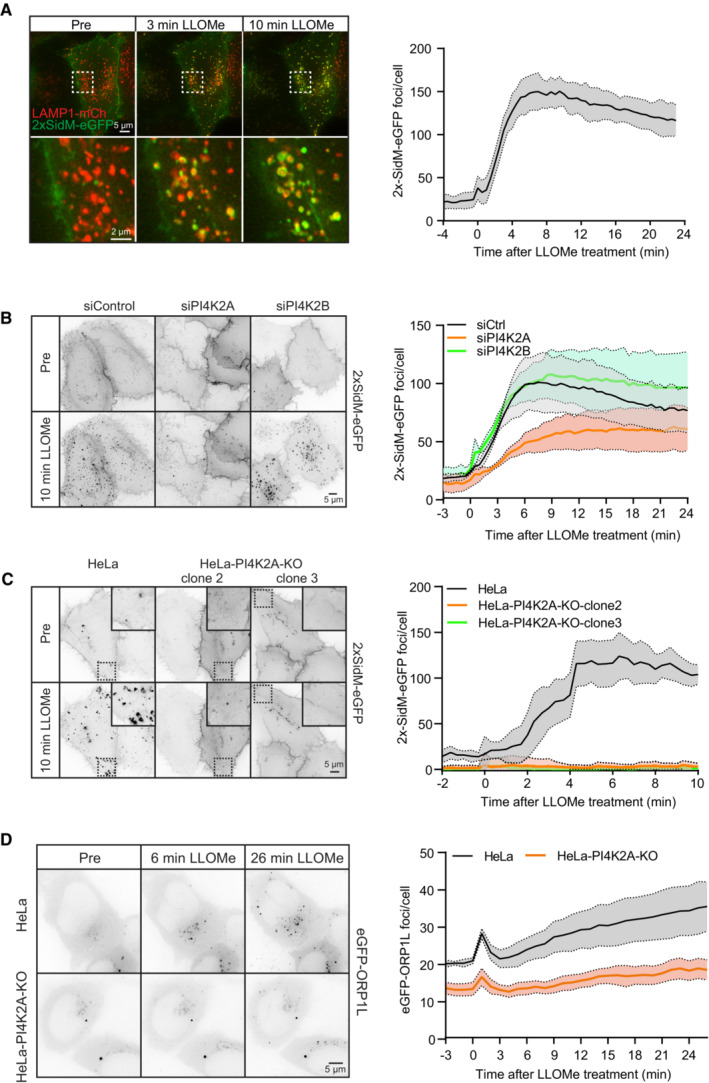
- Movie stills from a live‐cell imaging experiment showing rapid 2xSidM‐eGFP recruitment to damaged lysosomes upon 250‐μM LLOMe treatment (indicated in green). Lysosomes were visualized using transiently overexpressed LAMP1‐mCherry (indicated in red). Yellow spots show colocalized PtdIns4P probe (2xSidM‐eGFP) and LAMP1‐mCherry after 3 and 10 min of LLOMe treatment. The graph shows the quantification of 2xSidM‐eGFP foci per cell after LLOMe treatment. Error bars denote ± SEM from n = 3 independent live‐cell imaging experiments, > 20 cells were analyzed per experiment.
- HeLa cells were transfected with control siRNA or siRNA targeting PI4K2A or PI4K2B for 48 h (KD efficiency is shown in Fig EV5C) and transiently transfected with the 2xSidM‐eGFP probe to monitor PtdIns4P recruitment after LLOMe treatment by live‐cell imaging. Representative movie stills show the dynamics of 2xSidM‐eGFP recruitment in control, PI4K2A, and PI4K2B depleted cells. The graph represents the quantification of 2xSidM‐eGFP foci per cell after LLOMe treatment. Error bars denote ± SEM from n = 3 independent live‐cell imaging experiments, > 25 cells were analyzed per experiment for each condition.
- CRISPR‐Cas9‐mediated knockout of PI4K2A abolishes recruitment of the 2xSidM‐eGFP probe (PtdIns4P) to damaged lysosomes. Two different clones (clone 2 and clone 3) were tested. The graph represents the quantification of 2xSidM‐eGFP foci per cell after LLOMe treatment. Error bars denote ± SD from n = 2 independent live‐cell imaging experiments, > 20 cells were analyzed per experiment for each condition.
- Representative movie stills from live fluorescence microscopy showing reduced eGFP‐ORP1L recruitment in PI4K2A knockout cells compared to wild‐type HeLa cells upon treatment with 250‐μM LLOMe. The graph represents the quantification of eGFP‐ORP1L foci per cell after LLOMe treatment. Error bars denote ± SEM from n = 4 independent live‐cell imaging experiments, > 80 cells were analyzed per experiment for each condition.
Figure EV4. A targeted mini‐screen for recruitment of phosphoinositides to damaged lysosomes.
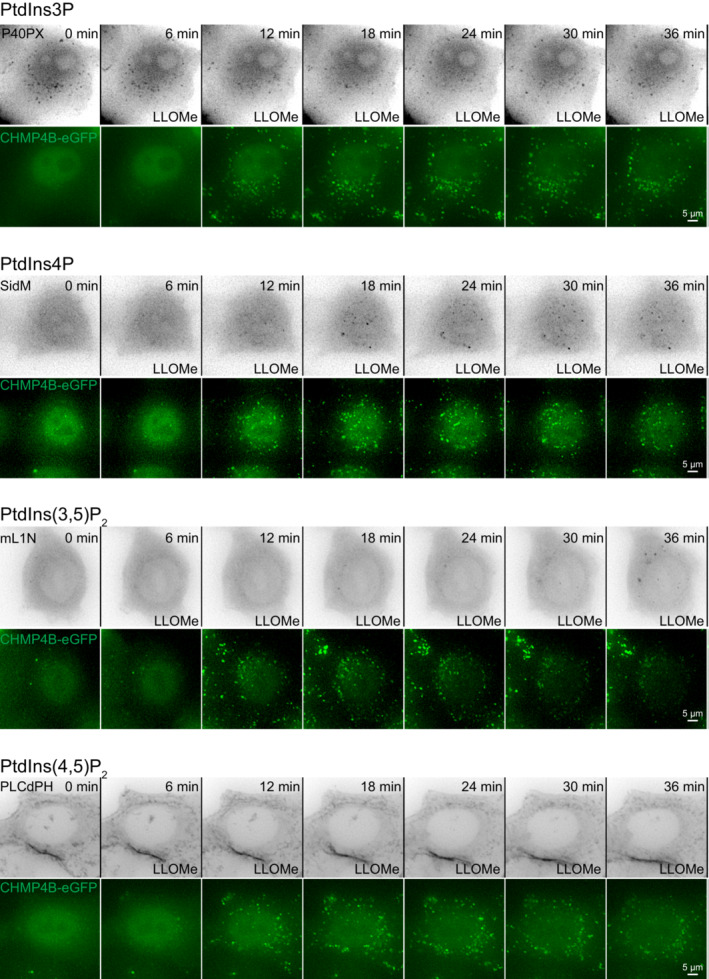
Representative movie montages of live‐cell imaging experiments using mCherry‐tagged lipid probes in HeLa cells stably expressing CHMP4B‐eGFP. P40PX was used for the detection of PtdIns3P, SidM for the detection of PtdIns4P, mL1N for the detection of PtdIns(3,5)P2, and PLCδ‐PH for the detection of PtdIns(4,5)P2. CHMP4B‐eGFP was used as a positive control for lysosomal damage. Scale bar: 5 μm.
PtdIns4P can be produced by four different PtdIns 4‐kinases (Balla, 2013), and we, therefore, examined which of these is responsible for the damage‐induced formation on lysosomes. For PI4K3A and PI4K3B, inhibitors are commercially available, and we incubated cells expressing 2xSidM‐eGFP with GSK‐A1 or PI3KIIIB‐IN‐10, inhibitors of PI4K3A and PI4K3B, respectively. Neither of these inhibitors had any effect on LLOMe‐induced PtdIns4P generation, suggesting that PI4K3A and PI4K3B are not involved (Fig EV5A and B). We next examined the possible involvement of PI4K2A and PI4K2B using specific siRNAs to knock down the expression of these kinases. Interestingly, whereas there was only a minor effect of siRNA against PI4K2B, siRNA against PI4K2A strongly inhibited LLOMe‐induced PtdIns4P accumulation on lysosomes (Figs 3B and EV5C), and CRISPR‐Cas9‐mediated knockout (KO) of PI4K2A abolished PtdIns4P recruitment as revealed with the 2xSidM probe (Figs 3C and EV5C). Importantly, exogenous expression of PI4K2A‐mNG in the PI4K2A knockout cells rescued the recruitment of PtdIns4P after LLOMe treatment (Fig EV5D). This indicates that PI4K2A is specifically activated to produce PtdIns4P on lysosomes in response to damage.
Figure EV5. PI4K3A and PI4K3B do not mediate PtdIns4P recruitment at damaged lysosomes.
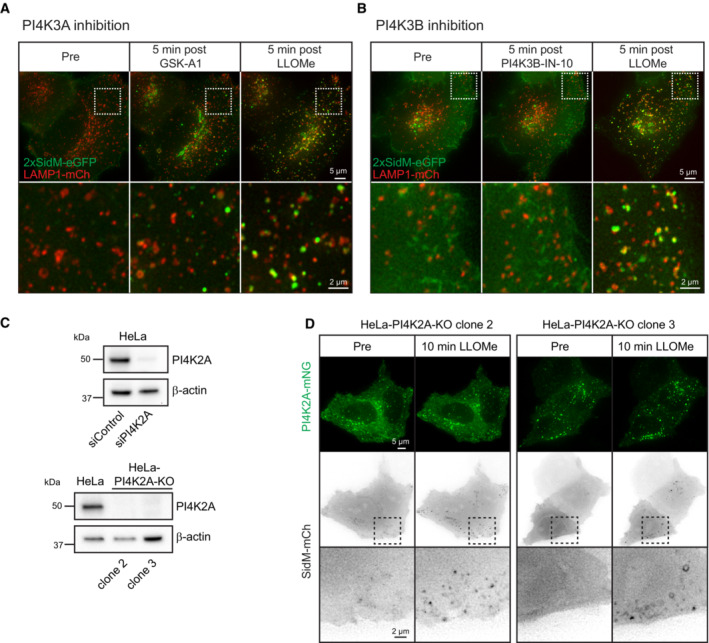
- Movie stills from live‐cell imaging experiments of HeLa cells transiently expressing the PtdIns4P probe 2xSidM‐eGFP and the lysosomal marker LAMP1‐mCherry. Cells were incubated with 10‐nM PI4K3A inhibitor GSK‐A1 for approximately 16 min before 250‐μM LLOMe was added.
- Movie stills from live‐cell imaging experiments of HeLa cells transiently expressing the PtdIns4P probe 2xSidM‐eGFP and the lysosomal marker LAMP1‐mCherry. Cells were incubated with 25‐nM PI4K3B inhibitor PI4K3B‐IN‐10 for approximately 30 min before 250‐μM LLOMe was added.
- Knockdown efficiency of siRNA against PI4K2A or CRISPR‐Cas9‐mediated knockout of PI4K2A (clone 2 and clone 3) as detected by western blot using an anti‐PI4K2A antibody. β‐actin used as a loading control.
- Exogenous expression of PI4K2A‐mNG and the PtdIns4P probe SidM‐mCherry in clone 2 and clone 3 of PI4K2A‐KO cells shows recruitment of SidM‐mCherry after incubation with 250‐μM LLOMe.
Source data are available online for this figure.
Knowing that PI4K2A produces PtdIns4P on damaged lysosomes and that the PH domain of ORP1L can bind PtdIns4P (Boutry & Kim, 2021), we investigated whether ORP1L recruitment is affected in PI4K2A knockout cells. Strikingly, live fluorescence microscopy showed that knockout of PI4K2A strongly reduced eGFP‐ORP1L recruitment (Fig 3D, Movie EV3). Thus, PI4K2A‐mediated formation of PtdIns4P provides a plausible mechanism for the recruitment of ORP1L to damaged lysosomes in order to form VAP‐containing contact sites with the ER.
PI4K2A mediates lysosome repair
The conspicuous PI4K2A‐mediated accumulation of PtdIns4P and ORP1L on damaged lysosomes begged the question whether PI4K2A is involved in lysosome repair. To address this, we used an assay based on a lysosomotropic fluorescent dye, LysoTracker™, which emits fluorescence when accumulating in acidic lysosomes. Upon damage, lysosome fluorescence ceases because of LysoTracker leakage and pH neutralization, and repair can be monitored with live fluorescence microscopy as recovery of fluorescence (Radulovic et al, 2018; Skowyra et al, 2018). Using this assay, we found that recovery of LysoTracker fluorescence in lysosomes after LLOMe treatment was strongly inhibited in cells lacking PI4K2A (Fig 4, Movie EV4). This indicates that PI4K2A mediates lysosome repair.
Figure 4. PI4K2A mediates lysosome repair.
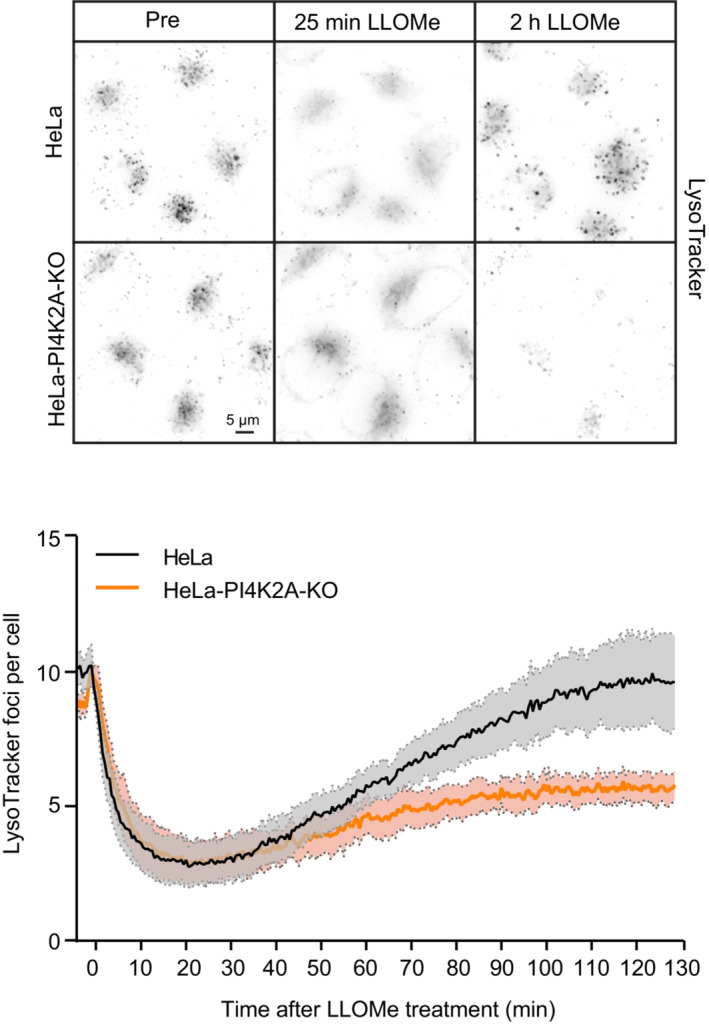
Representative movie stills from live‐cell imaging experiments monitoring Lysotracker recovery after induced lysosomal damage using 250 μM LLOMe. Cells were pre‐treated with 75‐nM Lysotracker Deep Red for 30 min before the addition of LLOMe. The graph represents the quantification of bright Lysotracker‐positive foci per cell. While the decrease in the number of Lysotracker spots is quickly recovering in HeLa cells, there is a severe impairment in lysosomal repair in HeLa‐PI4K2A‐KO cells. Error bars denote ± SEM from n = 3 independent experiments, > 217 cells were analyzed per experiment for each condition.
The PI4K2A‐mediated lysosome repair pathway is activated independently of ESCRTs
The rapid accumulation of PtdIns4P on lysosomes upon damage occurs with similar kinetics as ESCRT recruitment (Radulovic et al, 2018), and hence we asked whether activation of the two repair pathways is connected. Recruitment of the ESCRT‐III protein CHMP4B is strongly inhibited by siRNA‐mediated knockdown of the upstream ESCRT components ALIX and TSG101 (Radulovic et al, 2018), and we, therefore, monitored whether PtdIns4P accumulation is affected under these conditions. Live microscopy of cells expressing 2xSidM‐eGFP showed no effect of ALIX+TSG101 depletion on the lysosomal accumulation of the probe (Figs 5A and EV6, Movie EV5), indicating that PI4K2A activation is ESCRT independent.
Figure 5. The PI4K2A‐mediated lysosome repair pathway is activated independently of ESCRTs.
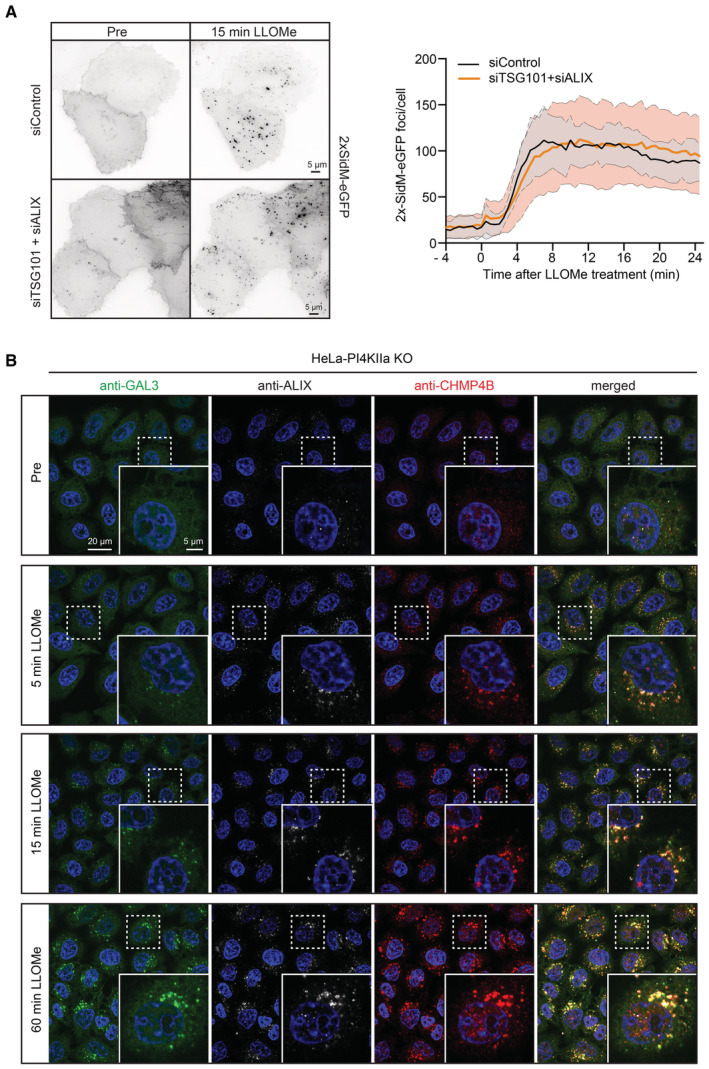
- HeLa cells were co‐transfected with siRNAs targeting ESCRT proteins TSG101 and ALIX (siTSG101 + siALIX) or with non‐targeting siRNA control (siControl) and incubated for 24 h before transfection with the PtdIns4P binding probe 2xSidM‐eGFP. Twenty‐four‐hour post‐transfection, lysosomes were damaged with 250‐μM LLOMe and PtdIns4P recruitment to damaged lysosomes was monitored using the 2xSidM‐eGFP probe in live‐cell imaging experiments. The graph shows the quantification of 2xSidM‐eGFP foci per cell in cells co‐depleted of TSG101 + ALIX and control (siControl) cells. Error bars denote ± SD from n = 2 independent live‐cell imaging experiments, > 27 cells were analyzed per experiment for each condition.
- Representative fluorescence micrographs of HeLa‐PI4K2A knockout cells treated with 250‐μM LLOMe or an equal volume of DMSO (Pre) for 5 min, 15 min, and 1 h before fixation and immunostained with Hoechst (blue), anti‐CHMP4B (red), anti‐GAL3 (green), and anti‐ALIX (white).
Figure EV6. Knockdown efficiency of TSG101 or ALIX.
Knockdown efficiency of siRNA oligonucleotides for TSG101 or ALIX as detected by western blot using anti‐TSG101 or anti‐ALIX antibodies. β‐actin used as a loading control.Source data are available online for this figure.
We next addressed whether ESCRT recruitment depends on PI4K2A. For this purpose, we incubated PI4K2A knockout cells with LLOMe, fixed the cells at various time points, and stained with antibodies against the lysosome damage marker GAL3 (Paz et al, 2010) as well as ALIX and the ESCRT‐III protein CHMP4B (Radulovic et al, 2018). Fluorescence microscopy showed that both ALIX and CHMP4B were rapidly recruited, whereas GAL3 recruitment occurred with slower kinetics as shown previously (Radulovic et al, 2018), but unchanged compared to the control (Figs 5B and EV7). This indicates that ESCRT recruitment occurs independently of PI4K2A.
Figure EV7. ESCRT and GAL3 recruitment to damaged lysosomes.
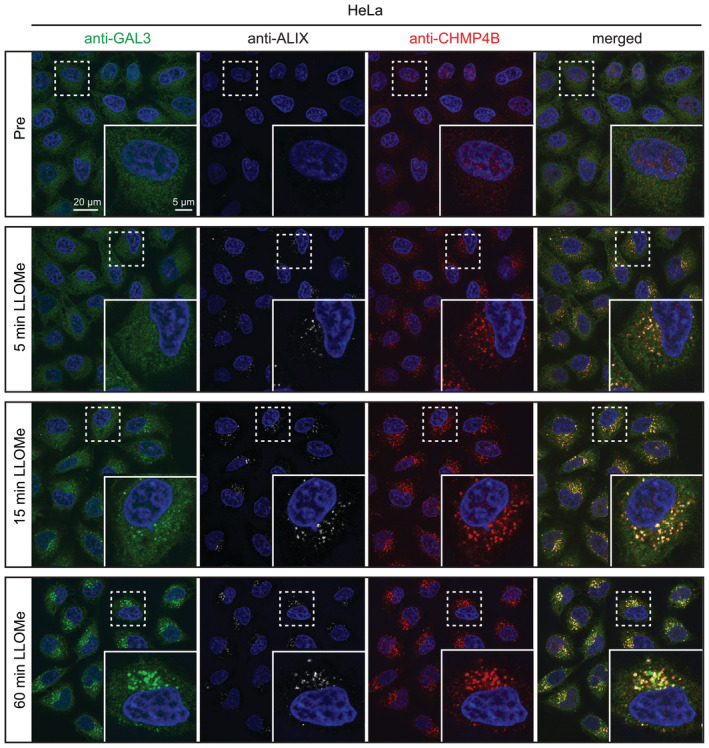
Representative fluorescence micrographs of parental HeLa cells treated with 250‐μM LLOMe or an equal volume of DMSO (Pre) for 5 min, 15 min, and 1 h before fixation and immunostained with Hoechst (blue), anti‐CHMP4B (red), anti‐GAL3 (green), and anti‐ALIX (white) (Fig 5B).
Cholesterol accumulation on damaged lysosomes depends on PI4K2A, VAPA/B, and ORP1L
Since our lipidomic analyses indicated increased cholesterol levels on lysosomes 45 min after damage, we monitored the temporal acquisition of cholesterol on lysosomes. To this end, we studied cells expressing an mCherry‐tagged cholesterol probe derived from domain 4 (D4) of the bacterial toxin Perfringolysin O (Maekawa, 2017). Interestingly, whereas PtdIns4P accumulated rapidly after LLOMe treatment as revealed with 2xSidM‐eGFP, there was a slow but steady accumulation of cholesterol as indicated by the mCherry‐D4 probe (Fig 6A, Movie EV6), consistent with the lipidomic data (Fig 1). The LLOMe‐induced accumulation of PtdIns4P and cholesterol was abolished in cells depleted for PI4K2A (Fig 6B), indicating that damage‐induced cholesterol recruitment requires PI4K2A and PtdIns4P. In VAPA/B knockout cells, PtdIns4P was still recruited, but cholesterol failed to accumulate after damage (Fig 6C). This is consistent with the recruitment of the VAP‐dependent cholesterol transporter ORP1L to damaged lysosomes (see Fig 2D), and we, therefore, investigated whether interference with ORP1L–VAP interactions would affect cholesterol accumulation. For this purpose, we studied cells expressing the cholesterol reporter mCherry‐D4 and an eGFP‐tagged ORP1L mutant incapable of binding to VAP (Vihervaara et al, 2011). Live fluorescence microscopy of these cells indicated that the VAP‐binding mutation of ORP1L strongly inhibited LLOMe‐induced accumulation of cholesterol on lysosomes (Fig 6D, Movie EV7). From these results, we conclude that cholesterol accumulation on damaged lysosomes requires both PtdIns4P and the formation of VAP‐ and ORP1L‐containing ER–lysosome contact sites.
Figure 6. Cholesterol accumulation on damaged lysosomes depends on PI4K2A, VAPA/B, and ORP1L.
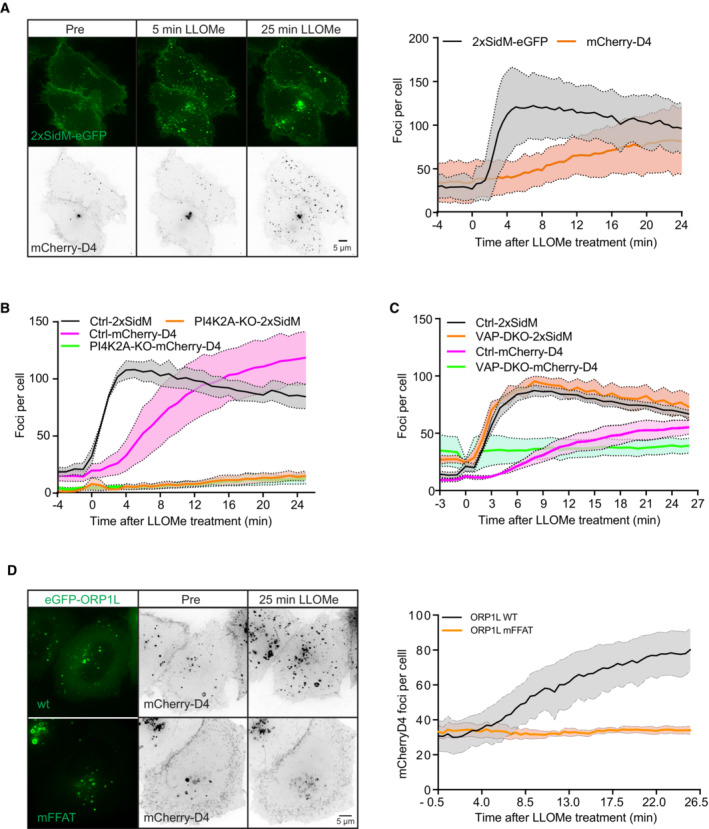
- Representative movie stills of a live‐cell imaging experiment illustrate faster recruitment of the PtdIns4P probe 2xSidM‐eGFP to damaged lysosomes when compared to cholesterol as indicated by the mCherry‐D4 probe. The graph shows the quantification of 2xSidM‐eGFP and mCherry‐D4 foci per cell. Error bars denote ± SD from n = 2 independent live‐cell imaging experiments, > 26 cells were analyzed per experiment for each condition.
- Graph showing quantification of mCherry‐D4 and 2xSidM‐eGFP foci per cell in parental HeLa cells (Ctrl) and PI4K2A knockout cells (PI4K2A‐KO). Error bars correspond to ± SEM from n = 3 independent experiments, > 30 cells per condition were analyzed.
- Quantification graph of 2xSidM‐eGFP and mCherry‐D4 foci per cell in HeLa and VAP double knockout cells. Error bars denote ± SEM from n = 3 independent experiments, > 30 cells per condition were analyzed.
- HeLa cells expressing an eGFP‐tagged ORP1L mutant incapable of binding to VAP (mFFAT) show no accumulation of the cholesterol reporter mCherry‐D4 upon lysosomal damage induced with 250‐μM LLOMe when compared to wild‐type (wt) eGFP‐ORP1L. The quantification graph shows D4‐mCherry foci per cell. Error bars denote ± SEM from n = 4 independent live‐cell imaging experiments, > 50 cells were analyzed per experiment for each condition.
Cholesterol protects lysosomes from damage
The correlation between cholesterol accumulation and lysosome damage raised the question whether cholesterol plays a role in the damage response. To investigate this, we took advantage of U18666A, an inhibitor of NPC1, a lysosomal membrane protein that mediates the export of cholesterol from lysosomes (Lu et al, 2015). U18666A causes accumulation of cholesterol in lysosomes, and we, therefore, monitored whether such accumulation affects the response to lysosomal membrane damage. Live cell microscopy of HeLa cells stably expressing CHMP4B‐eGFP showed a fast recruitment of CHMP4B to damaged lysosomes in control‐ and U18666A‐treated cells, indicating that LLOMe was able to induce an ESCRT‐dependent damage response under both conditions (Fig 7A, Movie EV8). The CHMP4B recruitment was, however, weaker in the U18666A‐treated cells, and using anti‐GAL3 as a damage marker, fluorescence microscopy showed that LLOMe‐induced lysosome damage occurred with strongly reduced kinetics in U18666A‐treated cells as compared with control cells (Fig 7B and C). This suggests that cholesterol accumulation prevents lysosome damage.
Figure 7. Cholesterol accumulation protects lysosomes from damage.
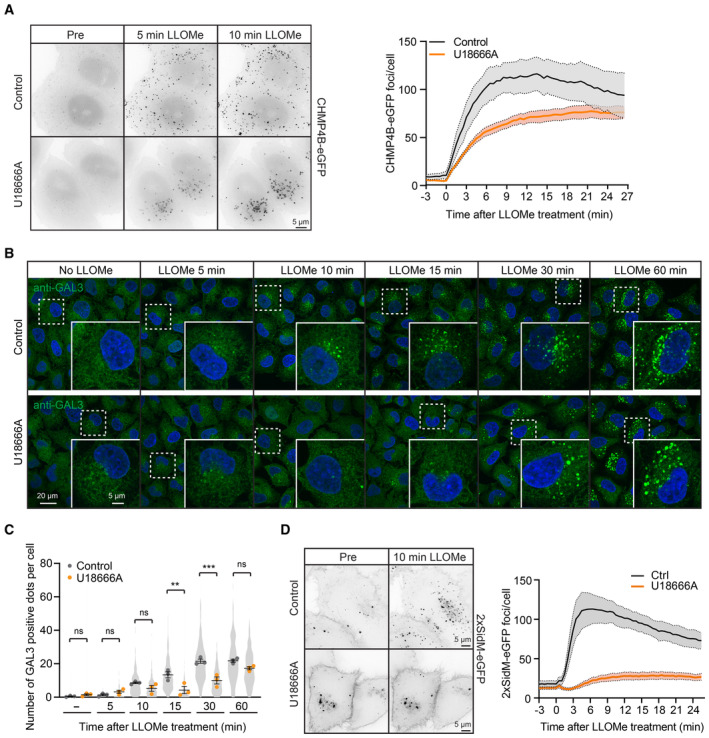
- Representative movie stills from live‐cell imaging experiments indicate recruitment of CHMP4B‐eGFP to damaged lysosomes in control and U18666A‐treated cells. HeLa cells stably expressing CHMP4B‐eGFP were treated with 2 μM U18666A for 17 h before treatment with 250‐μM LLOMe. The graph shows the quantification of CHMP4B‐eGFP foci per cell. Error bars denote ± SEM from n = 4, > 30 cells per experiment for each condition.
- Representative immunofluorescence images illustrate Galectin‐3 recruitment kinetics on the damaged lysosomes upon treatment with 2 μM U18666A for 17 h. HeLa cells were treated or not (Control) with U18666A and ± LLOMe for the indicated time points, fixed, and processed for immunofluorescence microscopy. Endogenous levels of Galectin‐3 were visualized.
- Number of Galectin‐3‐positive dots per cell was automatically quantified from the dataset described in (B). Error bars denote ± SEM from n = 3 independent experiments. > 225 cells were analyzed per condition, and distribution was visualized as violin plots. One‐way ANOVA, Tukey's multiple comparisons test, **P < 0.01, ***P < 0.001, ns = not statistically significant.
- Representative movie stills from live‐cell imaging experiments indicate recruitment of the PtdIns4P probe 2xSidM‐eGFP to damaged lysosomes in control but not in the U18666A‐treated cells. HeLa cells were treated with 2 μM U18666A for 17 h before treatment with 250‐μM LLOMe. The graph shows the quantification of 2xSidM‐eGFP foci per cell. Error bars denote ± SEM from n = 3 independent experiments, > 25 cells per experiment for each condition.
If cholesterol accumulation protects lysosomes from damage, one would expect less recruitment of PtdIns4P to lysosomes in U18666A‐treated cells in the presence of LLOMe. This was indeed found to be the case, as U18666A‐treated cells showed much weaker increase in lysosomal 2xSidM‐eGFP staining after LLOMe incubation than control cells (Fig 7D, Movie EV9). Thus, cholesterol‐rich lysosomes seem to be protected from LLOMe‐induced damage and are, therefore, refractory to the PI4K2A‐mediated damage response.
OSBP prevents hyperaccumulation of PtdIns4P on damaged lysosomes, and this promotes cell viability
Since the rapid PtdIns4P recruitment to lysosomes after damage was followed by decreased PtdIns4P levels at later time points (Figs 3A and 6A), there must be cellular mechanisms that remove PtdIns4P from lysosomes. Previous work has shown that the lipid transfer protein OSBP mediates exchange of PtdIns4P for cholesterol at ER–membrane contact sites, followed by dephosphorylation of PtdIns4P into PtdIns by the ER‐localized PtdIns4P phosphatase SAC1 (Mesmin et al, 2013) and that OSBP ensures a transient pool of PtdIns4P on endosomes (Dong et al, 2016). Like ORP1L, OSBP can interact with VAP proteins in ER membranes (Wyles et al, 2002; Rocha et al, 2009). To address whether OSBP might have a similar function at damage‐induced ER–lysosome contacts, we first monitored whether eGFP‐tagged OSBP is recruited to lysosomes after LLOMe‐induced damage. Live fluorescence microscopy showed that this was the case, as eGFP‐OSBP gradually accumulated on lysosomes after LLOMe addition (Fig 8A, Movie EV10).
Figure 8. OSBP prevents hyperaccumulation of PtdIns4P on damaged lysosomes, promoting cell viability.
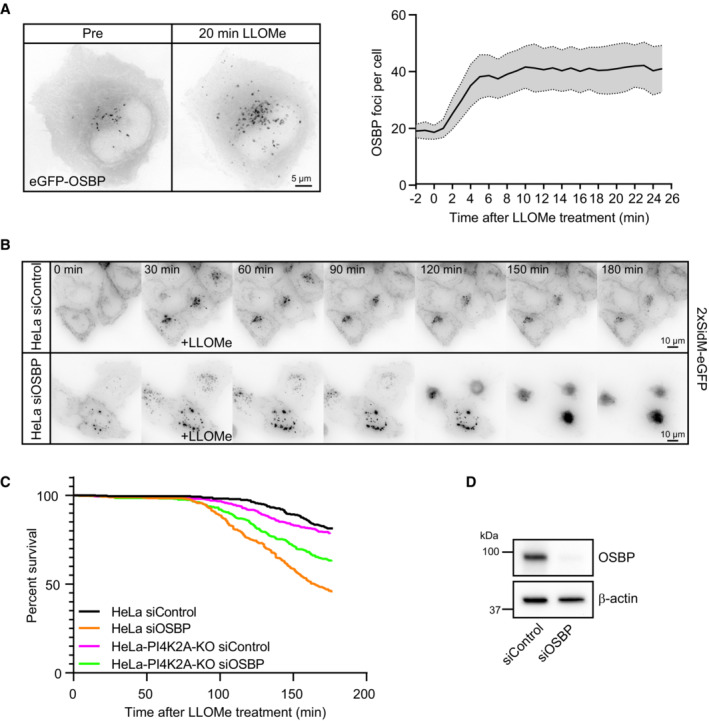
- Representative movie stills of a live‐cell imaging experiment show recruitment dynamics of transiently expressed eGFP‐OSBP in HeLa cells treated with 250‐μM LLOMe. The graph shows quantifications of OSBP foci per cell. Error bars denote ± SEM from n = 5 independent live‐cell imaging experiments, > 22 cells per experiment.
- Representative movie montage from a live‐cell fluorescence imaging experiment indicating accumulation of the 2xSidM‐eGFP probe and subsequent cell death in cells depleted of OSBP compared to control following treatment with 250‐μM LLOMe.
- Kaplan–Meier plot represents a decrease in cell viability in siRNA knockdown of OSBP (HeLa siOSBP) as compared to control cells (HeLa siControl). The rapid cell death observed in HeLa cells depleted of OSBP was ameliorated with PtdIns4P depletion (HeLa‐PI4K2A‐KO siOSBP and HeLa‐PI4K2A‐KO siControl). The graph represents data from n = 3 independent live‐cell imaging experiments, > 100 cells per experiment for each condition.
- Knockdown efficiency of siRNA depletion of OSBP in HeLa cells as detected by western blot. β‐actin used as a loading control.
Source data are available online for this figure.
In order to investigate whether OSBP is involved in PtdIns4P removal from damaged lysosomes, we used siRNA against OSBP to knock down its expression and used 2xSidM‐eGFP and live fluorescence microscopy to monitor PtdIns4P levels on lysosomes after LLOMe‐induced damage. Depletion of OSBP did in fact cause a strong accumulation of 2xSidM‐eGFP on damaged lysosomes, indicating that OSBP plays a role in removal of PtdIns4P. Surprisingly, the accumulation of PtdIns4P in response to OSBP depletion was accompanied by cell death as revealed by rounding and detachment of the OSBP‐depleted cells at about 120 min after LLOMe addition (Fig 8B–D, Movie EV11).
To study if cell death was caused by the accumulation of PtdIns4P, we monitored whether knockout of PI4K2A prevented the rapid cell death observed upon OSBP depletion. Indeed, although PI4K2A KO cells depleted for OSBP eventually died after LLOMe addition, as expected after lysosome damage, the rapid death observed with PtdIns4P depletion was diminished (Fig 8C). These results indicate that OSBP‐mediated removal of PtdIns4P after PtdIns4P‐mediated membrane repair is essential for cell viability.
Discussion
A number of recent publications have demonstrated that cells are equipped with alternative mechanisms to deal with damaged lysosomes (Hung et al, 2013; Maejima et al, 2013; Lopez‐Jimenez et al, 2018; Radulovic et al, 2018; Skowyra et al, 2018; Herbst et al, 2020; Jia et al, 2020a, 2020b; Niekamp et al, 2022). We have uncovered a new mechanism that initiates PI4K2A‐mediated generation of PtdIns4P on the membrane of the damaged lysosome. This is followed by the recruitment of ORP1L and VAP‐containing contacts between the damaged lysosomes and the ER. ORP1L transfers cholesterol to the lysosome membrane to augment its repair (Fig 9).
Figure 9. Model for ER‐mediated lysosome repair.
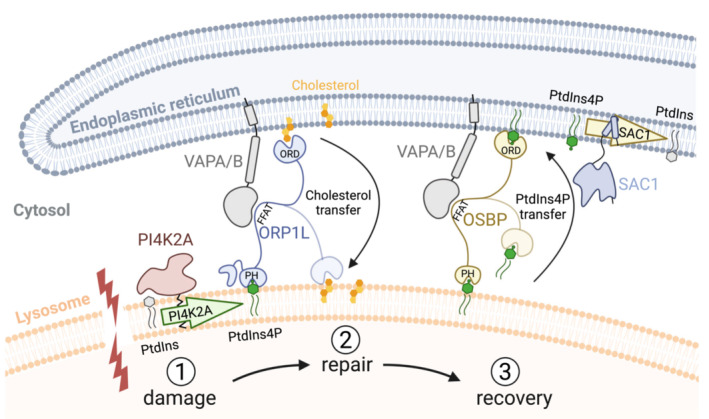
Lysosomal membrane damage (1) causes the activation of PI4K2A to produce PtdIns4P on the lysosome membrane. This causes the formation of contact sites via ORP1L, OSBP, and other PtdIns4P‐binding ORP proteins that bind to VAPA/B in the ER membrane via their FFAT motives and to PtdIns4P on the lysosome via their PH domains. Transfer of cholesterol from the ER, mediated by ORP1L, promotes membrane repair (2). Excess PtdIns4P is transported from the lysosome to the ER by OSBP (3) and thereafter dephosphorylated by SAC1 (the latter inferred from Mesmin et al, 2013). The lipid‐binding ORD domains of ORP1L and OSBP are indicated.
While this manuscript was under submission, another paper reporting the involvement of PI4K2A, PtdIns4P, and ER contacts in lysosome repair was published (Tan & Finkel, 2022). This paper identified three PS transfer proteins, ORP9, ORP10, and ORP11, as being recruited to damaged lysosomes by PtdIns4P, and also made the interesting observation that the lipid channel protein ATG2 is activated by PS for the repair of damaged lysosomes. The involvement of ORP9, ORP10, ORP11, and ATG2 provides a plausible explanation for the increased PS levels on damaged lysosomes that we detected in our lipidomic analyses. Here, we have provided evidence that ORP1L‐mediated cholesterol transfer is also important for repair. Our two studies nicely complement each other since the first used an unbiased proximity biotinylation approach to identify proteins recruited to damaged lysosomes, whereas we employed an unbiased lipidomic screen to identify changes in lysosomal lipid composition upon damage. While both studies have made extensive use of fluorescence microscopy to demonstrate the importance of PI4K2A‐mediated PtdIns4P production for lysosome repair, the first provided conclusive biochemical data based on in vitro assays whereas we have used electron microscopy to directly demonstrate an increase in ER–lysosome contacts upon damage. The first study found that an agonist of the lysosomal Ca2+ channel TRPML was sufficient to trigger PI4K2A accumulation on lysosomes, suggesting that Ca2+ release from damaged lysosomes could trigger ER‐mediated lysosome repair. Finally, whereas both studies found that OBSP is recruited to damaged lysosomes for (potential) cholesterol transfer, we also found that OSBP is essential for the removal of PtdIns4P.
The striking increase in ER–lysosome contact sites observed by electron microscopy shortly after LLOMe addition indicates that contact sites are rapidly formed upon lysosome damage. For this reason, we cannot formally exclude the possibility that detected changes in the lysosomal lipidome upon LLOMe might be, in part, due to contamination of the immunoisolated lysosomes with ER membranes. Indeed, the increase in PC levels detected at 10 min after LLOMe might be due to ER contamination since PC is a major lipid in ER membranes (Scrima et al, 2022), even though western blotting with ER antibodies revealed minimal contamination. However, the increase in PS and cholesterol levels at 45 min after lysosome damage cannot be explained by ER contamination, and the increase in cholesterol was verified independently with a cholesterol‐binding probe and correlates well with the acquisition of the cholesterol transporter ORP1L detected by live microscopy.
What is the role of PtdIns4P in lysosome repair? The most obvious function would be to recruit PtdIns4P‐binding proteins like ORP9, ORP10, ORP11, and ORP1L to establish contacts with the ER via the VAP‐binding domains of the ORP proteins. However, elegant work on ER–Golgi contacts has revealed an additional function for PtdIns4P in membrane contact sites, namely, in driving cholesterol transfer (Wenzel et al, 2022). In particular, OSBP has been shown to transfer cholesterol from ER to Golgi membranes in exchange for PtdIns4P, and dephosphorylation of PtdIns4P in the ER membrane serves to maintain a concentration gradient that drives the exchange (Mesmin et al, 2013). We found that OSBP depletion causes a strong accumulation of PtdIns4P on damaged lysosomes, suggesting that OSBP performs a similar function in ER–lysosome contact sites. Therefore, OSBP not only mediates cholesterol transfer as proposed (Tan & Finkel, 2022) but also serves to balance lysosomal PtdIns4P levels.
Our observation that lysosomal PtdIns4P accumulation in the absence of OSBP was accompanied by cell death illustrates the importance of PtdIns4P removal, although we still need to define why PtdIns4P accumulation is so hazardous to the cells suffering from lysosomal injury. PtdIns4P accumulation on lysosomes would induce increased and prolonged ER–endosome contact sites, since many contact site proteins depend on PtdIns4P, and this might affect lysosome function. In addition, the accumulation of endolysosomal PtdIns4P causes accumulation of membrane‐associated actin leading to impaired recycling from endolysosomal tubules (Dong et al, 2016).
It also needs to be explained exactly how the transfer of PS and cholesterol from the ER would mediate lysosome repair, and a relevant question is how the ER‐mediated repair relates to the previously identified ESCRT‐ and ceramide‐dependent repair mechanisms (Radulovic et al, 2018; Skowyra et al, 2018; Niekamp et al, 2022). The latter two mechanisms appear to be independent (Niekamp et al, 2022), and both we and others (Tan & Finkel, 2022) have found that activation of the ESCRT‐ and ER‐mediated repair occurs independently of each other. However, independent activation mechanisms do not exclude the possibility that the alternative repair mechanisms cross‐talk at some level. For instance, if ESCRTs and ceramide mediate inward budding of damaged membrane areas to be degraded in the lysosome lumen (Radulovic et al, 2018; Niekamp et al, 2022), the addition of more lipids would be required to maintain the area of the lysosome membrane, and ER‐mediated lipid transfer might play a role here. Further studies are needed in order to clarify these issues.
Materials and Methods
Reagents, cell culture, and stable cell lines
The lysosomotropic drug used in this study, L‐leucyl‐L‐leucine methyl ester (cat. no. 16008) was purchased from Cayman Chemical and used at 250 μM. LLOMe was dissolved in dimethyl sulfoxide (DMSO) and stored at −20°C. Other reagents used in this study are as follows: LysoTracker Deep Red (cat. no. 12492) for live‐cell imaging was purchased from Molecular Probes and was used at 75 nM; GSK‐A1 (cat. no. SML2453‐5MG) was purchased from Sigma‐Aldrich and was used at 10 nM; PI4KIIIB‐IN‐10 (cat. no. HY‐100198) was purchased from MedChemExpress and was used at 25 nM; and U18666A (cat. no. U3633‐5MG) was purchased from Merck Life Science (Sigma) and used at 2 μM.
Human HeLa “Kyoto” cells were maintained in DMEM (Gibco) medium, supplemented with 10% fetal bovine serum (FBS), 5 U/ml penicillin, and 50 μg/ml streptomycin. Cells were maintained at 37°C supplemented with 5% CO2. A stable HeLa cell line expressing CHMP4B‐eGFP was obtained from Anthony A. Hyman (Max Planck Institute for Molecular Cell Biology and Genetics, Dresden, Germany). The HeLa‐VAP‐DKO stable cell line was obtained from Pietro De Camilli (Yale School of Medicine, New Haven, USA).
CRISPR/Cas9‐mediated deletion of PI4K2A
CRISPR/Cas9‐mediated deletion of PI4K2A in HeLa “Kyoto” cells was introduced using CRISPR/Cas9. Guide RNAs were designed using Benchling software (www.benchling.com). For deletion of PI4K2A, the following guide was used gRNA: 5′‐TTAATCCTAAGTGGACCAAG‐3′. pX459‐derived plasmids encoding both Cas9 and the gRNA were transfected using Fugene 6 (cat. no. E2692 from Promega). Twenty‐four‐hour post‐transfection, puromycin was used to select transfected cells, these cells were further sorted to obtain single‐cell derived colonies, which were expanded and further characterized. Clones lacking PI4K2A were identified through western blotting.
siRNA transfections
Silencer Select siRNAs against TSG101 (#1, 5′‐CCGUUUAGAUCAAGAAGUA‐3′; #2, 5′‐CCUCCAGUCUUCUCUCGUC‐3′), ALIX (#1, 5′‐GCAGUGAGGUUGUAAAUGU‐3′; #2, 5′‐CCUGGAUAAUGAUGAAGGA‐3′), OSBP (predesigned, cat. no. 4392420), and nontargeting control siRNA (predesigned, cat. no. 4390844) were purchased from Ambion. ON‐TARGET plus Human PI4K2A siRNA (predesigned, cat. no. #1 J‐006770‐06‐0002, #2 J‐006770‐07‐0002) and ON‐TARGET plus Human PI4K2B siRNA (predesigned, cat. no. #1 J‐006769‐06‐0002, #2 J‐006769‐07‐0002) were purchased from Dharmacon. Cells at 50% confluency were transfected with 20–40 nM final siRNA concentration using Lipofectamine RNAiMax transfection reagent (Life Technologies) according to the manufacturer's instructions and harvested after 48 h.
Plasmid constructs
mCherry‐P4M‐SidM was a gift from Tamas Balla (Addgene plasmid # 51471; Hammond et al, 2014), GFP‐P4M‐SidMx2 was a gift from Tamas Balla (Addgene plasmid # 51472; Hammond et al, 2014), GFP‐C1‐PLCdelta‐PH was a gift from Tobias Meyer (Addgene plasmid # 21179; Stauffer et al, 1998), mCherry‐D4 was a gift from Matthew Ng. PI4K2A‐mNG was a gift from Kia Wee Tan. pLJM1‐FLAG‐GFP‐OSBP was a gift from Roberto Zoncu (Addgene plasmid # 134659; Lim et al, 2019), eGFP‐ORP1L was described previously (Johansson et al, 2007).
Immunofluorescence staining and antibodies
Cells seeded on coverslips were fixed in 4% EM‐grade formaldehyde in phosphate‐buffered saline (PBS) for 10 min. Cells were washed twice in PBS and once in PBS containing 0.05% saponin to permeabilize the cells before staining with the indicated primary antibodies for 1 h. Prior to staining with the secondary antibodies for 1 h, cells were washed three times in PBS containing 0.05% saponin. Cells were mounted in Mowiol containing 2 mg/ml Hoechst 33342 (Sigma‐Aldrich). Rabbit anti‐ALIX and rabbit anti‐CHMP4B antibodies were described previously (Christ et al, 2016). Human Galectin‐3 Alexa Fluor 488‐conjugated antibody (cat. no. IC1154G) was purchased from R&D Systems. Mouse anti‐LAMP1 (cat. no. H4A3) was purchased from Developmental Studies Hybridoma Bank, mouse anti‐β‐actin (cat. no. A5316) from Sigma‐Aldrich, and mouse anti‐TSG101 (cat. no. 612697) from BD Transduction Laboratories. Rabbit anti‐Calnexin antibody (cat. no. 2679S) was purchased from Cell Signaling Technology, and mouse anti‐TOM20 antibody (cat. no. 612278) was purchased from BD Biosciences. All secondary antibodies used for immunofluorescence studies and western blotting were obtained from Jackson ImmunoResearch Laboratories or from Molecular Probes (Life Technologies).
Western blotting
Cells were washed with cold PBS and lysed in 2× sample buffer (125 mM Tris–HCl, pH 6.8, 4% SDS, 20% glycerol, 200 mM DTT, and 0.004% bromophenol blue). Whole‐cell lysates were subjected to SDS–PAGE on 4–20% gradient gels (Mini‐PROTEAN TGX; Bio‐Rad). Proteins were transferred to polyvinylidene difluoride (PVDF) membranes (Trans‐Blot® Turbo™ LF PVDF, Bio‐Rad) followed by blocking in 5% fat‐free milk powder and antibody incubation in 5% fat‐free milk powder in Tris‐buffered saline with 0.1% Tween‐20. Membranes incubated with horseradish peroxidase‐conjugated antibodies were developed using Clarity Western ECL Substrate Solutions (Bio‐Rad) with a ChemiDoc XRS+ imaging system (Bio‐Rad).
Confocal fluorescence microscopy
Stained coverslips were examined with a Zeiss LSM 780 confocal microscope (Carl Zeiss) equipped with an Ar laser multiline (458/488/514 nm), a DPSS‐561 10 (561 nm), a laser diode 405‐30 CW (405 nm), and a HeNe laser (633 nm). The objective used was a Zeiss Plan‐Apochromat 63×/1.40 Oil DIC M27 (Carl Zeiss). Intensity settings for the relevant channels were kept constant during imaging. Image processing was performed with ImageJ software (National Institutes of Health).
Live‐cell imaging
Cells seeded in MatTek 35‐mm petri dish, 20‐mm Microwell No. 1.5 coverglass were imaged on a DeltaVision microscope (Applied Precision) equipped with Elite TruLight Illumination System, a CoolSNAP HQ2 camera, and a 60× Plan‐Apochromat (1.42 NA) lens. For temperature control during live observation, the microscope stage was kept at 37°C by a temperature‐controlled incubation chamber. Time‐lapse images (6 z‐sections 2.2 μm apart) were acquired every 1–3 min over a total time period of 4–6 h and deconvolved using the softWoRx software (Applied Precision). In addition, DeltaVision OMX V4 microscope equipped with three PCO.edge sCMOS cameras, a solid‐state light source, a 60× 1.42 NA objective, and a laser‐based autofocus was used. Environmental control was provided by a heated stage and an objective heater (20‐20 Technologies). Images were deconvolved using softWoRx software and processed in ImageJ/FIJI.
Image analysis and post‐processing
Images were analyzed in Fiji/ImageJ using custom python scripts. Briefly, images were filtered to remove noise, foci of different proteins were segmented by manual thresholding, and objects were then identified and scored by the “Analyze Particles” function of ImageJ. Manual thresholds were chosen to detect prominent LLOMe‐induced structures; therefore, only spots above a certain intensity were scored. For live‐cell movies, foci counts were normalized to the cell number. For fixed cell analysis, Hoechst‐positive nuclei and GAL3 puncta were automatically segmented and counted by Nis‐Elements, and all foci counts were normalized to the number of nuclei within a given field of view.
Transmission electron microscopy (TEM)
Cells were grown on glass coverslips (100,000 cells per well in 12‐well plates) and incubated with BSA‐gold for 16 h to identify lysosomes. They were fixed at different time points after LLOMe treatment, with 2% glutaraldehyde in 0.1 M PHEM buffer. Fixative was removed, coverslips were washed, and post‐fixation was done in 1% Osmium (EMS, 19134)/1.5% Ferricyanide/0.1 M PHEM for 1 h. Cells were thoroughly washed in water to remove Osmium residues. The cells were contrasted using 4% uranyl acetate (SPI, 02624‐AB) and dehydrated in a graded ethanol series, embedded in Epon, and polymerized for 48 h. Ultrathin sections of 100 nm were cut using an Ultracut UCT ultramicrotome (Leica, Austria) and transferred onto formvar/carbon‐coated grids. All prepared sections were observed at 80 kV in a JEOL‐JEM 1230 electron microscope and images were recorded using iTEM software with a Morada camera (Olympus, Münster, Germany).
Lysosome purification and lipidomics
Lysosomes were purified and their lipid contents were analyzed with quantitative mass spectrometry‐based shotgun lipidomics as described previously (Nielsen et al, 2020; Stahl‐Meyer et al, 2022). In brief, the post‐nuclear fractions prepared from the LLOMe‐treated or untreated cells were incubated with anti‐LAMP‐1 primary antibody (Abcam, #ab24170) or IgG control (Invitrogen, Waltham, MA, USA, #31235) and then with anti‐Rabbit IgG MicroBeads (Miltenyi Biotec, #130‐048‐602). The post‐nuclear fractions were loaded on magnetic columns (Miltenyi Biotec, Bergisch Gladbach, Germany, #130‐042‐401) to capture and elute lysosomes associated with MicroBeads. Aliquots of the eluates and the corresponding whole‐cell lysates were centrifuged at 21,100 g for 20 min, and the pellets were resuspended in 155 mM NH4HCO3 for shotgun lipidomics.
For shotgun lipidomics, a series of lipid internal standards in appropriate and known quantities (Stahl‐Meyer et al, 2022) were added to each sample prior to lipid extraction using a modified Bligh and Dyer protocol. The lipid extracts were mixed with positive (13.3 mM ammonium bicarbonate in 2‐propanol) or negative (0.2% (v/v) tri‐ethyl‐amine in chloroform:methanol 1:5 (v/v)) ionization solvents. The lipidomic analysis was performed on a quadrupole‐Orbitrap mass spectrometer Q Exactive equipped with a TriVersa NanoMate to allow direct infusion and ionization of the samples. The data were acquired through cycles of MS and MS/MS scans in both positive and negative ion modes. The acquired ion spectra were processed using a Python‐based software, LipidXplorer (Herzog et al, 2012), which reported the lipid species identified based on sets of criteria defined for the individual lipid classes as well as their m/z values and the intensities of associated precursor and fragment ions (Stahl‐Meyer et al, 2022). A subsequent sorting and quantification of lipid species were achieved using an in‐house built R‐based suite of scripts named LipidQ (https://github.com/ELELAB/lipidQ) and manual evaluation of the spectra. The absolute molar quantities were calculated based on the intensity values of sample‐derived lipids and their respective internal lipid standards.
Species of phosphatidylinositol phosphate (PIP) were identified in the negative ion mode using LipidXplorer based on the following criteria; MS1 detection (tolerance 6 ppm) of [M‐2H]2− precursor ion and MS2 detection (tolerance 0.0035 Da) of dehydrated phosphorylinositol headgroup, dehydrated glycerol phosphate, and a pair of carboxylate anions of fatty acids matching the sum composition of the associated precursor ion. Intensities of the precursor ions and the fragment ions were retrieved using LipidXplorer.
Lipidomic data analysis and statistics
The molar quantity of lipids was expressed relative to the sum of the molar quantities of all identified lipid species in the sample (mol%) unless otherwise specified. Lipid species were only included in the further analyses if the determined lipid species were detected in at least 75% of the replicates. Heatmaps were produced using the “pheatmap” package in R (version 4.0.3.) (Nielsen et al, 2020). For statistical analyses of lipid levels, t‐tests with Benjamini–Hochberg (BH) correction for multiple testing were performed in R and adjusted P‐values (q‐values) were determined. Lipids were considered to be at statistically significantly different levels between two sample groups if the adjusted P‐values were below 0.05. Lipid nomenclatures were as previously used (Nielsen et al, 2020).
Statistical analysis
The number of individual experiments and the number of cells analyzed are indicated in the figure legends. We tested our datasets for normal distribution and chose an appropriate test accordingly using GraphPad Prism version 5.01. The statistical tests performed for each experiment are specified in the figure legends. P‐values are indicated for each experiment, and P‐values below 0.05 were regarded as significant. In instances of multiple means comparisons, we used one‐way analysis of variance (ANOVA) followed by Tukey's or Dunnett's post hoc test to determine statistical significance.
Author contributions
Maja Radulovic: Conceptualization; data curation; formal analysis; validation; investigation; visualization; methodology; writing – review and editing. Eva Maria Wenzel: Data curation; formal analysis; validation; investigation; visualization; writing – review and editing. Sania Gilani: Investigation; visualization; methodology. Lya KK Holland: Data curation; formal analysis. Alf Håkon Lystad: Formal analysis; methodology. Santosh Phuyal: Investigation. Vesa M Olkkonen: Resources. Andreas Brech: Data curation; formal analysis; validation; investigation; visualization; methodology. Marja Jäättelä: Resources; methodology; writing – review and editing. Kenji Maeda: Data curation; formal analysis; validation; investigation; methodology; writing – review and editing. Camilla Raiborg: Data curation; formal analysis; validation; investigation; visualization; methodology; writing – review and editing. Harald Stenmark: Conceptualization; resources; supervision; funding acquisition; writing – original draft; project administration.
Disclosure and competing interests statement
The authors declare that they have no conflict of interest.
Supporting information
Expanded View Figures PDF
Movie EV1
Movie EV2
Movie EV3
Movie EV4
Movie EV5
Movie EV6
Movie EV7
Movie EV8
Movie EV9
Movie EV10
Movie EV11
Source Data for Expanded View
PDF+
Source Data for Figure 8
Acknowledgements
The Core Facilities for Advanced Light Microscopy and Advanced Electron Microscopy at Oslo University Hospital are acknowledged for providing access to and training on relevant microscopes. We thank the Lipidomics Core Facility at the Danish Cancer Society Research Center for providing access to instrumentations and materials. We thank Ulrikke Dahl Brinch and Catherine Sem Wegner for excellent technical help with sample preparation for electron microscopy. We thank Pietro De Camilli for kindly providing VAPA/B knockout cells and Matthew Yoke Wui Ng and Kia Wee Tan for providing plasmid constructs. AHL was supported by a Young Research Talents Grant from the Research Council of Norway (project number 325305). CR was supported by a grant from the Norwegian Cancer Society (project number 198140). HS was supported by the Norwegian Cancer Society (project number 182698), the South‐Eastern Norway Regional Health Authority (project number 2016087), the Research Council of Norway (project number 302994), and the European Research Council (Advanced Grant number 788954). MJ was supported by grants from the Danish National Research Foundation (DNRF125) and the Novo Nordisk Foundation (NNF17OC0029432), and KM was supported by the Independent Research Fund Denmark (6108‐00542B). This work was partly supported by the Research Council of Norway through its Centres of Excellence funding scheme (project number 262652). Figures were created using Adobe Illustrator CS6 and BioRender (https://biorender.com/).
Contributor Information
Maja Radulovic, Email: maja.radulovic@ous-research.no.
Harald Stenmark, Email: stenmark@ulrik.uio.no.
Data availability
This study includes no data deposited in external repositories.
References
- Balla T (2013) Phosphoinositides: tiny lipids with giant impact on cell regulation. Physiol Rev 93: 1019–1137 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ballabio A, Bonifacino JS (2020) Lysosomes as dynamic regulators of cell and organismal homeostasis. Nat Rev Mol Cell Biol 21: 101–118 [DOI] [PubMed] [Google Scholar]
- Bilgin M, Nylandsted J, Jaattela M, Maeda K (2017) Quantitative profiling of lysosomal lipidome by shotgun lipidomics. Methods Mol Biol 1594: 19–34 [DOI] [PubMed] [Google Scholar]
- Boutry M, Kim PK (2021) ORP1L mediated PI(4)P signaling at ER‐lysosome‐mitochondrion three‐way contact contributes to mitochondrial division. Nat Commun 12: 5354 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christ L, Wenzel EM, Liestol K, Raiborg C, Campsteijn C, Stenmark H (2016) ALIX and ESCRT‐I/II function as parallel ESCRT‐III recruiters in cytokinetic abscission. J Cell Biol 212: 499–513 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dong R, Saheki Y, Swarup S, Lucast L, Harper JW, De Camilli P (2016) Endosome‐ER contacts control Actin nucleation and retromer function through VAP‐dependent regulation of PI4P. Cell 166: 408–423 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dong J, Du X, Wang H, Wang J, Lu C, Chen X, Zhu Z, Luo Z, Yu L, Brown AJ et al (2019) Allosteric enhancement of ORP1‐mediated cholesterol transport by PI(4,5)P2/PI(3,4)P2. Nat Commun 10: 829 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ellson CD, Gobert‐Gosse S, Anderson KE, Davidson K, Erdjument‐Bromage H, Tempst P, Thuring JW, Cooper MA, Lim Z‐Y, Holmes AB et al (2001) Phosphatidylinositol 3‐phosphate regulates the neutrophil oxidase complex by binding to the PX domain of p40phox . Nat Cell Biol 3: 679–682 [DOI] [PubMed] [Google Scholar]
- Hammond GR, Machner MP, Balla T (2014) A novel probe for phosphatidylinositol 4‐phosphate reveals multiple pools beyond the Golgi. J Cell Biol 205: 113–126 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herbst S, Campbell P, Harvey J, Bernard EM, Papayannopoulos V, Wood NW, Morris HR, Gutierrez MG (2020) LRRK2 activation controls the repair of damaged endomembranes in macrophages. EMBO J 39: e104494 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herzog R, Schuhmann K, Schwudke D, Sampaio JL, Bornstein SR, Schroeder M, Shevchenko A (2012) LipidXplorer: a software for consensual cross‐platform lipidomics. PLoS One 7: e29851 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holland LKK, Nielsen IO, Maeda K, Jaattela M (2020) SnapShot: lysosomal functions. Cell 181: 748 [DOI] [PubMed] [Google Scholar]
- Hung YH, Chen LM, Yang JY, Yang WY (2013) Spatiotemporally controlled induction of autophagy‐mediated lysosome turnover. Nat Commun 4: 2111 [DOI] [PubMed] [Google Scholar]
- Ikonen E, Zhou X (2021) Cholesterol transport between cellular membranes: a balancing act between interconnected lipid fluxes. Dev Cell 56: 1430–1436 [DOI] [PubMed] [Google Scholar]
- Jia J, Bissa B, Brecht L, Allers L, Choi SW, Gu Y, Zbinden M, Burge MR, Timmins G, Hallows K et al (2020a) AMPK, a regulator of metabolism and autophagy, is activated by lysosomal damage via a novel galectin‐directed ubiquitin signal transduction system. Mol Cell 77: e959 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jia J, Claude‐Taupin A, Gu Y, Choi SW, Peters R, Bissa B, Mudd MH, Allers L, Pallikkuth S, Lidke KA et al (2020b) Galectin‐3 coordinates a cellular system for lysosomal repair and removal. Dev Cell 52: e68 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johansson M, Rocha N, Zwart W, Jordens I, Janssen L, Kuijl C, Olkkonen VM, Neefjes J (2007) Activation of endosomal dynein motors by stepwise assembly of Rab7‐RILP‐p150Glued, ORP1L, and the receptor betalll spectrin. J Cell Biol 176: 459–471 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lagace TA, Ridgway ND (2013) The role of phospholipids in the biological activity and structure of the endoplasmic reticulum. Biochim Biophys Acta 1833: 2499–2510 [DOI] [PubMed] [Google Scholar]
- Li X, Wang X, Zhang X, Zhao M, Tsang WL, Zhang Y, Yau RG, Weisman LS, Xu H (2013) Genetically encoded fluorescent probe to visualize intracellular phosphatidylinositol 3,5‐bisphosphate localization and dynamics. Proc Natl Acad Sci USA 110: 21165–21170 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lim CY, Davis OB, Shin HR, Zhang J, Berdan CA, Jiang X, Counihan JL, Ory DS, Nomura DK, Zoncu R (2019) ER‐lysosome contacts enable cholesterol sensing by mTORC1 and drive aberrant growth signalling in Niemann‐Pick type C. Nat Cell Biol 21: 1206–1218 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopez‐Jimenez AT, Cardenal‐Munoz E, Leuba F, Gerstenmaier L, Barisch C, Hagedorn M, King JS, Soldati T (2018) The ESCRT and autophagy machineries cooperate to repair ESX‐1‐dependent damage at the Mycobacterium‐containing vacuole but have opposite impact on containing the infection. PLoS Pathog 14: e1007501 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu F, Liang Q, Abi‐Mosleh L, Das A, De Brabander JK, Goldstein JL, Brown MS (2015) Identification of NPC1 as the target of U18666A, an inhibitor of lysosomal cholesterol export and Ebola infection. Elife 4: e12177 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maeda K, Anand K, Chiapparino A, Kumar A, Poletto M, Kaksonen M, Gavin AC (2013) Interactome map uncovers phosphatidylserine transport by oxysterol‐binding proteins. Nature 501: 257–261 [DOI] [PubMed] [Google Scholar]
- Maejima I, Takahashi A, Omori H, Kimura T, Takabatake Y, Saitoh T, Yamamoto A, Hamasaki M, Noda T, Isaka Y et al (2013) Autophagy sequesters damaged lysosomes to control lysosomal biogenesis and kidney injury. EMBO J 32: 2336–2347 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maekawa M (2017) Domain 4 (D4) of perfringolysin O to visualize cholesterol in cellular membranes‐the update. Sensors (Basel) 17: 504 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mesmin B, Bigay J, Moser von FJ, Lacas‐Gervais S, Drin G, Antonny B (2013) A four‐step cycle driven by PI(4)P hydrolysis directs sterol/PI(4)P exchange by the ER‐Golgi tether OSBP. Cell 155: 830–843 [DOI] [PubMed] [Google Scholar]
- Moser von Filseck J, Copic A, Delfosse V, Vanni S, Jackson CL, Bourguet W, Drin G (2015) Phosphatidylserine transport by ORP/Osh proteins is driven by phosphatidylinositol 4‐phosphate. Science 349: 432–436 [DOI] [PubMed] [Google Scholar]
- Niekamp P, Scharte F, Sokoya T, Vittadello L, Kim Y, Deng Y, Sudhoff E, Hilderink A, Imlau M, Clarke CJ et al (2022) Ca2+‐activated sphingomyelin scrambling and turnover mediate ESCRT‐independent lysosomal repair. Nat Commun 13: 1875 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nielsen IO, Groth‐Pedersen L, Dicroce‐Giacobini J, Jonassen ASH, Mortensen M, Bilgin M, Schmiegelow K, Jaattela M, Maeda K (2020) Cationic amphiphilic drugs induce elevation in lysoglycerophospholipid levels and cell death in leukemia cells. Metabolomics 16: 91 [DOI] [PubMed] [Google Scholar]
- Olkkonen VM, Ikonen E (2022) Cholesterol transport in the late endocytic pathway: roles of ORP family proteins. J Steroid Biochem Mol Biol 216: 106040 [DOI] [PubMed] [Google Scholar]
- Papadopoulos C, Meyer H (2017) Detection and clearance of damaged lysosomes by the endo‐lysosomal damage response and lysophagy. Curr Biol 27: R1330–R1341 [DOI] [PubMed] [Google Scholar]
- Paz I, Sachse M, Dupont N, Mounier J, Cederfur C, Enninga J, Leffler H, Poirier F, Prevost MC, Lafont F et al (2010) Galectin‐3, a marker for vacuole lysis by invasive pathogens. Cell Microbiol 12: 530–544 [DOI] [PubMed] [Google Scholar]
- Radulovic M, Schink KO, Wenzel EM, Nahse V, Bongiovanni A, Lafont F, Stenmark H (2018) ESCRT‐mediated lysosome repair precedes lysophagy and promotes cell survival. EMBO J 37: e99753 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rocha N, Kuijl C, van der Kant R, Janssen L, Houben D, Janssen H, Zwart W, Neefjes J (2009) Cholesterol sensor ORP1L contacts the ER protein VAP to control Rab7‐RILP‐p150 Glued and late endosome positioning. J Cell Biol 185: 1209–1225 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scrima S, Tiberti M, Campo A, Corcelle‐Termeau E, Judith D, Foged MM, Clemmensen KKB, Tooze SA, Jaattela M, Maeda K et al (2022) Unraveling membrane properties at the organelle‐level with LipidDyn. Comput Struct Biotechnol J 20: 3604–3614 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skowyra ML, Schlesinger PH, Naismith TV, Hanson PI (2018) Triggered recruitment of ESCRT machinery promotes endolysosomal repair. Science 360: eaar5078 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stahl‐Meyer J, Holland LKK, Liu B, Maeda K, Jaattela M (2022) Lysosomal changes in mitosis. Cell 11: 875 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stauffer TP, Ahn S, Meyer T (1998) Receptor‐induced transient reduction in plasma membrane PtdIns(4,5)P2 concentration monitored in living cells. Curr Biol 8: 343–346 [DOI] [PubMed] [Google Scholar]
- Tan JX, Finkel T (2022) A phosphoinositide signalling pathway mediates rapid lysosomal repair. Nature 609: 815–821 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thiele DL, Lipsky PE (1990) Mechanism of L‐leucyl‐L‐leucine methyl ester‐mediated killing of cytotoxic lymphocytes: dependence on a lysosomal thiol protease, dipeptidyl peptidase I, that is enriched in these cells. Proc Natl Acad Sci USA 87: 83–87 [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Meer G, Voelker DR, Feigenson GW (2008) Membrane lipids: where they are and how they behave. Nat Rev Mol Cell Biol 9: 112–124 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vihervaara T, Uronen RL, Wohlfahrt G, Bjorkhem I, Ikonen E, Olkkonen VM (2011) Sterol binding by OSBP‐related protein 1L regulates late endosome motility and function. Cell Mol Life Sci 68: 537–551 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watt SA, Kular G, Fleming IN, Downes CP, Lucocq JM (2002) Subcellular localization of phosphatidylinositol 4,5‐bisphosphate using the pleckstrin homology domain of phospholipase C delta1. Biochem J 363: 657–666 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wenzel EM, Elfmark LA, Stenmark H, Raiborg C (2022) ER as master regulator of membrane trafficking and organelle function. J Cell Biol 221: e202205135 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wyles JP, McMaster CR, Ridgway ND (2002) Vesicle‐associated membrane protein‐associated protein‐A (VAP‐A) interacts with the oxysterol‐binding protein to modify export from the endoplasmic reticulum. J Biol Chem 277: 29908–29918 [DOI] [PubMed] [Google Scholar]